
Abstract
Adaptation of the heart to intrinsic and external stress involves complex modifications at the molecular and cellular levels that lead to tissue remodeling, functional and metabolic alterations, and finally to failure depending upon the nature, intensity, and chronicity of the stress. Reactive oxygen species (ROS) have long been considered as merely harmful entities, but their role as second messengers has gradually emerged. At the same time, our comprehension of the multifaceted role of nitric oxide (NO) and the related reactive nitrogen species (RNS) has been upgraded. The tight interlay between ROS and RNS suggests that their imbalance may implicate the impairment in physiological NO/redox-based signaling that contributes to the failing of the cardiovascular system. This review initially provides basic concepts on the role of nitroso/oxidative stress in the pathophysiology of heart failure with a particular focus on sources of ROS/RNS, their downstream targets, and endogenous modulators. Then, the role of NO/redox regulation of cardiomyocyte function, including calcium homeostasis, electrogenesis, and insulin signaling pathways, is described. Finally, an overview of old and emerging therapeutic opportunities in heart failure is presented, focusing on modulation of NO/redox mechanisms and discussing benefits and limitations. Antioxid. Redox Signal. 14, 289–331.
Evidence for a Role of ROS in HF: From Acute Stressor to Signaling Pathways
Cardiomyocyte Energy Sources in the Healthy and in the Failing Heart
I. Introduction

An altered cellular production of ROS and/or reactive nitrogen species (RNS) is also related to insulin resistance (121). In fact, hindrance in insulin-related pathway is not restricted to the context of diabetes: recent research points to a pathogenetic role of insulin resistance in the progression of HF. Indeed, it is increasingly recognized that the maintenance of a balanced cardiac metabolism is a key protective factor in preventing progression of different cardiomyopathies toward HF (397).
Since HF is a systemic, multiorgan disease, the complexity of the scenario is further amplified by the involvement of several cell types—either cardiac specific or not. Cardiomyocytes (atrial, ventricular, and nodal cells), endothelial and smooth muscle cells, fibroblasts and intracardiac neurons, and other cell types undergo substantial alterations and contribute to cardiac electrical/mechanical failure and rearrangement of extracellular matrix and microcirculation. Therefore, none of these cells and tissues can be viewed as a bystander in the progression of disease toward overt failure. Notwithstanding these premises, for the sake of clarity and insightfulness, in this review our attention is focused on the cardiomyocyte unit when dealing with cellular pathological remodeling, while referring to specific reviews for other cell types (119, 152, 383).
In the first part of this review, we provide basic concepts on the role of NO/ROS and the consequence of their imbalance in the pathophysiology of HF with a particular focus on sources of ROS/NO, their downstream targets, and endogenous modulators. Then, the role of NO/redox regulation of cardiomyocyte function, including Ca2+ homeostasis, electrogenesis, and insulin signaling pathways, is described. Afterward, an overview of old and emerging therapeutic opportunities is presented; the selection was determined among several interventions on the basis of their possible modulating activity on the NO/redox balance, and discussed benefits and limitations. In this context, disappointing clinical studies led to the awareness that indiscriminate removal of oxidative stress is ineffective against cardiac detrimental process: indeed, NO/redox balance is a necessary adaptive mechanism for cell survival against noxious stimuli. Therefore, removal of ROS in a site-specific manner or inhibition of the source of injurious ROS without affecting nitroso/redox-sensitive survival signal transduction pathways or, on the other hand, increasing bioavailability of NO represents a promising approach for the development of new therapies for this debilitating condition.
II. Evidence for a Role of ROS in HF: From Acute Stressor to Signaling Pathways
A. Sources and species of ROS
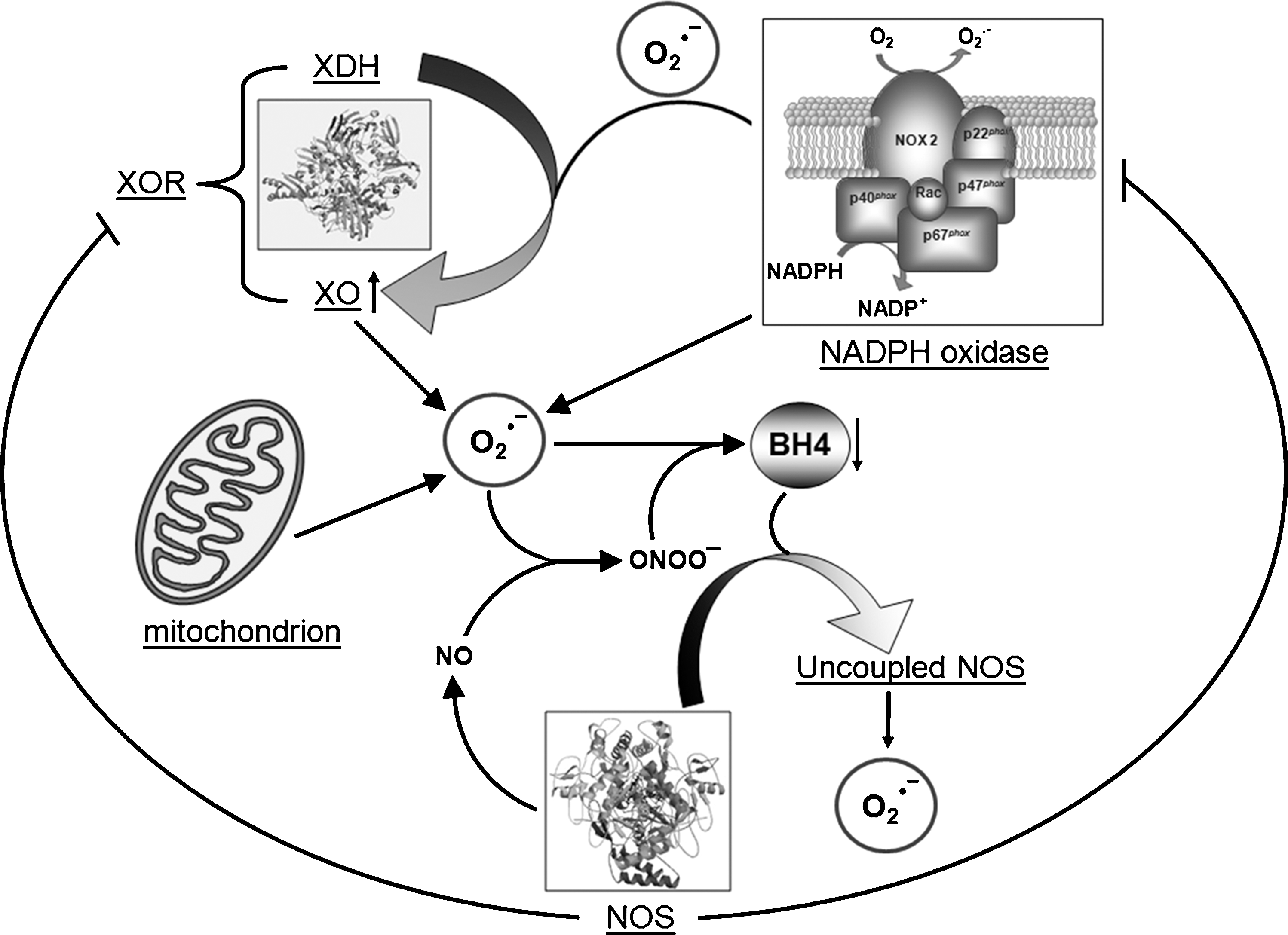
Growing experimental evidence documents that oxidative stress mediated by ROS plays a key role in the pathogenesis of HF (15, 63, 74, 102). ROS include free radicals, such as superoxide (O2 •−) and hydroxyl (·OH), and nonradical species such as hydrogen peroxide (H2O2). ROS generation occurs because the complete reduction of oxygen (O2) into two molecules of H2O requires four electrons and O2 accepts only one electron at a time. The addition of the first electron yields O2 •− that becomes H2O2 when one additional electron is added. ·OH could arise from electron exchange between O2 •− and H2O2 via the Haber–Weiss reaction. In addition, ·OH is also generated by the reduction of H2O2 in the presence of endogenous iron by means of the Fenton reaction. The cellular sources of ROS generation within the heart include cardiac myocytes, endothelial cells, vascular smooth muscles, fibroblasts, and infiltrating inflammatory cells of different types. In these cells ROS can be generated by mitochondria, NADPH oxidases, xanthine oxidase (XO), and uncoupled NO synthases (NOSs) (Fig. 2). Increasing evidence supports the view that NADPH oxidases play important roles as initiators and integrators of redox signaling via cross-talk with other ROS-producing systems (53). For example, exposure of endothelial cells to oscillatory shear stress leads to an NADPH oxidase-dependent activation of XO (252), whereas angiotensin II (AT-II) stimulation results in mitochondrial ROS production that is downstream of Nox activation (106). Endothelial (e)NOS uncoupling has also been shown to be a direct result of NADPH oxidase activation (212), and, in a recent review, a cross-talk between mitochondrial and NADPH oxidase-derived ROS is discussed (93). Amplification of ROS production may thus occur and may be important in affecting different signaling pathways. Such a novel aspect may have significant implication for understanding disease mechanisms and for designing rational therapeutic interventions aimed at restoring and resetting redox homeostasis.
1. Mitochondria
Cardiac myocytes have the highest density of mitochondria compared to most other cells to meet the demand for synthesis of ATP by oxidative metabolism (376). In cardiac myocytes, a large amount of ROS stems as a relevant by-product of electron flow through the respiratory chain. By using electron spin resonance spectroscopy, Ide et al. (173, 174) directly demonstrated that the inhibition of electron transport at the sites of complex I and III resulted in an enhanced generation of ROS in mitochondria from the failing myocardium. Electrons from complex I and III can convert O2 to O2 •−, which is then converted to H2O2 by spontaneous or enzymatic dismutation mediated by mitochondrial isoenzyme of superoxide dismutase. H2O2 can further react to form highly reactive ·OH radicals (Fig. 3). The generation of ·OH implies a pathophysiological significance of ROS in HF because ·OH radicals are the predominant oxidant species causing cellular injury (44). Further, the high reactivity of the ·OH and its extremely short physiological half-life of 10−9 s restrict its damage to a small radius from its origin since it is too short-lived to diffuse a considerable distance. Mitochondrial ROS generation is potentially significant in the setting of insulin resistance (279) and ischemia–reperfusion (140). In the latter context mitochondrial ROS production appears to be a dual effector responsible for both ischemia–reperfusion injury and cardioprotection (89). Ischemia causes immediate disturbance of mitochondrial function, including failure of ATP synthesis and drop in membrane potential that is accompanied by changes in cytosolic composition, like increased Ca2+, phosphate and fatty acids. This altered state is met during reperfusion by a large increase in ROS originating from the respiratory chain (111, 377). These factors promote opening of the mitochondrial permeability transition (MPT), a high-conductance pore in the inner mitochondrial membrane, which is the main cause of necrotic cell death in ischemia–reperfusion injury (103, 156, 391). It follows that any effort to protect the heart from these consequences must ultimately involve the prevention of MPT opening (391). The heart possesses self-defensive mechanisms able to reduce cell death and functional impairment after prolonged episodes of ischemia–reperfusion. Cardioprotective procedures include ischemic preconditioning, in which one or more periods of brief ischemia precede the index ischemia (270). Ischemic preconditioning protocol requires transient opening of the mitochondrial ATP-regulated potassium channel (mitoKATP) that is believed to be essential cardioprotective signal transduction against ischemia–reperfusion injury (Fig. 3) (89). In fact, mitoKATP opening induces a moderate increase in ROS production from Complex I; ROS produced by mitoKATP activity diffuses and inhibits MPT, thus reducing cell necrosis and infarct size (135).
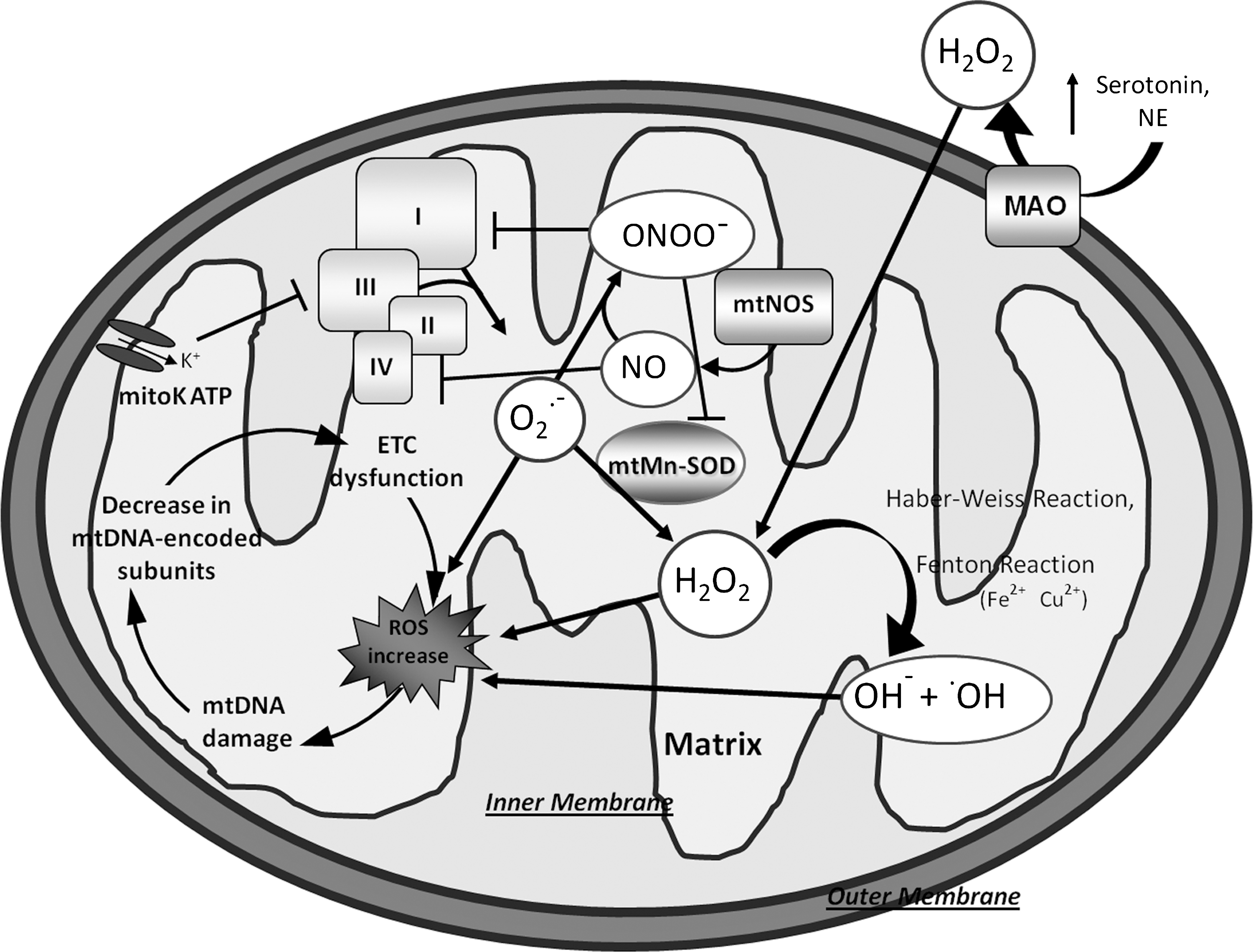
ROS are also produced within mitochondria at sites other than the inner mitochondrial membrane, for example, by monoamine oxidase (MAOs) activity (104, 258) (Fig. 3). MAOs are flavoenzymes located within the outer mitochondrial membrane, which catalyze oxidative deamination of catecholamines and biogenic amines such as serotonin. During this process, they generate H2O2 and thus can potentially be a source of oxidative stress in the heart, particularly under stress conditions. MAOs exist in two isoforms, MAO-A and -B, with a distinct substrate and inhibitor sensitivity (113). MAO-A, in particular, principally catabolizes serotonin, norepinephrine, and epinephrine and is inhibited by low concentrations of clorgyline. All of these neurotransmitters have major functional implications in the heart, especially in the modulation of cardiac inotropy. In contrast, MAO-B has a higher affinity for phenylethylamine and benzylamine, and is inhibited by selegiline (348, 411). Yet, although the role of MAOs in terminating neurotransmitter signaling in the brain is well established, little is known about its modulation of cardiac morphology and function. A tight regulation of serotonin to maintain normal cardiovascular activity has been demonstrated in different experimental models (148, 255). Pharmacological agents acting through serotonin-related pathways have been associated with a number of significant cardiovascular adverse effects, including neonatal death (20). Receptor-independent effects of serotonin, have been described recently. H2O2 produced by MAO-A dependent oxidative deamination of serotonin exerts hypertophic effects or aggravate ischemia/reperfusion injuries (41). A recent study of Kaludercic et al. (187) showed that in addition to serotonin, norepinephrine catabolism by MAO-A plays a prominent role in hypertrophy in vitro and in its progression toward HF in vivo through enhanced H2O2 production. Therefore, owing to metabolism of these amine, MAO-A activity may represent a check-point for the sympathetic/serotoninergic system but also a local producer of ROS, thus ascribing a role for MAO-A dependent catalysis in cell signaling (305). In cardiomyocytes catecholamines and serotonin also play a metabolic role; in fact, they show insulin-like effects by increasing glucose uptake with different mechanisms (129, 199). In this context, a modulated increase MAO-A activity might be of some beneficial on the metabolic de-arrangement of the failing heart (see below). To date, no evidence exists on the role of MAO in human HF. Preliminary results from our laboratory showed that in human end-stage failing hearts secondary to ischemic and dilated cardiomyopathies both MAO-A and MAO-B isoforms were present with total MAO activity (A+B) 10 times higher in the ischemic than in nonischemic cardiomyopathy. Moreover, activity of the two isoforms appeared different in the two ventricles with respect of the cardiomyopathy. Our results indicate that the evaluation of MAO activity, in particular MAO-A, might help to discriminate between nonischemic and ischemic cardiomyopathy associated with HF.
Although recently the question has become controversial, several earlier studies suggested the presence of NOS in mitochondria (mtNOS) (210). Local NO generates can decrease respiration and potentially trigger apoptosis or cell necrosis or both by inhibiting cytochrome oxidase (54). Moreover, in the presence of superoxide NO reacts very rapidly, generating peroxynitrite (ONOO_) radicals that can impair proteins involved in electron transport resulting in a further increase of ROS production (290) (Fig. 3).
The mitochondria would not only be the origin, but also the target of oxidative/nitrosative stress. The mitochondrial DNA (mtDNA) could be a major target for ROS-mediated damage for several reasons (Fig. 3). First, mitochondria do not have a complex chromatin organization consisting of histone proteins, which may serve as a protective barrier against ROS. Second, mtDNA has a limited repair activity against DNA damage. Third, a large part of O2 •−, which is formed inside the mitochondria, cannot pass through the membranes; hence, ROS damage may be contained largely within the mitochondria (376). In fact, mtDNA accumulates significantly higher levels of the DNA oxidation product (8-hydroxydeoxyguanosine) than nuclear DNA (141). A number of pathogenic mtDNA base substitution mutations, such as missense mutations and mtDNA rearrangement mutations (deletions and insertions), have been identified in patients with mitochondrial diseases (190, 226, 248, 390).
In HF, the centrality of the mitochondrion is not related only to its ROS generation and signaling, but also its role in bioenergetics' production, in particular at ATP levels that are critical for myocardial contractility and electrophysiology (see section VI. B).
2. NADPH oxidases
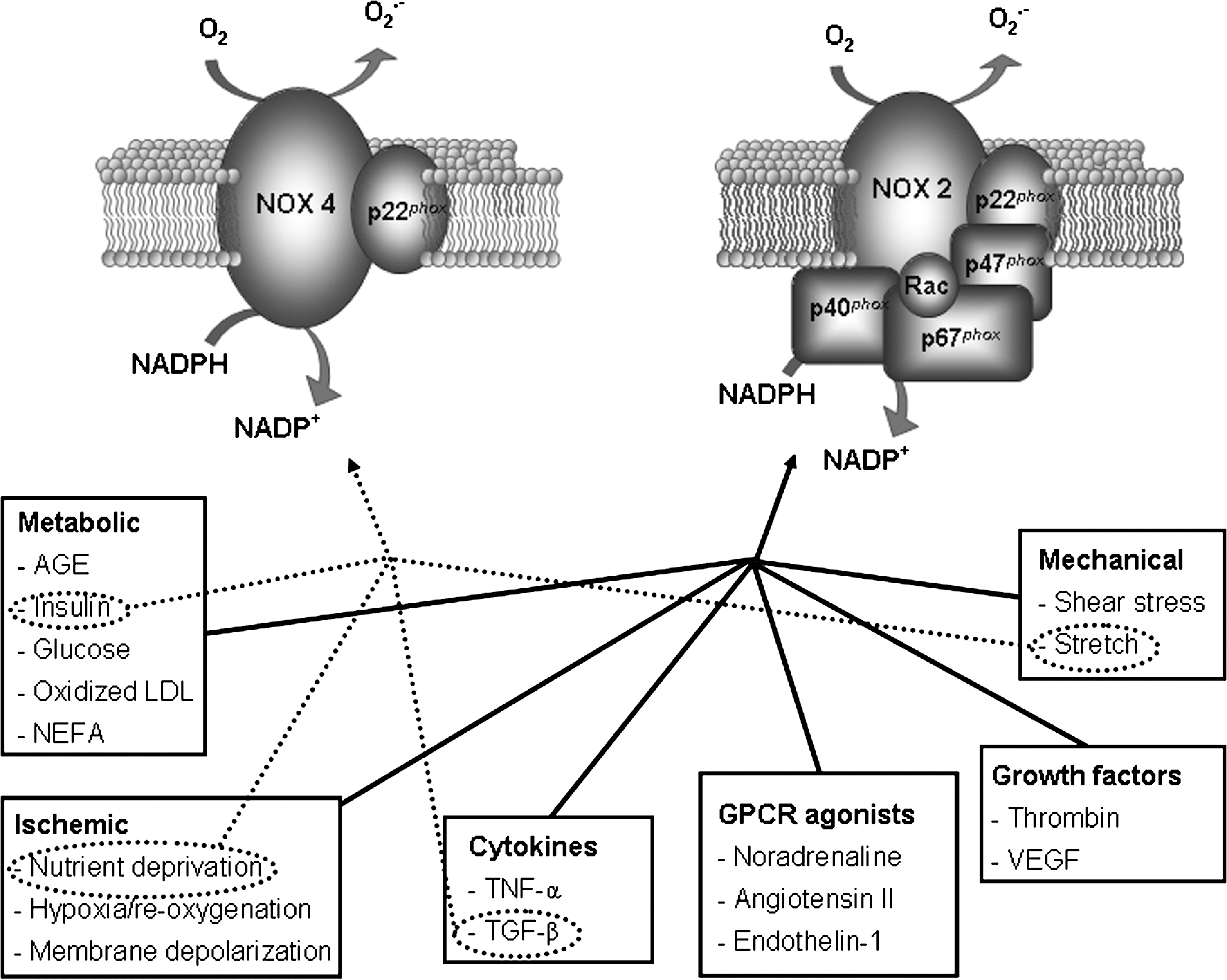
Another important source of ROS in the heart is the NADPH oxidase family of enzymes that generate O2 •− in a highly regulated manner by catalyzing electron transfer from NADPH onto molecular O2. NADPH oxidases appear to be the only enzymes whose primary function is ROS generation; they also appear to be especially important for redox signaling (211). The NADPH oxidase is a multicomponent enzyme complex, initially discovered in neutrophils, consisting of a membrane-bound flavocytochrome (which mediates the electron transfer process), a catalytic Nox subunit and a lower molecular weight p22 phox subunit. Five distinct Nox subunits (Nox 1–5,) each encoded by a separate gene and expressed in a tissue-specific pattern, have been identified (53). In the cardiovascular system, Nox1 is expressed mainly in ventricular smooth muscle cells (216). Nox2 (or gp91 phox , the original neutrophil isoform) is expressed in endothelial cells (144, 223), cardiomyocytes (34, 162, 402), fibroblasts (292), and some ventricular smooth muscle cells (375). Nox4 is expressed in endothelial cells (2), ventricular smooth muscle cells (159), cardiomyocytes (62, 224), and fibroblasts (91). Nox3 is not expressed in cardiovascular cells, while Nox5 has been reported in human endothelial cells and smooth muscle cells but is not found in rodents (33, 181). Multiple Nox subunits are expressed within a single cell at different levels and in different locations supporting the concept that individual Nox subunits have specifically delineated functions within the cell. Nox2 and Nox4 are the predominant isoforms in cardiomyocytes. These isoforms can be activated by a variety of stimuli, including agonists such as AT-II norepinephrine, endothelin, and insulin, inflammatory cytokines and growth factor, mechanical stretch, or hypoxia/reoxygenation (Fig. 4). Byrne et al. (62) reported a differential response of the Nox isoforms to pressure versus AT-II induced hypertrophy in that Nox2 was critical for the development of cardiac hypertrophy in response to AT-II, but pressure overload induced hypertrophy from other sources of ROS (possibly Nox4 oxidase) may be involved. A very recent study indicates that Nox2 and Nox4 exhibit distinct patterns of agonist-induced activation and downstream kinase activation, which could be attributable to specific compartmentation of redox signaling (15). Activation mechanisms for Nox2 involve complex formation with regulatory cytosolic subunits, namely, p47 phox , p67 phox , p40 phox , and Rac1 (7). Although oxidase activation is primarily driven by interactions with p67 phox and Rac1 subunit, other subunits (in particular p47 phox ) are important for translocation of the activating cytosolic subunits to the flavocytochrome and overall organization of the activated enzyme complex (32, 74). Posttranslational modifications of these subunits control the key steps, notably phosphorylation of p47 phox and geranyl-geranylation of Rac1, which are promoted by the above-described stimuli (223). However, Nox4 appears to be quite distinct in that the only subunit that appears to be necessary for its activity is p22 phox (12, 115, 246) (Fig. 4). Nox4 is an inducible Nox, and its activity is proportional to Nox4 protein expression alone. Heymes et al. (158) provided the first evidence that NADPH oxidase is expressed in human myocardium and specifically in cardiomyocytes. They also showed that NADPH oxidase activity is increased in end-stage failing human heart and that this activation was due to p47 phox translocation rather than increases in oxidase subunit expression. In line with this finding we (278) observed a coordinated increase of NADPH oxidase activity in the right (RV) and the left (LV) from failing end-stage hearts irrespective of the etiology of the disease (ischemic or dilated cardiomyopathy). We also found that increased Nox2 expression was not a prerequisite for increased Nox-dependent ROS production and, on the other hand, we observed both overexpression and membrane translocation of p47 phox the regulatory subunit of Nox2. The relative contribution of the two regulatory mechanisms to NADPH oxidase activation appeared different in the two ventricles (see below). The hypothesis that p47 phox translocation was a requirement for enzyme activation was reinforced by the finding of Borchi et al. (47) in which in human failing hearts Nox4 was not significantly increased with respect to the control ones. Moreover Maak et al. (238) showed that in patients with both ischemic and dilated cardiomyopathy, in addition to overexpression of p47 phox protein expression and translocation, an upregulation of Rac1 as well as increased rac1-GTPase activity was associated with increased NADPH oxidase. Treatment of HF patients with statins decreased Rac-1 activity in myocardium from these patients, possibly via statin effects on ROS activity (see below). In fact, statins, in addition to inhibiting cholesterol synthesis, downregulate Rac1-GTPase activity by reducing isoprenylation and translocation of Rac1 to the cell membrane (178).
3. Xanthine oxidase
XO and xanthine dehydrogenase (XDH) are both isoenzymes of xanthine oxidoreductase (XOR). They exist in two alternative forms deriving from the same 150-kDa gene product. The conversion of XDH to XO form may occur either irreversibly after limited proteolysis leading to a 130 kDa product or reversibly by phosphorylation or thiol oxidation of the 150 kDa protein. They differ in that XO only reduces O2 (thereby generating O2 •−), whereas XDH can reduce either oxygen or NAD+ but has greater affinity for the latter (36, 260). Both isoforms catalyze the conversion of hypoxanthine to xanthine and xanthine to uric acid, the terminal two reactions of the purine degradation pathway. Several studies have demonstrated an upregulation of XOR in animal models and in human dilated cardiomyopathy (36). XOR inhibition has been demonstrated to improve cardiac performance and induce reverse remodeling. XOR activity or protein content has been found to be elevated in the failing heart (99, 114, 205).
Doerries et al. (108) found that the increase in myocardial XO activity after myocardial infarction is dependent on the Nox system. This concept is strongly supported by the observation that XO activity was not increased in postmyocardial infarction p47 phox−/− mice, as suggested by spectroscopic analysis. Further, elevated levels of uric acid are associated with increased morbidity and mortality in HF (16, 122, 222). Of note, Cappola et al. (71) have recently reported that expression and activity of XO are both increased in the failing heart of patients with dilated cardiomyopathy. Nevertheless, the contribution of cardiac XOR to oxidative stress appears to be small in the human LV myocardium (100, 112, 180), as suggested by the fact that lack of effect in clinical trials of XOR inhibitors (e.g., allopurinol or oxypurinol) has been very successful in preventing or treating LV failure in rodents (116, 277, 358) but not in humans (191).
B. NO and RNS in HF
Accumulating evidence indicates that NO and its derivative RNS may also have important effects in the developing of HF (287, 290). NO is a lipophilic diatomic gas with a relatively small Stoke's radius, and this, in combination with its neutral charge, facilitates rapid membrane diffusion (143). The presence of an unpaired electron in NO supports its high reactivity with O2, O2 •−, transition metals, and thiols, which largely shape its cellular functions within the cell (287).
Biosynthesis of NO is dependent on enzymatic activity of NOSs, which catalyze the conversion of the amino acid L-arginine to L-citrulline in a reaction that requires O2 and a cofactor like as tetrahydrobiopterin (BH4), which is essential for the catalytic activity of all NOS isoforms. In fact, due to its oxidation and/or reduced synthesis BH4 depletion, can result in functional uncoupling of NOS (249, 357). Uncoupled NOS generates more ROS and less NO, shifting the nitroso-redox balance and having adverse consequences on the cardiovascular system (262).
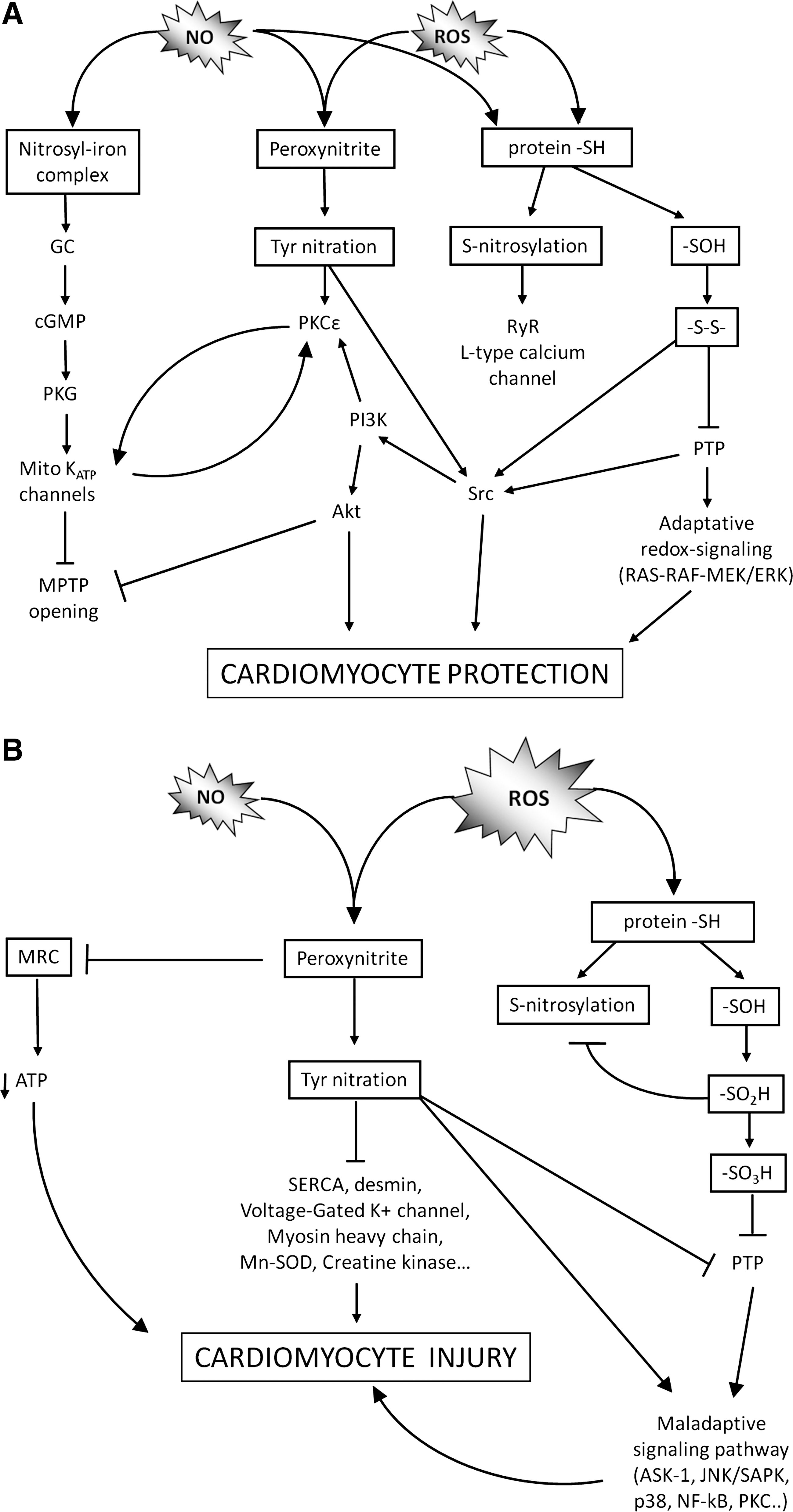
In the heart, NO is produced by constitutive NOS, eNOS, localized in endothelium and caveolae of cardiomyocytes, by neuronal NOS (nNOS), colocalized with ryanodine receptor (RyR) in cardiac sarcoplasmic reticulum (SR), and under pathological situations by inducible NOS (iNOS) in the sarcoplasm (339). The isoforms nNOS and eNOS are regulated by a variety of different stimuli in different cell types via Ca2+/calmodulin activation, as well as de novo synthesis (14, 51, 96). In addition to Ca2+/calmodulin activation, phosphorylation is also involved in the regulation of eNOS activity (171). In contrast, the activity of iNOS is regulated by expression in a Ca2+-independent manner (340). One of the major controversies surrounding NO in the heart derives from the observation that in HF and in cardiac injury due to myocardial infarction, NOS isoforms have been ascribed both protective and detrimental roles (327). The findings that eNOS-deficient mice develop more severe LV dysfunction and remodeling after myocardial infarction than do wild-type mice (330) and, vice versa, that endothelial overexpression of eNOS attenuates LV dysfunction in mice after myocardial infarction (183) suggest that NO was beneficial in HF. However, this effect is lost by the coexistence of oxidative stress from eNOS uncoupling that stimulates cardiac pathologic remodeling from chronic pressure load. In fact, in a transgenic eNOS knockout model with low ROS production, severely pressure-overloaded hearts developed only modest concentric hypertrophy with little fibrosis and without LV cavity dilation (366), indicating that eNOS activation becomes detrimental rather than beneficial in LV remodeling when ROS coexists and eNOS uncoupling occurs. Importantly, eNOS activation has been also proposed as a key mechanism for cardioprotection mediated by early ischemic preconditioning. In this setting, rise of intracellular Ca2+ stimulates eNOS function through activation of Ca2+/calmodulin-dependent protein kinase II (CAMK-II). Resultant increase in eNOS-derived NO promotes posttranslational modifications of proteins in cardiomyocytes as described in Figure 5A. The subsequent activation of cyclic guanosine 5′-triphosphate (cGMP)-dependent protein kinase and PKC-ɛ promotes opening of mitoKATP channels (see section II, A1) that inhibit MPT. The activation of PKC-ɛ also activates nuclear factor kappa B (NF-κB) and upregulates transcription of iNOS, which is an obligatory mediator of the late phase of ischemic preconditioning, and of manganese superoxide dismutase (Mn-SOD) (45). Recent evidence demonstrates that cardiomyocyte-restricted expression of iNOS is sufficient to confer chronic cardioprotection and that transgenic upregulation of cardiac iNOS decreases reperfusion-free radical generation, which in turn prevents MPT and chronically protects the heart against ischemia–reperfusion injury (394). It is an intriguing finding because numerous experimental and human studies have demonstrated overexpression and increased activity of iNOS in the myocardium of animals and patients with various forms of HF and benefits of iNOS inhibition on cardiac function (125, 204, 259) (see section II, D). Thus, the role of iNOS and NO in the development and progression of heart disease is a subject of recent debate, the preeminent hypothesis being that increased iNOS activity contributes to cardiac maladaptation. Nevertheless, it is tempting to speculate that increasing bioavailability of iNOS-derived NO concomitant to simultaneous inhibition of oxidative stress may become a novel strategy to treat HF failure (45, 110, 266).
At variance with eNOS and iNOS, the contribution of nNOS to myocardial pathophysiology has been undervalued for years. However, recent experimental studies point to an important role for myocardial constitutive NO production through nNOS in the regulation of basal and adrenergic-modulated cardiac function (58, 72, 228). The location of nNOS in the SR and/or sarcolemma suggests that ion channels and transporters involved in the regulation of Ca2+ cycling in the myocyte may be obvious targets for NO downstream signaling, as extensively described later. Emerging data showing colocalization of XOR and nNOS in the SR of rodents, and increased XOR activity in the nNOS−/− mice myocardium suggest that nNOS gene deletion may have wider implications on the myocardial redox state (196, 198).
In general, the documented nNOS-mediated inhibition of Ca2+ entry through L-type Ca2+ channels (337) is a likely mechanism through which nNOS-derived NO regulates Ca2+ cycling within intracellular Ca2+ stores and therefore excitation–contraction coupling mechanisms (see later on). Altogether, available evidence suggests that NO produced by the neuronal isoform may exert a negative feedback regulation on Ca2+ influx: any increase in intracellular Ca2+ would stimulate nNOS-mediated synthesis of NO, which in turn attenuates L-type Ca2+ current.
The discovery that eNOS and nNOS are expressed in distinct subcellular compartments in the cardiomyocyte (124, 396, 403) suggests that the two isoforms might couple to distinct effector molecules and elicit quite different results following enzyme activation. In fact, NO diffusion within cardiac myocytes is likely to be limited to a local environment by both a high cytoplasmic concentration of myoglobin (which has a high affinity for NO and acts as a scavenger) and, particularly in disease states, by an abundance of superoxide anions (which can react with NO to limit its bioavailability) (130). Although the NO effect is mainly compartmentalized, emerging evidence suggests that NO generated at one compartment can be converted to inert NO metabolites such as nitrite and nitrate and transferred to other compartment and then reduced back to NO in the presence of myoglobin (101). This finding has relevant consequences, since several studies in animal models demonstrated the ability of nitrite to provide potent cytoprotection against cardiac injury of the heart (101). For example, Hendgen-Cotta et al. (157) recently reported that during hypoxia nitrite can enter cardiomyocytes and be reduced to NO by endogenous myoglobin nitrite reductase activity, to inhibit mitochondrial respiration potentially by binding cytochrome c oxidase. The reversible slowing in the rate of mitochondrial respiration may allow preserving oxygen gradients during ischemia and favor a more controlled resumption of respiration during reoxygenation, minimizing free radical injury. Although the mechanisms of this phenomenon are still waiting complete characterization, the reproducibility of this effect in multiple animal models of ischemia–reperfusion suggests nitrite as a novel potential therapy for human ischemic diseases. Summarizing, in HF oxidant-producing enzymes are upregulated and NO-producing enzymes are altered in either their abundance or spatial localization. A relative NO deficiency may further promote oxidase activities, which suggests that NO may be a global modulator of O2–/ROS production (155). In particular, NADPH oxidase activities are increased in the failing hearts, at least in part due to increased levels of AT-II, which indicates a link between neurohormonal activation and NO/redox disequilibrium (263). Moreover, increased XO activity that directly reflected a dysregulation of NO signaling contributes to vasoconstriction and depressed cardiac function (324).
C. Signals activating ROS production
Among the stimuli that generate ROS are the following: peptide and amine hormones, such as AT-II, norepinephrine, endothelin, serotonin, and thrombin; cytokines, such as interleukin-1β, interleukin-6, and tumor necrosis factor-α; and mitogens, such as fibroblast growth factor and epidermal growth factor. These agonists bind to receptors of different classes that are coupled to multiple signal transduction cascades, including tyrosine kinases, mitogen-activated protein kinases (MAPKs), PKC, calcineurin, phosphoinositide 3-kinase (PI3-kinase)/Akt, and NF-κB (371). Mechanical stress is another important stimulus that generates ROS production (6, 418) as well as hypoxia-ischemia/reoxygenation (48).
AT-II, a small peptide and main component of the renin–angiotensin system (RAS), plays an important role in cardiovascular homeostasis (227), cell metabolism, and pathogenesis of cardiovascular diseases, including HF (19). Not only does AT-II bind to its cell surface receptors (angiotensin type 1 receptor antagonists [AT1] and AT2) and exert multiple effects by activating a number of intracellular signal transduction pathways through heterotrimeric G proteins, but it also interacts with receptor tyrosine kinases and cytokine receptors (139, 253, 381). It is well known that AT-II elicits in cardiomyocytes the generation of ROS, mainly due to NADPH oxidase, and that locally produced ROS acts as an initial mediator of AT-II-induced signal transduction, gene regulation, and phenotypic modulation in cardiomyocytes (9, 272, 349). During HF, cross-talk between some of these stimuli may affect many aspects of the adverse remodeling. For instance, in dilated cardiomyopathy, ventricular hypertrophy or dilatation elicits mechanical stress with concurrent changes of extracellular matrix and cytoskeleton (214, 329). By disarraying myocardial contraction and impairing cell metabolism, ischemia switches several ionic channels on/off, thus perturbing intracellular (and extracellular) ionic milieu (30, 280).
Mechanical stress also triggers the release of AT-II and endothelin, which in turn increases ROS production, stimulating the activity of NADPH oxidase (64, 97) via a PKC-mediated phosphorylation of the oxidase's p47 phox subunit, leading to its translocation to the membrane-bound cytochrome (32, 149, 263, 271). In ventricular myocytes, stretching (195, 406) and AT-II mediated pathways rapidly converge toward Rac1-GTPase activation (17), which likely plays an early role in mechano-transduction and neurohumoral signaling. Stretching depolarizes cell membrane during diastole changes the action potential profile and predisposes to arrhythmias, effects mediated at least in part by stretch-activated ion channels (170). Stretch-activated ion channels are mechano-sensitive molecules localized to specific regions of the cell such as the Z-disc, thus sensing changes in mechanical forces imposed by the hemodynamic load. Channel opening and consequent sodium/calcium flowing into the cell can influence electrophysiological properties and also trigger mechano-sensitive signaling cascades (355).
D. Downstream effectors and targets
The biological effects of ROS/RNS depend upon the specific moiety generated, its localization, and the relative balance between levels generated and the activity of antioxidant systems that reduce ROS levels. In the setting of NO/ROS imbalance where large amounts of radicals are generated, these may induce oxidation and damage of macromolecules, membranes, and DNA and thus be detrimental for cellular function and viability (155).
Accumulating evidence suggests that ROS/RNS are not only injurious but also essential participants in cell signaling and regulation (327, 339). The term nitroso/redox signaling describes a process by which milder physiological levels of ROS/RNS induce modifications to proteins that are discrete, site specific, and reversible. Nitroso/redox signals may abrogate or enhance activity of the target protein, and have been implicated in physiological signaling processes that include kinase signaling, channel function, apoptotic proteolysis, and regulation of transcription. The tenet of redox sensing is that certain proteins undergo reversible chemical changes in response to changes in local redox potential. Cysteine residues are the typical targets of redox modification, as they can adopt multiple oxidation states (133). The free thiol group of cysteine can undergo reversible or irreversible covalent modifications by ROS that can induce the sequential generation of reversible or irreversible oxidation products, such as sulfenic, sulfinic, and sulfonic derivatives. Additionally, ROS can induce disulphide formation. Blockade of cysteine -sulfenic derivate by disulphide formation (either with intramolecular cystine or with glutathione [GSH] binding) is the common mechanism of all redox regulated protein tyrosine phosphatases. This process leads to reversible inhibition of protein tyrosine phosphatases and prevents their further and irreversible oxidation. The same mechanism, on the contrary, induces activation of protein tyrosine kinases like Src (82).
At a molecular level, NO and RNS exert their biological actions by chemical modification of target molecules, preferentially interacting with transition metals, free radicals, and, similarly to ROS, with thiol groups (287). One well-known signaling pathway of NO is initiated by its reaction with heme iron of soluble GC, leading to increased cGMP accumulation and kinase activation. Binding of NO to other heme proteins, such as cytochrome c oxidase competes with that of oxygen and regulates oxygen consumption by the mitochondria (290) Another target is protein tyrosine residues, which can be modified to stable 3-nitrotyrosines. Posttranslational modification of tyrosine residues has been shown to play an important role in modulating the activity of several PKC isozymes, including PKC-ɛ and Src, which has consistently been implicated in the cardioprotective signal transduction (287), as described above. On the other hand, tyrosine nitration of several critical proteins (Ca2+-ATPase pump, sarco/endoplasmic reticulum calcium ATPase [SERCA], voltage-gated K+ channel, desmin, etc.) has been proposed as a major mechanism contributing to cardiomyocyte dysfunction (290). Another important signaling pathway of NO and RNS is represented by the reaction with thiols. Protein thiols and GSH react with NO derivatives to produce a range of products, including S-nitrosothiols (312). S-nitrosation of protein thiols has gained particular interest because it may modulate a fast reversible regulation of protein functions, including the cardiac L-type Ca2+ channel (168) and the RyR2 (228) as described in section V. Importantly, this process is sensitive to disruption by redox milieu. In this context, oxidative stress disrupts a physiologic signaling process driven by S-nitrosation and mediated by modification of effector proteins. In other words, the relative flux of NO and O2, depending on abundance and location of both NOS's and oxidases in cardiomyocytes, determines the chemical fate of their interactions. In fact, during physiologic situations when NO is higher than O2, S-nitrosylation is favored; instead, NO/O2 imbalance, characteristic of HF, favors oxidation reactions (155).
Intracellular GSH has a central role in cellular redox balance (250). Glutathionylation of proteins, through the formation of a mixed disulphide between one cysteine of glutathione and one cysteine of the other protein, constitutes an efficient mechanism to protect proteins from irreversible modifications. GSH also provides cellular protection against oxidative damage by reacting with RNS to form S-nitrosoglutathione. GSH thereby serves as an efficient endogenous scavenger of peroxynitrite and plays a major role in the cellular defense against this species (290). Accordingly, the susceptibility of cells to peroxynitrite toxicity largely depends on the amount of intracellular GSH.
Cellular GSH abundance, combined with the ready conversion of S-nitroso and sulfenic acid derivatives into S-glutathione mixed disulfides, strongly suggests that reversible protein S-glutathionylation is a central mechanism of redox signal transduction (342). The possible impact of S-nitrosation in mediating cardiac effects of NO in a situation of nitrosative stress is largely unknown. In circumstances of acute ischemia, sepsis, or HF, iNOS abundance may increase, leading to nitrosative stress, a pathophysiologic situation characterized by accumulation of S-nitrosylated proteins to hazardous levels (amount and/or spatio-temporal distribution) (154). Nitrosative stress may be exacerbated in situ by oxidants (oxidative stress); stimuli that lead to iNOS induction may also upregulate oxidases, and concomitant elevations in NO/RNS and O2–/ROS may lead to formation of higher amounts of peroxynitrite (291) This condition favors polynitrosylation and oxidation of cysteine thiols as well as nitration of tyrosines in proteins as reported above. This situation is relevant to the failing heart, in which the loss of spatial confinement of nNOS, which redistributes from SR to the plasma membrane, may be a proximate cause of oxidative/nitrosative stress both by relieving the local (SR) control of XO (196), and by altering NO/redox balance at the sarcolemma. At the molecular level, poly–S-nitrosylation, oxidation and nitration of the SERCA2a and RyR2 may ensue with adverse effects on Ca2+ homeostasis (404). Thus, a central pathophysiologic consequence of redox disequilibrium is the disruption of NO signaling by alteration of the occurrence or nature of the posttranslational modifications.
Multiple enzymatic systems regulate the functional changes induced by ROS/RNS, including glutaredoxin, thioredoxin (4), and glutathione S-transferase. Glutaredoxin acts as a specific and efficient catalyst of protein de-glutathionylation reactions (342). In addition to the glutaredoxins, thioredoxin can also reduce reversibly modified thiols. Some isoenzymes of the super family of glutathione S-transferases can also regulate MAPKs or facilitate protein S-glutathionylation (251).
Among the ROS, H2O2 is more stable than O2 •− and capable of crossing biological membranes. It makes H2O2 especially suitable as a signaling molecule. H2O2 is tightly regulated biologically by catalase (CAT), glutathione peroxidase (GPx), and peroxiredoxins, which convert H2O2 to water and other metabolites (53).
As illustrated in Figure 5 ROS/RNS may modulate diverse redox signaling pathways by activation of kinases and/or oxidative inactivation of protein phosphatases (82) resulting in increasing tyrosine and serine/threonine phosphorylation signaling events. These events are converged and integrated to induce various redox-sensitive transcriptional factors that are involved in regulating redox-sensitive gene expression, leading to various physiological and pathophysiological responses. Among the several downstream signaling pathways activated in cardiomyocyte the extracellular signal-regulated kinase, Jun N-terminal kinase (JNK), apoptosis signal-regulating kinase 1, and PKC (21, 179, 233), cyclic AMP-dependent protein kinase (PKA) (334), cGMP-dependent protein kinase (57), and Ca2+/calmodulin-dependent protein kinase II (118) are well established and characterized. Nonreceptor tyrosine kinases c-Abl and Abl-related gene (Arg), belonging to the so-called Abl family (313), have recently been added to the list on the basis of the latest observations by Borchi et al. (48), being able to modulate the activity of some antioxidant enzymes via posttranslational modification. Another important signaling pathways is phosphoinositide 3-kinase (PI3K)/Akt that plays an important role in myocardial metabolism (88, 207, 282), as central mediator of insulin's effects (see Section VI), and mechanotransduction (288, 361).
III. Endogenous Modulators of ROS Signals
The myocardium is equipped with a variety of endogenous nonenzymatic and enzymatic antioxidant systems that are sufficient to metabolize ROS generated during normal cellular activity. Nonenzymatic mechanisms include intracellular antioxidants such as the vitamins E, C, and β-carotene (a precursor to vitamin A), ubiquinone, lipoic acid, urate, and the above-described GSH. As for enzymatic antioxidant defenses, the best-characterized are CAT and GPx, which coordinate the catalysis of H2O2 to water, and the SODs, which facilitate the formation of H2O2 from O2 •− (22 –25).
A. Antioxidant enzymatic system
1. Cytosolic and mitochondrial SOD
SODs are metalloenzymes that catalyze the dismutation of O2 •− to molecular O2 and H2O2 (24, 414). They represent a crucial part of the cellular antioxidant defense mechanism. Three types of SODs have been described with regard to their metal content: copper–zinc, Mn, and iron SODs. In humans, the three forms of SODs are differently distributed in the cytosol, in mitochondria Mn-SOD, and in the extracellular space iron SOD (326). Copper–zinc SOD is a cytosolic homodimeric enzyme of 32 kDa. Mn-SOD is a homotetrameric enzyme of 25 kDa located in the mitochondrial matrix. The Mn2+ ion is coordinated to three histidine and one aspartate residues. Iron SOD is an extracellular homotetrameric enzyme localized both in the interstitial spaces of tissues and in extracellular fluids, accounting for the majority of the SOD activity in plasma, lymph, and synovial fluid (244, 362). The reaction catalyzed by SODs is extremely fast, with a turnover of 2 × 109 M −1 s−1, and the presence of sufficient enzymes amounts in cells and tissues typically keep the concentration of O2 •− very low (31). When SOD activity is either decreased or absent (i.e., SOD mutations) or NO concentration is increased (i.e., iNOS upregulation), NO outcompetes SOD for O2 •−, resulting in the formation of peroxynitrite. In the heart, Mn-SOD accounts for about 70% of total SOD activity; this percentage reaches 90% in cardiac myocytes (63). Induction of SOD gene expression is triggered in response to late phase of preconditioning throughout activation of transcriptional factors, NF-κB and activator protein -1, that are subjected to redox regulation. Superoxide dismutase may also catalyze S-nitrosylation reactions (347).
Little data are present in literature on SOD activity during the progression to overt HF. Experimental studies demonstrated that the activity of SOD was decreased in animal hearts during transition to failure (102, 161). Unlike animal models, studies in pathologic samples of patients with end-stage HF gave conflicting data, showing either unchanged (29, 47, 105) or decreased myocardial SOD activities (325).
2. Catalase
CAT is a tetrameric enzyme that catalyzes the reduction of H2O2 into H2O and O2. Being exclusively localized in the peroxisomes in mammalian cells, a major role of CAT is likely to remove H2O2 produced during β-oxidation of fatty acids in peroxisomes, particularly activated in insulin resistance (256). With the exception of rat myocardial cells, CAT is not detectable in the mitochondria (304). Further, despite its high turnover number, CAT is not efficient in eliminating low levels of H2O2 because it is difficult to saturate with H2O2 and its catalytic cycle requires the interaction of two H2O2 molecules with a single active site, which is less likely at low H2O2 concentrations. Therefore, CAT is not expected to play a significant role in eliminating low levels of H2O2 produced in response to receptor engagement. Nevertheless, Cao and colleagues (67 –70) showed that CAT is extensively regulated in responses to H2O2 concentration through the Abelson(Abl) family of nonreceptor tyrosine kinases. The Abl family members are c-Abl and Arg (the product of c-Abl-related gene) and they are activated in response to oxidative stress. Treatment of cells with H2O2 induces the binding between the protein kinase Cδ and c-Abl, followed by phosphorylation and activation of c-Abl (364). Activated c-Abl and Arg appear to regulate CAT by its phosphorylation at predominantly at Tyr231 and Tyr386, resulting in an increase of CAT activity.
3. Glutathione peroxidase
GPx reduces H2O2 to H2O through the oxidation of reduced GSH to its disulfide form. GPxs are obligatory tetramers, each containing one selenocysteine in its active site. There are at least five types of GPx in mammalian cells. There is present in the myocardium the classical GPx1 that is a more abundant ubiquitous protein (98) found mainly in the cytosol and in the matrix of mitochondria. Cao and colleagues (66) extended their studies on the role of c-Abl and Arg as antioxidant enzyme activators also to GPx1: they found that c-Abl and Arg also regulated GPx1 activity through its phosphorylation at Tyr96, again causing an increase of GPx activity.
B. Antioxidant enzymatic defense in HF
In various animal models of HF, enhanced oxidative stress may be combined with a decrease in scavenging enzyme activity, either as a cause or as a result. It remains unclear whether human endstage HF is also associated with altered myocardial antioxidant reserve (105). To date, a comprehensive view of changes in oxidative stress mechanisms and antioxidant enzyme activity in human HF is far from being achieved. Moreover, the currently available information is somehow contradictory. For example, CAT activity in the failing human ventricle was reported to be either decreased (29), upregulated (105) or unchanged (325) in different studies. Sam et al. (325) found a decreased Mn-SOD activity though in the presence of upregulated mRNA levels in human failing hearts, whereas Baumer et al. (29) showed Mn-SOD mRNA, protein levels, and activity unchanged in human hearts with idiopathic dilated cardiomyopathy, in accordance to Dieterich et al. (105). As for GPx, all these studies agreed for no change in activity of this enzyme. During pathophysiological conditions, several cellular ROS sources are activated which alter intracellular oxidative–antioxidant homeostasis in favor of the former. As an adaptive response, antioxidant defense systems are upregulated in response to increased ROS. Most antioxidant enzymes contain gene regulatory sequences in their promoter and intron regions that can interact with redox-sensitive transcription factors to trigger upregulation of gene expression (9). Thus, upregulation of gene expression via redox-sensitive signaling pathway becomes a necessary strategy to restore homeostasis. Mn-SOD is a well-known target gene for NF-κB activation and its expression has been shown to be upregulated by acute stimuli (166). A redox-dependent posttranslational modification, including protein phosphorylation/oxidation is another key signaling response.
A contribution to a possible mutual relationship between oxidant and antioxidant activities in human failing hearts was reported by our recent study (47). We found that despite a marked increase of CAT and GPx activities, mRNA expression and protein levels were unchanged for both enzymes. This finding supports the hypothesis that dissociation occurs between mRNA/protein expression and activity and suggests that the adaptive response to increased oxidative stress does not lead to increased transcription of these antioxidant enzymes but may reflect posttranslational modifications. In fact, a significant CAT and GPx Tyr phosphorylation was found in failing hearts when compared to the control ones, and these changes in Tyr phosphorylation go hand-in-hand with enzyme activities. Although clear evidence for a cause–effect relationship was prevented in end-stage hearts by obvious limitations, on the basis of Cao et al. (66 –68) and our (47) findings, we speculated that increased NADPH oxidase-mediated ROS production may activate c-Abl and Arg kinases, with subsequent stimulation of the CAT and GPx catalytic activities, a mechanism that may contribute to normalize the level of oxidative stress (Fig. 6).

IV. Differences Between RV and LV in HF
Knowledge about the role of the RV in health and disease traditionally has lagged behind that of the LV. Less muscular, restricted in its role to pumping blood through a single organ, and less frequently involved than the LV in diseases such as myocardial ischemia, cardiomyopathy, or valvulopathy, the RV has generally been considered a mere victim of pathological processes affecting the cardiovascular system (389).
The amount of literature on the RV and its influence on the pathophysiological processes is therefore much smaller. Nonetheless, there is a growing collection of evidence that the RV function is a powerful predictor of mortality in HF (52, 151, 389), but little is known about the concomitant cellular and molecular changes occurring in the RV associated with HF. Indeed, only some recent studies inferred that unlike mechanisms account for cardiac hypertrophy and failure in RV and LV (160, 231, 261). Modesti et al. (261) reported that in chronic aortocaval shunt the different experimental mechanical load and geometry of the two ventricles induces a distinct regional pattern of activation of cardiac growth factors, with overexpression of angiotensinogen and prepro-endothelin-1 restricted to the RV (facing both pressure and volume overload) and insulin-like growth factor-I gene expression activated in both ventricles. These different patterns of growth factor expression selectively regulate the adaptation of myocyte shape to mechanical load (insulin-like growth for cell length and prepro-endothelin-1 for cell diameter increase) and affect collagen deposition (angiotensinogen and prepro-endothelin-1). Differences between the two ventricles were also unraveled by the use of left ventricular assist device as a bridge to heart transplantation or as destination therapy in end-stage HF patients (95). In fact, despite the general clinical benefit of left ventricular assist device (26, 202) RV failure after device implantation emerged as a major postoperative problem and is associated with a high mortality rate (303). We recently reported that oxidative stress resulting from increased ROS generation by NADPH oxidase was present in both ventricles of end-stage human failing heart, with a significant correlation between the two chambers. Moreover, increased ROS generation by NADPH oxidase significantly correlated with enhanced lipid peroxidation, activation of redox-sensitive kinases, and of antioxidant enzymes activities. Our findings showed that these molecular and biochemical mechanisms, although qualitatively similar, were quantitatively different in the RV and the LV and suggest that the RV might be more vulnerable than the LV against oxidative stress. In fact, in the same failing heart, NADPH oxidase activity and lipid peroxidation are higher in the RV than the LV; on the other hand, the level of CAT activity appeared higher in the LV than the RV. In line with these findings, Liu et al. (231) observed that in isolated rat hearts subjected to retrograde perfusion in the presence of serotonin the RV and the LV differ in their responses to protein carbonylation mediated by this amine. Specifically, protein carbonylation was higher in the RV than in the LV and this increase might be due to a reduced RV MAO-A activity, and not to a difference in H2O2 scavenging activity in the two ventricles.
V. ROS/RNS and Functional Remodeling in Failing Cardiomyocyte: Focus on Excitation–Contraction Coupling and Arrhythmogenesis
A. NO/ROS and calcium homeostasis
A hallmark of HF is the diminished contractile capacity. Many culprits account for the malfunction: loss of viable myocytes due to necrosis and apoptosis (see above), their replacement by noncontractile substrate (fibroblasts and collagen), local release of mediators affecting inotropism (natriuretic peptides, purines), or desensitization to positive inotropic agents (e.g., loss of function of the β-adrenergic signaling pathway) and, not least, intrinsic cardiomyocyte remodeling. Indeed, even when isolated from cardiac tissue by enzymatic digestion, failing myocytes persistently exhibit depressed inotropic responses and altered intracellular Ca2+ homeostasis. To get insight into the mechanisms and role of redox imbalance in determining this maladaptive behavior, it is worthwhile to recall the key features of excitation–contraction coupling mechanisms (Fig. 7). Special attention will be paid to proteins, which are sensitive to the NO/redox environment: as above described, most of proteins contain free thiol groups of cysteine, making them susceptible to the redox action and nitrosylation and subsequent alterations in their properties. In Figure 7, proteins undergoing NO/redox control are marked by “*”: it is evident that most of Ca2+ handling proteins are potential substrates.

During the action potential plateau, L-type Ca2+ channels activation triggers a subsarcolemmal rise in Ca2+, which initiates the release of Ca2+ from SR mainly via the RyR2; a process termed Ca2+-induced Ca2+ release (CICR). The free Ca2+ in the cytosol increases from nanomolar to micromolar levels and binds to troponin C, thus producing a conformational change in the troponin I–tropomyosin complex, which permits the initiation of myosin-actin cross-bridge cycling and contraction. Recovery of intracellular Ca2+ concentration to diastolic level is mainly due to reuptake into SR via SERCA2 and, in order of relevance, to extracellular extrusion via sarcolemmal Na+–Ca2+exchanger (NCX), sarcolemmal Ca2+-ATPase, and mitochondrial Ca2+ uniporter (37). Amplitude of Ca2+-transients (e.g., as measured by Ca2+-sensitive dyes) is substantially decreased in HF and its kinetics slowed down (Fig. 7).
Redox-sensitive thiols are present in Ca2+ channel (65), which may account for the abrupt decrease in L-type Ca2+ current observed in isolated cardiomyocytes exposed to various ROS sources (75). The α1C subunit of the L-type Ca2+ channel contains 48 cysteine residues that are potential thiol redox-sensitive sites of the channel. While some of these residues are not available for oxidation or reduction, being within the plasma-membrane itself or already involved in disulphide bonds, others are likely target of redox or nitrosylation modification (168). However, Ca2+ current amplitude is not significantly diminished in failing myocytes, thus suggesting that downstream abnormalities are likely responsible, such as defective coupling between L-type Ca2+ channels and RyR2 and/or the contractile machinery. Underlying mechanisms, whose detailed description is beyond the scope of this review, have been extensively reviewed (42).
Reduced SR Ca2+ release is observed in end-stage HF as a consequence of diminished Ca2+ content (299); abnormalities in both release (e.g., RyR2) and uptake (SERCA) mechanisms cooperate to empty SR. Malfunction is caused by dysregulation of several pathways, namely, phosphorylation and redox pathways. RyR2 hyperphosphorylation by PKA (245) or Ca2+/calmodulin-dependent protein kinase II (5) in HF has been consistently observed; the consequence is an SR leak of Ca2+ during diastole because of enhanced Ca2+ sensitivity of the RyR2. Recently, the role of redox mechanisms as RyR2 regulator has emerged. Susceptibility of the RyR2 to reducing-oxidizing modifications is suggested by hyperactive cysteine residues, which may form disulfide bonds, thus altering RyR2 structure and function, including sensitivity to luminal or cytoplasmic concentration (123). In failing canine cardiomyocytes, altered redox balance was paralleled by reduced Ca2+ transients and enhanced leakage of Ca2+ from SR; treatment of HF myocytes with reducing agents (dithiothreitol and 2-mercaptopropionylglycine) led to normalization of Ca2+ cycling. At the same time, oxidizing agents converted normal cardiomyocytes to a failing phenotype (370).
Hyper-responsiveness to [Ca2+]i was also observed in dystrophic cardiomyocytes, a condition characterized by high ROS production, and could be normalized by the reducing agent mercaptopropionyl-glycine (378). A second modulator pathway consists in S-nitrosylation of RyR2, that is, the covalent attachment of a NO moiety to a reactive thiol side chain on a cysteine residue (228). Exogenous NO donors (S-nitroso-N-acetyl penicillamine, SNAP) increases RyR2 opening reversibly; on the other side, sulfhydryl reducing agents promote channel closure (356). This evidence, obtained in synthetic planar lipid bilayers, was confirmed in intact cardiomyocytes by Ziolo et al. (421); the picture emerging from these studies is a complex modulation of RyR2 by NO, which depends not only on NO concentration but also on the level of channel phosphorylation by PKA. Recent data indicate that the 1-electron reduction product of NO, nitroxyl, can potently activate RyR2 at picomolar concentrations (81).
Contrary to the diseased state, NO exerts a physiological role as endogenous and constitutive modulator of RyR2, as briefly mentioned in the previous sections (II, B). The nNOS immunoprecipitates with RyR2 (35, 94). Experiments in cardiomyocytes from nNOS knockout mice suggest that nNOS-derived NO modulate basic SR function by reducing release and accelerating reuptake, an effect that translates into a slight negative inotropic and positive lusitropic effect [as reviewed by Lim et al. (228)]. Finally, S-nitrosylation of several effectors of excitation contraction coupling mechanism (L-type Ca2+ channels, SERCA2) has been detected during ischemic preconditioning and appears to be involved in cardioprotection (363). Therefore, S-nitrosylation can be viewed as a physiological mechanism controlling beat-to-beat cardiomyocyte function; its rapid and reversible activity on several relevant effectors of excitation–contraction coupling is important for adjusting the contractile machinery to neurohumoral (e.g., beta-adrenergic stimulation) or metabolic state (e.g., oxygen supply). Likely, in chronic HF most of these regulatory and cardioprotective mechanisms are largely ruled out by irreversible post-transductional modifications, alterations in cell size, and compartmentation of subcellular structures.
Recent evidence points at the interplay between ROS and RNS species as a signaling axis (155). As reported above, NO and O2 − interact by forming the highly reactive species peroxynitrite, a reaction which—on one side—exhausts NO and—on the other side—scavenges O2 •−. Therefore, unscavenged O2 − could result in an RyR2 gain of function by irreversible activation consequent to oxidative stress and of RyR2 (404).
Being a highly reactive species, NO effect is extremely compartmentalized; that said, immunoprecipitation of RyR2 and nNOS points at a spatially circumscribed interaction. In HF, compartmentalization is grossly altered by multiple factors, such as changes in volume/surface ratio of hypertrophied cells and loss of T-tubules. nNOS does not colocalize with caveolin-3 in normal myocytes, since its sarcolemmal expression is very low. However, nNOS/cav3 complexes increase in HF suggest a redistribution of nNOS from SR to plasma membrane (35, 94), which might further lessen NO-mediated modulation of RyR2 in cardiac disease. Combined to increased NO-mediated depression of sarcolemmal L-type Ca2+ channels (337), this phenomenon may contribute to Ca2+-depletion in the SR. Inhibition of SERCA2 by ROS is another crucial checkpoint of Ca2+ homeostasis. The cysteine residue near the ATP binding site of SERCA2 represents a possible target for ·OH-induced loss of function (84). In addition, ·OH may directly affect alter Ca2+ sensitivity of sarcomeric proteins, as demonstrated for skeletal muscle (268). Altogether, disruption of the constitutive ROS/RNS control of SR function is likely to contribute to the phenotype of HF.
B. ROS and the arrhythmogenic hazard in HF
Besides impairing contractility, altered Ca2+ cycling and excitation–contraction coupling triggers a cascade of events such as nonuniform mechanical dysfunction, cardiomyocyte overstretching, and cell-to-cell uncoupling, which may predispose to arrhythmias; the interlink between Ca2+ and arrhythmogenesis has been reviewed elsewhere (369). However, ROS may directly affect electrophysiological properties of cardiomyocytes by modulating relevant channels. Figure 8 shows a schematic action potential and underlying ion currents, with emphasis on those modulated by radical species. Besides Ca2+ channels, already discussed, several other ion channels and pumps may be affected.
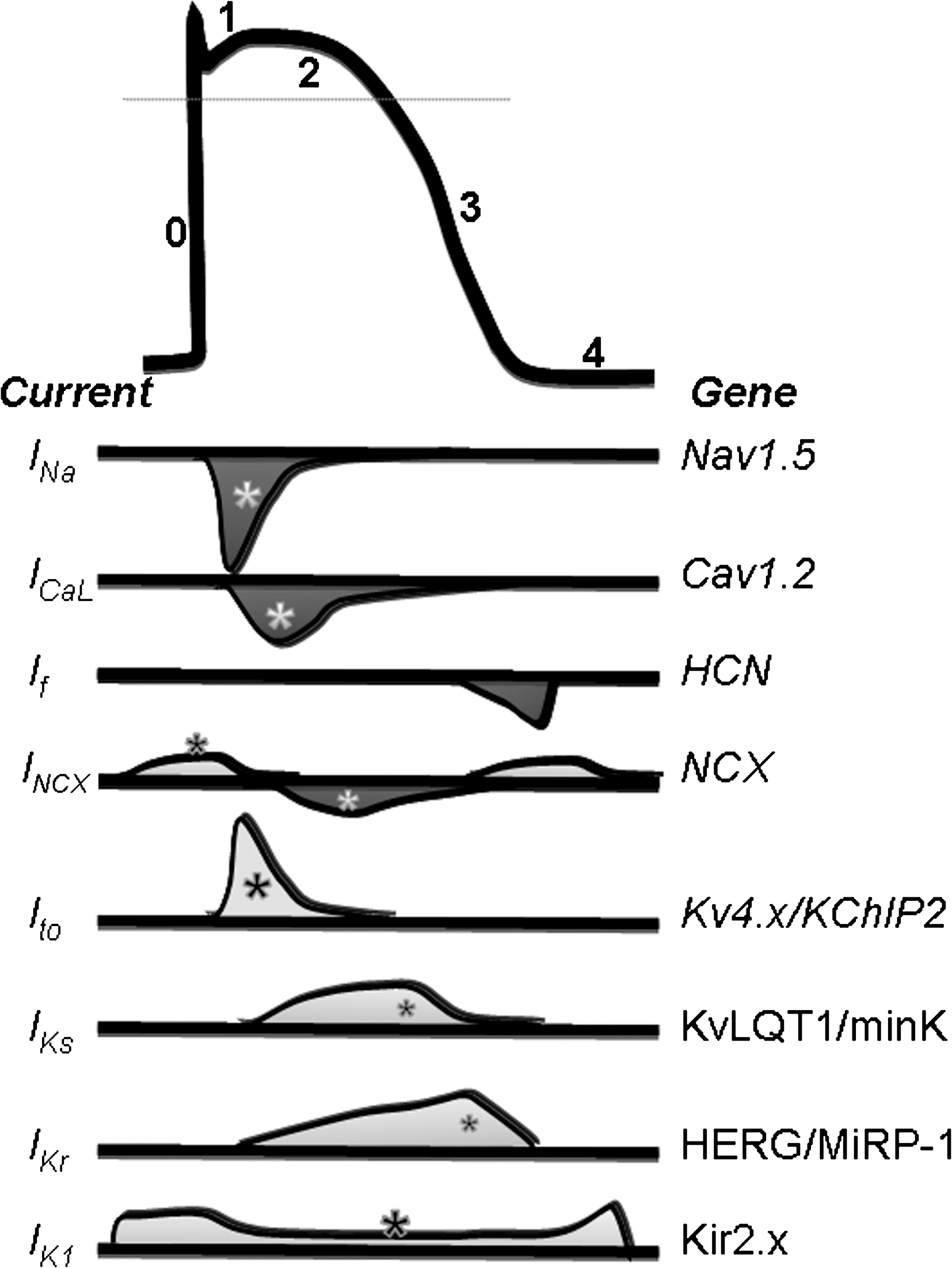
One of the first evidence of electrophysiological remodeling in human HF was the marked loss of function for the Ca2+-independent transient outward current (Ito). Channel activation/inactivation generates a fast, transient outward current that is responsible for the rapid repolarization phase of the ventricular action potential (phase 1) following the upstroke. The α- subunit is coded by a family of isoforms termed Kv1.4, Kv4.2, and Kv4.3 with different distribution among species. Downregulation of Ito expression and function has been documented in several animal models of myocardial hypertrophy and failure [see Nass et al. (274) for a review] and in diabetic cardiomyopathy (306). Examples of currents recorded in rat ventricular cardiomyocytes from control and hypertrophied or diabetic hearts are shown in Figure 9; current was restored by in-vivo treatment with AT-II receptor antagonists, thus suggesting a major role of redox-mediated mechanisms associated to RAS overactivation in this phenomenon (76, 306). A relevant transcriptional regulation has been proved, consisting in a reduction of mRNA levels and proteins coding for the fast Kv 4.2 and Kv 4.3 α subunits). On top of that, channel β subunits such as Kvβ1.2, nonenzymatic homologs of aldo-keto reductases (392), may confer redox-state sensitivity to the channel (296). Evidence of the redox control of potassium channels function comes from the pioneering work of Rozanski (318, 319). The reversible oxidative modification of proteins forming the TO-channel involves the reactivity of the free thiol group of cysteine residues, and may be viewed as a protective mechanism preventing other irreversible oxidative changes. Blunted expression (protein level) and function (current density) observed in rats with chronic myocardial infarction and diabetic cardiomyopathy were recovered by extracellular GSH or N-acetylcysteine, a putative GSH precursor (319, 405). In a rat model of impaired oxido-reductase due to inhibition of the thioredoxin/glutaredoxin systems, insulin significantly recovered Ito, an effect likely due to signaling control of intracellular redox state by the hormone (225). In general, the positive correlation between current density and protein level, more than with mRNA levels (225), suggests that redox mechanisms control sarcolemmal density of functional Ito channels at a posttranslational level, such as redox-mediated increase in the rate of channel degradation or decreased levels of chaperone β-subunits (K+ channel interacting protein 2). Notwithstanding the mechanisms, Ito reduction in HF is considered a proarrhyhmogenic mechanism that (i) destabilizes ventricular repolarization and, especially combined to chronic/acute fall of other potassium currents, predisposes to early after-depolarizations and torsade de pointes, (ii) alters the timing of early plateau phase and the kinetics of L-type Ca2+ current (25), (iii) affects crucial physiological processes such as action potential duration gradient within the ventricular wall (25) and the so-called cardiac memory (182).

Recently, redox-related modulation of the inwardly rectifying potassium channel IK1 has been demonstrated, with an unexpected high affinity of the channel for this gaseous mediator (142). Specific S-nitrosylation of Cys76 of Kir2.1, one of the proteins coding for the α-subunit of the channel, increases the opening frequency and therefore current amplitude. This feature may represent a relevant mechanism by which NO participates in the control of cardiac excitability under physiological conditions. In fact, IK1 is a key player for setting and maintaining diastolic membrane potential; therefore, any disturbance (i.e., impairment of NO signaling) would have profound effects on cardiac excitability and arrhythmogenesis.
Emerging evidence points to the sodium channel as a redox target leading to acute and chronic electrophysiological remodeling in HF. The sodium channel is responsible for the upstroke (phase 0) of the action potential; it generates a fast transient inward current (peak, INa,P) that inactivates almost completely within a few milliseconds. However, a noninactivating current persists and accounts for a net sodium influx during the plateau (phase 2).
Disruption of Nav1.5 (also known as SCN5A, sodium channel protein, type 5) inactivation increases the persistent Na+ current; well-known examples are mutations in the III–IV linker (Fig. 10) associated to the inherited LQT3 syndrome. The COOH terminal of the protein has binding sites for numerous regulatory proteins implicated in the modulation of Nav1.5 inactivation. The sodium channel is a highly modulated protein undergoing PKA and PKC phosphorylation, which causes, respectively, current gain and loss of function (419). In this respect, by activating PKC, ROS may exert an inhibitory yet indirect, activity on the channel. Recently, mutations in gene coding for glycerol-3-phosphate dehydrogenase 1–like have been shown to associate with the clinical phenotype of the Brugada Syndrome, a channelopathy usually due to mutations within the sodium channel (234). Being glycerol-3-phosphate dehydrogenase involved in NAD-dependent energy metabolism, this result was interpreted as a NADH-mediated regulation of human cardiac sodium channels (Nav1.5) whose dysregulation may predispose to arrhythmias (232). In the same line, increased intracellular NADH levels in HF may contribute to the reduced INa,p in this condition. Besides affecting the peak sodium current, redox modifications of the gating mode of the channel may lead to increased persistent or late sodium current flowing during the plateau phase (late sodium current [INa,L], Fig. 9). Increased INa,L might result from multiple reopening of a small fraction of noninactivating sodium channels (240) and is promoted by several factors such as hypoxia (184), and NO (3) H2O2 and ditiothreitol have opposite effects on peak and late sodium current, by respectively decreasing or increasing INa,p/INa,L ratio (237).

The role of INa,L in the pathogenesis of HF and associated drawbacks (arrhythmias and systolic/diastolic dysfunction) has emerged soundly in recent literature, and a comprehensive description is given elsewhere (242). Briefly, INa,L generates a net Na+ influx during the plateau phase with two major consequences: (i) unbalance between repolarizing and depolarizing currents, and (ii) altered intracellular Na+ ([Na+]i) level. The latter process is intimately linked to Ca2+ homeostasis, as increased [Na+]i negatively modulates the NCX activity (38) and causes cytoplasmic Ca2+ overload (Fig. 7). Indeed, increased INaL reportedly occurs in human ventricular cardiomyocytes from diseased explanted hearts (241) as well as in animal models of HF (380). In vitro models, such as H9c2 myocytes, showed that a 48-h exposure to AT-II increased INa,L, an effect that could be prevented by ROS scavengers or Nox inhibitors (341).
VI. Cardiomyocyte Energy Sources in the Healthy and in the Failing Heart
Cardiomyocytes need a great amount of energy to sustain their contraction, which is usually supplied by the oxidation of energy substrates, mainly fatty acids, and glucose. Increasing evidence confirms that a correct glucose/fatty acids balance is crucial for keeping the heart healthy conditions (353). Indeed, a shift in this ratio is recognized as a common determinant of multiple cardiac diseases and a hallmark of failing cardiomyocytes, which are wasteful producers of energy. In fact, in these cells some kind of shift in energy substrate choice is retained the inducer of a vicious, maladaptive cycle reducing myocardial ATP production, which dips cardiac mechanical efficiency (192, 193) and exposes to arrythmogenic hazard. However, the mechanistic link between the metabolic shift and the functional dysfunction associated with HF remains elusive (382).
Energy substrate utilization is governed by humoral factors, among which insulin plays a major role. Since the heart is highly sensitive to insulin, the most explored hypothesis is that alteration(s) of insulin signaling (a certain degree of local insulin resistance) may trigger those metabolic maladaptation found in the failing heart. This assumption is supported by several pieces of experimental and clinical evidence. First, insulin resistance is a predictor of HF (175). Second, diabetes is a strong negative prognostic factor, not only for the ischemic but also for nonischemic cardiomyopathy, and a dramatically increased prevalence of diabetes is observed in patients suffering from dilated cardiomyopathy (11). Third, cardiomyocyte fibrosis and hypertrophy are often triggered by increased ROS levels (consequence or the cause of insulin resistance (169, 316), and, finally, at least in the early phase of HF, the glycolytic flux (anaerobic) of glucose is increased. Whatever the first determinant is, unbalanced oxidative cell status is likely to lead to a crucial ethiopathogenic event in the development of the myocardial complications of insulin resistance (316). Evidence for a strong bidirectional link between ROS, cell metabolism, and functional cell features is reportedly emerging: metabolism ensures the maintenance of redox balance and, vice versa, basic metabolic pathways are thinly regulated by ROS (379). On the reverse, an altered flux of energy substrates may increase ROS levels, disrupt their compartimentalization, or reduce antioxidant species (415).
Overall, the insulin resistance hypothesis appears so compelling that someone refers to an oxidative stress and insulin resistance-based cardiomyopathy to describe nonischemic diseases associated with myocardial fibrosis and cardiomyocyte hypertrophy (397), paving the way to consider HF as a metabolic disease. However, such definition appears somehow conflicting with disappointing clinical results of the so-called metabolic drugs (including antioxidants) (see later). Indeed, as stated before, the mechanisms linking ROS/RNS pathways, insulin resistance, and cardiac disease are from being fully elucidated despite promising results in animal models with deficient cardiac antioxidant patterns (256). A similar consideration applies to the neurohumoral signal(s) that are found hyper-activated, among which sympathetic tone (adrenergic activation by desensitization of β1 cardiac receptors) and the RAS. In this respect, we have previously shown that in vivo treatment with losartan, an angiotensin type 1 receptor antagonist, leads to hand-in-hand recovery of electrophysiological properties (Fig. 9) and amelioration of insulin resistance in cardiomyocytes from type 1 diabetic rats (306). This and other studies have stated a strict link between cell metabolism and cardiac electrophysiology (408).
Cardiac metabolism in HF has been extensively reviewed elsewhere (176, 353), as well as the causal relationship between insulin resistance and ROS (315). From these studies it appears that the metabolic paradox in HF (353, 397) still represents a mystery to unravel and an enemy to be defeated as soon as possible. Here, we focus on modifications of glucose and fatty acid metabolism induced by insulin resistance and the relative impact on cell redox balance (Fig. 11).
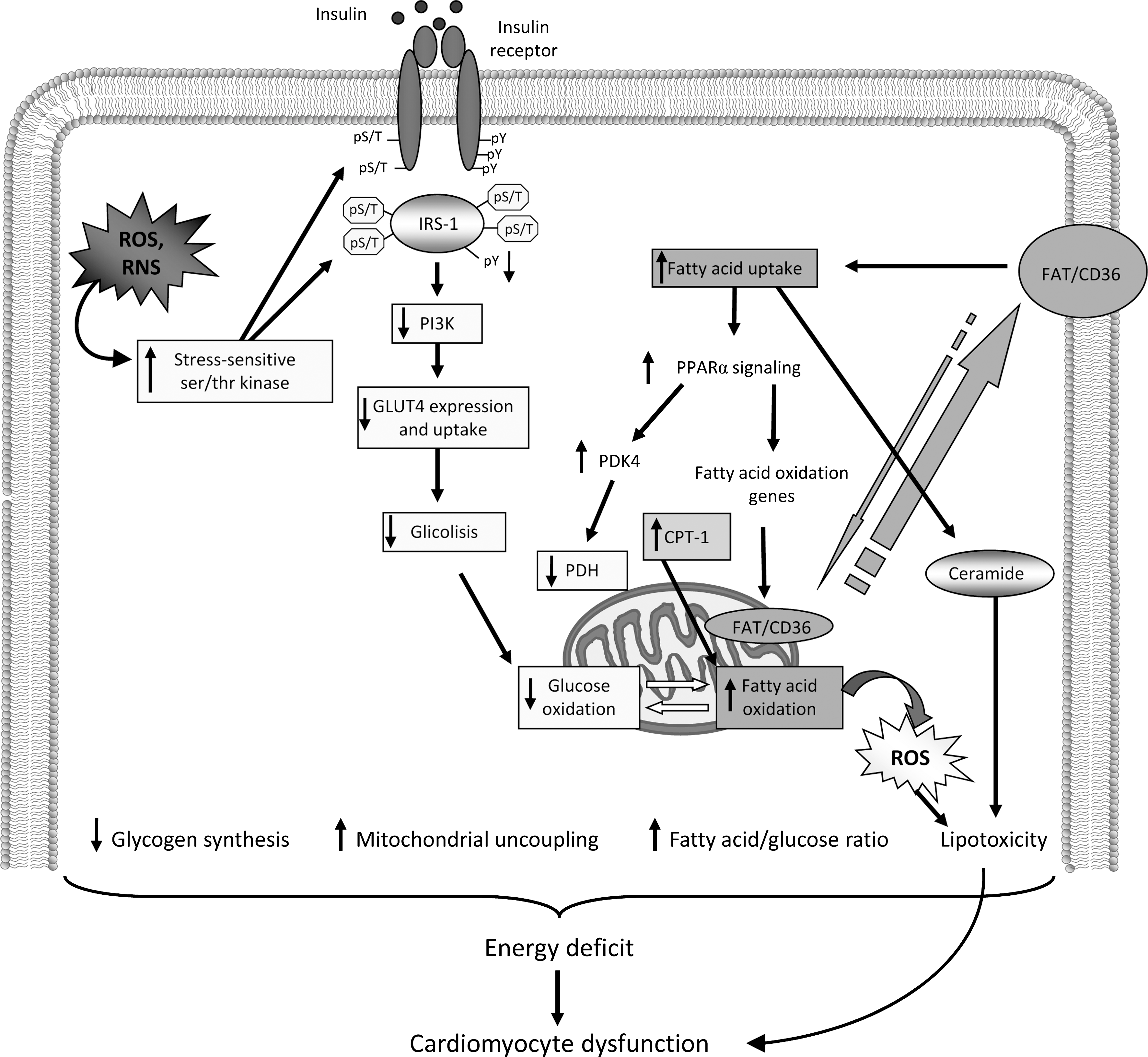
A. Energy substrate metabolism: the cytoplasmic branch
At least three main important steps can be identified in energy substrate metabolism: (i) intracellular transport, (ii) entering into mitochondria, and (iii) final oxidation. Each part of these steps cross-talks, but intracellular transport represents the limiting one.
Fatty acids are the main energy-producing substrates of the adult healthy heart. Due to their lipophylic nature, they cannot enter inside cells by simple diffusion but need to be transported. Proteins devoted to this affair constitute a family among which the fatty acid translocase (FAT/CD36) (78). FAT/CD36 is the only transporter sensitive to insulin and to contraction (236), and its transcription is stimulated by peroxisome proliferator-activated receptor α (PPARα), a key player in cardiac fatty acid metabolism (407). Both insulin and contraction, with nonexcluding mechanisms, induce FAT/CD36 translocation from an intracellular depot to the plasmalemma and its trafficking toward mitochondria, where it controls also fatty acid oxidation (77). Once having entered inside the cell and having been activated by acyl- CoA synthetase, fatty acids undergo two major fates: esterification, through glycerophosphate acyltransferase to triglycerides, or conversion by carnitine palmitoyltransferase I (CPT-I) into acylcarnitine (235). In the contracting heart, the bulk of long chain-CoA esters is taken up into the mitochondria via the carnitine shuttle and subsequently degraded in the β-oxidation pathway (353). The CPT-I represents a shuttle system for driving fatty acids from the outer to the inner membrane of mitochondria, but it does not represent the main limiting step in long chain fatty acid oxidation (46, 236, 336).
Glucose is the only substrate that can make energy without oxygen consumption; this peculiar feature makes glucose the preferential choice in conditions of reduced oxygen tension. Glucose enters the heart via two facilitative glucose transporters (GLUTs), GLUT1 and GLUT4. GLUT1 is a major mediator of basal cardiac glucose uptake and normally accounts for ∼30% of the total cardiac glucose transport in the adult heart. This contribution is higher in late fetal life and early neonatal life, whereas GLUT4 expression is typical of the adult heart which is, therefore, more insulin sensitive. Complete glucose oxidation ensures a huge amount of ATP through the glycolytic flux, which consists of a cytoplasmic (partial glucose oxidation) and a mitochondrial branch. Signaling downstream the insulin receptor is crucial to maintain glucose homeostasis.
Once activated and phosphorylated, insulin receptor stimulates a series of effectors, including the insulin receptor substrate-1 and Shc. This recruitment leads to the activation of two main pathways, the PI3K and the MAPK pathway, respectively. PI3K is a lipid kinase considered the main player of the metabolic action of insulin, whereas the MAPK pathway is involved in cell growth and differentiation. The increase in phosphoinositols at the plasma membrane induces the recruitment and colocalization of the phosphoinositide-dependent kinase 1 (PDK1), which activates protein kinase B (PKB)/Akt. Insulin is a very potent PKB/Akt activator in the heart (40). By this mechanism, insulin regulates GLUT4, translocation, and trafficking toward plasma membrane and activates the cardiac 6-phosphofructo-2-kinase isoform, the main enzyme-regulating glycolytic flux (314). Downstream to 6-phosphofructo-2-kinase is the glyceraldehyde-3-phosphate dehydrogenase activity, which catalyzes the conversion of glyceraldehyde 3-phosphate to 1,3-diphosphoglycerate, the major regulatory step in the glycolytic pathway. In fact, this enzyme is regulated by NADH/NAD+ and activity increases when NAD+ is high. Since, glycolytic enzymes are clustered near the sarcoplasmic reticulum and sarcolemma (117), it has been hypothesized that oscillations in this pathway might reflect a stress-induced need of good energy, as it occurs during acute myocardial ischemia when ATP/ADP ratio limited. Moreover, glycolytic-derived ATP would signal to mitoKATP triggering their opening (408), which is a typical survival mechanism against ischemic injuries. From this, it is not just a speculation the importance of maintaining a correct glycolytic flux to protect the heart against ischemia.
Once glycolysis has produced pyruvate, this substrate has to enter mitochondria, where it is phosphorylated by the pyruvate kinase, whose activity depends on NADH+ and acetyl-CoA coming from fatty acid β-oxidation. The activity of pyruvate dehydrogenase is regulated by a kinase whose phosphorylation reduces kinase activation. Experimental evidence suggests that activation of pyruvate dehydrogenase kinase is a cause of RV dysfunction and that reduction of such enzyme activity not only restores RV function but also enhances glucose oxidation (300). Insulin inhibits the pyruvate kinase activity. The relative pyruvate dehydrogenase/kinase activity can either determine or reflect fuel preference (carbohydrate vs. fat), occupies a pivotal position in fuel cross-talk permitting glucose oxidation, and allows the formation of mitochondrial intermediates (e.g., malonyl-CoA and citrate) that reflects fuel abundance. Fatty acid oxidation suppresses pyruvate dehydrogenase activity stimulating the pyruvate dehydrogenase kinases, which are regulated differentially by metabolite effectors with an NADH-dependent mechanism. By maintaining acyl-CoA removal by β-oxidation, upregulation of pyruvate dehydrogenase kinase facilitates the continuous uptake of long-chain fatty acyl-CoA into the mitochondria for oxidation, preventing accumulation in the cytoplasm, where it would exert deleterious effects on cardiac function (359).
In conclusion, any activator of fatty acid flux toward correct β-oxidation keeps the pyruvate dehydrogenase/kinase in homeostatic conditions. In this respect, a pivotal role might be played by PPARα (400) (Fig. 11).
FAT/CD36 is the only fatty acid transporter sensitive to insulin and to contraction (236) and its transcription is stimulated by PPARα, a key player in cardiac fatty acid metabolism (407). Both insulin and contraction, with not excluding mechanisms, induce FAT/CD36 translocation from an intracellular depot to the plasmalemma and its trafficking toward mitochondria, where it controls also fatty acid oxidation (77). In myocytes, contraction induces trafficking of vesicles containing GLUT4 or FAT/CD36 with a PI3/Akt-independent mechanism involving the activation of the AMP-activated protein kinase (AMPK) (150), retards GLUT4 endocytosis, and increases calcium, which activates the CAMK. This latter mechanism is a rapid response to energy stresses and a key reaction to stimuli requiring rapid energy supply (200). Events downstream of AMPK and CAMK lead to activation of various transcription factors. These include the nuclear respiratory factors 1 and 2, which promote expression of proteins of the respiratory chain; the metabolic regulator peroxisome proliferator-activated receptor-γ coactivator (PGC) PPAR-α, which upregulates the levels of enzymes of β oxidation; the mitochondrial transcription factor A, which activates expression of the mitochondrial genome; and the myocyte-enhancing factor 2A, the transcription factor that regulates GLUT4 expression (285). Activation of AMPK has also been shown to increase the phosphorylation and activity of endothelial NOS (eNOS) (171).
AMPK-dependent activation of GLUT4 appears to acts in synergism or in alternative to insulin, a finding of practical utility in type 2 diabetes, where physical exercise results as a potent stimulus counteracting insulin resistance. General strategies aimed at activating AMPK represent a promising approach for the treatment of HF being an energy-deficient state that gives rise to contractile dysfunction. Recent studies using metformin support this hypothesis (146) (see section VII).
B. Energy substrate metabolism: the mitochondrial branch
Mitochondrial metabolism of fatty acids accounts for approximately 60%–90% of total energy production (in the form of ATP), with carbohydrates contributing to the remaining 10%–40%. However, fatty acid metabolism requires approximately 10% more oxygen than glycolysis to produce an equivalent amount of ATP than glycolysis (59, 352). Despite its obvious benefits, β-oxidation has several drawbacks: (i) fatty acid oxidation requires more oxygen for producing energy than glycolysis; (ii) fatty acids uncouple the respiratory cycle increasing mitochondrial uncoupling proteins (UCP), making the respiratory cycle more ineffective (i.e., requiring more and more oxygen); and (iii) fatty acids can damage the plasma membrane and disturb ion channel behavior, thus favoring arrhythmogenesis (353). The NADH/NAD and NADPH/NADP from glycolysis and pentose phosphate shunt regulate the mitochondrial transmembrane potential (87). This looks of particular importance in HF, where oxygen supply is often limited and uncoupling protein, among which the cardio-specific UCP3 (126), levels change and fatty acid oxidation increases (80, 269).
Mitochondria present specific and autonomous DNA that allows self-generation biogenesis of proteins, a mechanism regulated by the function of DNA-binding transcriptional regulators often mediated by coactivators, among which the PPAR-γ coactivator-1α (PGC-1α) (220, 229, 401), which plays a pivotal role in fatty acid metabolism (activating PPAR-α, thus exerting a control on enzymes of fatty acid oxidation). Mitochondrial biogenesis and respiration are stimulated by PGC-1α through powerful induction of nuclear respiratory factor-1 and 2 gene expression. Moreover, PGC-1α increases nuclear respiratory factor transcriptional activities on the promoter of mitochondrial DNA maintenance factor, thus synchronizing activities of mitochondrial and nuclear genomes. Hearts of mice with cardiomyocyte-specific overexpression of PGC-1 α (myosin heavy chain-PGC-1 α) exhibit a marked increase in cardiomyocyte mitochondrial biogenesis and, ultimately, develop HF (219).
Recently, our knowledge of the control of some transcription factors has been further improved by the discovery of a novel system controlling their activation/deactivation (323). This system includes sirtuin1 (SIRT1), whose low SIRT1 mRNA expression in insulin-sensitive tissues reflects impaired regulation of mitochondrial function associated with insulin resistance in humans (323).
In HF, there is a downregulation of genes controlling mitochondrial biogenesis (257, 338) and of enzymes involved in fatty acid oxidation (136, 219). In our recent study (338) we documented that in different cardiomyopathies there is a specific metabolic gene expression profile caused by a different derangement of the mitochondrial energy production pathway and that in mitochondrial cardiomyopathy the metabolic gene expression profile is similar to that developing in diabetic cardiomyopathy (55, 127, 338). In this condition, increased circulating free fatty acids may lead to activation of a PPARα/PGC-1α complex, which in turn causes induction of mitochondrial biogenesis and fatty acid oxidation. Accordingly, we observed in mitochondrial cardiomyopathy, the induction of genes involved in fatty acid metabolism, glucose transport, and mitochondrial biogenesis. The latter change was consistent with a dramatic increase in mtDNA content per cell, as well as with histologic and ultrastructural features of marked mitochondrial proliferation. In contrast, downregulation of the PPARα/PGC-1α complex and its targets, as well as reduced expression of glucose transporters, was observed failing hearts with dilatative and ischemic cardiomyopathy, confirming previous observations (27, 309) (Fig. 12).
In the same study, we reported that in dilated and ischemic failing hearts an increased myocardial ROS production, mainly due to an increase of NADPH oxidase activity, was associated to a decrease of UCP expression. On the contrary, in mitochondrial cardiomyopathy, sharing similarities with diabetic hearts, a more marked increase in ROS production was observed compared to that found in cardiomyopathies with different etiology. Thus, in addition to NADPH oxidase, mitochondrial dysfunction may contribute to the increased oxidative stress both by mitochondrial-derived O2 − and by a reduction of ATP/AMP (adenosine monophosphate) ratio, which in turn activates the XO pathways. In mitochondrial cardiomyopathy, a high level of ROS was associated to increased expression of UCP2 and UCP3 that may lead to increased mitochondrial oxygen consumption, thus amplifying energy dysfunction. Moreover, overexpression of UCP in mitochondrial cardiomyopathy is insufficient to decrease ROS production through the mechanism of uncoupling respiration, and therefore high levels of ROS are maintained (338).
In summary, complete fatty acid oxidation supplies the acyl-CoA that enters in the citric acid cycle, thus linking fatty acid oxidation (β-oxidation) to complete glucose oxidation. A correct fatty acid oxidation throughout the respiratory chain supplies the intermediary products FADH2 and NADH linking electrons to CoQ and cytochrome-b producing ATP (Fig. 13).

C. Insulin resistance impacts glucose and fatty acid metabolism
Insulin signal dysregulation affects glucose and fatty acid metabolism deeply. It remains to be addressed whether insulin is the primer or the consequence of the shift in the energy substrate metabolism occurring in HF. In fact, on one hand, insulin resistance can be induced by a well-known pattern of inflammatory neurohumoral factors (409); on the other, by substrate excess as an increased availability of cardiac fatty acids (298), a condition that also induces the typical electrophysiological alterations observed in diabetic cardiomyocytes (153).
Insulin resistance is commonly defined as the inability of the hormone to activate its receptor-dependent cascade, which governs glucose and fatty acids intracellular metabolism in insulin-sensitive tissues. In this context chronic and/or increased production of ROS/RNS or a reduced capacity for elimination can cause the activation of multiple stress-sensitive serine/threonine kinase signaling cascades, which led to dysregulation in intracellular signaling, ultimately resulting in insulin resistance (121). The insulin signaling pathway offers a number of potential targets (substrates) of these activated kinases, including the insulin receptor and the family of insulin receptor subsstrate (IRS) proteins. As illustrated in Figure 11 activation of multiple stress-sensitive serine/threonine kinase signaling cascades, such as JNK/stress-activated protein kinase (SAPK) activity, increases the level of serine/threonine phosphorylation of the insulin receptor and IRS-1/2 and decreases the extent of tyrosine phosphorylation, resulting in the attenuation of insulin action. The serine/threonine phosphorylated forms of IRS molecules are less able to associate with the insulin receptor and downstream target molecules, especially PI 3-kinase, resulting in impaired insulin action, including PKB activation and glucose transport. More in general, the interruption of the main axes of the insulin receptor signals at the IRS-1-PI3K/Akt reducing GLUT4/GLUT1 and FAT/CD36 trafficking from endosomes to plasmalemma, producing a net but dynamic intracellular retention of GLUT4 and stabilization at plasmalemma of FAT/CD36. These modifications are responsible, at least in part, for the glucose and fatty acid metabolism impairment found in experimental (345) and human diabetes (298). On the reverse, the relationship between GLUT-mediated glucose transporters and cardiac phenotype is stressed further by results obtained in mice with cardiac knockout for GLUT4 or overexpressing GLUT1. While GLUT4 null mice exhibit cardiac hypertrophy and failure and show higher mortality (194) than wild-type mice, overexpression of GLUT1, enhancing glucose uptake and oxidation, in insulin-independent manner, reduces cardiac metabolic flexibility and makes the heart susceptible to contractile dysfunction. In particular, overexpression of GLUT1 does not allow upregulation of myocardial fatty acid oxidation when mice were fed a high-fat diet, resulting in increased oxidative stress, activation of p38 mitogen-activated protein kinase, and, more importantly, contractile dysfunction (Yan et al., 2009). It is well known that, in humans, insulin resistance at GLUT4 is associated with cardiac hypertrophy (295).
In HF there is a reexpression of fetal phenotype that includes overexpression of GLUT1 (309). A pivotal role in reexpression of GLUT1 might derive from altered expression/function of PPARα (27, 412). GLUT1 trafficking from recycling endosomes is increased by contraction and, to a lesser extent, by insulin (395). Experimental evidence confirms a role of GLUT1 as the bench warmer since its expression/activity increases when GLUT4 activity is reduced (194). Moreover, enhanced expression of GLUT1 has been associated with failure of postinfarcted rat hearts (354).
When insulin resistance emerges, glycolytic anaerobic flux interrupts at the cytoplasmic branch with accumulation of intermediary products and consequent activation of extra glycolytic pathways (i.e., polyols, pentose phosphates, and aldose reductase) (49) leading to modification of the lactate/pyruvate ratio (a measure of NADH/NAD+) and consequent reduction of protection against ischemic injuries (172).
Recent evidence suggests that activation of alternative glucose pathways also occurs in ischemic cardiomyopathy (Fig. 14). In particular, in the failing left ventricle of patients with ischemic cardiomyopathy, the activity of glucose-6-phosphate dehydrogenase was found higher than in normal donor hearts, thus enhancing NADPH levels, NADPH oxidase activity, and oxidative stress (147). Accordingly, inhibition of glucose-6-phosphate dehydrogenase activity might represent a further target of the metabolic therapy of HF (147). Another NADPH-dependent alternative pathway for glucose-6P oxidation is represented by the polyol pathway regulated by the activity of aldose reductase, which diverges glycolysis from to glyceraldehyde-3-phosphate transformation. Aldose reductase is a multifunctional enzyme with a broad substrate specificity, which overlaps with that of ubiquitous, structurally related aldehyde reductase, transforming aldehydes to the corresponding alcohol. Aldose reductase can lower intracellular NADPH levels, when it reduces glucose to sorbitol while increasing NADH (when sorbitol is oxidized to fructose). One of the main consequences of these changes is the depletion of key intracellular antioxidants (such as GSH), a finding that may expose cells to oxidative and nitrosative injury (92). The production of sorbitol from glucose increases osmotic stress, which has been implicated in the pathophysiology of diabetic complications, including cardiac disease. In this line, pharmacological inhibition of the aldose reductase exerts significant benefit against the development of various diabetic complications in animal models (10, 283). Instead, in virtue of its antioxidant features, from scavenging toxic aldehydes such as hydroxynonenale, aldose reductase activities considered a key protective component of late preconditioning against ischemia in nondiabetic rodents (343). Hence, a different role for aldose reductase activity is envisaged in normo- and hyperglycemic conditions.

By accumulating upstream glycolytic products, insulin resistance reduces glycolytic flux of glucose toward its mitochondrial oxidation (Fig. 11), thus impacting on cardiomyocytes oxidative/antioxidant status substrate phosphorylative oxidation other than mitochondrial biogenesis.
As mentioned above, the mitochondrial pyruvate dehydrogenase complex catalyzing the oxidative decarboxylation of pyruvate links glycolysis to the tricarboxylic acid cycle and mitochondrial ATP production. Adequate flux through the complex is important in tissues with a high ATP requirement as the heart, in lipogenic tissues (since it provides cytosolic acetyl-CoA for fatty acid synthesis), and in generating cytosolic malonyl-CoA, a potent inhibitor of CPT-I. Conversely, suppression of pyruvate dehydrogenase complex activity is crucial for glucose conservation when glucose is scarce. Insulin resistance upregulates PDK activity (360) in the heart. This defect in pyruvate metabolism triggers mitochondrial dysfunction by multiple mechanisms.
Boudina et al. (50) have recently demonstrated that a defect of cardiac insulin signal results in reduction of the pyruvate dehydrogenase activity, in stimulation of fatty acid, and in an increase of O2
Moreover, at mitochondria, insulin resistance impairs intracellular Ca2+ homeostasis (398) and belates the activation of protective mechanisms against ischemia-induced arrhythmogenesis (177), increasing heart susceptibility to ischemia (408). As mentioned above, modification of SERCA function is an early, crucial factor leading to diastolic dysfunction in type 1 and type 2 diabetic patients (164, 410).
A defective insulin signal greatly impacts on FAT/CD36 trafficking, by stabilizing the transporter at plasma membrane level and diverting it from mitochondria. We have previously demonstrated that exposure of immortalized cardiomyocytes, HL-1 cells, to AT-II increases oxidative stress and raises insulin resistance at glucose but also at fatty acids, by stabilizing FAT/CD36 at plasma membrane (8). Either oxidative stress or insulin resistance may be ameliorated by cell pretreatment with type 1 AT-II receptor antagonists. These results seem to confirm that FAT/CD36 is likely a target of AT-II-induced oxidative stress. An increased fatty acid uptake following insulin resistance at FAT/CD36 joined to their inefficient detoxification triggers the so-called lipotoxic effect (78), which provides the link between insulin resistance and loss of mitochondrial function (298). The pathophysiology of insulin resistance is, at least in part, related to the inability of mitochondria to oxidize fatty acids.
When long chain fatty acid oxidation increases, electron leakage from mitochondria also increases, favoring ROS production. Moreover, while a correct short, medium, and long chain fatty acid acyl-CoA dehydrogenase activity supplies mitochondria of reduced nucleotides (FADH2 and NADH, Fig. 12), their levels decrease in insulin resistant mitochondria, opening the way to ROS-induced mitochondrial dysfunction (279). Evidence demonstrates that UCP3 expression and activity are downregulated in hypertrophied and failing hearts (136, 219) but also in type 2 diabetic patients, suggesting that in the insulin-resistant heart, mitochondria are more exposed to ROS attack (80).
In conclusion, defects of the coordinated regulation of all metabolic pathways or of several pathways simultaneously could ultimately contribute in a meaningful way to the development of metabolic abnormalities, a base on which insulin resistant cardiomyopathy may develop with its pro-oxidant ground. Nevertheless, despite the aforementioned association between high fatty acid oxidation and cardiac diseases, interventions to sustain myocardial glucose oxidation and/or to prevent high fatty acid oxidation did not fulfill all the expectations in HF patients.
VII. Therapeutic Opportunities for HF
Prolonged activation of the sympathetic nervous system and of RAS axis, which initially compensates for depressed myocardial function, contributes to sodium and fluid retention, increased preload, and afterload damaging further myocardium. Improved understanding of this multifaceted pathophysiology has driven the development of treatment modalities, such as beta-blockers, angiotensin converting enzyme inhibitors, AT1, and diuretics, which are now mainstays of HF therapy. These strategies ameliorate HF symptoms and counteract metabolic and functional remodeling and, in turn, oxidative cell status. However, even if unbalanced oxidant/antioxidant is considered a crucial negative determinant for ventricular dysfunction, drug therapies based on the use of true antioxidants have, so far, produced disappointing results (120, 138, 372). Several causes might be involved in such failure, such as intrinsic features of many antioxidants, including diet-derived polyphenols (i.e., the low bioavailability and the little predictable pharmacokinetic) (208), which are crucial points to overt to customize antioxidants for HF.
Moreover, even if clinical evidence appears conclusive in some cases, antioxidant treatments should be reviewed in light of a personalized, redox-tailored therapy.
In this respect, it would be essential to assess, for example, the extent of the antioxidant reserves in plasma as well as of oxidative/nitrosative stress level of each patient. The availability of suitable biomarkers for risk stratification might help to (i) choose eligible HF patients, (ii) select appropriate drug therapy, and (iii) indicate the vulnerable time window for effective treatment.
In addition to pharmacological approaches, cell-based strategies are rapidly evolving and several aspects would deserve attention in the future. It is still uncertain, for example, how inflammatory and oxidative processes affect the self-regenerative potential of the cardiac muscle; to what extent the paracrine beneficial effects of resident or autologous stem cells are altered by the imbalanced redox environment; and whether current therapies beneficially or detrimentally influence cardiac regenerative strategies (83, 213). On-going preclinical and medium-sized clinical studies could help to answer some of these questions (86).
While new scenarios for HF treatments are opening, as compendium of the topic of this review we selected a limited choice of drugs currently used in clinical practice, focusing on their impact on ROS/RNS. In fact, even if many classes of drugs have proved as effective against HF in experimental animal models, very often they failed in clinical trials, suggesting that the translation from laboratory to bedside remains a difficult task.
The purpose of the first part of this section is to highlight the impact of standard HF therapies on ROS/RNS. In the second part, we speculate briefly on possible future therapies, which could found indication for HF.
A. The pharmacological management of HF includes indirect antioxidants
Conventional medical therapy for HF has been outlined extensively and several classes of agents are used, among which diuretics play a fundamental role in the relief of volume overload symptoms besides positively impacting the survival of patients with HF (301). Therefore, tolerance to loop diuretics may develop with a concomitant loss of function (308), making diuretic dosage and maintenance critical for HF control and management.
Even if some data sustain a certain degree of antioxidant capacity for furosemide (346), no clinica data, up to now, encourage the identification of furosemide antioxidant features as a part of its mechanism of action in controlling HF symptoms.
1. Angiotensin-converting enzyme inhibitors
Angiotensin-converting enzyme inhibitors (ACEi) represent the most beneficial, common, and recommended therapy for HF since their use is associated with reduced renal disease risk, and cardiovascular mortality and morbidity in various at-risk patient populations (1). Several studies have definitively demonstrated that the use of ACEi exerts beneficial effects on the cardiovascular system, which goes beyond the control of high systolic pressure. In fact, ACEi, by reducing AT-II production (local and systemic) opposite to cardiac hypertrophic remodeling, reduces catecholamine overload, thus exerting protective effects on cardiomyocyte metabolism and, directly and indirectly, on their redox state (Fig. 15). In fact, animal studies suggest that most of the deleterious effect of AT-II, including cardiomyocyte apoptosis (128), are ROS mediated (28). Moreover, the beneficial effects of ACEi on cardiomyocyte metabolism include the increase of insulin sensibility by several mechanisms: (i) the reduction of local AT-II production promotes FAT/CD36 recycling from plasma membrane and decreases AT-II-dependent activation of NADPHox and (ii) the increase of bradykinin tissue levels exerts positive effects on GLUT4 activity (302). For all these reasons ACEi are the drugs of choice in the prevention of target-organ damage (for example, cardiomyopathy and nephropathy) associated with diabetes, in reduction in the risk of new onset diabetes (243), the prevention of coronary artery disease in risk patients (The PEACE Trial Investigators, 2004), and in preventing the progression of coronary artery disease toward failure (The ONTARGET investigators, 2008).
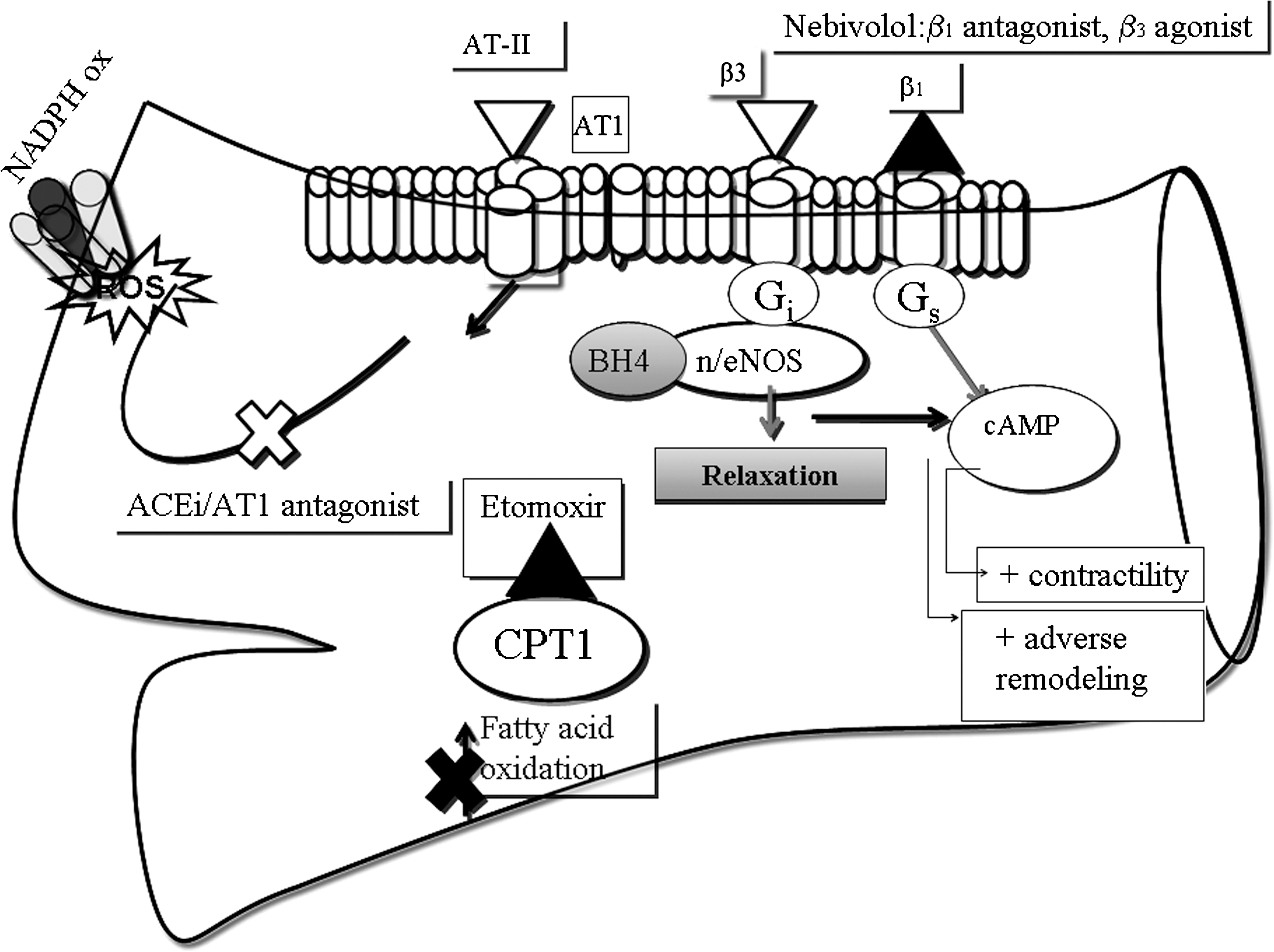
However, the developing resistance to ACEi or the occurrence of typical adverse effects is increasingly recognized as an emerging clinical problem that reduces their effectiveness. In such a case the use of AT1 antagonists or renin inhibitors (aliskiren) may represent a suitable alternative.
2. Angiotensin type 1 receptor antagonists: losartan
AT1 antagonists block AT-I type 1 receptor, the main receptor involved in AT-II-dependent negative cardiovascular effects. Similarly to ACEi, they reduce mortality in patients with arterial hypertension, chronic congestive HF, and acute myocardial infarction. In fact, clinical indications of AT1 antagonists mostly overlap with those of ACEi but differ in their collateral effects. Due to their chemical structure, AT1 antagonists also posses same PPARγ-stimulating activity (335). There is evidence suggesting that this feature might participate in their metabolic effects, including reduction of new onset of diabetes in risk patients and amelioration of insulin resistance (298).
Losartan is a second-generation AT1 antagonist presenting an interesting pharmacokinetic profile. Losartan is activated by first-passage metabolism in the liver into two active metabolites of which one is the AT1 antagonist (EXP3174). The other (EXP3179) is probably responsible from several ancillary effects attributed to losartan: such as the activation of eNOS, the inhibition of cyclooxygenase (thus, reducing formation of thromboxane A2), and, more importantly, in this setting, the negative modulation of NADPH oxidase activity by blocking PKC. Altogether, these properties may be indicative of peculiar antioxidant features of the drug (132) and account—at least partly—for its effectiveness against cardiac structural, electrophysiological, and metabolic remodeling observed in experimental models (8, 76, 306). Indeed, as extensively described in the previous sections, PKC is a hallmark of cardiomyocyte response to stress stimuli and an obligatory crossroad for several pathways linked to insulin resistance, hypertrophy, and transcriptional and posttranslational modulation of ion channels and pumps. However, to date, no clear evidence for losartan effects beyond AT-II receptor antagonism emerges from clinical trials carried out in patients with hypertension, HF, myocardial infarction, or diabetes mellitus (335, 393).
According to a recent study, losartan shows a clear dose-dependent effectiveness on clinical outcomes in patients with HF (206).
3. β-blockers: waiting for the third generation
Chronic overactivation of the orthosympathetic system in HF, though initially adaptive, is generally considered to be deleterious in the long term so that beneficial effects of adrenergic blockade in failing patients are strongly supported by clinical evidence. Most, if not all, the negative effects of sympathetic stimulation are mediated by β 1-adrenergic receptors, whereas stimulation of β 2 may have potentially beneficial effects, including inhibition of apoptosis and ventricle remodeling. β-Blockers are a large and heterogeneous class of drugs with different profile of agonism/antagonism at β 1 and β 2 receptors, even if selectivity, being dose dependent, decreases or disappears when used at higher posology.
β-Blockers of first and second generation (no vasodilating), although effective in lowering blood pressure and reducing cardiovascular morbidity and mortality in several conditions (including postmyocardial infarction and HF), have a major drawback: they exert negative metabolic effects, including reduced glycemic control, masking of hypoglycemia, worsened insulin resistance, and dyslipidemia.
Carvedilol and nebivolol belong to the third generation of β-blockers and possess direct vasodilator properties in addition to their adrenergic blocking characteristics. Nebivolol in particular is lipophylic and consists of a racemic mixture of D- and L-enantiomers. It has the highest β1 receptor affinity among β-blockers. The second major mechanism of action of nebivolol is vasodilatation, which, differently from carvedilol and labetalol, is not mediated by α1-adrenergic blockade, rather resides in its ability to stimulate the NO pathway (188).
This dual mechanism of action underlies many of the hemodynamic properties of nebivolol, which include reduction in heart rate and blood pressure, improvements in systolic and diastolic function, and a substantial amelioration of endothelial dysfunction joined to antioxidative properties. In addition, nebivolol has shown a better tolerability profile, particularly with respect to events commonly associated with β -blockers, such as fatigue and sexual dysfunction.
Nebivolol-dependent vasodilatation seems to be the result of activation of β 3 adrenergic receptors. In the peripheral vessels, β 3-adrenergic receptors are expressed in the endothelial cells where they stimulate eNOS with increased NO release (Fig. 15). Moreover, β 3 are also expressed in cardiomyocytes where they mediate negative inotropic effects by activating a G-α1 coupled with NOS (22) and modulate ion channel activation by interfering with cyclic nucleotide-mediated pathways (328). Cardiac NO production may have a profound impact on cardiac function through both vascular-dependent and vascular-independent effects. Vascular-dependent mechanisms include regulation of coronary vessel tone, thrombogenicity, proliferative and inflammatory processes, and angiogenesis. Direct effects of NO on myocardial contractility and diastolic function are mediated by changes in the excitation–contraction coupling (inhibition of the inotropic effects of α1- and α2-adrenergic receptor stimulation, modulation of calcium uptake, and release from SR; see above), presynaptic and postsynaptic modulation of autonomic signaling, and changes in mitochondrial respiration (73, 247).
β 3 Adrenoceptor mRNA expression and/or function was found to be increased in the ventricular myocardium in failing (dilatated and ischemic cardiomyopathy) cardiomyocytes (320) as well as in animal model of HF (328) and diabetes (107, 265). These observations and the characterization of the pharmacodynamic features of nebivolol led to propose this drug as a new and effective treatment of HF in senior patients (at least 70 years old) without preserved ejection fraction (whose survival may be as poor as in patients with a low ejection fraction) (384). However, according to the European drug agency note, recent clinical trials have not provided enough convincing evidence yet to implement current guidelines; additional work should warrant noninferiority of nebivolol not only with respect to placebo, but also versus other beta blockers. The lack of definitive proof has represented a strong limitation in the approval of nebivolol in HF, besides in hypertension, in Europe.
4. Nitrates: better not just alone but hybrid nitrates
Organic nitrates, among which nitroglycerine, are strong vasodilatator molecules effective for the acute treatment of stable-effort, mixed, and unstable angina acute myocardial infarction. These classes of drugs may be added to standard HF therapy even if their benefit should be reevaluated by dedicated clinical trials. Nitrates are potent relaxants of vascular smooth muscle cells in systemic veins but also cause arterial dilatation. This important hemodynamic feature reduces venous refill and decreases afterload of the heart, on which it might have direct protective effects too. Vasodilatation is the result of local increase of NO following nitrate bioconversion (90). Unfortunately, clinical effectiveness of nitrates decreases after long-term use due to the onset of tolerance to hemodynamic effects so that their loss of function remains a strong clinical concern and is suspected to be responsible for the increased cardiovascular mortality associated to long-term treatment of postinfarcted patients (189). Even if definitive identification of the mechanisms of nitrate tolerance remains to be addressed, one of the most explored hypothesis is the so-called free radical hypothesis. According to such a theory, nitrates would induce self-tolerance, extended to NO-donor drugs, activating a vicious cycle involving ROS/RNS. In particular, three mechanisms would be involved: (i) nitrate-dependent activation of a specific PKC that triggers intracellular events, leading to eNOS uncoupling, activation of vascular NADPH oxidase, thus increasing O2_production, and, in turn, peroxynitrite formation; (ii) oxidation-mediated inhibition of aldehyde dehydrogenase-2 (ALDH-2), an enzyme involved in scavenging toxic aldehydes and products of lipid metabolism; (iii) activation of COX2. Increased vascular peroxynitrite formation may affect the proper function of eNOS and thus induces endothelial dysfunction oxidizing the eNOS cofactor BH4 to dihydrobiopterin via intermediate formation of trihydrobiopterin radicals. Provided that dihydrobiopterine reductase activity is not sufficiently high, the resulting intracellular BH4 deficiency may lead to dissociation of the dimeric functional eNOS or mislead electron flow to molecular oxygen, resulting, again, in O2 − formation. ALDH-2 is among the enzymes involved in nitrates bioactivation (163) and its levels are reduced in nitrate-tolerant patients. Moreover, peroxynitrate negatively influences the NO/cGMP signaling and reduces prostacycline formation. While low levels of nitrates are activated by ALDH-2, at high levels they inhibit ALDH-2, through oxidation of the cofactor of this enzyme resulting in toxic accumulation of aldehydes and isoprostanes, which are sensitive markers of lipid peroxidation (185). In turn, inhibition of ALDH-2 also produces accumulation of nitrate in mitochondria, an event that uncouples oxidative phosphorylation generating more ROS and mitochondrial dysfunction, amplifying an ROS/RNS-mediated deleterious mechanism. Irrespective of the precise sequence of events, antioxidants may have new therapeutic implications in counteracting nitrate tolerance. The Veterans Heart Failure Trials (V-HeFT) have stated the beneficial effects on LV function, exercise capacity, and most notably on survival in a large patient population with severe HF of a combination of hydralazine and isosorbide dinitrate (368).
Hydralazine, an old vasodilator drug, has recently been reconsidered new wave in virtue of its potent antioxidant features. Hydralazine reduces O2 •− generation by XO and NADPH oxidase (267), and blocks iNOS and COX-2 gene expression (221). Moreover, since it also stimulates the transcription of proangiogenic genes, hydralazine effect may be of great beneficial in ischemic heart disease (203). For all these features, the combination nitrates/hydralzine may represent the prototype of antioxidant hybrid drugs.
B. Metabolically active antioxidants: from a failure new successes?
1. Inhibitors of fatty acid oxidation: beyond etomoxir
As discussed in section VI, CPT-1 plays a regulatory role in controlling trafficking of fatty acids from cytoplasm to mitochondrial final metabolism. When fatty acid load increases, β-oxidation also accelerates, causing an excess of electron flux in the mitochondrial respiratory chain, which, as already stated, results in increased ROS generation (165). The latter is among a major contributor to JNK activation, which plays a culprit role in raising insulin resistance. In addition, cardiac metabolism exclusively based on fatty acid oxidation is dangerous being associated, as in diabetes, with important changes in calcium homeostasis and impaired systolic and diastolic function (388). In the healthy heart, UCP3 protects mitochondria against lipid-induced oxidative damage by facilitating the transport of nonmetabolized fatty acids and lipid peroxides from the mitochondrial matrix. Patients with HF, with or without type 2 diabetes, have reduced UCP3 content (333). Therefore, these patients are indeed less protected against lipid-induced damage, which ultimately led to mitochondrial damage at lower drug dosages.
Then, a reduction of fatty acid flux toward mitochondria might theoretically ameliorate insulin-resistance forcing cardiomyocytes to use more glucose. Among drugs that might show beneficial effects, when myocardial functions and glucose metabolism abnormalities coexist, are inhibitors of fatty acid oxidation, among which etomoxir (an inhibitor of CPT-1), trimetazidine (specifically a 3-ketoacyl coenzyme A thiolase inhibitor), and ranolazine (a partial inhibitor of fatty acid oxidation and of the late sodium current). Etomoxir is an irreversible and stereospecific inhibitor of mitochondrial CPT-1, originally developed for the treatment of diabetes mellitus (399) to switch energy metabolism from fatty acids to glucose oxidation (Fig. 15). Since etomoxir was found to influence gene expression of SERCA2 and myosin isoenzymes (322) and to induce an upregulation of UCP3 in human muscle (333), its use was thought it might have some usefulness in counteracting HF-associated malfunctions (321, 413). Experimental evidence demonstrates that etomoxir did reduce long chain fatty acid flux throughout mitochondria exerting protection against raise of ROS and that it ameliorates insulin resistance (273). A first small clinical trial assaying etomoxir safety in HF patients for a 3-month period (80 mg/day, oral application) was performed by Schweda and Holubarsch (332) and subsequently (40 and 80 mg/daily) in a placebo-controlled trial in 2007 (167). Unfortunately, those studies were interrupted prematurely, because unacceptably high liver transaminase levels were detected in four patients taking etomoxir, thus revealing that the risk-to-benefit ratio for the individuals did not justify their continuation. Etomoxir hepatic toxicity is likely based on the promotion of mitochondrial dysfunction, essentially mitochondrial enlargement, which results in a decrease of ATP and GSH levels producing overall decline of cell energy and increase of oxidative stress. Concurrent with mitochondrial dysfunction is an increased apoptosis, evident by increased caspase activity, and the activation of a tissue inflammatory response (386). The etomoxir lesson teaches us that its mechanism of action likely goes beyond the reduction of fatty acid oxidation and toxicities are often multifaceted, involving several cell types and biochemical networks of cell–cell and cell–matrix interactions. Moreover, etomoxir has other metabolic effects, including stimulation of food intake in humans (186). Since it ameliorates ATP production from the oxygen-efficient conversion of glucose rather than from fatty acid metabolism, it might hold the promise of antianginal effect in ischemic heart disease (218), provided that dosage is carefully titrated and side effects monitored. These latter beneficial effects have paved the way to the exploration of other fatty acid inhibitors as potential antianginal agents. Patients with diabetes mellitus and coronary artery disease more frequently develop HF and have a greater amount of myocardial ischemia than patients without diabetes (353); a major contributor to this increased susceptibility derives from the exclusive use of fatty acid as energy source. Partial inhibitors of myocardial fatty acid oxidation such as trimetazine are effective at increasing exercise time and reducing the frequency of anginal attacks when combined with traditional antianginal drugs. Besides, it has been found that treatment with trimetazine significantly decreased the number of anginal attacks and improved myocardial ischemia and exercise capacity in patients with diabetes mellitus (365). There is substantial positive data on the use of trimetazidine that demonstrates metabolic and clinical benefit with almost no side effects, but data from a large outcome trial are lacking (286). Instead, ranolazine effectiveness as an antianginal drug has been stated (79). However, ranolazine antiischemic effect does not relate with fatty acid metabolism but to inhibition of the late Na+ current (310) (see section VB). A recent experimental evidence indicates that, since the late Na+ current is activated by oxidative stress, ranolazine profile of activity might also include an amelioration of cardiac oxidative stress during stress conditions (417). Ranolazine has not been tested in human HF yet.
2. Incretin mimetics
Even if conceptually valid, the intensive control of fatty acid metabolism (and of glucose) by insulin has been proved to be harmful rather than beneficial in treating HF (145). On this basis, to circumvent insulin receptor activation, two drug therapies are now possible: thiazolidindiones and modulators. Due to the scientific debate around thiazolidindiones and cardiovascular risk (420), we have chosen to talk about glucagon-like peptide-1 (GLP-1) as a prototype of drug modulating the incretin system.
Incretin modulators include GLP-1, GLP-1 analogs (exenatide and liraglutide), and dipeptidyl dipeptidase-4 (DPP-IV) inhibitors. GLP-1 is a hormone of the incretin family produced by the intestine in response to nutrients and to neurohumoral factors (331). It is well established that incretins represent an important complementary system to the pancreas in controlling postprandial glycemia and, GLP-1 is of particular effectiveness in reducing glycemia in experimental diabetes (109) and in type 2 patients (276), where it reduces HbA1 and body weight (60, 61, 275). During a meal, the incretin system mediates insulin release even before blood glucose increases. In addition, GLP-1 reduces gastric emptying (331), amplifies nutrient-induced insulin response, reduces hepatic glucose output (215), and likely promotes the maintenance and regeneration of pancreatic β-cells (137).
In type 2 diabetic patients, the incretin system is dysregulated (254) and this is thought to represent a pathogenetic milestone in the history of pancreatic dysfunction first and insulin resistance thereafter. On this basis, new therapeutic approaches of type 2 diabetes include GLP-1 analogs (GLP-1 has the disadvantage of negligible oral bioavailability) and inhibitors of the enzyme scavenging GLP-1, the DPP-IV inhibitors, which increase GLP-1 half-life but also exert ancillary actions such as the increase of the other incretin GP1. Both strategies show substantial overlapping effects in reducing glycemia (mainly postprandial) and glycated hemoglobin, thus ensuring cardiovascular protection and controlling weight gain; moreover, DDP-IV inhibitors do not present hypoglycemia as adverse effect. GLP-1 analogs and DPP-IV inhibitors await clinical evidence supporting their effectiveness in reducing the incidence of stroke in type 2 patients.
GLP-1 analogs and DPP-IV inhibitors are not indicated as monotherapy yet. Even if the lack of oral bioavailability and the short half-life remain critical points, GLP-1-based therapy is now possible, thanks to the presence on the market of analogs (exenatide and the synthetic extendin-4, a GLP-1 receptor agonist) more resistant to the action of the DPP-IV, whose use has been associated with the improvement of several cardiac and vascular risk factors (134, 201). Recent evidence suggests that GLP-1 pharmacokinetic features allow its penetration into the central nervous system, where it shows some effectiveness in treating neurodegenerative diseases, including Alzheimer's and Parkinson's diseases (39, 297).
Exenatide and DPP-IV therapies are indirect antioxidants since their use produce the reduction of glycated hemoglobin (43, 56, 294). However, in vitro studies suggest that GLP-1 might have direct antioxidant features. In human umbilical vein endothelial cells GLP-1 protects against H2O2-induced cell senescence and is able to attenuate oxidative stress-induced DNA damage by activating PKA and inducing the oxidative defense genes heme oxigenase-1 (284). Moreover, in neuroblastoma cells GLP-1, activating the survival kinase, the PKB, restores redox balance and protects from advanced glycated end products-induced apoptosis (197). All these neuroprotective effects propose this hormone as a potential therapeutic strategy against diabetes-induced encephalopathy.
Up to now, no evidence is available on the possible direct antioxidant features of DPP-IV inhibitors; more in general, it is unknown whether exenatide (or exendin-4) and DPP-IV inhibitors ameliorate insulin-resistance at muscle and adipose cells in diabetic patients (317, 385).
Several experimental studies suggest that GLP-1 may have important cardiac effects. Notwithstanding GLP-1 increases cAMP, it reduces heart contractility likely decreasing myofilament Ca2+ responsiveness resulting from intracellular acidification (387). In addition, GLP-1 has shown protective effects on normoglycemic rat hearts and in diabetic cardiac myocytes subjected to ischemia and reperfusion (281, 351). All these features encouraged studying GLP1 and its analog in HF. Preliminary clinical studies indicate that short-term GLP-1 treatment (3 days) improves the LV contractile function (both systolic and diastolic) in a small number of type 2 diabetic patients with chronic HF (373) and that a longer treatment (5 weeks) significantly improved the LV ejection fraction reducing oxygen consumption in both diabetic and normoglycemic chronic HF (New York Heart Association class III and IV) with no effects in patients with normal cardiac function (350). In contrast, the only double-blind clinical study, carried out on 20 nondiabetic HF patients (treated for 48 h with GLP-1) with ischemic heart disease, showed no major cardiovascular effects of GLP-1. Hence, further preclinical research is needed, aimed at exploring the mechanism underlying GLP-1 (or analogs) beneficial cardiovascular effects that might go beyond GLP-1 receptor activation (23).
3. Metformin
Metformin is a biguanide of natural origin, which represents a milestone in the treatment of type 2 diabetic/obese patients. Metformin, together with incretin mimetics or analogs, represents an attempt to cure diabetes rather than control hyperglycemia. Metformin is an antihyperglycemic agent but also has ancillary features that may account for part of its clinical effectiveness. The antihyperglycemic effect of metformin does not involve insulin receptor cascade activation but the muscle-specific pathway AMPK, which plays a fundamental role in contraction-induced GLUT4 recruitment at plasmalemma. Metformin, together with incretin analogs, attempts to interrupt the vicious cycle between insulin and glucagon: it opposes insulin resistance in liver, muscle, and adipose tissue level, but also shows ancillary protective features to the cardiovascular system (367). By virtue of these features, metformin is the only oral antidiabetic medication that has been shown to decrease diabetic cardiovascular complications in large-scale clinical trials (85).
Recent evidence suggests that metformin also has antioxidant capacities. Metformin, via AMPK, upregulates expression/activity of thioredoxin, a potent antioxidant which reduces, for instance, ROS produced by high fatty acid load (344). Moreover, thioredoxin, by binding and inhibiting apoptosis signal-regulating kinase-1, an upstream kinase of the JNK and p38 pathways, also prevents an excessive stress response, and unmasks a novel and important protective mechanism of metformin against cardiovascular complications of hyperglycemia. Thus, AMPK pathway can be a target of ROS but also a way to reduce ROS levels (209, 289, 416).
Metformin also reduces body weight gain likely increasing GLP-1 bioavailability (293). Recent studies suggest that metformin effectiveness might go beyond the treatment of type 2 diabetes. In particular, in a murine model of HF following ischemia–reperfusion, metformin has showed a significant cardioprotection with preservation of LV function (146). In any case, prospective controlled trials with metformin in human HF are still lacking: since it is not a hypoglycemic agent, metformin could represent a suitable metabolic therapy for nondiabetic patients with HF.
4. Statins: lights and shadows
Statins are a large class of hypolipidemic drugs that found indication in the secondary prevention of cardiovascular risk patients. In these patients the beneficial effects of statins are now recognized to extend well beyond their lipid-lowering properties. Statins confer significant protection of the vasculature through a combination of both distinct and interdependent effects on endothelial cell Rho GTPase regulation, NADPH oxidase activity, NO bioavailability, and differential gene expression (antioxidants). Abundant in vitro data, in addition to myriad reports relying on a range of animal models, now firmly support the idea that these drugs may serve as novel and effective therapeutic agents in a variety of disease states characterized by vascular dysfunction (178). In fact they have become a mainstay of the primary and secondary prevention of coronary artery disease, the most common etiology of HF in industrialized nations. Further, patients with nonischemic cardiomyopathy may have comorbidity or risk factors such as diabetes, which may affect their cardiovascular risk. Thus, a large proportion of patients with HF appear to be candidates for statin therapy. Unfortunately, the cardiovascular effects of statins depend on the molecule used. It is in fact rather well known that statins differ by their pharmacokinetics features, among which volume of distribution. The effect of the statins (if any, which one?) in HF patients is still largely debated. In fact, beneficial or harmful effects have been described. In particular, as a part of their beneficial effect, statins, as stated in experimental models (see Raina et al. for a review) (307), reduce cardiovascular oxidative stress and inflammation independently on their lipid-lowering efficacy.
In this context, as there is ample literature available, only recent clinical and experimental studies have been used as they highlight new antioxidant features of two statins. For example, pravastatin would protect against cardiac ischemia/reperfusion injuries activating CAT, via phosphorylation of extracellular signal-regulated kinase, thus preserving mitochondrial membrane potential and likely activating mitoKATP, which, in turn, moderately raises ROS levels (374). Instead, short-term treatment of HF patients with rosuvastatin has revealed the effectiveness of the drug in reducing, more than allopurinol, myeloperoxidase activity, considered a marker to follow to monitor HF progression and severity (13).
On the reverse, rosuvastatin therapy would lower plasma total CoQ10, which is, together with cholesterol, a product of the mevalonate biosynthetic pathway (131). It is recognized that CoQ10 deficiency, an essential cofactor for mitochondrial electron transport and myocardial energy supply, occurring in HF (264) contributes to compromise mitochondrial function. It is therefore plausible that CoQ10 depletion is a part of the worsening outcomes following rosuvastatin therapy of HF patients (18).
Statins also have harmful effects interfering with LDL metabolism. Moreover, since (i) patients with HF have been largely excluded from major randomized clinical trials of statins and (ii) the effect of statins has been tested by using surrogate markers of cardiovascular risk, the clinical safety and efficacy of statins in patients with HF remain, so far, to be clearly addressed (307).
In this setting, three important multicenter randomized trials (all of them rosuvastatin vs. placebo) are ongoing (and partially closed): GISSI-HF, which included both ischemic and nonischemic cardiomyopathy patients with normal ejection fraction and low or normal level of cholesterol; UNIVERSE, aimed to elucidate whether the ability of statins to impact cytokine production and cardiac remodeling observed in animal and in vitro studies translates clinically to human subjects; and CORONA, whose primary end point was a composite end point of time to first cardiovascular death, nonfatal myocardial infarction or nonfatal stroke in chronic symptomatic HF patients.
When this section was written, results from CORONA and GISSI–HF showed the beneficial effect of rosuvastatin in terms of reduction of the atrial fibrillation occurrence in patients with new diagnosis of HF (239) but, also, no evidence underneath rosuvastatin addition in nonischemic HF.
Overall, even if the effect of statins in HF might be largely influenced by the molecule (230) and the dosage used, clinical data, up to now, point to null effects of statins on the progression of HF and patients' survival (217).
5. Tetrahydrobiopterin
BH4 is the cofactor of monooxygenases, enzymes that incorporate one atom of O2 into the substrate and reduce the other atom to H2O. BH4 availability represents the limiting step to the synthesis of tyrosine but, from the 1980s it was recognized as the cofactor also for the three isoforms of the NOS. Therefore, BH4 has been hypothesized to represent a rate-limiting cofactor in NO biogenesis and a suitable therapeutic agent for all the diseases characterized by reduced levels of BH4 and NO dysfunction such as hypertension, diabetes, and, more recently, HF. Supporting this view is the fact that BH4 acts as an antioxidant agent able to stabilize the catalytic site of the NOS enzymes (311).
However, the employment of BH4 as a drug is limited by its chemical features and its variable pharmacokinetic, which make bioavailability unpredictable following administration. These problems have been partially overcome by the preparation of a thermo- and photostable tablet form (6R-BH4) or by testing the effectiveness of folic acid and its active metabolite, the 5-methyltetrahydrofolate. Although both strategies led to positive results in experimental animal models, the only clinical trial using 6R-BH4 (5 mg/kg) versus placebo failed to yield positive results in hypertensive patients.
Of course, there might be several reasons for such disappointing results that should prompt some general consideration. There is no doubt that HF and other cardiovascular diseases are characterized by dysfunction of NO system, as extensively described in the previous sections. Cardiac NOS isoforms are, however, tightly compartmentalized (72) and exogenous administration of a reduced active component of the system (L-arginine, BH4) might not ensure recovery of appropriate NO levels at specific intracellular sites. This might be a general limitation to the use of exogenously administered antioxidants, including BH4.
VIII. Conclusions
The past few years have been characterized by a tendency toward a unifying hypothesis for the pathogenesis of HF. After many proposals and retractions, the assumption that pathways regulating redox balance in the cardiomyocyte are the sole or major culprit for function derangement in the failing heart would appear—by far—oversimplified. Such a conclusion would also arise from disappointing outcomes of clinical trials employing rescue antioxidant therapies. Remarkably, most drugs able to restore mechanical or metabolic function and reshape ventricular chambers in animal models of HF failed to meet major outcomes in the clinical arena.
However, a mass of thorough studies allowed the unraveling of crosslinks and checkpoints among processes—Ca2+ homeostasis and electrogenesis and metabolic pathways—where ROS and RNS act as fine modulators. Thanks to their unique short-life and hyper-reactivity, these radical species exert spotted and limited effects in time and space but, paradoxically, represent a widespread chemical communication in the cellular syntax. Overall, multifaceted evidence led to two general and remarkable consequences: first, the awareness that gross rescue interventions by antioxidant might be ineffective or disproportionate; second, the appraisal that drugs effective against HF possess a redox-added value. Reinterpreting available medicines and supplementations in the light of novel knowledge on ROS/RNS signaling would help to design not just an antioxidant strategy, but a redox-tailored intervention.
Footnotes
Acknowledgments
This work was supported by grants from Ministero of Education, University and Research and Ente Cassa di Risparmio di Firenze.
Abbreviations Used
Reviewing Editors: Cynthia A. Carnes, Torsten Doenst, Hyoung-Gon Lee, Hajime Otani, and W.H. Wilson Tang