
Abstract
Dr. Joseph Loscalzo (M.D., 1978; Ph.D., 1977) is recognized here as a Redox Pioneer because he has published two articles in the field of antioxidant/redox biology that have been cited more than 1,000 times and 22 articles that have been cited more than 100 times. Dr. Loscalzo is known for his seminal contributions to our understanding of the vascular biology of nitric oxide. His initial discovery that the antiplatelet effects of organic nitrates are potentiated by thiols through a mechanism that involved metabolism to S-nitrosothiols was followed by the demonstration that S-nitrosothiols are formed endogenously through S-transnitrosation, stabilize nitric oxide, and facilitate the transport and transfer of nitric oxide between and within cells of the vessel wall. These properties led to the development of S-nitrosothiol–containing pharmacotherapies to treat disease states characterized by nitric oxide deficiency. Dr. Loscalzo's other scientific contributions include identifying the vascular functional consequences of genetic deficiencies of antioxidant enzymes that decrease nitric oxide bioavailability, collectively termed the “oxidative enzymopathies,” and demonstrating the role of mitochondria in modulating the disulfide subproteome, and in redox signaling in hypoxia. He has received numerous awards and honors for his scientific contributions, including election to the Institute of Medicine of the National Academy of Sciences. Antioxid. Redox Signal. 13, 1125–1132.
Educational and Professional Training
Summary of Dr. Loscalzo's Top Contributions
Organic nitrates and nitric oxide (NO•) donors were known to limit platelet aggregation; however, the mechanism underlying this response remained unknown for more than a decade. In 1985, Dr. Loscalzo discovered that the antiplatelet effects of organic nitrates are dependent on their reaction with thiols to form S-nitrosothiols. He demonstrated that S-nitrosothiols are a stable reservoir of NO•, facilitate NO• transport within and between cells, and promote the vascular biologic effects of NO•. His research increased our understanding of NO• signaling and metabolism in the vasculature and the pathophysiologic significance of NO•-deficiency states.

Background, Development, and Training
Joseph Loscalzo was born in 1951 in Camden, New Jersey, where he spent his childhood with his three siblings and a large extended family. His great-uncle, a pediatrician who served on the first board of the American Academy of Pediatrics, was a significant influence on Dr. Loscalzo's decision to become a physician. Family has always been important to Dr. Loscalzo, and he and his wife Anita are the parents of two highly accomplished grown children, Julie and Alex, and the proud grandparents of Charlotte.
Dr. Loscalzo joined the faculty of Harvard Medical School at Brigham and Women's Hospital in 1984, after completing his training, and remained on staff until 1994. During this time, he was appointed Chief of the Cardiology Section at the Brockton/West Roxbury VA Medical Center, a Harvard Medical School affiliate institution, and Director of the Center for Research in Thrombolysis and of the Center for Research in Vascular Biology.
In 1994, Dr. Loscalzo moved to Boston University School of Medicine to assume the roles of Chief of Cardiology and Director of the Whitaker Cardiovascular Institute. In 1997, he was named the Wade Professor and Chairman of the Department of Medicine. While there, he expanded the Whitaker Cardiovascular Institute by recruiting a number of talented investigators. In 2005, he returned to Harvard Medical School and Brigham and Women's Hospital as the Hersey Professor of the Theory and Practice of Medicine and Chairman of the Department of Medicine.
Area of Interest in Redox Biology
Mitochondrial ROS and the disulfide subproteome
Loscalzo's group found that mitochondrial ROS were involved in de novo disulfide bond formation, a key event in protein synthesis and function, by demonstrating that global disulfide bond formation paralleled mitochondrial ROS levels. Disulfide bond formation influenced the activation and cell-surface translocation of selected disulfide-containing proteins such as insulin growth factor-1 receptor, indicating the existence of a mitochondrial ROS-dependent redox-sensitive disulfide subproteome (44). Sparsely cultured endothelial cells were found to have a lower protein disulfide content compared with confluent cells, associated with decreased mitochondrial membrane potential, superoxide production, and increased levels of reduced glutathione, indicating a more reductive state. This, in turn, was associated with diminished ligand-induced phosphorylation of insulin growth factor-1 receptor, suggesting that differences in receptor function observed in sparsely cultured cells may result from insufficient oxidative potential (44). Other studies revealed that GPx-1 regulates epidermal growth factor receptor signaling, in part, through a similar mechanism. Cells that overexpressed GPx-1 were found to have decreased epidermal growth factor receptor–mediated activation of Akt, resulting in decreased proliferation, owing to a reduction in global disulfide bond formation, mitochondrial membrane potential, and ATP production (14). These studies highlight the importance of mitochondrial ROS and intracellular reductive potential for protein disulfide formation, select disulfide-containing cell-surface receptor expression, and growth factor receptor–mediated signaling (Fig. 1).
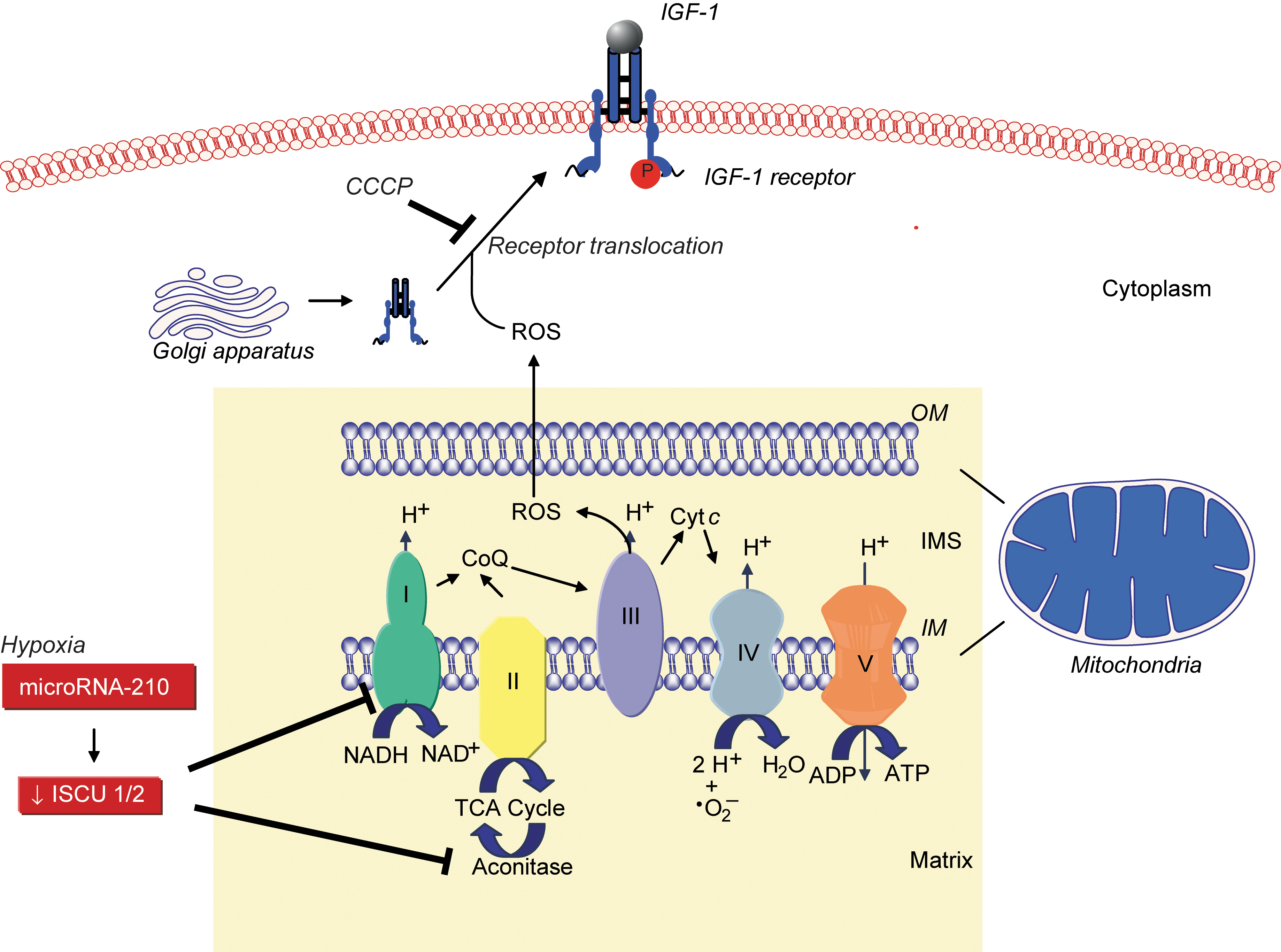
Hypoxia, endothelial cells, and microRNA-210
Hypoxia is associated with inflammation and ischemia, and, under these conditions, NO• levels may be markedly elevated, rendering vascular endothelial cells susceptible to apoptosis and cell death. Loscalzo's laboratory demonstrated that this occurred as a result of increased peroxynitrite formation that activated the mitochondria-independent pathway of apoptosis, as well as induced mitochondrial dysfunction and cytochrome c release to activate caspase-9 (39).
Under hypoxic conditions, repression of mitochondrial electron transport and oxidative phosphorylation in favor of glycolysis is known as the “Pasteur effect.” In endothelial, vascular smooth muscle, and transformed cells, as well as in vivo, this metabolic switch was shown by Dr. Loscalzo's group to be regulated by hypoxia-mediated upregulation of microRNA-210 to decrease the expression of iron-sulfur cluster assembly proteins 1 and 2. These proteins facilitate the assembly of iron-sulfur clusters, which are necessary for Complex I and aconitase activity to facilitate electron transport and mitochondrial oxidation–reduction reactions. Thus, microRNA-210 is a key regulator of the cellular adaptation to hypoxia (4). These data suggest that microRNA-210 functions as a master microRNA under hypoxic conditions and modulates disease states characterized by hypoxia, such as ischemia and tumorigenesis (3) (Fig. 1).
Description of Key Finding 1
S-nitrosothiols and the biologic effects of NO•
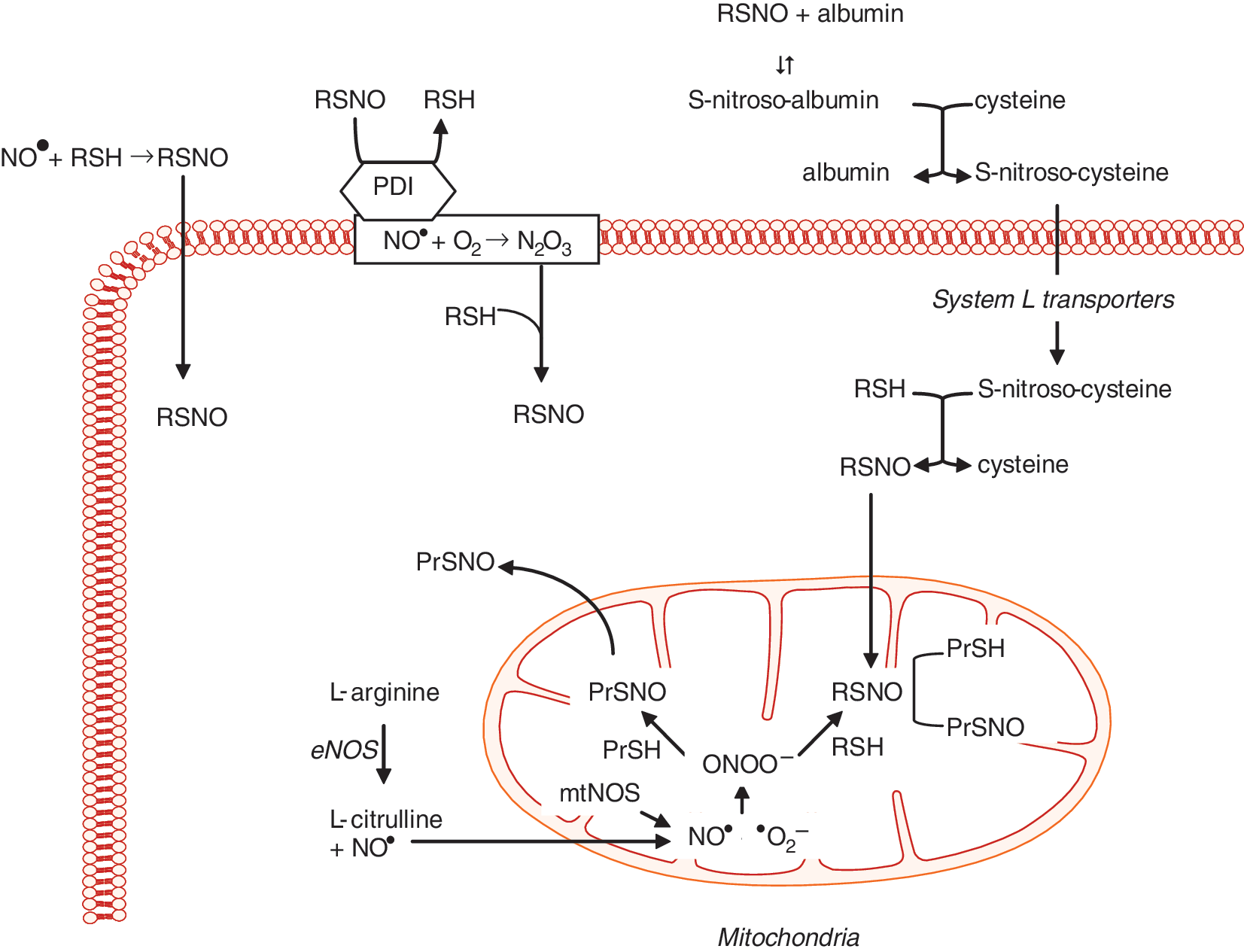
Dr. Loscalzo first demonstrated that the antiplatelet effects of organic nitrates or endothelium-derived NO• were dependent on thiols to generate S-nitrosothiols, and he confirmed this finding by showing that S-nitrosoproteins, such as S-nitroso-albumin, were potent antiplatelet agents (22, 29). This discovery by Dr. Loscalzo established the fundamental role of S-nitrosothiols in modulating the biologic effects of NO•. Subsequently, S-nitrosoglutathione was shown to enhance platelet formation from megakaryocytes by inducing apoptosis (1, 2). Loscalzo's group demonstrated that S-nitrosothiols are formed naturally in vivo and serve as a stable reservoir of NO• (28, 33). Subsequently, they found that de novo formation of S-nitrosothiols occurred through several mechanisms, including S-transnitrosation (Fig. 2) (34, 35). This process was shown to be catalyzed by the cell-surface protein disulfide isomerase, which, through S-transnitrosation, facilitated the transfer of NO• from the extracellular to the intracellular compartment (45). In vivo, S-nitrosoproteins with limited intracellular access were shown to exert their biologic actions by undergoing thiol-S-nitrosothiol exchange with low-molecular-weight thiols to form low-molecular-weight S-nitrosothiols (28). Loscalzo's group demonstrated further that endogenous NO• or exogenous S-nitrosothiols promoted the formation of S-nitrosoproteins in endothelial cells. These S-nitrosoproteins were localized principally to the mitochondria and the perimitochondrial compartment, with a half-life of ∼1 h, and their formation paralleled eNOS activity and mitochondrial function. Mass spectrometry revealed that a limited repertoire of S-nitrosoproteins exists in resting endothelial cells, with GAPDH being the most abundant, suggesting that NO• may play a role in regulating glycolysis by this mechanism (43).
Description of Key Finding 2
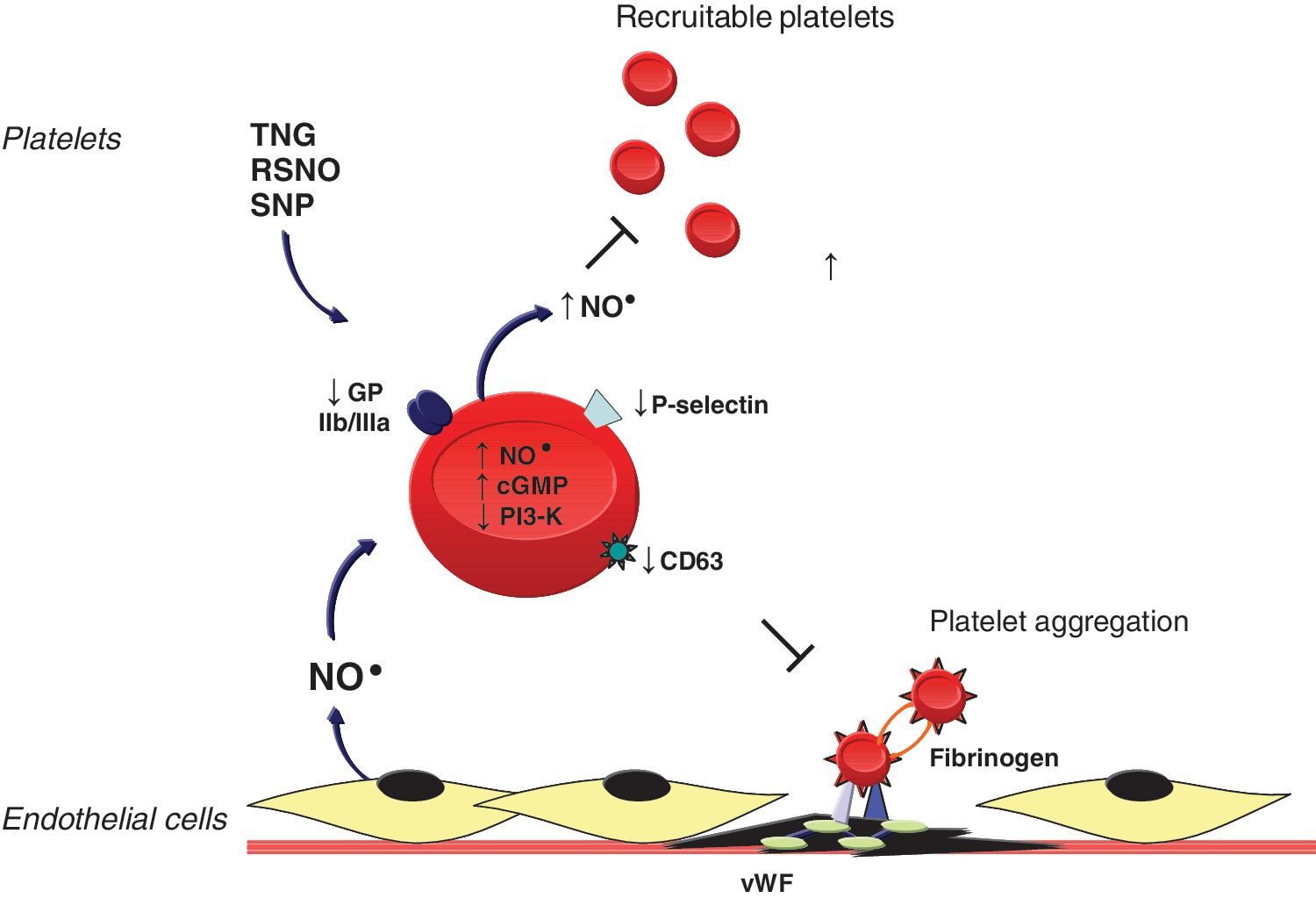
Nitric oxide and platelet activation
Dr. Loscalzo's laboratory has shown that endogenous or exogenous NO• modulates platelet activation, recruitment, and aggregation (Fig. 3). Endothelium-derived NO•, in the presence of N-acetylcysteine, inhibited ex vivo aggregation by increasing platelet cGMP levels (33). In vivo, in a coronary artery stenosis model, NO• inhibited periodic platelet thrombus formation through a similar mechanism (6, 27). Nitroglycerin acts synergistically with tissue plasminogen activator or the platelet inhibitory prostaglandins I2 and E1 to promote platelet disaggregation, whereas inhibiting NO• with N
G-mono-methyl-
Description of Key Finding 3
Oxidative enzymopathies
Loscalzo's laboratory found that inherited or acquired deficiency of glucose-6-phosphate dehydrogenase or the antioxidant enzyme glutathione peroxidase(s) is associated with increased oxidant stress, decreased bioavailable NO•, and vascular dysfunction (Fig. 4). Deficient glucose-6-phosphate dehydrogenase activity increased ROS levels (19); inhibited endothelial cell proliferation, migration, and tube formation in vitro, as well as angiogenesis in vivo (21); impaired vascular reactivity to endothelium-dependent and -independent vasodilators (19, 20); and mediated the adverse effects of aldosterone on vascular function by contributing to endothelial dysfunction and oxidative posttranslational modification of soluble guanylyl cyclase (20, 23).
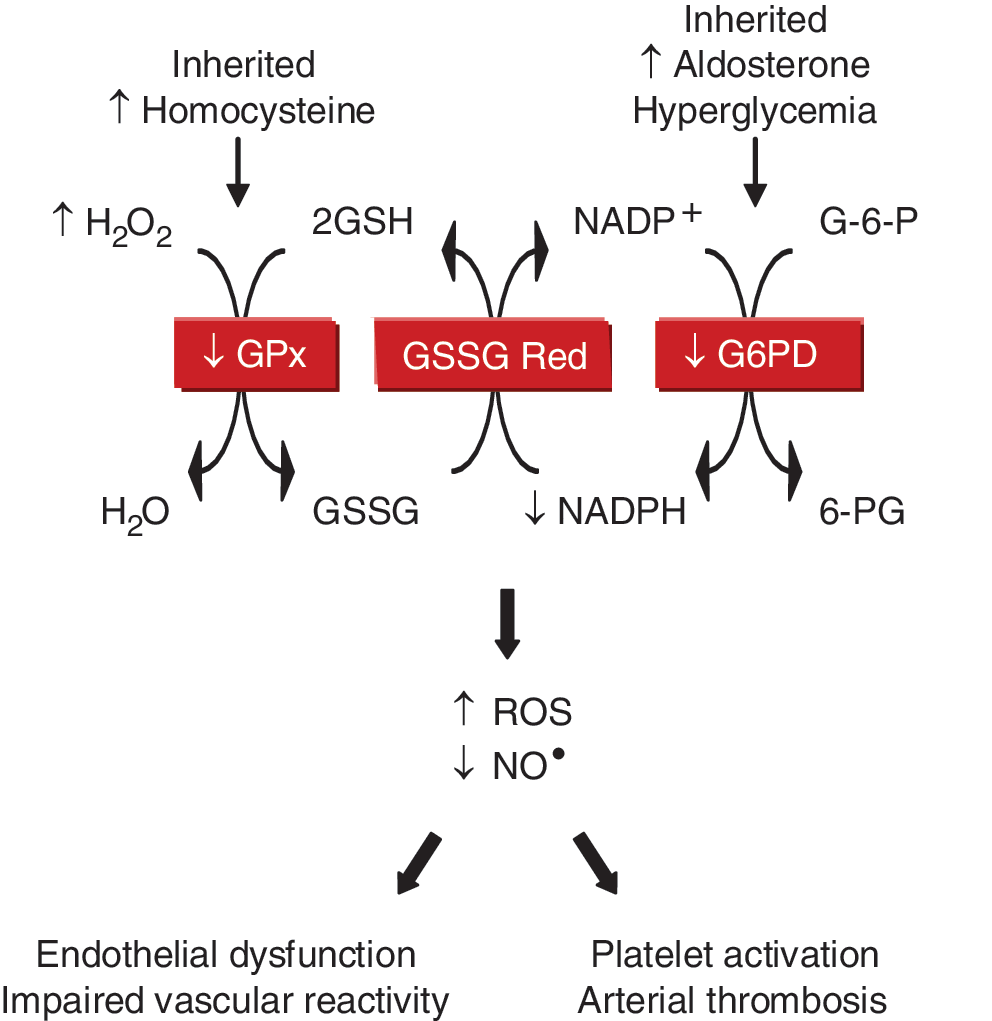
They also found that GPx-1 deficiency was associated with endothelial dysfunction and vascular structural abnormalities, including increased matrix deposition and intimal thickening (7, 8). Deficiency of plasma GPx, or GPx-3, promoted platelet insensitivity to NO•, thrombosis, and was associated with stroke (9, 11). This led to the identification of a unique promoter haplotype (H2) of seven tightly linked polymorphisms in the GPx-3 gene that is overrepresented in young patients with arterial ischemic stroke and central venous thrombosis (37, 38).
In related studies, Loscalzo's laboratory demonstrated that mild-to-moderate hyperhomocysteinemia was associated with endothelial dysfunction, enhanced lipid peroxidation, and impaired vasodilation (5, 17, 41). These effects occurred, in part, as a result of homocysteine-mediated inhibition of GPx-1 translation (16), and overexpression of GPx-1 rescued the adverse vascular phenotype (42). Elevated levels of homocysteine also were shown to underlie the negative effects of
Other Achievements
5-Lipoxygenase and pulmonary hypertension
In pulmonary artery endothelial cells and animal models of pulmonary hypertension, Dr. Loscalzo's group demonstrated the adverse biologic effects of 5-lipoxygenase, a nonheme iron-containing dioxygenase that generates the biologically active leukotrienes and 5-hydroxyeicosatetraenoic acid. Increased 5-lipoxygenase expression decreased bioavailable NO• and cGMP levels in pulmonary artery endothelial cells, whereas inhibition of 5-lipoxygenase limited cell proliferation by causing a cell-cycle block at G0/G1 (40, 46). In the monocrotaline-rat model of pulmonary hypertension, pulmonary overexpression of 5-lipoxygenase increased right ventricular systolic pressure, hypertrophy, inflammation, and muscularization of small- and medium-sized pulmonary vessels (18). Bone morphogenetic protein receptor-2 (BMPR2) mutations have also been linked to pulmonary arterial hypertension. In BMPR2 heterozygous mutant mice, pulmonary vascular overexpression of 5-lipoxygenase increased pulmonary artery pressures, vascular remodeling, and thromboxane A2 production compared with unstressed mice (32). Challenge with 5-lipoxygenase and monocrotaline resulted in early and severe increases in pulmonary pressures, vascular endothelial injury, and perivascular infiltration of inflammatory and immune cells. This work suggests that increased 5-lipoxygenase expression enhances the susceptibility to pulmonary hypertension when the BMPR2 mutation is present (31).
Current Position
Dr. Loscalzo is the Hersey Professor of the Theory and Practice of Medicine at Harvard Medical School, Vice Director of the Biomedical Research Institute, and Chairman of the Department of Medicine at Brigham and Women's Hospital, a department of approximately 900 full-time clinicians and researchers. Dr. Loscalzo's research laboratory comprises 15 members currently, and his scientific discoveries have led to 30 patents for his work. He currently holds several NIH awards, including a Method to Extend Research in Time (MERIT) Award from the NHLBI. He is a member of the Council of Councils at the NIH and an elected member of the American Society of Clinical Investigation and the Institute of Medicine of the National Academy of Sciences.
Dr. Loscalzo is the Editor-in-Chief of Circulation; an Associate Editor for the Interactive Medical Case Series at the New England Journal of Medicine; a Consulting Editor for the Journal of Clinical Investigation, Circulation Research, and Arteriosclerosis, Thrombosis and Vascular Biology; and is a member of the editorial board of 16 journals. He is also a senior editor of Harrison's Principles of Internal Medicine.
Dr. Loscalzo has a profound commitment to teaching and has trained more than 50 investigators in his laboratory, many of whom have gone on to successful careers in biomedical research and leadership positions at prominent institutions. According to Dr. Loscalzo, “Biomedical research is an extraordinarily gratifying enterprise. Solving a perplexing problem in an innovative and rigorous way, creating new knowledge in the process, and using that knowledge to (re)define an underlying mechanism are each highly rewarding aspects of the research process. Perhaps the most rewarding feature, however, is the ability to influence the next generation of scientists through the impact of one's scientific contributions, and the guidance and advice given to one's trainees. Those of us who have succeeded in developing biomedical research careers have been given a great and special opportunity for which I, for one, am most grateful.”
Footnotes
Acknowledgments
Professor Joseph Loscalzo thanks the many extremely talented individuals with whom he has had the great good fortune to work with over the past 30 years, including his mentors and collaborators, but especially his former and current trainees. “It is their intellectual contributions, commitment, and hard work that form the basis for the recognition I have received. I wish to thank them deeply for what they have taught me, and what we have scientifically contributed together.”
This work was supported by NIH grants HL81110 and HL70819.
Abbreviations Used
Reviewing Editors: Dipak K. Das, Aron Fisher, Santiago Lamas, Elizabeth Murphy, and Pasquale Pagliaro
Author note: I met Dr. Loscalzo in 1994 as a Cardiology Fellow at Boston University School of Medicine and joined his research laboratory. Since then, we have been collaborating on studies that examine the pathophysiologic consequences of perturbations of redox homeostasis and nitric oxide deficiency on vascular function.
For a list of frequently cited articles published by Prof. Loscalzo, see Supplemental Tables 1 and 2, available online at
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.