
Abstract
The brain is a metabolically active organ exhibiting high oxygen consumption and robust production of reactive oxygen species (ROS). The large amounts of ROS are kept in check by an elaborate network of antioxidants, which sometimes fail and lead to neuronal oxidative stress. Thus, ROS are typically categorized as neurotoxic molecules and typically exert their detrimental effects via oxidation of essential macromolecules such as enzymes and cytoskeletal proteins. Most importantly, excessive ROS are associated with decreased performance in cognitive function. However, at physiological concentrations, ROS are involved in functional changes necessary for synaptic plasticity and hence, for normal cognitive function. The fine line of role reversal of ROS from good molecules to bad molecules is far from being fully understood. This review focuses on identifying the multiple sources of ROS in the mammalian nervous system and on presenting evidence for the critical and essential role of ROS in synaptic plasticity and memory. The review also shows that the inability to restrain either age- or pathology-related increases in ROS levels leads to opposite, detrimental effects that are involved in impairments in synaptic plasticity and memory function. Antioxid. Redox Signal. 14, 2013–2054.
I. Introduction
Although changes in redox status are often linked to age-dependent declines in synaptic plasticity and cognitive function, a growing body of evidence from both neuronal and nonneuronal cells suggests that ROS also can function as small physiological molecules involved in functional and structural changes necessary for synaptic plasticity. ROS have been implicated as modulators of hippocampus-dependent and hippocampus-independent memory formation (78, 134, 138). ROS also have been shown to regulate synaptic plasticity-related signaling molecules, receptors, and channels, including N-methyl-
Investigating the role of ROS in synaptic structure and function reveals a very interesting yet complex role for these molecules. ROS undoubtedly are very important physiological mediators of plasticity and signaling, but they can become detrimental to neuronal function when they accumulate excessively in the brain. The fine line of role reversal from good molecules to bad molecules of these very highly reactive players is far from being fully understood. This review briefly outlines the important components of learning and memory, and then focuses on identifying the multiple sources of ROS in a mammalian nervous system and on presenting evidence for the critical and essential role of ROS in synaptic plasticity and memory. The review also shows that the inability to restrain age- or pathology-related increases in ROS levels leads to opposite, detrimental effects that are involved in impairing synaptic plasticity and memory function.
II. Basic Components of Learning and Memory
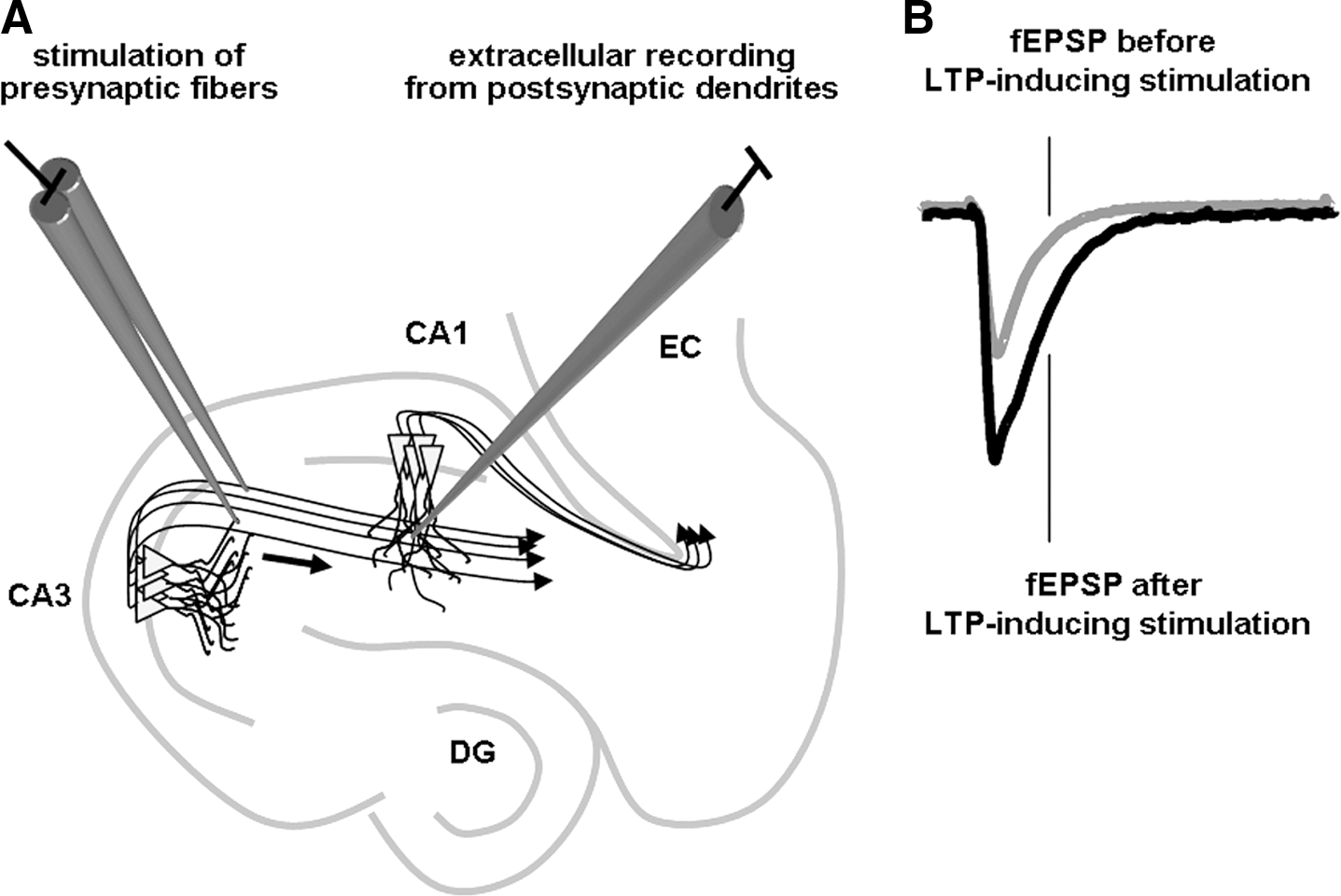
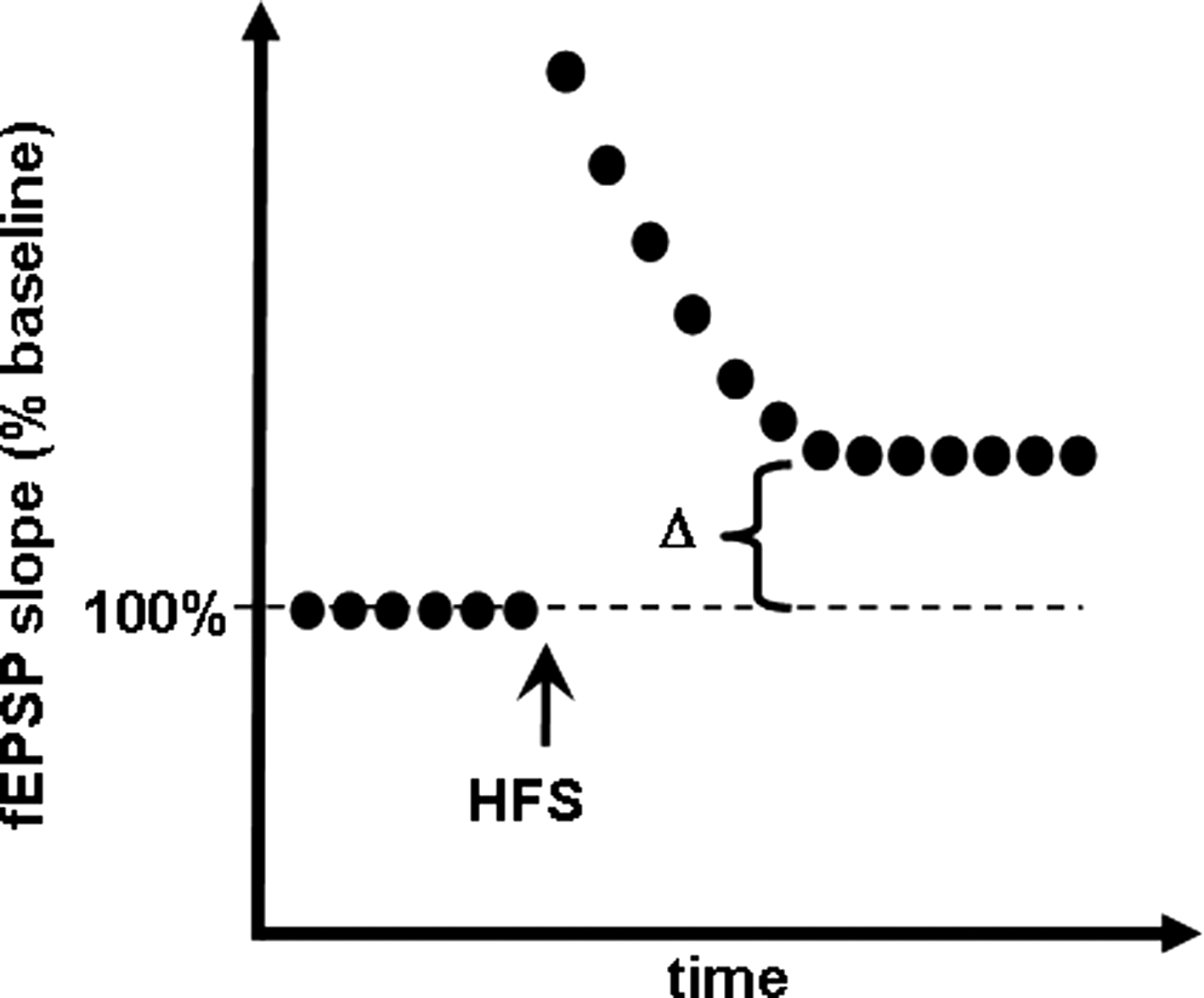
Learning and memory can be grossly defined as the process through which information is obtained, stored, and then retrieved for use by the brain. Much progress has been made in the past two decades toward understanding the molecular mechanisms governing the formation of memories. Multiple publications review these mechanisms, and the reader is referred to these reviews for further details (3, 18, 55, 176, 187, 217, 224, 230, 295, 304, 353, 428, 444). Here we briefly summarize key components of learning and memory processes, as a reference to the subsequent examination of the effect of ROS on such processes (Fig. 3).

Memory formation begins at the neuronal plasma membrane at synapses. At excitatory synapses, stimulation of presynaptic neurons causes the release of glutamate, which activates a variety of glutamate receptors, ionotropic and metabotropic, located on the postsynaptic spines. During periods of robust synaptic activity, enough glutamate is released to cause a large postsynaptic depolarization after activation of the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) subtype of glutamate receptors. The postsynaptic depolarization activates the NMDA subtype of glutamate receptor, which allows massive Ca2+ influx into the postsynaptic neuron. These are hallmarks of the induction of LTP, the most intensely studied cellular substrate for memory. Ca2+ influx leads to the production of small messenger molecules such as cAMP, cyclic guanosine monophosphate/nitric oxide (NO), and arachidonic acid. Ca2+ also binds to calmodulin, inducing a conformational change that permits interaction with and activation of other molecules. The small messengers, along with the activated Ca2+/calmodulin complex, activate multiple protein kinase signaling pathways, including CaMKII, cAMP-dependent protein kinase (PKA), protein kinase C (PKC), the Ras pathway, and ERK. One of the downstream targets of PKC is neurogranin/RC3, which is a Ca2+-sensitive calmodulin-binding protein whose calmodulin-binding affinity is attenuated by phosphorylation via PKC and oxidation via NO, thus prolonging the availability of calmodulin long after Ca2+ influx has subsided (33, 192, 250, 261, 386). Neurogranin has been shown to play a critical role in synaptic plasticity and memory. It was shown to be phosphorylated by PKC after induction of LTP (84, 193), and antibodies against the neurogranin phosphorylation domain prevented the induction of LTP (127). Additionally, mouse transgenic knockouts lacking neurogranin have been shown to have deficient spatial learning in the Morris water maze (322), as well as changes in the induction of hippocampal LTP and short-term plasticity (445). The combined activation of these signal transduction cascades ultimately results in the phosphorylation of CREB, which in turn recruits multiple transcription coactivators to initiate a wave of transcription, followed by translation. The newly formed proteins modulate synaptic strength and efficacy via altering the electrical properties of membranes, increasing glutamate receptor expression, changing synaptic morphology, increasing the number of synapses, etc., all of which are necessary for long-lasting LTP and the formation and consolidation of long-term memories.
The formation of memory is therefore dependent on the coordination of complex intracellular signaling pathways. Of particular interest for this review, ROS have been shown to act as small signaling molecules, necessary to modulate the aforementioned cascades. However, as a consequence of the complexity of memory formation, minor disruptions in one or more of the involved signaling cascades, including ROS, can be deleterious to memory formation. In the sections that follow, we will explore the involvement of ROS in memory formation, both under physiological and pathological conditions.
III. Sources of Reactive Oxygen Species
ROS can be produced at multiple sites in a mammalian cell. Mitochondria are the energy machinery of the cell and produce the largest amount of ROS. Nonetheless, other sources, such as monoamine oxidase (MAO) and NO synthase (NOS), produce ROS amounts large enough to participate in a myriad of pathophysiological cellular processes. ROS have been shown to be critical signaling molecules that are essential for the proper induction of synaptic plasticity and memory formation. However, the specific sources of ROS with regard to learning and memory have not been distinctly identified. In this section, we will discuss the potential sources of ROS involved in the physiological signaling role of these molecules. Later in this review, we will address the involvement of ROS and their sources in pathological conditions.
A. The mitochondrial respiratory chain
Similar to every other cell in mammalian systems, neurons use adenosine triphosphate (ATP) as a major energy source to drive cellular processes necessary for its function (426). ATP is produced mainly in mitochondria by the process of oxidative phosphorylation, of which the major byproduct is superoxide (426). Superoxide often plays an important role in modulating signaling pathways, but is relatively short-lived and its usual fate is rapid dismutation to H2O2 by the mitochondrial SOD (SOD-2 or manganese [Mn]-SOD). Given the extremely high metabolic rate of the brain, neurons produce very large amounts of ROS compared to other organs (164). Although the production of ROS ensures the induction of physiological events such as synaptic plasticity and memory, it also puts the brain at a higher risk for oxidative stress, leading to the impairments of the very functions that ROS are necessary for. Thus, the balance between ROS production and clearance is the critical factor in determining whether the beneficial effects of ROS are outweighed by their ability to cause oxidative stress.
Mitochondrial ROS generation occurs mainly through the leaking of superoxide at complexes I (NADH ubiquinone oxidoreductase) and III (coenzyme Q, bc1 complex, ubiquinone/cytochrome c reductase) of the mitochondrial respiratory chain (398) (Fig. 4).

1. Complexes I and III
Complex I, also known as NADH ubiquinone oxidoreductase, is a trans-mitochondrial membrane complex that oxidizes a previously reduced NADH using coenzyme Q10 as the electron acceptor (398). Complex I is the major site of entry of reducing equivalents, followed closely by complex II (398). It is coupled to proton pumping, specifically pumping four protons across the inner mitochondrial membrane, from the matrix to the intermembrane space, thus contributing to the proton gradient that will later fuel the ATP synthase (239). Complex I is the major site of the premature leak of electrons to oxygen, thereby creating the free radical superoxide (469).
Complex III contains two pools of ubiquinone: Qi faces the mitochondrial matrix and Q0 faces the intermembrane space (93, 394). Complex III catalyzes the reduction of ubiquinone in a stepwise fashion. First, two electrons are removed from ubiquinone (Q0) and are passed on to two molecules of cytochrome c to eventually reach complex IV. In a second step, two other electrons are passed on to the Qi site to reduce quinone to quinol. In total, this sequential reaction releases four protons (398). Complex III participates in the generation of the proton gradient across the mitochondrial membrane by an asymmetric absorption/release of protons rather than pumping protons through the membrane. In terms of ROS production, complex III can leak electrons and participate in superoxide formation (394). It is believed that if the electron transfer from ubiquinone to cytochrome c is delayed for any reason, ubiquinone tends to auto-oxidize and release an electron that is free to attack oxygen for superoxide formation (146).
Animal models carrying mutations in the various components of the electron transport chain, as well as pharmacologic inhibitors, have proven very useful in coupling effects with sources of ROS. We focus the remainder of this section on discussing complexes I and III as potential sources of superoxide involved in modulating learning and memory.
2. Mitochondrial superoxide in learning and memory
Several studies have suggested that ROS of mitochondrial origin might impact signaling during synaptic plasticity and memory, although the studies are at best suggestive. The exact source of ROS during synaptic plasticity has yet to be determined. For example, treatment of rat isolated mitochondria with elevated levels of sodium and Ca2+ (thus mimicking the events happening during depolarization of neurons and synaptic transmission of neuronal signals) leads to increased production of superoxide (116). In addition, application of glutamate to cultured forebrain neurons activates NMDA receptors and induces an increase in ROS production that can be blocked by the NMDA receptor channel blocker MK801 (362). NMDA receptor-dependent superoxide production also occluded further superoxide production after uncoupling of mitochondrial proton transport with the inhibitor p-trifluoromethoxy carbonyl cyanide phenyl hydrazone (51). Although the authors of the latter study concluded that their data provided evidence for mitochondrial superoxide production after NMDA-receptor activation, one should not rule out the possibility that p-trifluoromethoxy carbonyl cyanide phenyl hydrazone could be uncoupling proton transport through NADPH oxidase as well (4). Other investigators (113) have used specific inhibitors of complexes I (rotenone) and III (antimycin A) to demonstrate that NMDA receptor-dependent ROS production in cortical neurons originates in the mitochondrial electron transport chain. Although these studies suggest a link between mitochondrial superoxide and synaptic transmission that is essential for plasticity, they have not directly investigated the involvement of mitochondrial ROS in synaptic plasticity.
Perhaps the most direct evidence linking mitochondrial superoxide to synaptic plasticity and memory comes from studies demonstrating that large stimulus-induced increases in cytosolic Ca2+ result in production of superoxide by mitochondria (181, 183). This superoxide acts as a modulator of two kinases, CaMKII and PKA, both known to be involved in the induction of synaptic plasticity. On another hand, in the course of investigating the function of mitochondria in CD8+ T cell functioning, it was demonstrated that incubation with rotenone, a widely used inhibitor of complex I, leads to decreased production of H2O2, Ca2+ influx, and ERK phosphorylation (457), another kinase known to be involved in synaptic plasticity (194, 210).
A counter argument to the suggestion that mitochondrial ROS may be involved in synaptic plasticity comes from behavioral and electrophysiological studies in transgenic mice overexpressing SOD-2. These mice have lower levels of mitochondrial superoxide; however, the mice do not exhibit deficits in either LTP or in learning and memory (188), suggesting that mitochondrial superoxide does not contribute in a major way to synaptic plasticity and memory formation under normal physiological conditions. In contrast, mitochondrial superoxide plays a major role in memory dysfunction associated with neuropathological conditions such as aging and AD (115, 277), both discussed later in the review.
Thus, the evidence at this time suggests that there is minimal involvement of mitochondrial ROS in synaptic plasticity and memory, and that other sources of ROS are likely to be involved in these processes.
B. Monoamine oxidase
MAOs are flavoproteins located in the outer mitochondrial membrane (426) (Fig. 5). They catalyze the oxidative deamination of monoamine substrates producing H2O2 as a byproduct of the reaction (426), which is widely thought to contribute to oxidative stress resulting in neuronal degeneration. Two subtypes of MAO have been identified, MAO-A and MAO-B (426), and implicated with redox state modulation of both neurons and glia. The role of MAOs as redox modulators of learning and memory has been studied with the use of specific MAO inhibitors. Total inhibition of MAO-A by clorgyline (an MAO-A specific inhibitor) and pargyline (nonselective MAO inhibitor) induced increased locomotion, but no facilitation of spatial memory in the adult rat. In addition, inhibition of MAO-A combined with inhibition of MAO-B with deprenyl did not enhance spatial memory, suggesting that ROS produced by MAO do not alter normal memory function (28). In contrast, deprenyl was shown to improve spatial memory deficits in aged rats (221), where the effect of deprenyl was improved by the addition of estradiol (222). In a model of ischemia/reperfusion, deprenyl significantly reduced oxidative stress and prevented spatial memory deficits when administered before ischemia (220). Deprenyl also has been shown to protect aged, but not young, dogs from spatial memory deficits (173), supporting the notion that MAO-B-induced ROS production does not affect memory under normal conditions, but does contribute to memory-impairments during aging. Similar results were found where deprenyl protected rats against age-related short- and long-term memory impairments (101), iron-induced cognitive impairments (102), ischemia-induced memory deficits (262), and HIV-associated reduced verbal memory (366).

Two studies have assessed the effects of coadministration of deprenyl with donepezil, an acetylcholinesterase inhibitor used in the treatment of AD, on cognitive function. Administration of either drug alone improved spatial and associative memory. Both drugs used together at doses that are not individually efficacious improved memory, indicating that deprenyl potentiated the effect of donepezil on cognition (407, 418). Additional studies have shown that deprenyl has long-term beneficial effects in AD on memory modalities that require prefrontal areas of the brain, which are rich in dopamine receptors. In these studies, the effects of deprenyl were delayed, suggesting that the mechanism for memory improvement is through either neuronal rescue or neuroprotection (131). Studies in PD patients failed to demonstrate that deprenyl improved PD-related cognitive dysfunction, consistent with the idea that different mechanisms underlie cognitive impairments in PD and AD (97, 215).
C. NOS: NO (and related gases)
NOS is a flavin-containing Ca2+/calmodulin-dependent enzyme requiring NADPH, tetrahydrobiopterin, and molecular oxygen as cofactors (155) (Fig. 6A, B). The primary function of NOS is to generate NO gas from
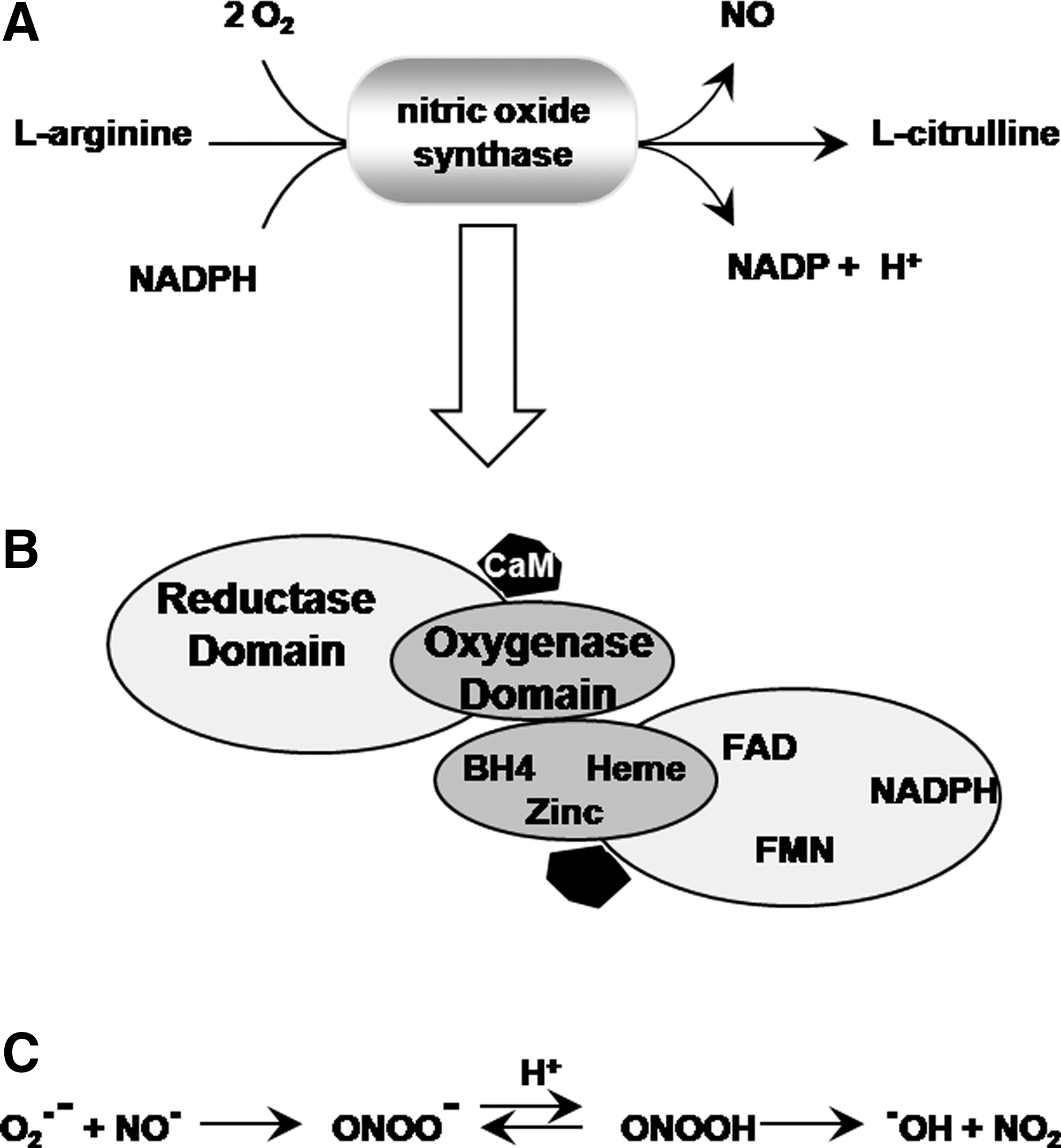
An interesting enzymatic feature of NOS is that if its substrate
NOS also has been shown to act as an NADPH:oxygen oxidoreductase, catalyzing the formation of H2O2 at suboptimal concentrations of either the substrate
These NOS-induced ROS (either superoxide or H2O2) may contribute to the events underlying LTP and memory formation. Their role in synaptic plasticity, however, as opposed to NO, has not been investigated. The main reason for the poor understanding of the role of NOS-induced ROS is that most studies using pharmacological inhibitors of NOS fail to discriminate between the two enzymatic activities of NOS (NO-producing versus superoxide/H2O2-producing). Thus, in such experiments, disruptions in learning and memory occurring after inhibition of NOS are always attributed to the decrease in NO production. No specific role of NOS-induced superoxide is addressed.
The role of NOS-generated superoxide has been studied by cotransfecting NOS with SOD. NOS expression leads to increased ERK activation, which suggests a role for NOS in synaptic plasticity and learning (434). This role could be attributed to either NO or NOS-generated superoxide. Cotransfection of SOD resulted in the inhibition of the NOS-mediated ERK activation, suggesting that either superoxide or H2O2 (generated by the dismutation of superoxide by SOD) contributes to synaptic plasticity via ERK activation. Mutation studies in the NOS enzyme helped to address this ambiguity. Mutations in NOS that silence the NO-producing domain did not bear any consequence on the NOS-induced ERK activation (434), suggesting that it is a superoxide-mediated event. This is supported by experiments using NOS mutants with an incompetent NADH domain that abolish NOS-mediated ERK activation (434). Collectively, these results suggest that NOS might contribute to synaptic plasticity and memory formation not only via the production of NO, but also via NADPH oxidation and production of superoxide.
Whereas the role of NO in learning and memory processes has been investigated extensively, research investigating the role of endogenous carbon monoxide (CO), a diatomic radical structurally similar to NO, has lagged behind. Although CO is traditionally regarded as an environmental toxic pollutant, it has been shown to be produced endogenously, usually via a reaction catalyzed by the enzyme heme oxygenase (HO) (241, 416). Endogenous CO functions as a signaling molecule in the nervous system and is involved in the regulation of neurotransmitter release [reviewed in ref. (446)]. Similar to NO, CO has been documented to have vasorelaxant properties, and it has been shown to activate guanylate cyclase (397), which is relevant for synaptic plasticity and memory. For example, several studies suggest a role for CO in LTP and memory. Various protoporphyrin inhibitors of HO were shown to block the induction of LTP in a dose-dependent manner and attenuate preexisting LTP in rodent hippocampal slices (396, 467, 468). These effects were also shown to be distinct from the effect of NO on LTP (467). Using similar inhibitors, CO has also been shown to play a role in Ca2+-dependent release of glutamate (388), which is an important component of LTP. On another hand, two studies using HO knockouts (341) or HO inhibitors (286) challenged these results and showed no clear effect of CO on LTP. Instead, the authors suggested that the observed effects of one HO inhibitor (chromium mesoporphyrin IX) may be due to a secondary action on NOS (286). These contradictory results may be in part due to the different concentrations of inhibitors used in the different studies. CO also has been implicated in memory formation, most notably tasks of spatial or avoidance learning (43, 52, 96, 132). On the other hand, no effect of CO has been observed on long-term depression (396), a form of synaptic plasticity manifested as a long-lasting decrease in synaptic strength. For further details, the reader is referred to two reviews covering the physiological role of CO andits role in learning and memory (95, 446). Although these findings suggest an important role for CO produced by HO in synaptic plasticity and memory, further studies are needed to determine whether CO is as important as NO for memory function.
In recent years, interest in other naturally occurring gases as signaling molecules has been revived, most notably hydrogen sulfide (H2S) [reviewed in ref. (219)]. Although H2S is not a radical itself, it is involved in redox signaling because of its connection to cysteine biology. H2S is produced by cystathionine β synthase, cystathionine γ lyase, or 3-mercaptopyruvate sulfurtransferase with cysteine aminotransferase in the brain, cardiac muscle, and vascular endothelium. It acts as a neuromodulator as well as vasodilator. Although its effect on learning and memory is poorly understood, it has been shown to enhance the activity of NMDA receptors and facilitate the induction of LTP. It has also been shown to protect neurons from oxidative stress. Recent progress in the studies of the physiological functions of H2S in neurons can be found in a recent review (219).
D. NADPH oxidase
NADPH oxidase is a large enzymatic complex whose primary function is the oxidation of NADPH with concomitant production of superoxide (Fig. 7). Although NADPH oxidase is better characterized as an enzyme of the immune system, it recently emerged as a neuronal enzyme with a potential role in synaptic plasticity and memory, which will be discussed in the following section.
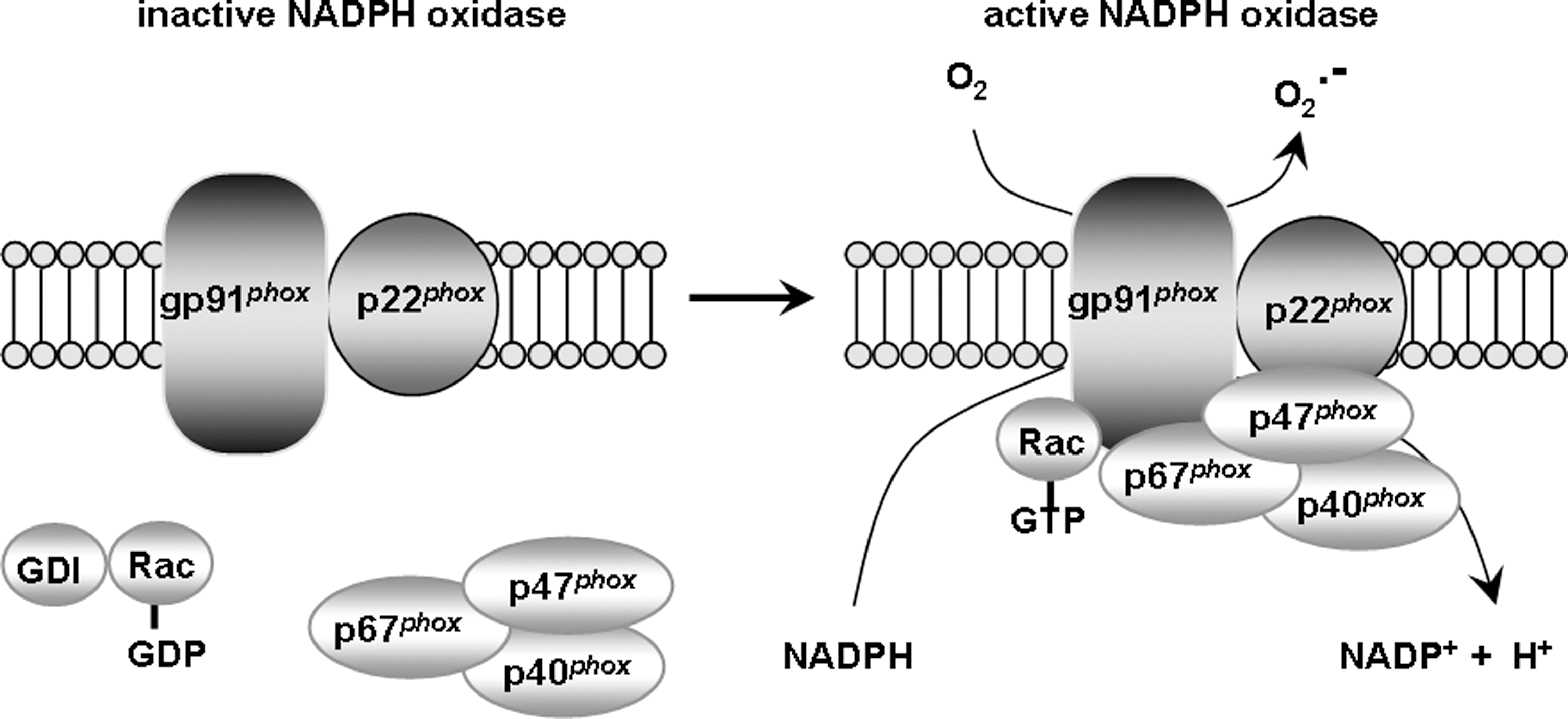
1. Structure and regulation of the NADPH oxidase
NADPH oxidase first was identified as a membrane-bound enzymatic complex of the phagosome (24, 240) (Fig. 7). It is made up of six subunits: One Rho-GTPase (usually Rac 1 or Rac 2) and five “phox” subunits (phox stands for phagocytic oxidase): gp91 phox (aka NADPH oxidase 2 [Nox2]), p22 phox , p40 phox , p47 phox , and p67 phox . In its latent status, gp91 phox and p22 phox are membrane-bound, whereas the four remaining subunits are cytosolic (24, 240). Upon stimulation, the cytosolic subunits translocate to the membrane to form a complex with the membrane-bound components (240, 404). The assembly of all the subunits yields an active enzyme that catalyzes the oxidation of NADPH into NADP+, releasing an electron in the process (240, 404). The electron is coupled to oxygen to generate superoxide. gp91 phox is the catalytic core of the enzyme, responsible for the transmission of an electron to oxygen (240, 404). However, the presence and correct positioning of all subunits is needed for this transfer to occur. p67 phox and Rac act as the activators of gp91 phox , whereas p47 phox acts as the organizer, ensuring that all subunits are properly aligned for optimal function (240, 404). Thus, the regulation of NADPH oxidase occurs via complex interactions between the various subunits, and proper activation and alignment of all subunits is required for full function of the enzyme.
NADPH oxidase also is regulated by multiple signaling pathways. Many of these regulatory pathways also are important for synaptic plasticity and memory (83, 98). Most notably, Ca2+ influx, particularly store-operated Ca2+ entry, has been shown to play a critical role in the activation of NADPH oxidase and the subsequent production of superoxide in neutrophils (62). Given the important role of Ca2+ in synaptic plasticity and memory, Ca2+-dependent NADPH activation and superoxide production could very likely be occuring at synaptic terminals, especially since NADPH oxidase recently was found to be localized to neuronal structures in multiple brain areas, including the hippocampus (216, 373, 415). Such regulation and localization suggest a direct role of NADPH oxidase in synaptic plasticity and memory and will be discussed in further depth in the following two sections.
2. NADPH oxidase in the brain
NADPH oxidase is classically linked to respiratory bursts of superoxide serving as host defense against bacteria in polymorphonuclear neutrophils (24, 240). This function and localization of NADPH oxidase have been studied extensively since it was first described over 40 years ago. However, recent findings indicate that this classical view of NADPH oxidase being exclusively an immunological enzyme as being outdated. Instead, in the past decade substantial evidence has accumulated indicating that NADPH oxidase has nonphagocytic functions. These extra-phagocytic functions of NADPH oxidase include regulating cellular growth and death, regulating endothelial function, and mediating intracellular signaling (154, 356).
The first nonphagocytic NADPH oxidase was discovered in 1999 in vascular smooth muscle cells and was named Nox1. This discovery was followed during the next 10 years by a large body of research investigating the existence and function of nonphagocytic Nox (402). We now know of the existence of at least seven different Nox isoforms (Nox 1–5 and Duox 1 and 2). Although structurally very similar and related, each of these isoforms is expressed in different locations and serves a different cellular function. Of particular interest to this review is the discovery that NADPH oxidase can be localized to multiple brain structures, most notably the hippocampus and amygdala (216, 373), which have primary roles in the formation and maintenance of memory.
NADPH oxidase in neurons was first demonstrated by Tammariello and colleagues, who showed that Nox2 subunits were expressed in rat isolated sympathetic neurons in culture. Expression of Nox2 was shown to be necessary for a proper apoptotic response after insult by nerve growth factor withdrawal (409). Shortly after this study, two immunohistochemical studies in mouse and rat tissue extensively characterized the distribution of various NADPH oxidase subunits in the central nervous system (216, 373). Using a variety of antibodies to stain mouse brain sections, widespread distribution of p40 phox , p47 phox , p67 phox , p22 phox , and gp91 phox was observed in neurons in all regions of the neuraxis. Particularly prominent regions included the cortex, striatum, thalamus, hippocampus, and amygdala, suggesting that NADPH oxidase may play a role in both normal neuronal function as well as neurodegeneration in the brain. Similar experiments in rat brain tissue yielded results showing strong p47 phox and Nox2 immunoreactivities in the cortex and hippocampus (373). In contrast, no particular localization of NADPH oxidase subunits was observed in either the thalamus or the amygdala, but some immunoreactivity was observed in cerebellar neurons. Nox4 and Nox5 were also found localized to the brain, particularly to the cortex, cerebellum, and hippocampal pyramidal cells (424). Nox3 was found highly expressed in the inner ear (27), and finally, a fully functional NADPH oxidase was observed in the lens epithelium (354). Besides their localization to the brain, little is known about the regulation of these different Nox isoforms and whether they are involved in ROS signaling in the brain.
Tejada-Simon and colleagues further characterized the mouse hippocampal NADPH oxidase and found that all of its subunits (including Nox2) are present in cell bodies and dendrites of hippocampal neurons. They also found that these subunits were enriched in synaptoneurosome preparations and that p67 phox colocalized with synaptophysin, suggesting that NADPH oxidase is localized at synaptic sites (415). Finally, the authors also showed that hippocampal NADPH oxidase is fully functional, producing superoxide in response to stimulation with phorbol esters (415).
Taken together, these studies clearly demonstrate localization of a functional NADPH oxidase in hippocampal neurons, suggesting that this enzyme might be the source of superoxide required for LTP and memory formation, a subject addressed in the following section.
3. NADPH oxidase in synaptic plasticity
We have argued that given its localization to key brain structures and to its functionality, NADPH oxidase is a prominent candidate as a source for ROS production during synaptic plasticity and memory formation. Another important feature is that NADPH can produce large amounts of superoxide in a very well-controlled manner (349). Thus, ROS production by NADPH oxidase is a punctuate event that can be turned on or off rapidly and specifically in response to particular extracellular stimuli and subsequent signaling events. Signaling events of particular interest to this review include the NMDA receptor-dependent activation of ERK. This type of signaling has been extensively shown to be involved in various forms of synaptic plasticity and memory (340, 405). Inhibiting NADPH oxidase with diphenylene iodonium (DPI) resulted in the inhibition of NMDA receptor-dependent ERK activation, suggesting a direct role for NADPH oxidase in this type of signaling that is associated with synaptic plasticity and memory (225). This observation was supported by studies in transgenic mice lacking the p47 phox subunit. These mice do not have a functional NADPH oxidase and do not exhibit NMDA receptor-dependent ERK activation (225). The p47 phox knockout mice, along with mice lacking the gp91 phox subunit of NADPH oxidase are typically used as models of chronic granulomatous disease, a genetic condition characterized by phagocytic dysfunction (199, 336). In addition, they can be used to study the effect of selective inhibition of NADPH oxidase activity on LTP induction and memory formation. Using these NADPH oxidase mutant mice, it was demonstrated that ablation of NADPH oxidase activity leads to obstruction of the early-phase LTP (223). Nox2 knockout mice also exhibited deficits in posttetanic potentiation, which is a form of short-term synaptic plasticity that is independent of NMDA receptors (223). These studies are unique in that they demonstrate a direct role for NADPH oxidase in LTP induction. This suggests that ROS involved in synaptic plasticity and memory (as will be described later in section IV b) could be generated by the large enzymatic NADPH oxidase complex.
IV. Physiological Roles of ROS
A. Synaptic signaling and LTP
Synaptic plasticity can be defined as the altering of the strength of preexisting synapses or the formation of new synapses. It is thought to be the physiological process that underlies learning and memory at the cellular level (117). One common form of synaptic plasticity is hippocampal LTP, which is defined as a long-lasting increase in synaptic strength and is widely used as a substrate of learning and memory. A wealth of research has been performed to clarify the role played by ROS in LTP induction. Evidence implicating ROS in the proper induction of LTP came from experiments involving the exogenous application of ROS to hippocampal slices, and the use of transgenic mice and pharmacological approaches to block the activity of ROS. Two main types of ROS have been examined, namely, superoxide and H2O2.
LTP studies in the rodent hippocampus have revealed that superoxide accumulates in hippocampal slices after NMDA receptor activation, which is a critical event in the induction of LTP (51). In addition, superoxide was shown to regulate the activities of ERK (209) and PKC (232), both of which are essential for normal LTP (Fig. 8). In hippocampal slices, scavenging superoxide with manganese porphyrin compounds that mimic SOD function blocked high-frequency stimulation–induced LTP (228). On the other hand, as mentioned above, reduction of superoxide production by genetic deletion of various NADPH oxidase subunits results in deficient LTP (223). In vitro production of superoxide via exogenous application of xanthine/xanthine oxidase (X/XO) to hippocampal slices resulted in a transient depression of synaptic transmission that ultimately recovered and resulted in the induction of LTP (231). This late-forming LTP was inhibited by the application of SOD, indicating that it is superoxide dependent (231). In line with these studies, mice overexpressing SOD, the main superoxide scavenger in mammalian cells, exhibit differential effects on LTP formation and maintenance depending on the SOD isoform expressed. Three SOD isoforms are present in cells and there are transgenic mice that overexpress each of them (Fig. 9A). SOD-1 (also known as Cu/Zn-SOD) and the extracellular SOD (EC-SOD) are responsible for scavenging the cytosolic and extracellular pools of superoxide, respectively, whereas SOD-2 (or Mn-SOD) is responsible for scavenging the mitochondrial superoxide pool. Using these mice, it was shown that both EC-SOD and SOD-1 transgenic mice exhibit deficits in LTP (205, 417), but the mechanisms responsible for these deficits are different. EC-SOD transgenic mice have deficient LTP because there is too little superoxide present in the hippocampus (417), whereas the SOD-1 mice have a deficient LTP because of increased H2O2 produced as a result of superoxide dismutation (205). Surprisingly, transgenic mice that overexpress SOD-2 exhibit normal LTP (188), indicating that the mitochondrial pool of superoxide probably does not play a role in LTP induction. This could perhaps be due to the nature of the mitochondrial membrane, which is composed of a phospholipid bilayer that hinders the free diffusion of mitochondrial superoxide to the cytosol (460). Because overexpression of SOD-2 should result in increased H2O2 production, it appears that mitochondrial H2O2 does not contribute to LTP, contrary to its cytoplasmic counterpart, which as discussed later likely plays a complex role in learning and memory. In contrast to superoxide, H2O2 has been recently shown to cross membranes. Specifically, recent evidence indicates that select aquaporin homologs from plants and mammals (aquaporin 8) have the capacity to channel H2O2 across membranes (48). The mammalian nervous system contains predominantly aquaporins 1, 4, and 9 (14, 25, 306) and brain inner-mitochondrial membranes have been shown to specifically contain aquaporin 9 (13). Considering that the permeability of different aquaporin subtypes to H2O2 has not been investigated, to the best of our knowledge, no direct in vivo or in vitro evidence is available to suggest that the brain mitochondrial membrane is permeable to H2O2. Therefore, the idea that excessive superoxide dismutation inside the mitochondria could increase cytoplasmic H2O2 is possible, but highly speculative at this point.
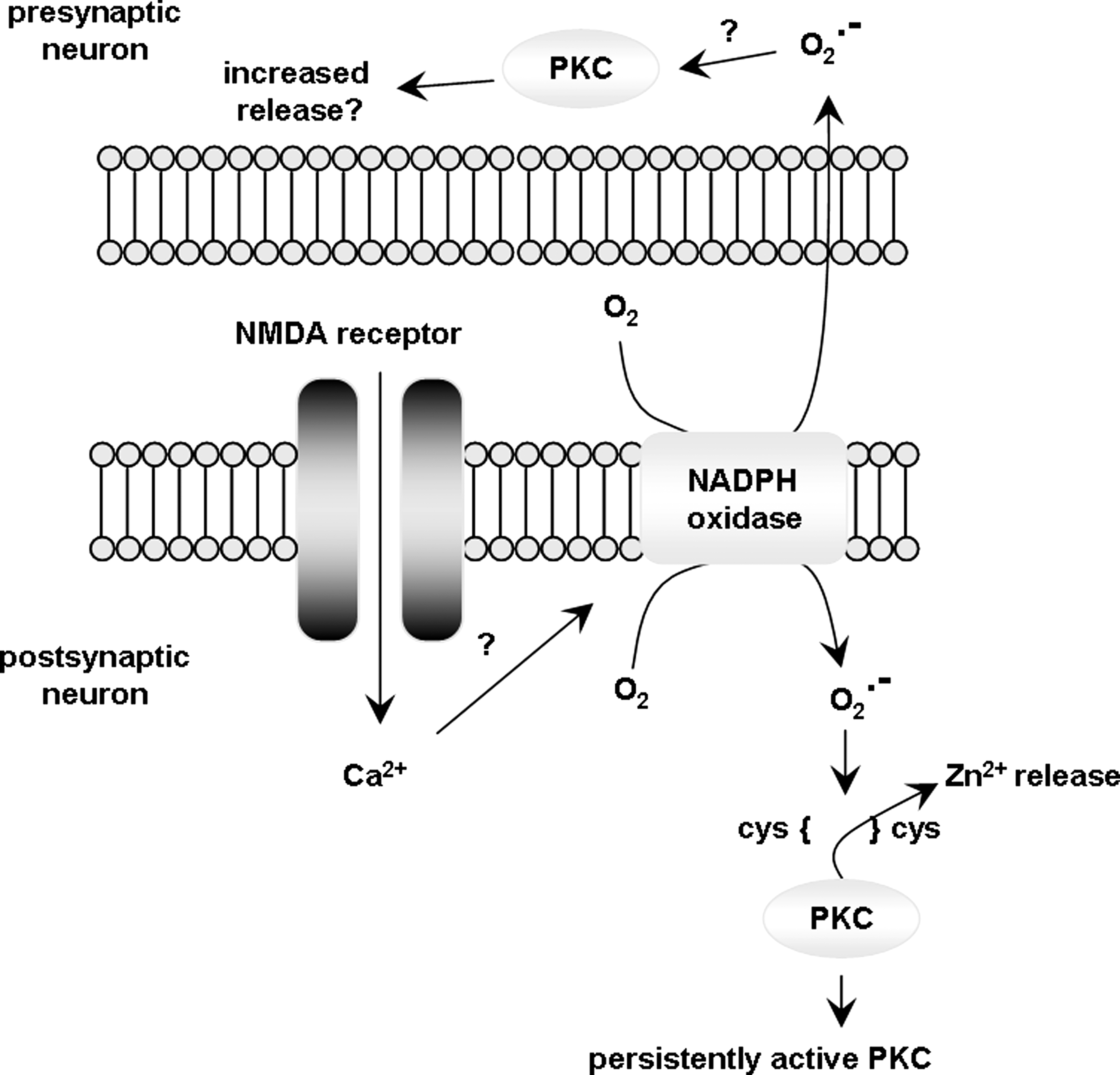
In studies where superoxide was shown to modulate LTP, the addition of catalase to quench the superoxide dismutation product H2O2 (Fig. 9B) results in an attenuation of LTP, indicating that H2O2 also plays a role in LTP (229). However, investigations of the role of H2O2 in the induction of LTP have produced divergent results. On one hand, studies in rat hippocampal slices demonstrate that H2O2 inhibits LTP and causes a long-lasting depression of population spikes and excitatory postsynaptic potentials (EPSPs) (211). On the other hand, H2O2 causes a significant increase in EPSPs in sympathetic preganglionic neurons (252). The apparent discrepancy in these reports can be explained by the large differences in the doses of H2O2 used in the different studies (mM vs. μM). Indeed, Kamsler and Segal have shown that H2O2 can either potentiate or depress high-frequency stimulation–induced LTP in a concentration-dependent manner (204). They also observed a dose-dependent effect of H2O2 on long-term depression. In a subsequent study (205), these authors proposed a model by which H2O2 modulates synaptic plasticity based on the background concentration of H2O2 present in the system at the time of stimulation: in young wild-type mice, the background levels of H2O2 are very low, and therefore stimulation-induced transient increases in H2O2 facilitate the induction of LTP. Exogenous addition of H2O2 pushes the system to a threshold that is detrimental to LTP induction. SOD-1 transgenic mice have a higher ambient H2O2 concentration and maintain an intracellular redox milieu of proteins adapted to functioning in higher H2O2 levels, and is therefore desensitized to the small rise in H2O2 that follows stimulation, resulting in deficient LTP. In such an instance, addition of exogenous H2O2 boosts the small stimulation-induced H2O2 increase to a level sufficient for LTP induction. During aging, the opposite effects occur, as will be discussed later.
In summary, how ROS contribute to LTP depends on the identity of the specific ROS, the location of ROS production, and the concentration of the ROS. Along the same lines, induction of oxidative stress in honeybees by injections of ferrous ammonium citrate causes learning and memory impairments in a dose- and time-dependent manner (125).
LTP is the leading candidate for the cellular basis of learning and memory (117). Hence, in addition to its role in modulating synaptic plasticity at the cellular level, ROS play a prominent role in the formation and maintenance of memory, a subject discussed in the next section.
B. Learning and memory
In the previous section, we discussed the role of ROS in the induction of LTP, a commonly studied cellular substrate for learning and memory (117). The role of ROS also has been studied from a behavioral point of view, especially with respect to the limbic system and cognitive function. These studies show that, similar to their effect on the neuronal circuitry and synaptic plasticity, ROS act as a double-edged sword with respect to memory function. On one hand, ROS have been shown to be an essential signaling component for memory formation; on the other hand, they also have been shown to impair the same neuronal networks necessary for memory function. The dichotomy of ROS function is a recurrent theme in redox regulation of learning and memory and will be further discussed in this section.
ROS have long been linked to impaired learning and memory processes, mainly via their toxic effect on the neuronal circuitry necessary for memory formation (see section V of this review). The beneficial role of ROS on the formation of memory has been implied from LTP studies described in the above section. However, their involvement in memory function was not directly demonstrated until the late 1990s with the generation of transgenic mouse models of the various SOD isoforms, and with mice with genetic deletions for NADPH oxidase subunits.
The earliest evidence for the involvement of ROS, specifically superoxide, in learning and memory came from the study of transgenic mice overexpressing SOD-1 (143). Utilizing a water maze paradigm designed to assess spatial learning and memory, it was shown that SOD-1 transgenic mice display impaired hippocampus-dependent spatial memory function (143). As discussed earlier, hippocampal slices prepared from SOD-1 transgenic mice exhibit deficient LTP (143), indicating that superoxide is necessary both for memory formation and its cellular substrate at the synaptic level. SODs are responsible for dismutating superoxide into H2O2, which is then transformed into water by catalase. Therefore, mice overexpressing SODs have decreased levels of superoxide, but also exhibit increased H2O2 as a byproduct of the dismutation reaction. It is therefore conceivable that at least part of the observed effect on LTP and cognition could be due to increased H2O2 as opposed to the exclusive removal of superoxide. In the same study described above, hippocampal slices from SOD-1 transgenic mice were treated with either an antioxidant spin-trapping agent or catalase and the LTP deficits were reversed, indicating at least a partial role for H2O2 in the LTP impairment (143). The same effect on behavior has not yet been investigated, so it is not clear whether the behavioral deficits observed in mice overexpressing SOD-1 are solely due to superoxide or involve other ROS. EC-SOD mice also have been investigated in the context of learning and memory. Two groups have shown that young EC-SOD mice exhibit impaired memory function (247, 417). Mice overexpressing EC-SOD had impairments in the consolidation of contextual fear memory as assessed by a hippocampus-dependent fear conditioning paradigm (417). In addition, the EC-SOD transgenic mice exhibit significant impairments in the acquisition of spatial memory using the radial arm maze, which also is a hippocampus-dependent task (247). In the latter study, the authors showed that EC-SOD overexpression also impacted other brain regions. They found that learning in the young EC-SOD mice is highly dependent on the motivational state of the mouse as induced by food restriction. Mice with low motivation have impaired learning, whereas mice with high motivation are able to learn, though at a slower rate, and express normal long-term memory. These studies indicate that ROS also affect brain regions distinct from the hippocampus (247). EC-SOD overexpression also was investigated in pharmacological studies where hippocampal slices from the EC-SOD transgenic mice were treated with the copper chelator diethylcarbamate with the intention of lowering EC-SOD activity by neutralizing its cofactor. Such treatment resulted in a recovery of the LTP impairments observed in young EC-SOD transgenic mice (417), but has not been tested in behavioral experiments.
Mice overexpressing SOD-2 have been reported to have no significant behavioral phenotypes as far as learning and memory is concerned (188), which is consistent with the lack of LTP phenotypes displayed by these mice (188). This observation reinforces the idea introduced above that mitochondrial superoxide might be restricted due to sequestration in the mitochondrial membrane. However, we will discuss later that quenching mitochondrial superoxide with SOD-2 is beneficial to AD-related cognitive dysfunction (115, 277).
The source of ROS required for normal learning and memory function has been investigated with NADPH oxidase mutant mice that were described earlier. In addition to its involvement in LTP, NADPH oxidase is required for hippocampus-dependent memory. Specifically, gp91 phox mutant mice have mild deficits in spatial memory formation as measured by the Morris water maze task and that p47 phox mutant mice have deficits in associative memory as measured with a contextual fear conditioning paradigm (223). These mice also exhibit deficient performance in the rotating rod and open field tests, indicating that other brain areas also may be affected by impairing NADPH oxidase function (223).
In summary, in this section we have described numerous experiments directly linking superoxide and H2O2 in memory acquisition and consolidation in young, healthy mice. In addition to the modulatory role of ROS in the signaling events underlying LTP and memory formation in the young adult mice, ROS also play an important role in the impairment of memory in the aged and diseased brain. In the next section, we will discuss the importance of redox regulation of memory function in the context of physiological aging, neurodegenerative diseases, and ischemic insult.
V. Pathological Release and Effects of ROS
Although ROS contribute to the regulation of long-term functional changes in neurons and appear to be required for normal learning and memory in healthy, young adult animals, the opposite appears to be true of the aged and diseased brain. The high energy demand of the brain, together with its high level of ROS production, places it at risk during conditions of increased stress, such as the ones occurring during aging, in neurodegenerative disorders, and after either traumatic or ischemic insults. Altered ROS levels in the brain are compounded by aging-related reductions in antioxidant defense, leading to a disruption of the fragile ROS balance. In other words, when ROS levels become too high, their ability to trigger oxidative stress outweighs their role as signaling molecules, and the outcome shifts from positive modulation of synaptic plasticity and memory to impairments in these processes. In the following sections, we will discuss the toxic effect of ROS on cognition in the context of physiological aging and oxidative stress conditions such as ischemia and neurodegenerative diseases.
A. ROS in physiological aging
Oxidative stress has long been linked to aging by the free radical theory of aging proposed by Harman in 1956 (169). This theory postulates that the age-dependent accumulation of oxidative damage to cellular macromolecules causes a progressive functional deterioration of cells, tissues and organ systems, which then exhibit functional senescence and ultimately death (169). A variety of functions are altered in an aging individual, including locomotor, reproductive, sensory, immune, and memory. In most cases, there is evidence that oxidative damage contributes to the aging-related decline in these functions. This section will be dedicated to reviewing the evidence implicating ROS and oxidative damage in age-related cognitive decline.
Age-related cognitive decline includes functions such as short-term memory, problem-solving abilities, information processing speed (87), and the cellular mechanisms underlying these mechanisms such as LTP.
LTP impairments have been observed in aged animals (31) and specific deficits in LTP in area cornu ammonis (CA)1 and the dentate gyrus of the hippocampus have been attributed, at least in part, to the rising levels of ROS. Consistent with the oxidative damage theory of aging, overexpression of either EC-SOD or SOD-1 throughout the lifetime of mice protected against age-related LTP deficits (190), and improved performance in spatial memory (203, 247). As mentioned earlier, overexpression of SOD-2, the mitochondrial isoform of SOD, had no effect on either age-related LTP deficits or memory impairments associated with aging (188), suggesting that the mitochondrial superoxide pool does not contribute to these events, perhaps due to limited diffusion across the mitochondrial membrane. A pharmacological approach using antioxidants also has been used to assess the role of oxidative damage in senescence of brain function. Continuous systemic administration of two SOD/catalase mimetics (EUK compounds) in aged mice reduced the age-related oxidative damage to protein, lipids, and DNA, and resulted in better memory performance in a fear conditioning paradigm (255). Administration of a carboxyfullerene SOD mimetic to wild type mice, starting middle age, resulted in improved age-related performance in the Morris water maze and increased the life span of these mice (347). Other dietary antioxidants also have been used to examine synaptic plasticity and memory in aged animals, including vitamin E, coenzyme Q, vitamin C, lipoic acid, berries, the nitrone spin trap α-phenyl-N-tert-butyl nitrone (PBN), and various combinations of these compounds (see section V for details and references). Studies with these compounds largely show decreased markers of oxidation as well as improved synaptic plasticity and memory function in aged animals, generally in spatial and working memory tests. Further, combinations of various antioxidants improve cognition better than larger doses of a single antioxidant, suggesting that synergism may be taking place (281). Also, large doses of single antioxidants such as vitamin E have been associated with toxicity in human patients with chronic diseases (292). Therefore, a combination of multiple antioxidants and supplements might be a better approach for the prevention of age-associated memory dysfunction.
Because oxidative damage accumulates with age, an important prediction of the oxidative stress theory of aging would be that the severity of cognitive decline should correlate with the amount of accumulated oxidative damage (392). This prediction was confirmed in a study where the investigators subjected young (4-month old) and aged (22-month old) mice to a battery of behavioral tests and then measured the extent of oxidative damage in multiple brain regions. In support of ROS involvement in aging-related cognitive decline, they found that age-related spatial memory deficits were directly correlated with the amounts of oxidized proteins in the cortex (134).
Data from clinical trials in humans support the animal research, and show variable antioxidant benefits on memory function depending on the compound and dose used. For example, serum levels of the antioxidants vitamin C, vitamin A, carotenoids, and selenium do not correlate with poor memory performance assessed by a delayed recall test in an elderly multiethnic sample of individuals. Only vitamin E showed benefit in this particular setting (328). There is other research showing similar findings (162, 329). Additional data have attributed beneficial effects on cognitive function to a combination of vitamin C, vitamin E, and carotene (438). These results are supported by studies showing that vitamin E and vitamin C supplementation, and/or a multivitamin regimen results in an improvement in aging-related cognitive decline and vascular cognitive impairment (278). Clinical trials in elderly men receiving β-carotene supplementation suggest that short-term antioxidant supplementation had no effect on cognitive dysfunction, whereas long-term supplementation positively impacted it (156). In a randomized, controlled trial of multivitamin supplementation to an aging population, it was reported that there was no significant effect on age-related cognitive dysfunction. However, the authors did notice a trend toward improved cognition in a particular subgroup of their study population: those older than 75 years were more susceptible to vitamin deficiency (284). Of all the trials assessing the link between antioxidants and cognition, very few measure cognitive decline as opposed to cognitive dysfunction. Of particular interest, a study of antioxidant supplementation in aged, free-living adults showed a 36% reduction in the rate of cognitive decline over 3 years for individuals that fall in the highest to lowest quintiles of vitamin E intake (296). This study suggests that perhaps antioxidants can slow the rate of cognitive decline, as opposed to totally eradicating cognitive dysfunction.
Interpretation of all of the clinical trials collectively becomes quite complicated, especially when some show cognitive benefits of a certain antioxidant versus others not showing a cognitive benefit. The discrepancies could arise from several reasons, such as the presence of confounding factors that were unaccounted for. For example, people who eat diets rich in antioxidants or take vitamin supplements generally tend to lead healthier lifestyles, and therefore the effects of antioxidants in such individuals may be potentiated by other factors such as lower caloric intake and/or physical exercise. Another possibility for the differential results of these studies could be the different duration of antioxidant treatment. This argument is supported by a study showing that long-term β-carotene is beneficial for age-related cognitive dysfunction, whereas short-term treatment is not (156). Other possibilities may include suboptimal dosage and reduced bioavailability of synthetic compounds. Despite the range of variability in these clinical studies, it remains clear that ROS participate in the events leading to cognitive dysfunction through damaging oxidizing effects on protein, lipids, and DNA. Whether dietary antioxidants constitute appropriate therapy remains open for debate.
B. ROS in AD
AD is a progressive neurodegenerative disorder characterized by cognitive dysfunction, memory decline, speech loss, and drastic personality changes (414). The disease progression phenotype varies greatly amongst individuals based on general health and social status. This heterogeneity makes it difficult to pinpoint specific clinical determinants for the onset and progression of AD. Increasing evidence, however, both from animal models as well as human patients, implicates oxidative damage in AD-related cognitive dysfunction (175, 273, 391). For example, subjects with established AD-related cognitive dysfunction have been shown to have an imbalance in oxidant/antioxidant levels (390). Also, Aβ, the major pathological determinant of AD, has been shown extensively to act as an oxidizing molecule (40, 68). It has been linked to increased H2O2 production and decreased cytochrome C oxidase activity in the Tg2576 mouse model of AD (266). Both Aβ and its precursor amyloid precursor protein (APP) have been shown to enter the mitochondria and compromise its function through energy processing dysfunction, causing the release of large amounts of ROS (15, 267). Superoxide has been specifically implicated with Aβ vascular deposition and cerebral hypoperfusion (166), which also are thought to be an integral part of the disease and its ensuing cognitive dysfunction. In addition, superoxide has been shown to mediate cell death by enhancing the impact of presenilin-1 mutations, specifically mitochondrial Ca2+ accumulation and membrane depolarization after exposure to Aβ42 (157). Aβ also can initiate a signaling amplification cascade that culminates in the inactivation of SOD-2, leading to further accumulation of mitochondrial superoxide (16). One particular study demonstrated a global increase in SOD-2 in AD patients, presumably as a compensatory mechanism to the increased oxidative stress originating from mitochondrial superoxide production (272). This increase was less pronounced in hippocampal area CA1 (272), which suffers the most damage during AD (439) and incidentally happens to be one of the major areas involved in the learning and memory processes (439). In addition, the same SOD/catalase mimetics used in the aging studies described above proved effective against Aβ toxicity in cell culture (65). Finally, we have shown that Aβ-induced ERK phosphorylation in organotypic hippocampal cultures was mediated by redox signaling through NADPH oxidase, suggesting similar mechanisms occur during AD (372). Because ERK plays a critical role in LTP (194, 210), the most probable cellular substrate for learning and memory (117), this study indirectly implicated ROS produced by NADPH oxidase in the learning and memory dysfunction associated with AD.
Despite the overwhelming evidence for the pro-oxidant role of Aβ, several other studies demonstrated free radical involvement in AD before Aβ pathology (236, 399), and also have implicated ROS in increases in Aβ levels. In addition, transgenic AD model mice with reduced SOD-2 activity exhibit increased amyloid plaque burden (249) and increased tau phosphorylation (287), both of which are implicated with AD-related cognitive dysfunction. SOD-2 deficiency also has been shown to accelerate the onset of a number of behavioral deficits in the hAPP mouse model of AD (123). Recent studies have further explored the involvement of mitochondrial superoxide in AD-related cognitive dysfunction. We and others concurrently found that overexpression of SOD-2 in the Tg2576 AD mouse model reduced the levels of hippocampal superoxide, presumably from mitochondrial origin, and led to the prevention of both associative and spatial memory deficits as measured by a fear conditioning paradigm and the Morris water maze, respectively (115, 277). Our data are in agreement with several studies that demonstrate the involvement of mitochondrial superoxide, through SOD-2 deficiency, in increasing AD-related cognitive dysfunction and correlating reduced mitochondrial ROS with improved cognition (112, 123, 249, 287). Of particular interest are two studies by Dumont and associates showing that AD-related increases in mitochondrial ROS and impairments in memory function were exacerbated by deficiency in the α-ketoglutarate dehydrogenase mitochondrial enzyme complex (114) and improved by SOD-2 overexpression (115).
In recent years, an increasing number of studies have been focused on characterizing antioxidant therapies to help prevent and/or treat the declining cognitive functions of affected individuals. Intrahippocampal injections of a lentiviral vector carrying the nuclear factor E2-related factor 2 (Nrf2) transcription factor improves spatial learning in the APP/PS1 mouse model of AD concomitant with a change in Aβ solubility consistent with reduced Aβ toxicity (207). Nrf2 binds to and activates the antioxidant response element (ARE) enhancer sequence, which upregulates a cassette of antioxidant enzymes, thereby constituting an important oxidative stress response (305). Nrf2/ARE pathway activity is reduced during AD; therefore, boosting its activity via agents such as tBHQ (118) or tert-butylhydroquinone (208) may prove beneficial for AD-related memory dysfunction. In addition, animal models have demonstrated that dietary supplementation with antioxidant vitamins can decrease oxidative stress and either prevent or reverse the age-related changes in cognitive functions (198, 314, 352, 451). Clinical trials, however, have produced conflicting results concerning the therapeutic efficacy of antioxidant supplementation during AD (121, 122, 258). Similar to the conflicting results of antioxidant therapy during physiological aging, this discrepancy may be due to multiple factors such as reduced bioavailability and/or poor specificity of these treatment regimens. As described above, we and others have shown specific involvement of mitochondrial superoxide in AD-related cognitive dysfunction in mouse models, and therefore targeted mitochondrial antioxidant therapy may prove more useful in the treatment of AD-related cognitive dysfunction.
C. ROS during hypoxia/ischemia and traumatic brain injury
It has long been established that ROS are produced in association with traumatic brain injury (TBI), during ischemia and hypoxia, as well as during the reperfusion and reoxygenation after these events (23, 75, 420, 425). Ischemia is characterized by a transient block in blood flow that leads to decreased glucose and oxygen perfusion to the brain, resulting in energy failure, neuronal dysfunction and death, and impairments in cognitive functions, especially if the focal point of the ischemia was in learning and memory regions of the brain. After an ischemic insult, a core of tissue is usually severely injured and is surrounded by an area called the penumbra, where neurons are impaired, but sufficiently active to maintain membrane potentials (185). Although the precise timing and mechanisms by which ischemia causes neuronal dysfunction and death are not fully understood, there seems to be a consensus that three main events are involved: (a) oxidative stress, (b) glutamate-induced excitotoxicity, and (c) apoptotic-like cell death (111, 257). These three events together lead to the activation of the same cellular machinery involved in the induction of synaptic plasticity. After oxygen and glucose deprivation, neurons fail to generate sufficient ATP leading to loss of ionic gradients and release of glutamate, which activates NMDA receptors that results in excessive Ca2+ entry into neurons. Although NMDA receptor activation (54, 72, 264) and Ca2+ entry (57, 265) are necessary for the induction of LTP, the excessive activation of NMDA receptors leads to toxicity and impairment of LTP. At the same time, the failing of mitochondrial energy metabolism coupled to the rush of oxygen during reperfusion results in increased production of ROS, which adversely affects synaptic plasticity, therefore compounding the effect of ischemia on these events. Given the established critical role of oxidative stress in the events shortly after ischemia, a large body of research effort has been focused on assessing the efficacy of a variety of antioxidants at quenching ROS after ischemic events and at improving ischemia-induced learning and memory impairments. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a free radical scavenger, has been shown to protect the rat hippocampus from ischemia-induced LTP impairments as well as increases in hydroxyl radical, with a specific very early therapeutic time window (day 0–1 postischemia) (309). In a separate study, edaravone was found to afford significant protection against learning and memory impairments and altered neuronal morphological integrity when administered for 2 days in the acute phase after ischemia (317). Both of these studies suggest that free radical formation after ischemia/reperfusion is a pivotal trigger of brain dysfunction after global ischemic stroke. MAO-B has been shown to participate in the generation of hydroxyl radicals during ischemia (370). Hence, several groups investigated the effects of deprenyl, the MAO-B irreversible inhibitor, and found that it reduced ischemia-induced oxidative stress and prevented spatial memory deficits and apoptotic neuronal cell death (220, 262). One of these studies administered deprenyl before ischemia (220) and the other administered it right after the ischemic insult (262), in the early stages of dysfunction. These results indicate that ROS play a role in ischemia-related cognitive deficits and that therapeutics should be targeted to the time window immediately surrounding the ischemic injury to quench oxidative stress before the occurrence of irreversible damage. In addition, idebenone, a coenzyme Q analog, also ameliorated memory impairment induced by cerebral vascular disturbance in rats (453). Several other antioxidants of either chemical or plant origin (including, but not limited to, L-NAME, flavonoids, and carnosine) also have been used and produced very similar results in terms of improvement of memory function as well as reduction of oxidative stress after ischemia (294, 326, 344, 351, 376), strengthening the postulate that ROS play a critical role in the cognitive impairments associated with ischemia and reperfusion. In addition to ischemia, ROS have been shown to contribute to the secondary injury process after TBI. ROS scavenging compounds have been shown to have neuroprotective properties in various models of experimental brain injury, including TBI. For example, nitrone spin traps such as NXY-059 (91), PBN, and its sulfo-derivative, 2-sulfo-PBN (274), administered after moderate to severe lateral fluid percussion injury in rats have been shown to be neuroprotective and to improve cognition. Vitamin E also has been shown to protect against oxidative damage and learning disability after mild TBI in rats by affecting molecular systems involved in the maintenance of synaptic plasticity, such as brain-derived neurotrophic factor and the silent information regulator 2 (443). Sulforaphane, a naturally occurring compound found in cruciferous vegetables, has been used after TBI, and was demonstrated to reduce cerebral edema (463) and attenuate blood–brain barrier permeability (464) via induction of the Nrf2/ARE pathway, which is primarily a free radical scavenging system. Sulforaphane also has been shown to improve performance in the Morris water maze after TBI (99). These studies suggest a major role for ROS in the learning and memory pathology associated with TBI.
D. ROS in multiple disease states
ROS have been implicated with the etiology of multiple disease states, such as PD, diabetes, and homocysteinuria. In the following section, we will address the involvement of oxidative stress specifically in the learning and memory deficits accompanying each of these diseases.
1. Parkinson's disease
PD is a neurodegenerative disease of the basal ganglia, characterized by dopaminergic neuronal loss, mainly in the substantia nigra, and results in bradykinesia, tremor, and postural instability. Although the mechanisms of cell death in PD are not yet fully understood, oxidative stress is known to play an important role. In fact, the oxidation of dopamine during PD generates toxic semiquinones and the accelerated metabolism of dopamine by MAO-B induced the production of excessive H2O2 and hydroxyl radicals (313). Further evidence for the role of oxidative stress in PD comes from studies showing that the two main misfolded proteins of PD, parkin and α-synuclein, are a source of oxidative and nitrative stress (59). PD was found to be associated with increased oxidative damage to DNA (38), lipid peroxidation (107, 108, 458), and protein carbonylation (10, 150). Other studies showed increased brain levels of iron (105, 151) and reduced levels of ferritin (106), which creates a source of free radicals that potentiate the oxidative damage, especially in the substantia nigra. Although there is a large body of literature discussing the beneficial effect of antioxidant therapy for PD, no specific reports directly discuss the involvement of oxidative stress in PD-related learning and memory deficits. In a recent study investigating the effect of deprenyl and tocopherol on the PD-related cognitive dysfunction, the authors reported an improvement in the cognitive functions of the group treated with antioxidants. However, the authors do mention that this could be due to recruitment bias and/or limitations of the mini mental state examination (MMSE) test used to assess mental function (423).
2. Diabetes
Growing data report learning and memory problems (135, 161, 441) as well as LTP (21, 202) deficits associated with diabetes mellitus. Expression of neuronal NOS mRNA and protein in the hippocampus of diabetic rats is decreased (357), suggesting a critical role for NO in synaptic plasticity associated with diabetes. Supporting studies, using the active avoidance learning test in rats, demonstrated the involvement of NO imbalance in the occurrence of diabetes-induced learning deficits (234). Multiple studies using a variety of antioxidants to treat and/or prevent the cognitive dysfunction associated with diabetes also demonstrated an involvement of ROS in these events. For example, melatonin and vitamin E have been shown to improve diabetes-induced spatial memory impairments (421). Lycopene also has been shown to decrease markers of oxidative stress associated with diabetes and to attenuate spatial memory deficits (235). Thus, ROS likely play a role in cognitive dysfunction associated with diabetes.
3. Homocysteinuria
Homocysteinuria is a metabolic disorder characterized by deficiency of the enzyme cystathione β-synthase activity leading to accumulation of homocysteine. Homocysteine causes free radical formation and severe neurological dysfunction, which is thought to be due to the increased oxidative stress (263). Administration of homocysteine to adult rats caused severe memory impairments in the step-down inhibitory avoidance test that was prevented by pretreatment with vitamins C and E, suggesting that ROS play a critical role in the onset of memory dysfunction during homocysteinuria (358). Using the same model system it was shown that homocysteinuria caused severe learning and memory deficits in the passive avoidance and Morris water maze tests, and that these deficits were reversed by chronic melatonin treatment, which also significantly reduced the levels of lipid peroxidation (35). Taken together, these findings suggest that homocysteinuria causes cognitive deficits via increased oxidative stress.
VI. Antioxidant Defenses Against Pathological ROS
In this review we have discussed extensively the important role played by ROS both as cellular messengers in physiological events such as learning and memory and as toxic molecules in pathological events such as ischemia, aging, and neurodegeneration. Because of this dichotomy in the function of ROS in mammals, the balance between ROS formation and scavenging is of utmost importance for proper neuronal function. Multiple sources of ROS were discussed earlier in the review. The following section will be dedicated to presenting the variety of antioxidant mechanisms, which constitute first line of defense when ROS start exceeding their physiological levels. Antioxidant mechanisms include a variety of enzymes, such as SOD, catalase, glutathione peroxidase (GPx), and glutathione reductase (GR). Organisms can also synthesize nonenzymatic antioxidants such as glutathione (GSH), coenzyme Q, and uric acid. In addition, antioxidants can be obtained through the diet, including vitamins and minerals (such as ascorbate, carotenoids, tocopherol, flavonoids, selenium, manganese, copper, and zinc), and a variety of herbs (such as curcumin, gingko, and grape-seed extract).
A. Antioxidant enzymes
1. Superoxide dismutases
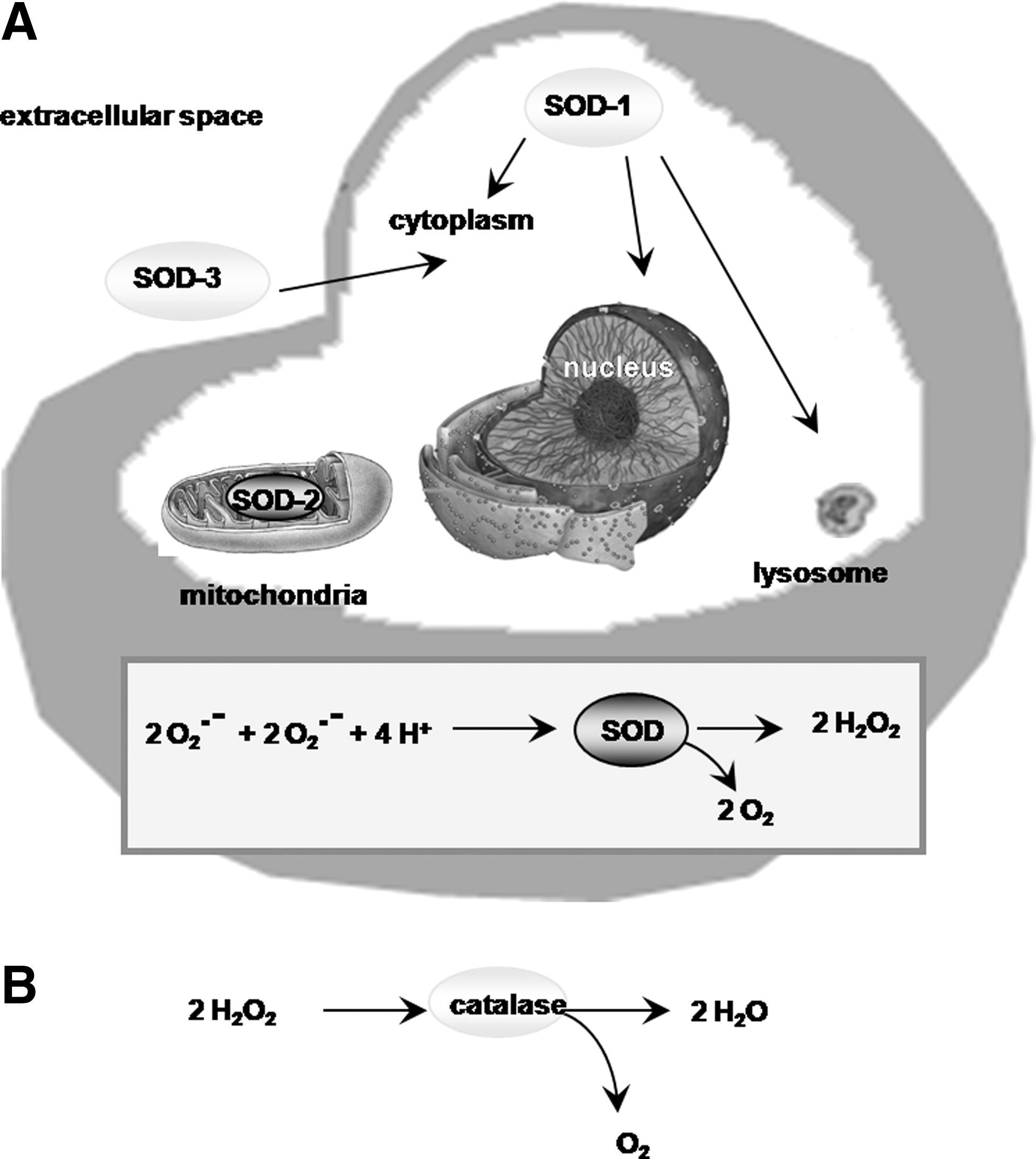
SODs are a class of metalloenzymes involved in catalyzing the dismutation of the superoxide radical into oxygen and H2O2 and, as such, constitute a very important element of the antioxidant defenses of organisms (136) (Fig. 9A). In mammals, including humans, there are three isoforms of the enzyme that are encoded by three different genes. The SOD isoforms differ with regard to their localization as well as the metal bound to them. SOD-1 is a cytoplasmic enzyme with copper and zinc acting as cofactors, and therefore also is known as Cu/Zn-SOD. SOD-2 is a mitochondrial enzyme with manganese at its core and hence, is also referred to as Mn-SOD. SOD-3 also is a Cu/Zn enzyme, but is localized extracellularly and is often called EC-SOD. The three SOD isoforms have some structural differences, with SOD-1 being functional as a dimer, whereas SOD-2 and EC-SOD are tetramers. The physiological importance of all three SODs is illustrated by the multiple pathologies exhibited in mutant mice lacking each isoform. SOD-1 knockout mice develop hepatocellular carcinoma, age-related mass loss, increased formation of cataracts, and a reduced life span (119, 298). SOD-2 knockout mice survive only a few days postbirth and then die of severe oxidative stress (251). EC-SOD knockout mice do not exhibit an obvious major phenotype, but do develop hypersensitivity to free radical-induced injury (371). The physiological importance of the SODs is also illustrated by studies demonstrating that upregulation of SODs serve to attenuate neuropathological conditions such as ischemic injuries (212) and AD (115, 277).
The use of SOD transgenic mice and/or pharmacological mimetics has been instrumental in determining the functions of these enzymes and the role of superoxide in synaptic plasticity and memory. In line with the dual effect of superoxide and H2O2 as crucial signaling molecules in healthy organisms, but involved in oxidative stress in the aging and/or diseased brain, the effects of overexpressing SODs were found to be dependent on the age of the animals.
a. Synaptic plasticity and memory in young mice
LTP studies in hippocampal slices from young EC-SOD transgenic mice revealed impairments in LTP, without affecting normal synaptic transmission (417). The LTP impairments persisted even after treatment with catalase to quench excessive H2O2, indicating that superoxide is crucial for LTP under physiological conditions (417). In line with its effect on LTP induction, significant deficits in the acquisition of spatial memory were observed in mice overexpressing EC-SOD (247). These mice also are deficient in the consolidation of contextual fear memory, which requires the hippocampus (190). LTP impairments in slices from EC-SOD transgenic mice can be reversed by treating the slices with the copper chelator diethylcarbamate, raising the possibility that targeting the cofactor of EC-SOD modulates its action on memory formation (417). Similar to the studies of EC-SOD transgenic mice, SOD-1-overexpressing mice, which have a higher level of H2O2, have normal baseline synaptic physiology, but are deficient in LTP and spatial memory formation (143). The effects of SOD-1 overexpression on LTP were reversed by H2O2 quenching antioxidants such as catalase and spin trap reagents (143). Whether the same H2O2 antioxidant treatments improve the impaired memory in the SOD-1 mice has not been investigated.
The differential impact of catalase on the actions of SOD-1 and EC-SOD indicates that these enzymes are involved in synaptic plasticity and memory formation using different mechanisms; SOD-1-induced LTP impairments are due to high ambient H2O2 levels, whereas EC-SOD-induced LTP impairments are due to decreased levels of superoxide. Despite these mechanistic differences, the conclusion remains the same in that free radical balance, under physiological conditions, is crucial for proper synaptic plasticity and memory formation.
Whether mitochondrial superoxide contributes to LTP and memory formation is debatable, especially since it has been suggested that the mitochondrial membrane may not allow the free diffusion of mitochondrial superoxide (460). SOD-2 transgenic mice have been characterized extensively and, contrary to its cytoplasmic and extracellular counterparts, SOD-2 overepxression does not affect either LTP or memory formation in healthy young mice (188).
Collectively, these studies indicate that the different SOD isoforms, and consequently the different pools of superoxide they act on, affect synaptic plasticity and memory via different mechanisms, but lead to the same conclusion that free radicals are important for synaptic plasticity and memory formation under physiological conditions.
b. Synaptic plasticity and memory in the aged and diseased brain
It appears that high levels of SOD enzymes are detrimental to normal synaptic function in young animals, but that they act in an opposite manner in the aged or diseased brain. During aging there are significant impairments in LTP that are accompanied by deficits in cognitive abilities and memory formation (31). Overexpression of EC-SOD improved LTP in aged mice and catalase treatment did not alter this improvement, indicating that EC-SOD improves LTP in aged mice via quenching superoxide independently of H2O2 (190). EC-SOD overexpression also improved age-related spatial memory deficits. Associative memory measured by a fear conditioning paradigm was not improved in the aged EC-SOD transgenic mice (190). Other studies have shown that age-related deficits in associative memory may not present themselves unless the interval between training and testing is greater than 24 h (152), which may explain why associative memory was not improved in the aged EC-SOD transgenic mice. In addition to improving aging-related spatial memory impairments, EC-SOD also has been shown to be beneficial to TBI-related cognitive dysfunction (332).
The level of key synaptosomal proteins in the hippocampus is significantly reduced in mice overexpressing SOD-1 (140, 387), suggesting that the beneficial quenching of superoxide may be outweighed by the predominant presence of H2O2, which has been shown to possess neurotoxic effects. This idea is supported by studies of SOD-1 overexpression during AD or TBI. These studies specifically showed that AD-related cognitive dysfunction and loss of memory after TBI not only were exacerbated by SOD-1 overexpression (41, 170), but also could be reduced by SOD-1 deficiency (41, 140). These results are surprising in light of the findings that SOD-1 overexpression can reverse the age-related decline in LTP (205). Using a theta-burst protocol, which should represent more closely the physiological processes during learning and memory, improvements in LTP have been reported in slices from aged mice overexpressing SOD-1, possibly due to increases in H2O2 in these mice (205). The authors of this study proposed a model by which the redox milieu present in SOD-1 transgenic mice has been adapted to working at high H2O2 levels and, as such, the combined increase in H2O2 from superoxide dismutation and from age-related mitochondrial leakage, while detrimental to a normal system, becomes beneficial to this system (205). Consistent with this notion, aged EC-SOD transgenic mice exhibit better spatial memory than wild-type littermates (203). Thus, excessive production of H2O2 might improve memory in aged mice.
Surprisingly, there was no effect of overexpression of SOD-2 on synaptic plasticity and memory in aged mice (188). SOD-2 overexpression appears to be beneficial under conditions of either acute toxicity or increased oxidative stress. For example, SOD-2 overexpression was shown to protect against 6-hydroxydopamine- and methamphetamine-induced brain injury (74, 271). In addition, SOD-2 overexpression reverses the AD-related cognitive deficits (115, 277). This will be discussed in more detail later in the review.
Taken together, these studies show that the functional involvement of SODs in synaptic plasticity and memory depends on the age of the animal as well as the isoform of SOD. Another important factor may be the nature of oxidative insult initiating the events, as well as the area of the brain affected.
2. Catalase
Catalase is an antioxidant enzyme found in nearly all living organisms. Its main function is to catalyze the decomposition of H2O2 to water and oxygen (82) (Fig. 9B). Catalase is an iron enzyme; it contains four porphyrin heme groups that allow it to interact with H2O2. Studies investigating the role of catalase in the mechanisms underlying memory function have done so in relation to H2O2 metabolism. For example, LTP in hippocampal slices from adult or aged wild-type mice was examined in the presence or absence of an enzymatic treatment of either catalase or SOD-1. In line with Kamsler and Segal's model of H2O2 modulation of synaptic plasticity (205), increased SOD-1 had no effect on LTP from young adult mice, but made LTP impairments worse in old mice (436). It was also found that catalase inhibited LTP in slices from the young adult mice, but reversed age-related LTP deficits in older mice, indicating that in wild-type mice, H2O2 plays a critical role in synaptic plasticity at young ages, but becomes detrimental during aging (436). This study supported earlier findings that demonstrated a dual role for H2O2 in the regulation of LTP that is mediated by the protein phosphatase calcineurin. H2O2 was found to be necessary in young animals, but became detrimental in old animals (204). Several studies investigated the contribution of brain catalase to ethanol-induced learning reinforcement. It was found that catalase is involved in the acquisition of ethanol-induced conditioned place preference (133), as well as learning in the social recognition test (270). Additional supporting evidence of the involvement of catalase in ethanol-induced behavioral reinforcements came from studies of conditioned taste aversion combined with a catalase inhibitor (aminotriazole). These effects are thought to be mediated by acetaldehyde, which is an ethanol metabolite directly produced in the brain by the catalase system (20, 308, 346). In studies directly relevant to aging and neurodegeneration, it was shown that aging-related cognitive dysfunction can be reversed by treatment with synthetic SOD/catalase mimetics, further supporting the idea that free radicals are involved in learning and memory (90, 255).
3. GPx, glutathione reductase, and related enzymes
GPx is a general nomenclature attributed to a group of antioxidant enzymes dedicated mainly to reducing H2O2 to water and lipid hydroperoxides to their corresponding alcohol. GPx reduces H2O2 to water by oxidizing GSH. Then, the oxidized glutathione disulfide (GSSG) is regenerated back to its reduced form by the action of GR (Fig. 10). GR is constitutively active and inducible upon oxidative stress; therefore, GSH is found almost always in its reduced form, ready to counteract the deleterious oxidant effects (26).
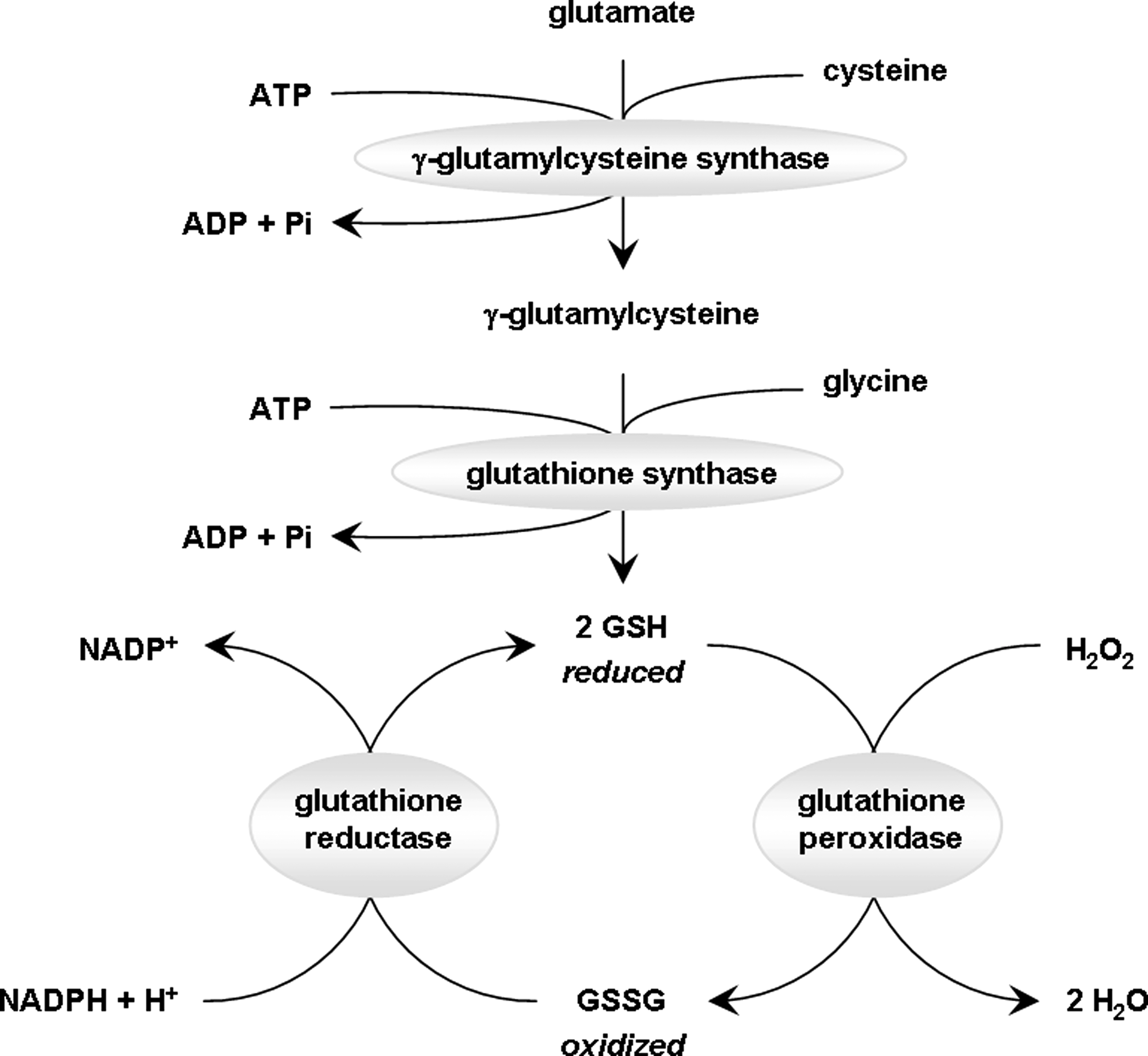
Up to eight isoforms of GPx have been identified in humans thus far, with specific antioxidant mechanisms attributed to each. For example, the cytoplasmic homotetrameric GPx1, the most abundant of these peroxidases, has been shown to preferentially quench H2O2, whereas the ubiquitous but less abundant monomeric GPx4 plays an important role in detoxification of membrane lipids. With the exception of increased cataract occurrence, GPx1 knockout mice are phenotypically normal, indicating that this enzyme is not crucial for survival. However, genetic deletion of GPx4 is embryonic lethal, illustrating the different function of the different GPx isoforms (297). GPx levels and/or activity are used widely as an index of oxidative stress and assessment of therapeutic efficacy. In studies of oxidative stress-induced synaptic plasticity and memory impairments, impaired cognition is typically found to be correlated with reduced levels of GPx that are brought back to normal upon certain antioxidant treatments [examples in refs. (86, 172, 301)]. This is in agreement with the evidence discussed in this review, implicating ROS dysfunction with cognitive diseases.
Additional studies using GPx transgenic mice have addressed more directly the role of this antioxidant enzyme in oxidative stress-induced cognitive deficits. For example, moderate increases in GPx1 were shown to reverse hypoxia-induced functional damage to hippocampal slices. Control slices subjected to hypoxia suffered an irreversible loss of EPSPs with an inability to generate LTP. GPx overexpression resulted in a significant recovery of synaptic transmission and LTP (141). In addition, GPx overexpression has been shown to be beneficial to TBI-induced cognitive deficits in the absence of any effect on the lesion anatomy and brain volume. Further, GPx protected hippocampal neurons via its antioxidant effect on early oxidative stress (419). The effect of GPx and GR as antioxidants cannot be dissociated from the main precursor for the system, namely GSH (discussed in details later in this review). In fact, boosting GSH levels can result in increasing the activity of both GPx and GR, as well as other antioxidants (300). In addition, the effectiveness of these enzymes in neuroprotection and improvement of cognitive function depends on the availability of both GSH and GSSG as reducing equivalents (110, 283). In certain instances, increased activity of GPx has been shown to correlate positively with memory impairments, which appears counterintuitive (66). However, the levels of GPx are a better indicator of oxidative stress-induced impaired memory function, which also resulted in the activation of antioxidant defenses, including the increased activity of GPx.
A group of enzymes functionally related to the GSH system are thiol-disulfide oxidoreductases that are primarily involved in the catalysis of thiol-disulfide interchange reactions. This group includes thioltransferase (glutaredoxin [GRX]), thioredoxin (TRX), and protein disulfide isomerase (437). Whereas TRX and protein disulfide isomerase have broad substrate specificities (289), GRX specifically reduces GSH-containing mixed disulfides with greater efficiency than TRX (153, 289). The TRX system contains, aside from TRX, an NADPH-dependent TRX reductase, which reduces the oxidized form of TRX. The GRX system uses GSH as a reducing agent, which then enters the GSH system and is regenerated with GR [reviewed in ref. (201)]. Two gene isoforms of each GRX and TRX have been identified in mammals and a summary of their function can be found in the following review (179). Both GRX and TRX are endogenous antioxidants that contribute to protection against oxidative stress, specifically the formation of intra- and intermolecular disulfide bonds in proteins. As such, both of these systems may modulate memory formation. However, studies specifically investigating the direct involvement of GRX and TRX in synaptic plasticity and memory are limited. TRX upregulation, along with increased ERK-mediated Nrf2 phosphorylation and a reduction in free radical levels, contributes to the beneficial effects of acetyl-L-carnitine on hypoxia-induced spatial memory impairment (29). Additionally, GRX1 and TRX1 are deregulated in AD and contribute to Aβ-induced neurotoxicity (9). However, these studies did not specifically address the involvement of GRX1 and TRX1 in memory dysfunction associated with AD. GRX also was shown to be essential for maintaining the integrity of complex I of the mitochondrial respiratory chain after toxic insult by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a neurotoxin that produces PD-like symptoms through inhibition of complex I (214). This finding suggests that GRX also may be involved in PD pathology; however, its involvement in learning and memory can only be inferred.
Another group of related enzymes, peroxiredoxins (PRXs), is involved in the decomposition of H2O2, organic hydroperoxides, and peroxinitrite (79). PRXs are related to TRX because distinct isoforms of TRX are cofactors for PRX. PRX has been shown to contribute its antioxidative capability in the realm of aging- and AD-related cognitive dysfunction. Specifically, PRX II-deficient mice were shown to have increased mitochondrial ROS generation and pronounced age-related LTP impairments compared to their wild-type littermates (218). These mice also exhibited an impairment in the activation of synaptic plasticity-related signaling pathways involving CREB, CaMKII, and ERK, indicating that PRX II is necessary for maintaining redox homeostasis (218). Dietary vitamin E alleviated a number of PRX II deficiency-related impairments, including a restoration of the cognitive decline in aged PRX II-deficient mice (218), suggesting that PRX II helps maintain hippocampal synaptic plasticity against age-related oxidative damage. In a differential proteomic analysis, PRX 1 from aged rat temporal cortex tissue is carbonylated, suggesting that this enzyme contributes to the decline in cognitive function that occurs during aging (431). PRX II also has been shown to be upregulated in response to increase Aβ levels during AD, with the purpose of protecting neurons against Aβ-induced toxicity (456). Conversely, reducing the levels of Aβ in the brains of senescence accelerated mice resulted in reduced PRX II levels (338). Although these findings do not directly link PRX II to AD-related cognitive dysfunction, they do support the idea that PRX II plays a crucial antioxidant role during aging and AD.
This section reviewed evidence for the involvement of the glutathione system, including GSH, GPx, GR, and related enzymes, in reducing oxidative stress and improving cognitive dysfunction, providing more evidence that oxidative stress is associated with neurotoxicity and memory impairment.
4. Metallothionein
Metallothionein (MT) is a family of cycteine-rich low-molecular-weight proteins that are ubiquitous in eukaryotes. Their primary function is to bind essential physiological (copper, zinc, and selenium) and nonessential xenobiotic (cadmium, mercury, silver, and arsenic) heavy metals via their cysteine residues. A significant literature is available (167, 177, 200, 243, 310, 325, 368) describing the physical structure, biochemistry, and molecular biology of MT, as well as their role in protecting cells and organisms from environmental toxicant damage (metal toxicity), but also from oxidative stress. Specific evidence documenting the involvement of MT in synaptic plasticity and memory is also available (248, 459); however, it is not clear whether this involvement is related to their antioxidative capabilities and therefore will not be discussed here.
B. Antioxidant molecules (nonenzymatic)
Although behavioral outcomes of increased brain oxidative stress are usually discussed with reference to enzymatic dysfunctions in the antioxidant system, several molecules with redox-dependent activity are involved in the synaptic plasticity that plays a role in learning and memory. These nonenzymatic molecules are discussed in this section.
1. Ascorbate (vitamin C)
Ascorbate (vitamin C) is a potent antioxidant that can neutralize free radicals by donating a hydrogen atom. Vitamin C itself becomes a free radical in the process. However, this does not pose a major concern, as the vitamin C radical is very stable (nonreactive) due to its resonance structure (Fig. 11). Moreover, the reduced form of vitamin C can be readily regenerated by the action of NADH and NADPH-reductases. Excess vitamin C radical becomes a problem in the presence of metal ions such as iron (Fe) or copper (Cu), where it can turn into a powerful pro-oxidant. Normally ferrous iron (II) (Fe2+) is oxidized by H2O2 to produce ferric iron (III) (Fe3+), along with the highly reactive hydroxyl radical and a hydroxyl anion. This is known as the Fenton reaction (Fig. 12). After the Fenton reaction, Fe3+ can be reduced back to Fe2+ by the action of H2O2 to produce a peroxyl radical and a proton in the process. The presence of ascorbate can accelerate the latter free radical producing reaction by acting as an electron acceptor for Fe3+, instead of H2O2 (Fig. 11). Organisms have developed defenses against this process, such as having iron bound to the transport proteins ferritin and transferritin. However, the exogenous introduction of metal ions from environmental pollutants can cause failure in this defense mechanism.
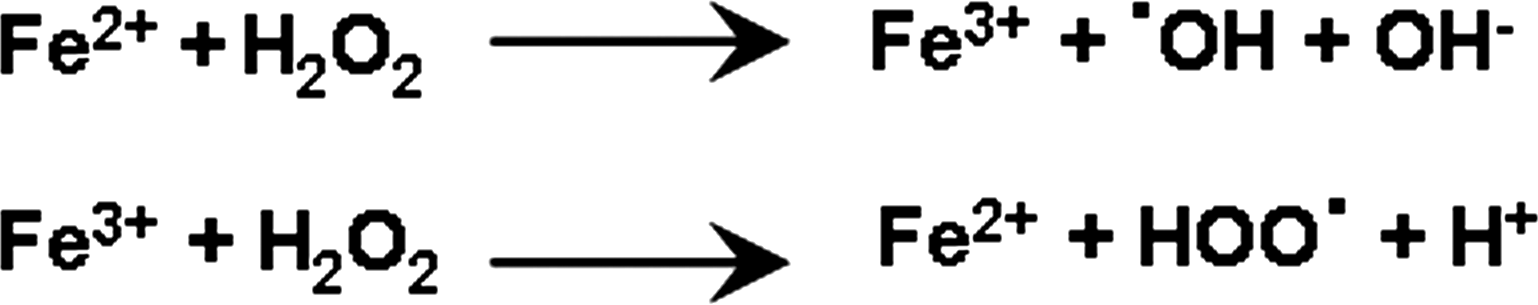
Vitamin C is an essential dietary supplement because it is required for collagen synthesis and primates have lost the ability to synthesize it. However, its antioxidant properties make it an increasingly desired dietary addition. These antioxidant properties have been explored both in vitro and in vivo and their effect on protein oxidation, lipid peroxidation, and DNA damage have been well characterized. In the interest of this review, we will focus on the role vitamin C plays in the learning and memory processes.
The vast majority of studies investigating the role of vitamin C in learning and memory have done so in the setting of memory dysfunction. In other words, these studies investigate the usefulness and effectiveness of vitamin C in reversing memory deficits induced by various oxidative stress-related conditions, ranging from chemical induction to disease states. Very few studies have addressed the role of vitamin C in normal learning and memory. One study from the early 1980s compared short-term and immediate memory performance of 20 individuals at times when their plasma level of vitamin C was high (owing to supplementation) versus times with low plasma levels of vitamin C (5). This study found no differences in the memory performance, nor did it find any differences in alertness, concentration, or mood. The purpose of this study primarily was to determine the effect of vitamin C on alertness, and therefore the memory test used was a simple procedure consisting of repeating the last five digits of a series of numbers played through headphones. This task was designed to test immediate short-term memory, stemming from concentration as opposed to actual learning (5). Two more recent studies addressed the antioxidant role of vitamin C in learning and memory (374, 422). It was demonstrated that although acute injections of ascorbate had no effect on the learning ability of rats measured with passive avoidance test, short- and long-term supplementation with vitamin C facilitated acquisition and retrieval of learning and memory in rats (374). Tveden-Nyborg et al. went a step further and investigated the effect of chronic vitamin C deficiency on spatial memory as measured by the Morris water maze and also counted hippocampal neurons to assess whether ascorbate had an effect on neurodegeneration (422). In this study, two groups of newborn guinea pigs were randomly assigned to either a vitamin C-sufficient diet or a deficient diet (minimally adequate to prevent scurvy) for 2 months. The objective of the study was to assess the effect of vitamin C deficiency on the neonatal brain, because it is more susceptible to oxidative damage than the adult mature brain. The results showed a significant reduction in spatial memory, compounded by a lower number of neurons in all regions of the hippocampus (422). Collectively, these animal studies indicate that ascorbate, at least partially, contributes to protection of neurons from free-radical-induced memory impairments. Clinical trials investigating the benefits of vitamin C on cognition have yielded more complicated results and are discussed in section V under physiological aging and AD.
2. Tocopherol (vitamin E)
Vitamin E is a generic term referring to a group of tocopherols and tocotrienols, of which α-tocopherol is the best studied. Vitamin E is a fat-soluble vitamin known for its role as an antioxidant, involved in stopping ROS from forming in membranes undergoing lipid peroxidation chain reactions. The role of vitamin E as a systemic antioxidant has been studied extensively, with conflicting results as to its beneficial effects. Similar to ascorbate, the involvement of vitamin E in learning and memory has been for the most part investigated under therapeutics for either ROS-induced disease states or physiological aging (discussed above in section V). Very few studies have addressed the role of vitamin E in normal memory function. In the early 1970s, it was reported that there was no significant effect of vitamin E deficiency on the learning ability of rats, although they needed more trials to acquire a conditioned response (238). The most striking effect of vitamin E deficiency in these experiments was the impaired retention of a one-trial experience, a task dependent on reference memory (238). In similar experiments, the effect of vitamin E deficiency on reference and working memory were subtle at best (367). Studies examining the effect of long-term vitamin E deficiency and vitamin E supplementation in rats found that it had no effect on the acquisition and maintenance of memory tasks, but did impact their learning (195). Collectively, the results of these studies have not conclusively demonstrated whether vitamin E acts as an antioxidant in learning and memory, which may seem surprising given that memory processes depend on the redox status of neurons. In fact, two more recent related studies (449, 450) show a direct role of vitamin E in the modulation of LTP, which is the substrate of learning and memory formation (117). In these studies (449, 450), vitamin E deficiency resulted in LTP impairments and that vitamin E induced a slowly developing long-lasting increase in EPSPs that was independent of NMDA receptor activation. The same effect was not observed with ascorbate. Although these studies clearly argue for a role for vitamin E in LTP (and thus memory formation), the ascorbate study suggests that this effect may not be mediated by ROS (449, 450). Collectively these studies demonstrate that the involvement of vitamin E as an antioxidant in the learning and memory process is not clear and is at best inferred.
3. Glutathione
Glutathione (GSH,
With regard to learning and memory, the role of GSH has mainly been investigated after chemical depletion and its effect discussed in the context of schizophrenia, where a deficiency in GSH is common and is associated with cognitive decline. It was demonstrated that low levels of GSH negatively impact LTP and paired-pulse facilitation, indicating that low GSH content can impair short- and long-term forms of synaptic plasticity (11). The effect of GSH depletion in the presence of dopamine, which is a potent pro-oxidant, was studied by inducing GSH depletion in rats by injecting buthionine sulfoximide (BSO) either before or after dopamine administration, and then assessing the spatial memory of rats using the Morris water maze. These investigators found that GSH depletion before the oxidant insult introduced by dopamine led to significant impairments in memory, indicating that GSH is a necessary component of the antioxidant machinery dedicated to maintain a healthy balance of free radicals necessary for proper memory function (389). A specific role for GSH in the acquisition, but not retention, of spatial memory in maze tasks was demonstrated by inducing GSH depletion in rats with systemic diethylmaleate injections and then examining metabolism and cognitive performance in the passive avoidance and Morris water maze tests. These studies showed that GSH depletion resulted in reduced levels of GSH and GPx activity in the hippocampus. It also resulted in reduced acquisition of spatial memory when the drug was administered before training, but did not affect retention of already acquired memory when the drug was administered after memory acquisition (94). More complex assessments of spatial and working memory were performed after transitory GSH depletion by BSO injection in either young normal or mutant rats (osteogenic disorder Shionogi [ODS]—incapable of synthesizing ascorbate). Ascorbate is known to compensate for GSH depletion, and therefore the ODS group served to dissect the direct effect of GSH on cognitive abilities. BSO-ODS rats exhibited impaired spatial memory whether they were tested at a young age, immediately after GSH restoration to physiological levels, or at an older age, long after treatment cessation. In contrast, BSO-normal rats did not exhibit memory impairments at an early age, possibly due to ascorbate compensation, but they did exhibit selective spatial memory impairments at an advanced age when the task required integration of multimodal cues, such as visual and olfactory cues. Although the results of this study were exclusively discussed in the context of schizophrenia, they clearly show that GSH depletion causes complex behavioral impairments associated with increased brain oxidative stress (70). In a more recent study, GSH depletion was induced with 2-cyclohexene-1-one both in rats and mice and then their memory was assessed in the Y-maze test. In both species, GSH depletion caused a significant impairment in short-term spatial recognition memory, suggesting that GSH is necessary for proper spatial memory function, possibly explaining its involvement in psychiatric diseases such as schizophrenia (104). Collectively, these studies outline an important role for GSH in spatial memory formation. On a related note, administration of reduced GSH into the antennal lobes of honeybees prevented oxidative stress-induced impairments in olfactory learning and memory (125), indicating that GSH plays a critical role in learning not only in mammals, but also in invertebrates.
4. Coenzyme Q
Coenzyme Q, also known as coenzyme Q10, ubiquinone, ubidecarenone, or simply Q, is a benzoquinone found in most eukaryotic cells, primarily in the mitochondria. It participates in the electron transport chain in its capacity of electron acceptor/donor and essentially functions as an electron transferring molecule (426) (Fig. 4). Electron transferring ability means that coenzyme Q can act as an antioxidant. Because of this antioxidant ability, coenzyme Q is widely used as a dietary supplement taken to enhance bioenergetics and ameliorate all side effects of increased oxidative stress during aging and a multitude of pathological conditions. Coenzyme Q has been explored as a therapeutic avenue for heart failure (327), mitochondrial dysfunction (160), migraine headaches (47), and neurodegenerative disorders associated with cognitive dysfunction (39). Idebenone, a benzoquinone commercially marketed as a synthetic analog of ubiquinone, also has been extensively studied for its antioxidant properties. We review here the role of both in learning and memory mechanisms.
A general statement that seems to be true of the involvement of quinone in learning and memory is that these compounds do not really affect cognitive abilities under physiological conditions, but rather improve cognitive dysfunction induced by a variety of pathological conditions. One study of the late 1980s directly linked quinones to synaptic plasticity by determining a role for idebenone in enhancing LTP in hippocampal slices (196). Two different concentrations of ubiquinone were tested on several physiological functions that are known to be impaired during aging, including cognitive dysfunction. These investigators supplemented their mice with coenzyme Q at a young age and then assessed their spatial learning and memory at several age points. Although the amount of ubiquinones was increased in the cerebral cortex of treated mice, the low dose treatment did not alter ubiquinone levels. Surprisingly, at higher doses, age-related decreases in learning and memory were made worse, indicating that low coenzyme Q levels had no discernable impact on cognitive dysfunction, whereas higher levels impaired cognitive function (403). These results suggest that under physiological conditions ubiquinone supplementation is not beneficial, but rather can be detrimental to memory. In a similar study investigating the role of coenzyme Q, vitamin E, or both on brain function of aged mice, it was observed that only mice treated with both coenzyme Q and vitamin E exhibited memory improvements (281). Similar experiments with higher doses of coenzyme Q were conducted, and unlike the previous study, there was no adverse effect on memory. On the other hand, the investigators also did not observe any memory improvements. They concluded that coenzyme Q is only beneficial for memory when paired with vitamin E, perhaps because they work in concert, possibly via mutual regeneration of antioxidant function (281). Similar to previous studies, rats fed a ubiquinone-rich diet did not exhibit improvement in the early stages of the Morris water maze test, but did exhibit improvements in the later stages of the test (312). After hyperoxia to induce oxidative stress, rats fed coenzyme Q showed improved memory function over control rats. Repeating the same studies in vitamin E-deficient rats or rats supplemented with vitamin E showed the same pattern of improvement and indicated no synergistic effect of coenzyme Q with vitamin E (312), in contrast to previous studies that demonstrated that coenzyme Q can regenerate the antioxidant powers of vitamin E (281).
Two other conditions in which ubiquinones proved to be beneficial for learning and memory were after ischemia and scopolamine-induced amnesia, which was studied in the context of cholinergic dysfunction during AD. Idebenone ameliorated memory impairment induced by cerebral vascular disturbance in rats. More specifically, the authors demonstrated that idebenone treatment after the acquisition trial of passive avoidance learning caused shortening in the latencies of the retention test trial, that is, idebenone reversed the retrograde amnesia caused by cerebral ischemia (453). In a different study, in embolized rats, idebenone improved learning and memory abilities in the radial arm test (227), as well as passive avoidance test (226), indicating that quinones have a positive effect on learning impairments caused by brain hypofunction.
Perhaps the largest body of literature investigating the effect of quinones in the mechanisms governing memory function is in studies related to AD (AD). Idebenone has long been investigated as a possible therapy for AD with mild, but generally positive, results, suggesting that quinones are involved in preserving the oxidant balance necessary for proper cognitive function. Several groups have studied scopolamine-induced amnesia, which induces deficits in the cholinergic system as seen in AD patients. Idebenone treatment improved scopolamine-induced spatial memory deficits (256) and working memory deficits (452). Using a delayed alternation task in rats, it was shown that idebenone treatment improved short-term memory dysfunction induced by scopolamine, but it did not alter memory in control rats (454). These observations were extended to a serotonin-deficient rat model, where idebenone improved the retardation of discrimination learning induced by central serotonergic dysfunction (455). Because nerve growth factor (NGF) is necessary for the maintenance of cholinergic neurons, its depletion mimics cholinergic deficiency as observed in AD. Unfortunately, NGF cannot be used directly as a therapy because of its inability to cross the blood brain barrier. However, idebenone administration stimulated NGF synthesis in vivo, resulting in an amelioration of the behavioral deficits induced by lesions in the basal forebrain cholinergic system. In these studies, idebenone was beneficial as an NGF stimulator rather than as an antioxidant (307).
An interesting question in the AD field is whether oxidative stress contributes to learning and memory deficits caused by Aβ42. Rats given continuous intracerebroventricular infusion of Aβ42 were treated with either idebenone or vitamin E and then their memory was assessed in the Y-maze, the Morris water maze, and the passive avoidance test. It was observed that both idebenone and vitamin E improved the performance of the Aβ42-treated rats in the Y-maze and Morris water maze, but not in the passive avoidance test (451). These results suggest that quinones might be effective antioxidants to improve learning and memory deficits associated with AD. Finally, streptozotocin (STZ)-treated rats display markers of oxidative damage (increases in thiobarbituric reactive substance, GSH, protein carbonyls, and the activities of the GSH peroxidase and reductase enzymes) and a decline in the level of ATP in the hippocampus and cerebral cortex. Supplementing STZ-infused rats with coenzyme Q reversed all the aforementioned markers, indicating that ubiquinone has neuroprotective effects on cognitive impairments and oxidative damage associated with STZ toxicity (197). In a multicenter, randomized, double-blind, placebo-controlled study in AD patients, treatment with idebenone was effective in delaying memory impairments associated with AD, with no significant side effects (42). In a more recent study, treatment of AD patients with idebenone in the predementia stage resulted in short- and long-term memory improvements in 37% of the patients (427). On a concluding note, although the precise mechanism of action of ubiquinone remains unknown and it does not appear to alter learning and memory under normal physiological conditions (147), it appears to play a central role as an antioxidant molecule involved in restoring the redox balance necessary for improvement of cognitive dysfunction associated with a multitude of pathological conditions, especially AD.
5. Carotenoids
Carotenoids are phytochemicals produced in plants, with β-carotene and lycopene being the most investigated (321). Many epidemiologic studies have made the association of high dietary carotenoid intake with a lower incidence of chronic disease, but the biological mechanisms underlying such protection are not understood. There are several theories that have been put forward to explain the protective effect of carotenoids (321): (a) they can be converted to retinoids and thus acquire pro-vitamin A activity, (b) they can act as potent antioxidants, (c) they can modulate the function of lipooxygenases, and (d) they can modulate expression of genes involved in cell–cell interactions. In the interest of this review, we will focus on the role of carotenoids as antioxidants with an emphasis on their involvement in the regulation of learning and memory. The antioxidant properties of carotenoids lie in their ability to physically quench singlet oxygen and to trap peroxyl radicals. Singlet oxygen is not a free radical but it has electrons in an excited state that may adversely affect molecules containing double bonds. Carotenoids are the most effective singlet oxygen quenchers (109), which results in the formation of excited carotenoids. The excited carotenoids have the capacity to rapidly dissipate newly acquired energy through a series of rotational and vibrational interactions with the solvent, resulting in regeneration of the unexcited carotenoid that can be reused for further cycles of singlet oxygen quenching (67). Several carotenoids, with β-carotene in particular, have the capability of scavenging peroxyl radicals by the formation of a transitory carotenoid adduct radical (67). This radical is highly stable and undergoes decay to a nonradical species, thereby terminating the actions of harmful peroxyl radicals. This latter process destroys carotenoids (442). The antioxidant properties of carotenoids vary depending on the system being used to study them as well as the concentration used. For example, carotenoids can protect cells against oxidant-induced lipid peroxidation in a pro-vitamin A-independent fashion (275). In transformed thymocytes, carotenoids act as potent antioxidants when oxygen tension is low, but turn into pro-oxidants when oxygen tension is high (324). In addition, supraphysiological concentrations of carotenoids may result in pro-oxidative effects (145). These pro-oxidative effects can be prevented by the coadministration of other antioxidants such as vitamin E (323). Many clinical studies have been undertaken to assess the antioxidant ability of carotenoids and produced conflicting results. It was demonstrated that a diet deficient in carotenoids, but otherwise adequate for all other nutrients, resulted in diminished markers of antioxidant capacity in the blood (315). Conversely, supplementing the diet with β-carotene has been shown to increase plasma levels of antioxidant molecules (288). In one study using the oxygen radical absorbance capacity test, no positive role for carotenoids on plasma levels of antioxidants was demonstrated (76). Another report supported this finding by showing that there was no effect of carotenoids on the total antioxidant capacity of the plasma (58). The differences in results between these studies could be due to either different types of supplementation or different age groups and genders of the study individuals. Humans have the capacity to absorb intact carotenoids from the intestines; however, such absorption may differ depending on the type of supplementation offered (within the food as opposed to capsules). Certain forms may be more readily bioavailable than others. For example, lycopene has been shown to be better extracted from heated tomatoes as opposed to raw ones (395). Thus, special care should be put into selecting the right concentrations and form of carotenoids for each particular study setting.
Carotenoids have been investigated as an antioxidant in various pathological conditions associated with learning and memory dysfunction. One carotenoid, retinoic acid (an essential growth factor derived from vitamin A), has been extensively studied for its modulation of the signaling events involved in learning and memory. In fact, retinoic acid activates gene transcription through nuclear receptors, thereby influencing LTP and neurogenesis, particularly in the hippocampus (279, 293). This suggests a role for retinoic acid in learning and memory processes, but it appears to be distinct from the ability of carotenoids to act as antioxidants. Because this review is dedicated to the redox regulation of memory mechanisms, we will limit the discussion in this section to studies specifically addressing the antioxidant function of carotenoids.
A number of epidemiological studies have been undertaken to assess the association between carotenoid levels and either age- or disease-related declines in cognitive function. With the exception of one study, which delineated a pro-oxidant effect of vitamin A in the substantia nigra and striatum at clinical doses (103), all of the other studies showed either neutral or positive correlations with cognitive dysfunction. Engelhart et al. examined whether high plasma levels of vitamins A and E together were associated with lower prevalence of cognitive decline. They performed a cross-sectional study within another AD study and found that if a univariate model was used, high plasma vitamin A and E could be correlated to lower cognitive decline. However, when adjustments were made with respect to age, gender, and total cholesterol, the correlation weakened, and it was concluded that there was no association between plasma levels of vitamin A and E and cognitive decline (122). The same question was examined in a cohort of elderly woman and no correlation between plasma carotenoids and tocopherols and cognitive deficits was observed (206). The same group explored the effect of β-carotene supplementation on cognitive function in men and found that short-term supplementation (mean duration of 1 year) did not affect cognition, whereas long-term supplementation (mean duration of 18 years) provided cognitive benefits (156). The latter study illustrates that careful consideration should be given to every study parameter (such as treatment duration) before conclusions can be drawn.
Five additional studies have described beneficial effects of carotenoids on cognitive deficits. The interaction between carotenoids and the AD susceptibility gene apolipoprotein E was investigated and it was found that among high-functioning older persons, β-carotene supplementation offered protection from cognitive decline, even in persons with greater susceptibility evidenced by the presence of the APO-E4 allele (191). The relationship between plasma carotenoid levels and the MMSE was examined and it was found that lower levels of carotenoids correlated with cognitive impairments (7). As part of the cache county study, high antioxidant intake from food combined with supplementation with vitamin E, vitamin C, and carotene may delay cognitive decline in the elderly (438). Although this study did not dissect a specific role for carotenoids in this protection, it did suggest that carotene is potentially a beneficial antioxidant involved in protection of cognitive abilities. In addition, a significant association between higher carotenoid levels and docosahexanoic acid with higher MMSE scores has been reported, supporting a protective role for both nutrients in aging and AD-related cognitive impairments (433). Finally, the involvement of lycopene in diabetes-associated cognitive decline in rats was examined, and this particular carotenoid has a significant therapeutic potential in diabetes-induced learning and memory impairments (235).
A large body of research has centered on the interesting and widely beneficial effects of a naturally occurring carotenoid chemical called crocin. Crocin is found in the flower Crocus sativus L., more commonly known as saffron. Crocin is the ingredient primarily responsible for the color of saffron. It is a diester formed from a disaccharide (gentiobiose) and the dicarboxylic acid crocetin. Aside from being a potent antioxidant (311, 465), it also has been reported to have anticarcinogenic (1, 88), antidepressant (8), and aphrodisiac properties (184). Given its potent antioxidant capacity, a role for crocin in modulating learning and memory has been proposed. Recent behavioral and electrophysiological studies have shown a beneficial effect of crocin on ethanol-induced LTP deficits and learning and memory impairments. In several separate studies, Saito and associates have demonstrated that crocin can prevent ethanol-induced impairment of hippocampal synaptic plasticity, both in vitro in rat slices (401) and in vivo in anesthetized rats (400). They further investigated the underlying mechanism of these effects and found that crocin specifically antagonized the inhibitory effect of ethanol on NMDA receptor-mediated responses in hippocampal neurons (2). Using the same ethanol-induced impairment model, they also demonstrated that although crocin had no effects on memory registration, consolidation, or retrieval in normal mice, it did improve the ethanol-induced impairments of memory function in all of its constituents (461). In a more recent study (333), crocin counteracted delay-dependent recognition memory deficits in the normal rat, suggesting that this carotenoid can modulate storage and/or retrieval of information. It also attenuated scopolamine-induced spatial memory deficits in the radial arm maze. This study not only demonstrated the beneficial effect of crocin on learning and memory impairments induced by ethanol, but it also suggested that crocin is a compound that can modulate the mechanisms underlying normal recognition and spatial memory (333).
In conclusion, although carotenoids have well-documented antioxidant effects, there continues to be a great deal of uncertainty as to their beneficial role in the mechanisms underlying learning and memory deficits. Depending on the conditions of their use (administration, absorption, duration of administration, etc.), carotenoids appear to offer protection against learning and memory deficits associated with physiological aging as well as a multitude of pathological conditions. The mechanism responsible for the beneficial effects of carotenoids on cognitive function remains to be elucidated.
6. Melatonin
Melatonin is primarily a pineal hormone derived from its precursor serotonin. Its main function is in sleep cycle regulation. Melatonin also is a very powerful scavenger of hydroxyl radicals, making it a potent antioxidant molecule (168). Unlike vitamin C and GSH, which are only effective in aqueous phase, and vitamin E, which is only effective in lipid phase, melatonin can be functional in both phases (412). Melatonin also easily crosses cell membranes and the blood–brain barrier (360), making it a powerful candidate for protecting nuclear and mitochondrial DNA and the central nervous system from damage induced by oxidative stress (359). Many biological effects of melatonin are produced through activation of melatonin receptors (61), so it is important to distinguish its action as an antioxidant on learning and memory.
The discovery of melatonin as an antioxidant was made in 1993 (412). Melatonin is a direct scavenger of hydroxyl and peroxyl radicals. It can also neutralize superoxide, singlet oxygen, H2O2, and hypochlorous acid (335). Melatonin also inhibits peroxynitrite formation by inhibiting NOS in brain tissue (245). Melatonin differs from other antioxidants in two important aspects. First, melatonin does not undergo redox cycling; that is, once oxidized, it cannot be regenerated to its reduced form. This happens because melatonin forms stable intermediates upon reacting with free radicals, which makes it a terminal antioxidant (410). Second, the antioxidant action of melatonin involves the donation of two electrons, not one electron, thereby ensuring that melatonin does not become a free radical in the process (410). Melatonin is a much more potent antioxidant than many traditionally used antioxidants. For example, it is twice as effective as vitamin E in protecting cell membranes from lipid peroxidation (330), five times more potent than GSH in scavenging hydroxyl radicals (331), and 60 times more effective than vitamins C and E in providing DNA protection (345). Besides its own ability to scavenge a variety of free radicals, several of its metabolites are themselves potent antioxidants (361, 411). Melatonin can also boost expression and/or activity of other antioxidants, most notably GSH peroxidase (up to eightfold increase), SOD, and catalase (233, 276).
Because the brain has lower GSH concentrations than other organs (164), melatonin plays a particularly important antioxidant role in the central nervous system. In fact, melatonin receptors have been shown to play an important role in the mechanisms of learning and memory in mice (242), and melatonin has been shown to alter the cellular processes associated with memory, such as LTP.
a. Melatonin and LTP
Using rat hippocampal slices, it was shown that melatonin (100 μM) has the ability to block the induction of LTP in an NMDA receptor-independent manner (92). In addition, in studies in the suprachiasmatic nucleus, melatonin prevented LTP induction via an inhibitory effect on CaMKII autophosphorylation (139). Moreover, a specific role for the circadian clock, and therefore melatonin levels, in modulating synaptic plasticity in the hippocampus has been demonstrated (81). Another group showed that melatonin inhibits hippocampal LTP via a mechanism involving MT2-receptor-mediated regulation of the adenylyl cyclase-cAMP-PKA pathway (430). Finally, it has been reported that melatonin significantly alters synaptic transmission and LTP in area CA1, but has only modest actions in area CA3 in mouse hippocampal slices (318). Taken together, these findings suggest that melatonin inhibits hippocampal LTP.
In addition to its impact on hippocampal synaptic plasticity, melatonin was shown to prevent the induction of neocortical LTP and impair spatial performance in the radial maze (393). Neonatal rats given low doses of melatonin exhibited impaired hippocampal LTP and displayed learning and memory deficits. Melatonin also exacerbated LTP impairments as well as learning and memory deficits induced by lead (77). Finally, in a study designed to assess if dark rearing (i.e., high levels of melatonin) affects hippocampal synaptic plasticity, melatonin administered early postnatally prevents the induction of LTP (408).
There seems to be a consensus that melatonin inhibits LTP induction. This may seem surprising as it might be expected that an antioxidant should improve cognition rather than making it worse. However, these studies (77, 92, 139, 318, 393, 408, 430) were performed in the absence of excessive oxidative stress. Going back to the first part of this review, we described numerous studies indicating that ROS are important signaling molecules necessary for proper LTP induction, and therefore scavenging radicals with melatonin likely explains the detrimental effects of melatonin on synaptic plasticity.
b. Melatonin and learning and memory
Similar to what has been observed in LTP studies, behavioral studies indicate that melatonin has a detrimental effect on learning and memory performance (242, 355, 375, 466). On the other hand, in studies performed under situations of oxidative stress, the effect of melatonin on learning and memory deficits is for the most part positive. This is consistent with the idea that melatonin quenches bad ROS under stress situations to produce a positive effect on learning and memory. The studies describing the beneficial effects of melatonin and memory function can be largely classified in four major subcategories, depending on the pathological condition causing the oxidative stress-induced learning and memory deficits: Alzheimer's disease, alcohol poisoning, excitotoxicity/trauma/ischemia, and other oxidative stress conditions.
1. Alzheimer's disease
The effect of melatonin on AD-related decreases in learning and memory function has been studied using multiple animal models of the disease and various types of behavioral tests, as well as measurements of oxidative stress markers. These animal models include intracerebroventricular injections of either STZ (377, 379) or amyloid β peptides (381 –383), transgenic APP695 mice (128, 144), transgenic APP/PS1 mice (314), and scopolamine-induced cognitive dysfunction (6). Behavioral tests included passive avoidance (6, 128, 379, 382), shuttle box (382), water maze (144, 253, 314, 381 –383), open field activity (144, 314), radial arm water maze (314), Y maze (314), elevated plus maze (314), and circular platform (314). Oxidative stress markers included malondialdehyde (379), GSH (379, 383), lipid peroxidation (383), SOD (253, 314, 383), choline acetyltransferase activity (128), acetylcholinesterase (6), GPx (314), catalase (314), MAO (253), and ER stress-related proteins including BiP/GRP78 and CHOP/GADD153 (253). With the exception of one study (413), all studies uniformly showed that melatonin has a positive effect on memory performance and affords protection against oxidative stress-induced damage associated with AD.
2. Alcohol poisoning
It has been shown that either aged or chronic ethanol-treated mice exhibit poor retention of memory in step-down passive avoidance and in elevated plus-maze task (352). Chronic melatonin treatment reversed cognitive deficits in aged and ethanol-intoxicated mice, but failed to modulate the retention performance of young mice, both effects being consistent with the antioxidant properties of melatonin (352). In another study of the effect of melatonin supplementation on chronic ethanol-induced learning and memory dysfunctions and markers of oxidative stress (37), it was found that melatonin significantly reduced lipid peroxidation and elevated GSH levels with a concomitant improvement in learning (Morris water maze and passive avoidance) that was more substantial in older as opposed to younger rats (37). The effects of melatonin on acute ethanol-induced increases in oxidative stress (lipid peroxidation, GPx, SOD) and impairments in learning and memory (Morris water maze) have been studied. These investigators found that melatonin reversed increases in the markers of oxidative stress without a significant effect on spatial memory (148). Surprisingly, in this study melatonin alone had a positive effect on water maze performance (148).
3. Excitotoxicity/trauma/ischemia
Trauma and excitotoxi-city induce cognitive dysfunction resulting from hippocampal damage. Melatonin exhibited a protective effect against head trauma-induced hippocampal damage and spatial memory deficits in immature rats (319). It also prevented learning disabilities associated with ibotenate (a glutamate analog)-induced excitotoxicity (60). During ischemia and cerebral hypoperfusion, melatonin plays a similar protective role (100, 246), highlighting its important antioxidant protective role against learning and memory deficits induced by oxidative stress.
4. Other
Melatonin reversed oxidative stress and improved the learning and memory deficits induced by either ovariectomy (129, 130) or hyperhomocysteinemia (34, 35) in adult rats. Melatonin also reduced memory impairments and oxidative damage in mouse models of senescence (85, 384) and diabetes (421). Lastly, melatonin improved stress-related declines in learning and memory (363), as well as deficits associated with exposure to the neurotoxic industrial solvent thinner (36).
In conclusion, melatonin is a potent antioxidant that shows significant positive effects on learning and memory performance under oxidative stress conditions. Its negative effects on learning and memory under normal physiological conditions recapitulate a recurring theme in this review: ROS are important signaling molecules for synaptic plasticity, and therefore quenching balanced physiological levels of ROS may lead to detrimental rather than beneficial effects with respect to memory function. Moreover, care should be taken in selecting the proper concentration for treatment as it has been shown that blood concentrations which are 10 times normal levels can cleave heme molecules to liberate iron and induce oxidative stress (89). Even higher levels of melatonin concentrations can deplete reduced glutathione levels and promote fas-induced cell death (440).
7. Lipoic acid
Lipoic acid is an organosulfur compound that was first postulated to be an effective antioxidant because it prevented symptoms of vitamin E deficiency (334). The mechanism of action for lipoic acid to act as an antioxidant is not well understood. It has been shown that dihydrolipoic acid (the reduced form of lipoic acid) can regenerate oxidized antioxidants, such as GSH, vitamin C, and vitamin E (50, 320), thereby permitting them to enter the circulation to quench more free radicals. Lipoic acid also has been shown to possess the ability to scavenge ROS in vitro; however, there is no clear evidence that this actually occurs in vivo. Recent findings suggest that lipoic acid can boost mitochondrial membrane potential and oxygen consumption, while decreasing mitochondrial production of oxidants (280). This occurs by lipoic acid amplifying key antioxidant protective mechanisms via PPARγ coactivator-1α (PGC-1α) mediation (280).
Lipoic acid has been studied extensively as a therapeutic antioxidant to protect against the damaging effects of oxidative stress on synaptic plasticity and memory. Studies have shown that lipoic acid significantly reversed oxidative stress-induced deficits in LTP (282, 429) and learning and memory in a variety of pathological conditions, such as aging (126, 254, 291, 339), radiation poisoning (268, 269), AD (348, 377, 380), and diabetes (49). In an attempt to identify the underlying mechanism of lipoic acid protection of cognitive functions, α-lipoic acid enhances Ca2+ entry through presynaptic N- and P/Q-type channels to cause an increase in evoked glutamate release from rat cerebrocortical synaptosomes (432). This facilitation of glutamate release by α-lipoic acid was found to be due, at least in part, to activation of PKA and PKC (432). Collectively, these studies support the hypothesis that ROS are involved in the modulation of learning and memory processes.
C. Minerals
Given that they are essential cofactors for antioxidant enzymes, minerals such as selenium, zinc, and manganese play an important antioxidant role, especially in the mechanisms underlying learning and memory processes.
1. Selenium
Although it is toxic in large doses (462), selenium is an essential micronutrient for animals. In humans, selenium is a trace element nutrient that functions as cofactor for multiple enzymes, most notably GSH peroxidases and certain forms of TRX reductase found in animals and some plants (178, 365). This confers to selenium an indirect role as an antioxidant and neuroprotectant mineral. In fact, selenium has been shown to protect the brain from oxidative damage in various models of neurodegeneration. For example, rats treated with intracerebroventricular STZ are a model for oxidative damage and sporadic AD, and display severe deficits in the passive avoidance and Morris water maze tests that is accompanied by high levels of lipid peroxidation, protein carbonylation, reduced GSH, GPx, GR, and ATP in the hippocampus and cortex. Selenium, in line with its powerful antioxidant function, prevented all of the alterations induced by STZ, including the cognitive deficits and the oxidative damage (198). Selenium also has been shown to be neuroprotective and beneficial for memory deficits induced by lead poisoning (237) and psychosis, without affecting baseline behavior (260). The beneficial effects of selenium also were explored in fluoride toxicity (462) and ischemia models (158), where it was shown to enhance ROS scavenging and improve short-term memory impairments. In addition, organic selenium compounds enhanced long-term memory in a novel object recognition test (364). However, memory in the active avoidance shuttle box test was normal in rats that were selenium and vitamin E deficient (32). In a more recent study, using the same model it was shown that the absence of selenium can cause changes in synaptic function in area CA3 of the hippocampus, and inferred that it must play a role in learning and memory (435). These studies suggest that the effects of selenium as an antioxidant beneficial to learning and memory is dependent on the task, as well as the concentration used. Consistent with this idea, it was demonstrated that at higher concentration, selenium could cause toxicity (462).
2. Zinc
After iron, zinc is the second most abundant metal in humans. Zinc is an essential micronutrient for a wide variety of biological functions and has been shown to be a component of more than 1200 enzymes, either acting as a cofactor or as a structural element (137). It is actually the only metal found in all enzyme classes. In the brain, zinc is packaged into vesicles of glutamatergic neurons (53) and plays a critical role in brain excitability (165) as well as synaptic plasticity and learning (303). Similar to selenium, zinc has antioxidant functions, but not without controversy (290). Selenium is an integral component of GSH peroxidases and therefore acts as a general antioxidant, whereas zinc is an antioxidant only at specific sites (45). Although zinc is an integral component of the antioxidant enzyme SOD-1, it plays a structural role as opposed to being a cofactor. In support of this notion, zinc deficiency does not affect SOD-1 activity. On the contrary, SOD-1 activity is depressed with high zinc intake (45). The antioxidant properties of zinc stem mainly from competition with iron and copper for binding to certain proteins, thereby displacing redox-active metals. Zinc also can bind the sulfhydryl groups in proteins, thereby protecting them from oxidation (45). There is a large body of literature discussing the involvement of zinc in synaptic plasticity and memory (44, 303), but this is beyond the scope of this review. To our knowledge, none of these reports have discussed the role of zinc in learning and memory in the context of its antioxidant abilities.
3. Manganese
Manganese is an essential trace element (120) acting as a cofactor for multiple classes of enzymes, including, but not limited to, oxidoreductases, transferases, and hydrolases, most of which are involved in redox homeostasis. The best known manganese-containing enzymes are SOD-2 and arginase (406). As discussed earlier, SOD-2 is the main superoxide scavenger of mitochondria and as such is one of the most characterized antioxidant enzymes. The involvement of manganese in oxidative stress-induced synaptic plasticity and memory stems mainly from its association with SOD-2, which was described earlier in the review.
VII. Conclusions
In this review we have provided extensive evidence demonstrating a dual role for ROS in the brain (Fig. 13). Under physiological conditions, ROS act as essential signaling molecules, necessary for the proper formation of learning and memory processes. However, during aging, ischemia, trauma, or neurodegenerative diseases, the levels of ROS increase to levels higher than the antioxidant machinery of the cells can handle, and therefore their beneficial signaling role becomes outweighed by the ambiance of oxidative damage that they create. We also have reviewed evidence showing that the specific species of ROS act differently in various systems. For example, both cytosolic and extracellular superoxide have been shown to be involved in synaptic plasticity and memory. Mitochondrial superoxide, however, failed to show any involvement in age-dependent synaptic plasticity and memory dysfunction, perhaps due to limited diffusion of mitochondrial superoxide across the mitochondrial membrane. Mitochondrial superoxide proved to be a key player in conditions of extreme oxidative stress such as occurring during AD. This is not surprising, in light of the wealth of literature documenting mitochondrial dysfunction (and hence increased probability for leakage of mitochondrial free radicals through damaged membranes) as an integral part of AD (Fig. 14).
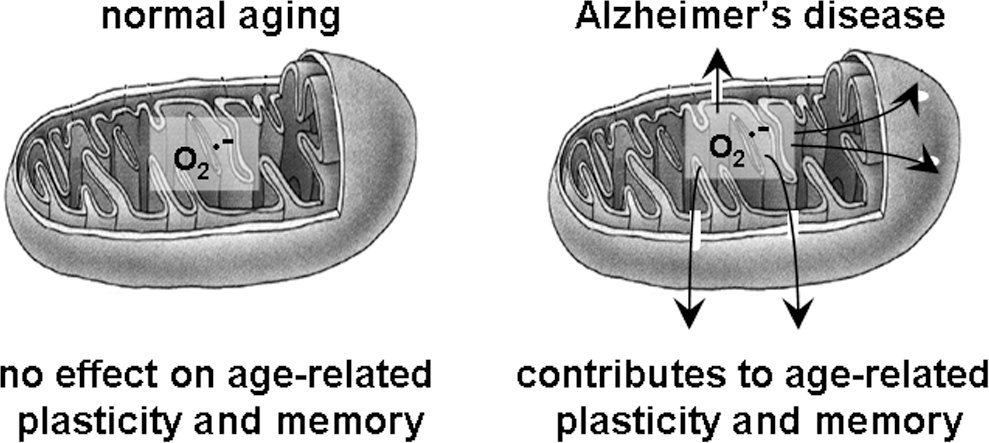
Footnotes
Acknowledgments
C.A.M. is supported by a fellowship of the American Health Assistance Foundation, Alzheimer's Disease Research. This work also was supported by NIH grant NS034007 and Alzheimer's Association Investigator Initiated Research Grant 09-131756 (to E.K.). The authors apologize to their fellow scientists whose work could not be cited due to space limitations.