
Abstract
Cancer chemoprevention is a process of using either natural or synthetic compounds to reduce the risk of developing cancer. Observations that NF-E2-related factor 2 (Nrf2)-deficient mice lack response to some chemopreventive agents point to the important role of Nrf2 in chemoprevention. Nrf2 is a member of basic-leucine zipper transcription factor family and has been shown to regulate gene expression by binding to a response element, antioxidant responsive element. It is generally believed that activation of Nrf2 signaling is an adaptive response to the environmental and endogenous stresses. Under homeostatic conditions, Nrf2 is suppressed by association with Kelch-like ECH-associated protein 1 (Keap1), but is stimulated upon exposure to oxidative or electrophilic stress. Once activated, Nrf2 translocates into nuclei and upregulates a group of genes that act in concert to combat oxidative stress. Nrf2 is also shown to have protective function against inflammation, a pathological process that could contribute to carcinogenesis. In this review, we will discuss the current progress in the study of Nrf2 signaling, in particular, the mechanisms of Nrf2 activation by chemopreventive agents. We will also discuss some of the potential caveats of Nrf2 in cancer treatment and future opportunity and challenges on regulation of Nrf2-mediated antioxidant and antiinflammatory signaling in the context of cancer prevention. Antioxid. Redox Signal. 13, 1679–1698.
Introduction
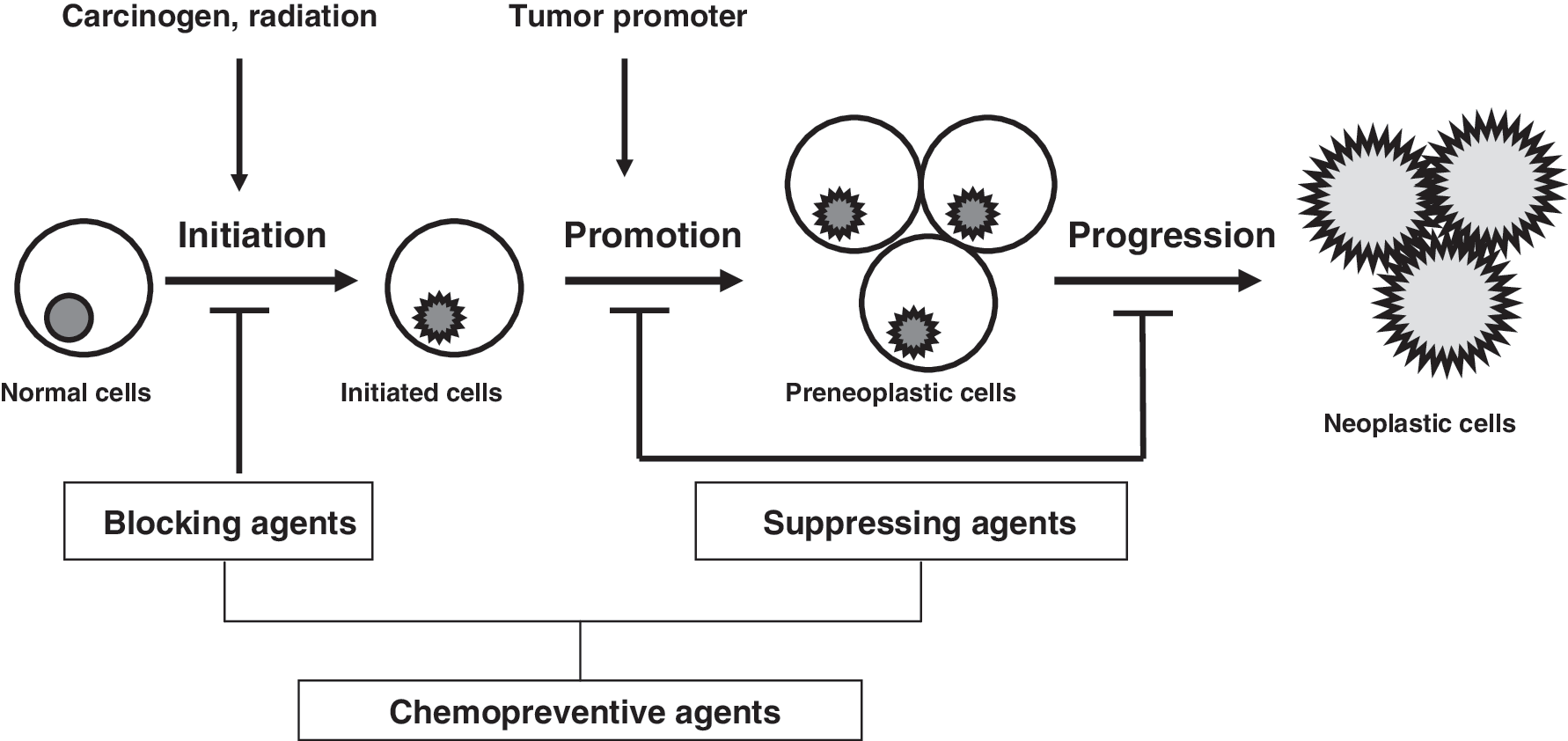
Most of the chemical substances used in cancer chemoprevention studies are natural phytochemicals found in food (143). On the basis of the inhibition stages, Wattenberg has classified chemopreventive agents into two categories—blocking agents and suppressing agents (161). Blocking agents act by preventing carcinogens from reaching the target sites, from undergoing metabolic activation, or from subsequently interacting with crucial cellular macromolecules such as DNA, RNA, and proteins at initiation stages. Suppressing agents, on the other hand, inhibit the malignant transformation of initiated cells at either the promotion or the progression stage. Some agents may work on all three stages of carcinogenesis, and are hence classified into both categories (Fig. 1).
Many different animal models and cancer cell lines have been used to evaluate the chemopreventive values of phytochemicals, and have led to the discovery of new classes of chemopreventive agents, such as isothiocyanates from cruciferous vegetables, polyphenols from green and black tea, and flavonoids from soybeans (143) (Fig. 2). Progress has been also made in understanding the mode of action of newly identified chemopreventive agents. Exposure to the chemopreventive agents produces certain level of reactive oxygen species (ROS) or electrophiles, and causes mild oxidative/electrophilic stresses in cells (20, 84, 125). Such mild oxidative stresses are sufficient to initiate the signaling pathways that, in turn, can activate a variety of cellular events, such as induction of phase II detoxification enzymes and antioxidant enzymes, expression of tumor-suppressor genes, and inhibition of cell proliferation and angiogenesis (51). Although the signal transduction pathways in response to oxidative and/or electrophilic stress have been studied extensively for many years, our understanding in the cutting-edge area of redox signaling and cancer chemoprevention is still in its infancy. In this review, we will discuss the current progress in study of NF-E2-related factor 2 (Nrf2) signaling, in particular, the mechanisms of Nrf2 activation by chemopreventive agents. Future challenges on regulation of Nrf2-mediated antioxidant and antiinflammatory signaling in the context of cancer prevention will also be discussed.
Kelch-Like ECH-Associated Protein 1-Nrf2-Antioxidant Responsive Element Signaling
To survive under a variety of environmental or intracellular stresses, eukaryotic cells have developed elaborate yet highly efficient cyto-protective machinery to protect themselves from oxidative or electrophilic challenges (57, 92). Proteins that comprise phase II detoxification and antioxidant enzymes provide an enzymatic line of defense against electrophiles and ROS. Among the family of phase II and antioxidant enzymes are glutathione S-transferase (GST), UDP-glucuronosyltransferase, heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO1), and glutamate cysteine ligase (gamma glutamylcysteine synthetase). Induction of phase II and antioxidant enzymes are coordinately regulated through a consensus cis-element at the 5′-flanking promoter region, named antioxidant responsive element (ARE) (33, 136) or electrophile response element (34) (for simplicity, we will use ARE from here on). ARE-mediated gene expression plays a central role in the cellular defense against cellular oxidative damage. Ample experimental evidence also supports the view that induction of ARE-mediated cytoprotective enzymes is a critical and sufficient mechanism to enable protection against carcinogenesis provoked by environmental and endogenous insults.
One of the key ARE-binding transcription factors is Nrf2 (108). Induction of cytoprotective enzymes in response to ROS, electrophiles, and chemopreventive agents is a cellular event that is highly dependent on Nrf2 protein. By activating Nrf2 signaling, chemopreventive agents are able to increase cellular detoxification and antioxidant enzymes, thereby enhancing removal of reactive carcinogens and blocking carcinogenesis. This hypothesis has been tested in many studies. For example, when investigating the chemoprotective effect of oltipraz in human subjects having a high incidence of liver cancer, Kensler's group found that oral administration of oltipraz significantly enhanced the urinary excretion of the phase II conjugation products of the carcinogen aflatoxin (77). Another study with sulforaphane (SFN), an isothiocyanate present abundantly in cruciferous vegetables, has shown that oral administration of SFN could effectively block benzo[a]pyrene-induced forestomach tumors in ICR mice and this protective effect of SFN was abrogated in the Nrf2 knockout (Nrf2 KO) mice (31), supporting a critical role of phase II detoxification and antioxidant enzymes in prevention of carcinogenesis by chemopreventive agents.
Given the different chemical nature of Nrf2 inducers (128), a mechanism of activation requiring the interaction of Nrf2 inducers with a structurally complementary receptor appears to be quite unlikely. However, many Nrf2 inducers and chemopreventive agents exhibit prooxidant and electrophilic property and may generate different levels of oxidative stress, depending on their doses. It would be reasonable to speculate that a mild oxidative stress generated by chemopreventive agents at appropriate concentrations is sufficient to activate Nrf2 signaling, but not strong enough to cause any adverse effect such as DNA damage or cells death. Activation of Nrf2 signaling further induces detoxification and antioxidant enzymes, thus protecting cells from damage by more active oxidants or electrophiles, such as carcinogens.
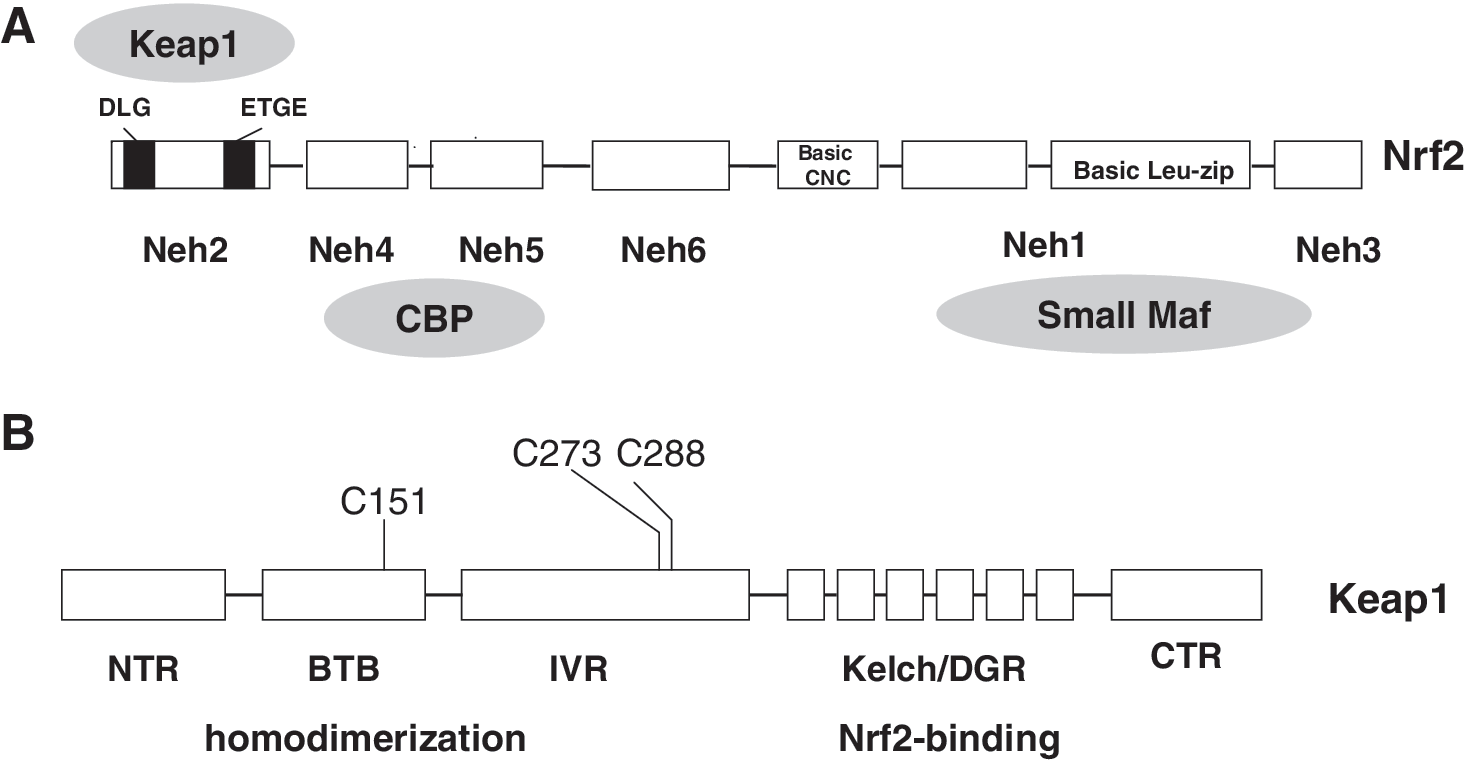
Nrf2 was initially discovered in a screen that utilized a tandem repeat of the consensus AP-1 and NF-E2-binding sequences identified in the 5′ locus control region of β-globin gene (108). Nrf2 belongs to basic-leucine zipper (bZip) transcription factor family and has six highly conserved domains called Nrf2-ECH homology (Neh) domains (Fig. 3A). The Neh1 contains a bZip DNA binding motif that enables Nrf2 to interact other bZip transcription factor and form a heterodimer on ARE (109). The Neh3 domain has been shown to function as a transactivation domain and might be involved in interaction with components of transcriptional apparatus (118). The Neh6 domain may contain a degron that contributes to Nrf2 degradation (107). Neh4 and Neh5 domains are found to cooperatively bind with the KIX and CH3 domains of coactivator CBP and to enhance Nrf2 transcription activity (74). The Neh2 domain located at the N-terminus mediates binding with Kelch-like ECH-associated protein 1 (Keap1), a cytosolic protein that inhibits Nrf2 signaling by promoting Nrf2 degradation through ubiquitin/proteasome pathway (73, 109). Similar to many other transcription factors, Nrf2 is subject to regulation by cellular localization. Under basal condition, majority of Nrf2 molecules are found to be in the cytoplasm. Upon exposure to the oxidative stresses, Nrf2 can quickly translocate into the nucleus (60, 123, 164) and form a heterodimer with small musculoaponeurotic fibrosarcoma (Maf ) protein (59), which then binds to ARE and enhances transcription of a group of genes that encode phase II detoxification and antioxidant enzymes (28, 121, 126). Many years of research establish that a signal transduction pathway consisting of Keap1-Nrf2-sMaf-ARE is a primary sensor of the signaling generated by electrophiles and chemopreventive agents, and is essential for elicitation of the antioxidant response.
Keap1
Keap1 plays a central role in the regulation of Nrf2 activity. Keap1 was isolated as a Nrf2-associating protein by using a yeast two-hybrid screening system (35). Knockout of Keap1 resulted in constitutive activation Nrf2 signaling (121, 156, 157). Keap1 is a 69-kDa cytosolic protein with high homology to Drosophila actin-binding protein Kelch (61). Primary sequence and alignment analysis reveals that human Keap1 consists of five domains: (i) the N-terminal region (a.a. 1–66), (ii) broad complex, tramtrack, and bric a brac domain (BTB, a.a. 67–178), (iii) an cysteine-rich intervening region (IVR, a.a. 179–321), (iv) double glycine/Kelch repeat region (DGR a.a. 322–608), and (v) C-terminal region (CTR, a.a. 609–625) (29) (Fig. 3B). Keap1 was observed to locate mainly in the cytoplasm, presumably by association with F-actin or myosin VIIa through the Kelch domain (61, 69, 70, 153). Accordingly, Keap1 is hypothesized to be a cytosolic anchor protein that sequesters Nrf2 in cytoplasm under the unstimulated condition. Site-directed mutagenesis of a conserved serine (S104A) within the Keap1 BTB domain revealed that Nrf2 is indeed sequestered in cytoplasm by Keap1 (182). A recent study using transgenic animal-originated cells demonstrated that direct interaction between the Neh2 domain of Nrf2 and Kelch repeat domain of Keap1 is essential for retaining of Nrf2 in the cytoplasm by Keap1 (70). Keap1 also contains a strong nuclear exporting signal (NES) sequence in the IVR domain, which may play a major role in determining the subcellular distribution of Keap1 (72, 142, 154). Keap1 is identified to be associated with Cullin-3, a scaffold protein responsible for formation of an ubiquitin ligase E3 complex (25, 36, 87, 177). It is now clear that Keap1 may not only dictate Nrf2 localization, but also actively targets Nrf2 to degradation.
Due to its high cysteine content, Keap1 was proposed to be the sought-after sensor of ARE inducers soon after its discovery as a suppressor of Nrf2 transcriptional activity (128). Convincing evidence has since accumulated that the cysteine residues in Keap1 indeed play an important role in sensing the presence of ARE inducers and oxidative stress. Several independent studies have demonstrated that the sulfhydryl groups of several Keap1 cysteine residues can be directly modified by oxidation, reduction, or alkylation. Among these cysteine residues, Cys151, 273, and 288 appear to be essential for regulating Nrf2 function (90, 102, 168, 176). Mutation of Keap1 Cys151 to Ser did not affect Nrf2 degradation by Keap1, but abolished response to electrophiles and oxidants (176). The role of Cys151 in repression of Nrf2 ubiquitination by electrophiles or oxidants has been verified in vivo (168). Mutation of either Cys273 or Cys288 residues prevented Keap1 from degrading Nrf2 protein, pointing to a critical role of these cysteine residues in Keap1-mediated Nrf2 ubiquitination (150, 176). A recent study confirmed that the Cys273 and Cys288 amino acids are essential for Nrf2 degradation by Keap1; however, mutation of these two cysteine residues does not affect interaction of Nrf2 and Keap1 (88). Further, both C273A and C288A mutants are able to complement each other in degradation of Nrf2 protein when coexpressed in cells (156). However, later, the same laboratory has reported that simultaneous expression of C273A and C288A mutant proteins in mice could not rescue Keap1 null mice from juvenile lethality and repress Nrf2 at all (168). Putting together, these studies demonstrate the significance of multiple Keap1 cysteine residues in sensing the presence of oxidants and electrophiles, although it is not clear how the modification of those cysteine residues alters Keap1 function.
Like Nrf2, Keap1 also was reported to be subject to ubiquitination, but its degradation was proteasome independent, which may play a role in activation of Nrf2 signaling by ARE inducers (178). However, different ARE inducers appear to have very different effects on Keap1 ubiquitination and stability. Zhang et al. first proposed that tert-butylhydroquinone (tBHQ)-induced ubiquitination of Keap1 results in increased degradation of Keap1 by a proteasome-independent mechanism. They observed that, unlike tBHQ, SFN treatment did not cause any Keap1 ubiquitination and degradation. They also noted that the increased Keap1 degradation occurred through a pathway independent of C151, which has been shown to be essential for regulation of Nrf2 degradation. Other groups also observed effects of ARE inducers on Keap1 ubiquitination and stability. For example, Hong et al. (48) noted that electrophilic adduction to Keap1 cysteine residues coincided with the increase of Keap1 ubiquitination at K298 and stabilization of Nrf2 protein in the cells exposed to N-iodoacetyl-N-biotinylhexylenediamine. Treatment of HepG2 cells with quercetin was found also to decrease endogenous Keap1 levels, although change in Keap1 ubiquitination was detected (145). These studies suggest that ubiquitination and subsequent degradation of Keap1 might be a mechanism of Nrf2 induction after exposure to certain ARE inducers. However, further studies are needed to clarify whether the different findings are consequence of using different cell lines or different ARE inducers. Studies are also needed to determine whether medication of Keap1 cysteine residues play the role in Keap1 ubiquitination.
The “hinge and latch” two-site binding model
Keap1 is characterized as a substrate adaptor protein for a cullin-3-based ubiquitin E3 ligase and is essential for ubiquitination of Nrf2 (25, 36, 87, 177). Keap1 has two functional domains: the Kelch/DGR domain, which binds Nrf2, and the BTB domain, which is responsible for homodimerization and interaction with the N-terminus of cullin-3 (25, 36, 87, 177). On the basis of sequence homology and mutation analyses (73, 89, 107), it was reported that Nrf2 has two Keap1 binding motifs in the Neh2 domain: an ETGE motif, which locates at the N-terminal region and is important for ubiquitination of Nrf2 (73, 107), and a DLG motif, which is essential for interacting with Keap1 (89) (Fig. 3A). The ETGE motif and the DLG motif mediate a cooperative binding to Keap1. Deletion of the ETGE motif significantly weakened Keap1 binding mediated by the DLG motif; therefore, the ETGE motif appears to play a permissive role for the DLG motif. Recently, isothermal calorimetry measurement also detected a two-phase binding between Keap1 and Neh2. The Keap1 binding affinity of the ETGE motif is almost two orders of magnitude stronger than the DLG motif (148). In addition, seven lysine residues of the Neh2 domain, which reside upstream of the ETGE motif, have been shown to be indispensable for Keap1-dependent poly-ubiquitination and degradation of Nrf2 (177).
A “hinge and latch” two-site binding model is proposed (149). Two Keap1 molecules form homodimer via their BTB domain (182). The high affinity ETGE motif functions as a hinge to pin down Neh2 to Keap1, while the low affinity DLG motif functions as a latch. Linking the hinge and the latch motif is a lysine-rich central α-helix. In this nine-turns α-helix, six of seven lysine residues are located on one side of the helical surface (148). Since these lysine residues are targets for Keap1-mediated ubiquitination by E3 ligase (177), these lysine residues are speculated to be biologically important for the regulation of Nrf2 stability. When the latch is in position, it helps to set the central α-helix in an adequate orientation to expose these lysine residues (177).
The two-site binding is relatively unstable. The low affinity latch binding may be more vulnerable to the conformation change of Keap1 dimer resulted from oxidative modification. It is proposed that the DLG motif may possess the signaling switch for the repressive regulation of Nrf2 via the Keap1-dependent ubiquitination and proteasomal degradation. The switch from two-site binding to one-site binding may explain the observed facts that the inhibition of Nrf2 degradation is not at the expense of Keap1/Nrf2 binding. Only recently, the X-ray crystallography structure of mouse Keap1 homodimer was reported. Three-dimensional reconstruction at 24-A resolution revealed two large spheres attached by short linker arms to the sides of a small forked-stem structure, resembling a cherry-bob. Each sphere has a tunnel corresponding to the central hole of the β-propeller domain, as determined by X-ray crystallography. The IVR domain appears to surround the core of the β-propeller domain. The unexpected proximity of IVR to the β-propeller domain suggests that any distortions generated during modification of reactive cysteine residues in the IVR domain may send a derepression signal to the β-propeller domain and thereby stabilize Nrf2. This study thus provides a structural basis for the two-site binding and hinge-latch model of stress sensing by the Nrf2-Keap1 system (120).
Nrf2
In human Nrf2 protein, one bipartite nuclear localization signal (NLS) was identified in the basic region, named bipartite nuclear localization signal at the basic region (bNLS) (62, 100). In addition, a monopartite NLS was characterized at the amino-terminus (NLSN) and another monopartite NLS was identified at the carboxyl-terminus (NLSC) of human and murine Nrf2 (146). One NES was characterized in the ZIP dimerization domain, named NESzip (62, 100). Recently, another NES was characterized in the Neh5 transactivation (TA) domain, named NESTA (103). In full-length wild-type (WT) Nrf2, the combined nuclear exporting activities of NESTA and NESzip appear to be able to counter balance the nuclear importing activities mediated by bNLS, NLSN, and NLSC motifs. We have shown that when expression of a green fluorescence protein (EGFP)-tagged Nrf2 was enhanced in HeLa cells, a heterogeneous distribution pattern of EGFP-Nrf2 was observed. Nearly 60% cells showed whole cell distribution, 30% cells showed nuclear distribution, and 12% cells showed cytosolic distribution (103). Mutation of single key leucine residue in the NES motif, such as L184 in the NESTA motif and L544 in the NESzip motif, was sufficient to abolish their nuclear exporting activity (100, 103). Mutation of either the L184 residue or the L544 residue in full-length Nrf2 was sufficient to convert the heterogeneous distribution of EGFP-Nrf2 to the homogeneous nuclear distribution pattern (103), suggesting that both the NESTA and NESzip motifs are indispensable for balancing the effect of bNLS, NLSN, and NLSC motif. We also found that the NESTA motif was redox sensitive (103), but not the bNLS motif and the NESzip motif (100). The redox sensitivity of the NLSN and NLSC motif has not been determined and was speculated to be redox insensitive, since no cysteine residue exists in these two NLS motifs (102).
As discussed above, Nrf2 contains a redox-sensitive NES motif; it is, therefore, reasonable to assume that Nrf2 by itself can sense and transducer redox signaling. We speculate that, under homeostatic conditions, both the NLS and the NES counteract each other and reach a dynamic balance; as a result, Nrf2 exhibits whole cell distribution. The presence of Nrf2 in nuclei may account for the basal or constitutive Nrf2 activities. When challenged with oxidative stress, the redox-sensitive NESTA is disabled, whereas the redox-insensitive NLS remains functional (15, 100), leading to predominant nuclear distribution of Nrf2. Nrf2 consists of a constitutively active NESzip bNLS-NLSN-NLSC tandem and a conditional NESTA motif. The NESTA motif may function as a redox-sensitive switch to determine Nrf2 localization under different redox conditions.
A redox-signaling model of the antioxidant response
Accumulating evidence showed that Keap1-mediated Nrf2 ubiquitination is highly sensitive to redox status, although Keap1/Nrf2 dissociation is relatively insensitive. Nrf2 nuclear translocation also has high redox sensitivity. Accordingly, we propose a redox-signaling model for activation of Nrf2 signaling by oxidative stress (Fig. 4). There are two pools of Nrf2 proteins in cells: the free-floating Nrf2 (fNrf2) and the Keap1-binding Nrf2 (kNrf2). Under homeostatic condition, kNrf2 are constantly degraded through ubiquitin/proteasome pathway, and cells will maintain a relatively small size of fNrf2 pool when the equilibrium is reached between Nrf2 synthesis and its degradation. However, upon exposure to oxidative stress, the Keap1-mediated redox-sensitive Nrf2 ubiquitination and degradation is impeded, and the pool of fNrf2 proteins begins to expand (129, 175, 178). The fNrf2 proteins can further response to the redox signals and translocate into the nucleus. On the basis of this model, the size of fNrf2 pool is believed to be a key determinant of the magnitude of antioxidant response (170). If cells have more fNrf2, same amount of ROS may elicit more Nrf2 influx into the nucleus and consequently induce a stronger antioxidant response. Thus, it would be more reasonable to hypothesize that both Keap1 and Nrf2 are functioning as redox sensors in response to oxidative stress.
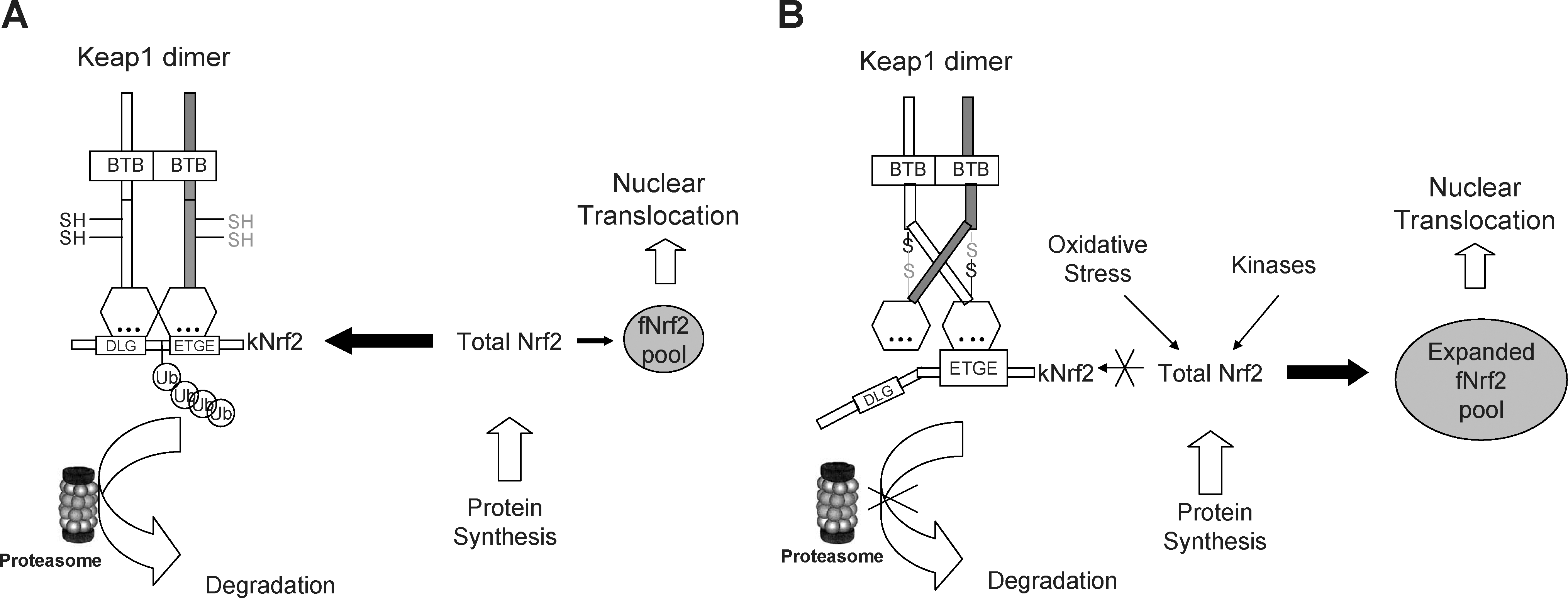
This model may explain the chemoprotective effect of phase II enzyme inducers. Treatment with a low dosage of phase II enzyme inducers can impede Keap1-mediated ubiquitination of Nrf2 and expand the fNrf2 pool. As such, the phase II inducers exert a priming effect to tune up the overall redox sensitivity of the cell. Once the cell confronts oxidative stress, it can readily initiate an effective antioxidant response to neutralize the oxidative toxicity and restore redox homeostasis.
Regulation of Nrf2 signaling by other pathways
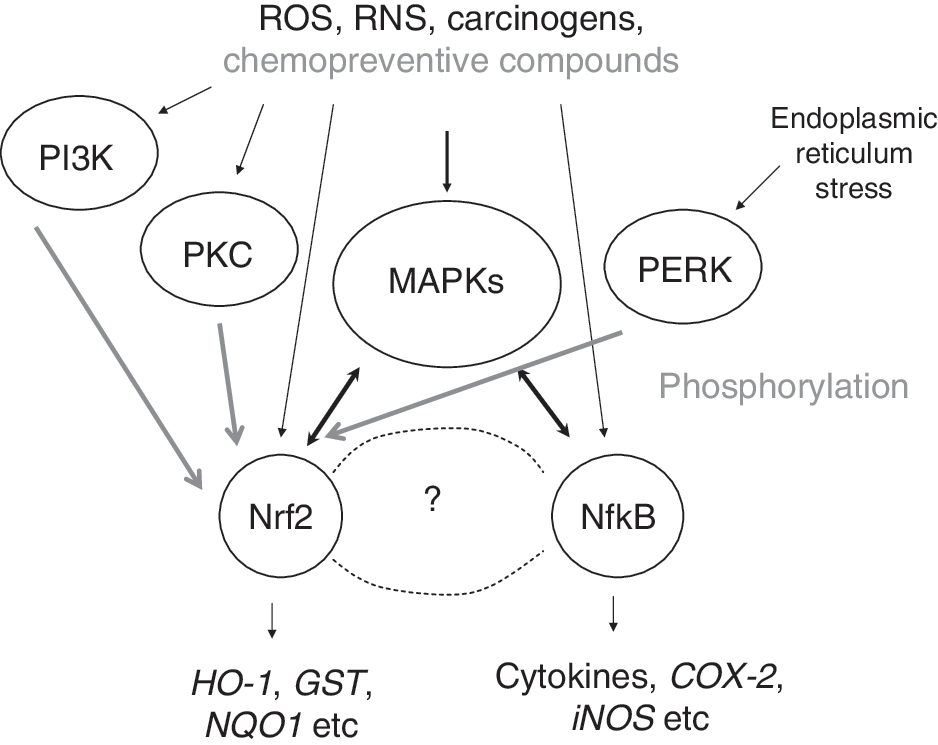
Other signaling pathways have been reported to influence Keap1-Nrf2-sMaf-ARE signaling, among which are several kinases, such as protein kinase C (PKC), mitogen-activated protein kinases (MAPKs), phosphatidylinositol 3-kinase (PI3K), and PKR-like endoplasmic reticulum kinase (PERK) (24, 53, 99, 170). Activation of the extracellular signal-regulated protein kinase 2 (ERK2) and c-Jun NH(2)-terminal kinase-1 (JNK1) pathway appeared to enhance Nrf2 signaling (6, 137, 167, 170, 171, 180, 181). In contrast, the activation of the p38 MAPK pathway appeared to inhibit Nrf2 signaling (79, 137, 172, 181). Most recently, it was found that phosphorylation of Nrf2 by p38 increases Keap1/Nrf2 binding (79). This finding suggests that p38 may inhibit Nrf2 signaling by enhancing Keap1 sequestering of Nrf2 in cytoplasm. By using a cell-free system, Huang et al. have demonstrated that PKC can directly phosphorylate Nrf2 at Serine 40 (52), thereby promoting its dissociation from Keap1 (16, 53, 119). PI3K is also reported to play a role in Nrf2 activation (68, 69, 99, 113). In response to oxidative stress, PI3K signaling is activated and results in depolymerization of actin microfilaments, which somehow facilitates Nrf2 nuclear translocation (69). PERK has been shown to phosphorylate Nrf2 and enhance its nuclear translocation by disrupting Keap1 binding (24).
Several coactivators, like p160 and CBP/p300, are shown to interact with the Nrf2–Maf–ARE complex and enhance Nrf2 transcriptional activity (74, 179). A recent study shows that activation of ERK and JNK pathways promotes recruitment of coactivator to the transcription initiation complex and upregulates Nrf2 transcriptional activity (137).
Nrf2 and Chemoprevention
Cancer has been reported to be a leading cause of death in the United States (64). As discussed earlier, carcinogenesis is a multistep process, thus providing numerous opportunities for cancer prevention. At this time, prevention could be very costly, but it could be the best approach toward cancer managements. It is estimated that it would cost clinical prevention in the United States from under $US2,000 to over $US6,000,000 per life-year saved (54). Even so, American society would appear to be willing to pay for such costly cancer prevention programs since the disease inflicts great pains and the burden for treatment is overwhelming. The good news is, cancer is a preventable disease. After going through the review of previous section on Keap-Nrf2-ARE signaling, it is timely to enter into the topic on regulation of Nrf2 signaling for cancer chemoprevention.
Current studies have shown that some chemopreventive compounds are not only effective in animal models, but also promising in the on-going clinical trials (50). In general, chemopreventive compounds may work via multifaceted molecular pathways (4, 46, 66), such as by suppressing inflammation, cell proliferation, invasion, and angiogenesis of tumor cells (5), and by enhancing the antioxidant response (102, 174). A growing body of evidence justifies that targeting the Nrf2 pathway is a viable approach in cancer prevention (82, 83, 94, 112).
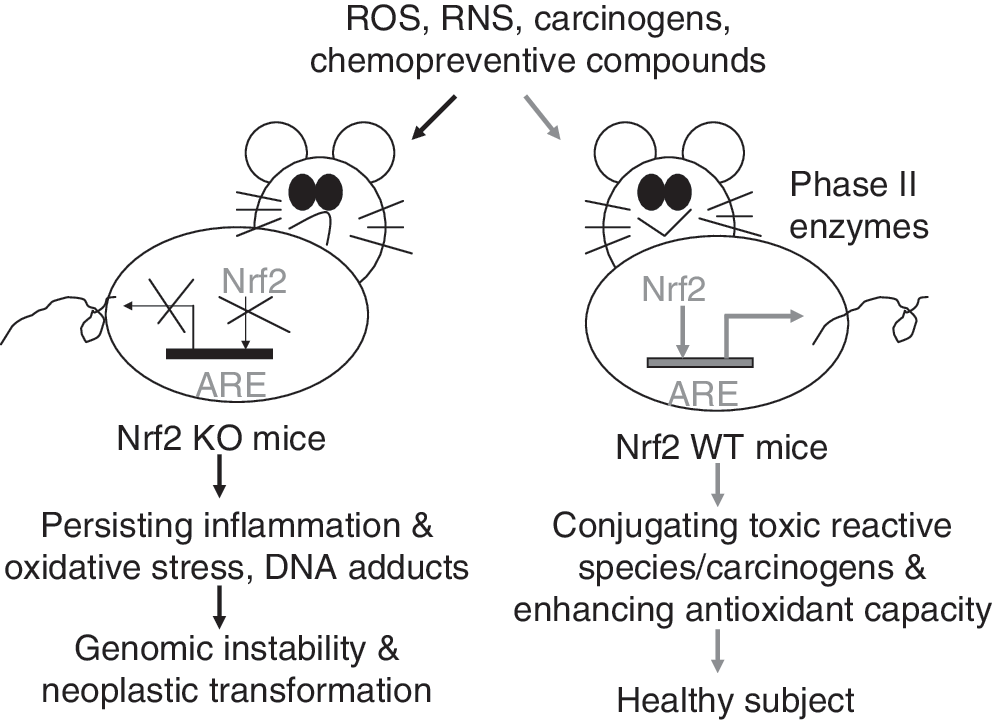
The first in vivo evidence showing a critical role of Nrf2 in induction of phase II detoxification enzymes was published in 1997 (58). By using Nrf2-deficient (Nrf2 KO) mice, Ito et al. found that the magnitude of GST and NQO1 induction was significantly lower in the Nrf2-deficient mice than that in Nrf2 heterozygous mice. Such a critical role of Nrf2 in activation of phase II antioxidant genes has been further confirmed in many studies using Nrf2-deficient mice (Table 1). An immediate application of this finding in cancer prevention is to test the anticarcinogenic activity of chemopreventive compounds in Nrf2 KO mice. Figure 5 has shown how the Nrf2 pathway protects mice from carcinogen-induced neoplastic transformation. As Nrf2 is disrupted, induction of cytoprotective phase II enzymes is impaired. When Nrf2 KO mice are exposed to carcinogens, the uncontrollable oxidative stress generated by carcinogens would damage DNA and induce persistent inflammation, which, in turn, increases genomic instability and enhance neoplastic transformation. In comparison with Nrf2 KO mice, the WT mice maintain normal induction of phase II detoxification enzymes, and hence limit the cytotoxic effect of carcinogen-generated oxidative stress. Many chemopreventive compounds have been shown to induce phase II detoxification enzymes through a mechanism dependent on Keap1-Nrf2-ARE signaling (Fig. 2). Thus, it has been hypothesized that chemopreventive agents may act as oxidants or electrophiles to modify several key cysteine residues of Keap1, thereby preventing Nrf2 degradation; meanwhile, chemopreventive-agent-generated redox signal may directly act on Nrf2 molecules and enhance their nuclear localization. Given the important role of Nrf2 in protection against carcinogenesis, attempts have been made to develop more effective chemopreventive agents by targeting Nrf2 pathway.
ACF, aberrant crypt foci; AOM, azoxymethane; BaP, benzo[a]pyrene; BBN, N-nitrosobutyl(4-hydroxybutyl)amine; D3T, 3H-1,2-dithiole-3-thione; DMBA, 7,12-Dimethylbenz(a)anthracene; DSS, dextran sulfate sodium; gpt, guanine phosphoribosyltransferase; GST, glutathione S-transferase; HO-1, heme oxygenase-1; IQ, 2-amino-3-methylimidazo[4,5-f]quinoline; K14, keratin 14; Nrf2, NF-E2-related factor 2; KO, knockout; NQO1, NAD(P)H:quinone oxidoreductase 1; SFN, sulforaphane; tBHQ, tert-butylhydroquinone; TPA, 12-O-tetradecanoylphorbol-13-acetate; UGT, UDP-glucuronosyltransferase; WT, wild type.
Nrf2 and Antiinflammation–Nrf2 KO-Dextran Sulfate Sodium and Azoxymethane-Dextran Sulfate Sodium Colon Model
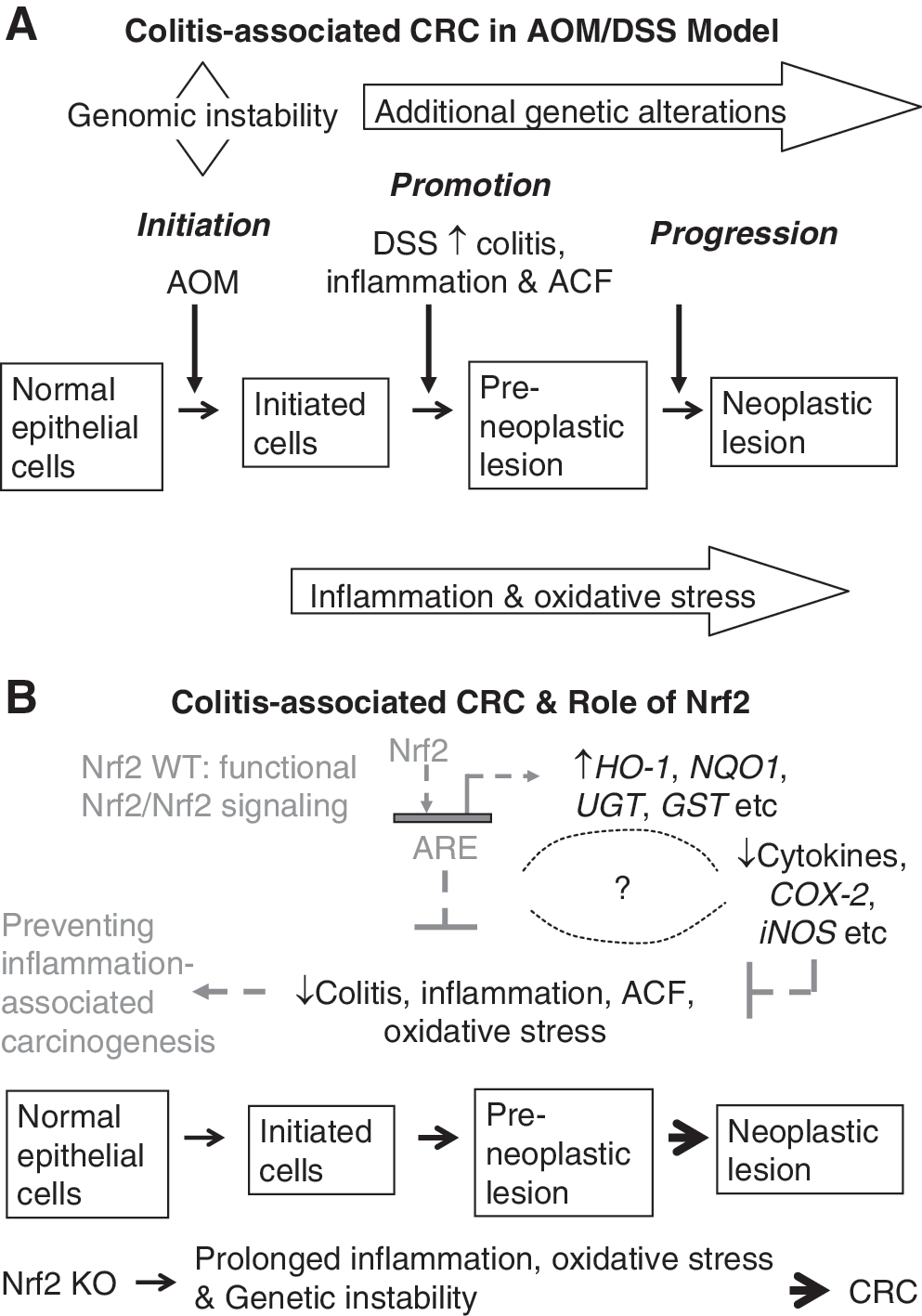
Current studies have linked chronic inflammation to cancer development (105). Colorectal cancer (CRC) is one of the classic examples that demonstrate a close relationship between chronic inflammation and carcinogenesis (106) (Fig. 6A). Treatment of animals with dextran sulfate sodium (DSS) has been shown to induce colitis, a chronic inflammatory disease, whereas treatment of animals with both azoxymethane (AOM) and DSS induces CRC. Using Nrf2 KO mice, we found that disruption of Nrf2 gene renders animals more susceptible to DSS-induced colitis and to AOM-DSS-induced colon carcinogenesis (80, 81). Similar results were obtained by Kensler's group when investigating the role of Nrf2 in prevention of inflammation-associated aberrant crypt foci (ACF) formation (122). They noted that Nrf2 KO mice developed significantly higher incidence of colonic tumor than WT mice. Further, AOM-DSS-treated Nrf2 KO mice had more severe colitis and higher incidence of prolapsed rectum and bleeding than WT mice. Therefore, Nrf2 pathway appears to mediate a strong antiinflammatory response, besides induction of detoxification and antioxidant enzymes as described above.
Regulation of Nrf2-Target Genes in Transgenic Adenocarcinoma Mouse Prostate Mice Prostate Cancer Model
Prostate cancer as a major health concern
Prostate cancer (PCa) remains the top leading cancer types responsible for 25% of new cancer cases with an estimated 192,280, and is second leading cause of cancer death in men, estimated 27,360 deaths in the United States in 2009 (64). Presently, metastatic PCa is not curable and is associated with a mean survival of 2–3 years. Development of PCa is often characterized by a relatively long latent period from the precursor lesions, called prostatic intraepithelial neoplasia (PIN), to the invasive carcinoma and metastasis that is typically associated with age and elderly men (1, 26, 86, 91, 96, 115 –117, 124, 165). Therefore, to decrease the incidence of PCa, or to delay the neoplastic development, or to slow the progression of PCa to late stage aggressive diseases achieved through either dietary or chemical intervention would be logical and would be tremendous clinical benefit to hundreds of thousands of men (10). Further, chemoprevention of PCa will be more cost effective than therapeutics designed to treat/cure the disease.
The transgenic adenocarcinoma mouse prostate mouse model for prostate neoplasia
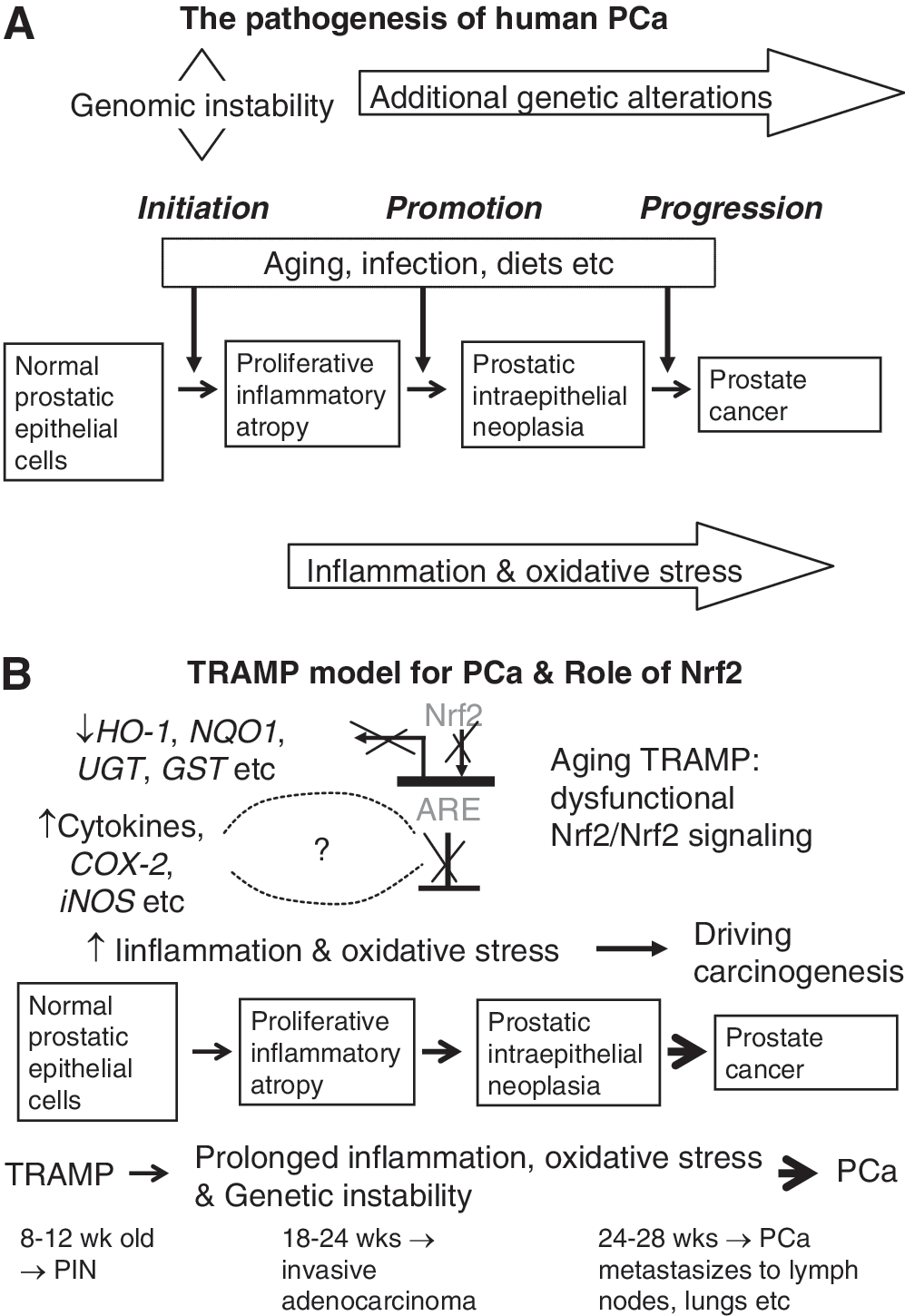
Development of effective chemopreventive compounds against these malignancies is important and requires conclusive evidence from animal models that emulate human cancers. The transgenic adenocarcinoma mouse prostate (TRAMP) mouse is an autochthonous transgenic animal model of PCa that recapitulates the whole spectrum of human prostate tumorigenesis from the earliest PIN lesions to androgen-independent disease (71).
The TRAMP model was originally developed in Norman Greenberg's laboratory using the minimal prostate specific rat probasin promoter to drive expression of the simian virus 40 (SV40) early region tumor antigen-coding region (T, t; large T and small t antigens) specifically in prostatic epithelium (40). The tumor (T) antigens have the ability to induce transformation in vivo (19) and have been used successfully in transgenic mice to induce a transformed state in a variety of systems, including pancreas (45), mammary gland (152), and others [reviewed in Adams and Cory (2)]. The large T antigen acts as an oncoprotein through interaction with and inhibition of retinoblastoma (Rb) and p53 tumor-suppressor gene products, whereas the small t antigen interacts with a protein phosphatase (40). At first glance, it appears that this artificially expressed SV40 trangene oncogene may not be an appropriate model for human PCa; however, given that the loss of WT p53 and Rb has been implicated in the development and progression of PCa (17, 135, 139, 160), this would seem logical. The TRAMP mice express the T antigen oncoprotein by 8 weeks of age and develop distinct pathology in the epithelium of the dorsolateral prostate by 10 weeks of age. TRAMP mice are first observed to develop mild epithelial hyperplasia between 8 and 12 weeks of age. Lymph node metastases can be detected between 18 and 24 weeks of age, and pulmonary metastases were frequently identified by 24 weeks of age. By 28 weeks of age, 100% of the TRAMP mice harbor metastatic PCa in the lymph nodes or lungs (39).
Therefore, the TRAMP model may provide a consistent source of primary and metastatic tumors for histopathobiological and molecular analysis to further define the earliest molecular events involved in the genesis, progression, and metastasis of PCa (38). Further, as seen in human PCa, TRAMP mice develop androgen-independent PCa after castration (38). Overall, the TRAMP model exhibits many similarities to human PCa, including epithelial origin, progression from the PIN stage to adenocarcinoma, and metastasis by a transgene that is hormonally regulated by androgen (38 –41), so that it serves as a suitable model (114) (Fig. 7A). PCa, just like CRC, has a critical component of inflammation in the prostate carcinogenesis as well (116, 124). Specifically, for example, immunotherapy consisting of irradiated tumor cell vaccine and anti-CTLA-4 antibodies has been shown to markedly decrease the incidence of prostate tumors in TRAMP mice (55). CTLA-4 is a T cell surface antigen that plays an important role in attenuating T cell activity, and mice treated with anti-CTLA-4 antibodies were found to display a robust immune response to a variety of antigens (98). The same group has continued this work and found that indeed TRAMP mice allowed the functional identification of immunogenic prostate tumor antigens with relevance for human immunotherapy (32). Further studies with the TRAMP model by Gupta et al. have demonstrated a remarkable reduction in the rate of prostate tumorigenesis and metastasis formation between TRAMP mice fed a control diet and those fed a diet supplemented with the COX-2 inhibitor celecoxib (100% vs. 25% and 65% vs. 0%, respectively) (42). These findings suggest a critical role of proinflammatory enzyme COX-2 in PCa development and progression in the TRAMP model (Fig. 7B).
Nrf2 and GSTs are downregulated in human and TRAMP PCa and regulation of Nrf2-target genes in TRAMP mice by chemopreventive compounds
Importantly, it has been found that from a meta-analysis on 10 human PCa gene expression studies (35), Nrf2 and members of GSTmu are extensively decreased statistically. Using the TRAMP transgene and Rb and Nrf2 KO mouse models, the same group has also demonstrated that the loss of Nrf2 initiates a detrimental cascade of reduced GST expression, elevated ROS levels, and ultimately DNA damage associated with tumorigenesis. Our laboratory has also found that tumor progression in TRAMP occurred with a significant suppression of antioxidant enzymes such as catalase, superoxide dismutase, glutathione peroxidase, HO-1, and phase II detoxification enzymes, supporting the relevant use of TRAMP in examining Nrf2-related pathways in human PCa (12). Curcumin and phenethyl isothiocyanate alone or in combination significantly decreased the incidence of PCa tumor formation (11), by downregulating Akt signaling pathway, which later was also found in the TRAMP mice treated with broccoli sprout, a rich source of SFN (78). Again, the important role of Nrf2 pathway for effectiveness of SFN in preventing PCa is confirmed in TRAMP mice by inducing expression of Nrf2 and HO-1, along with other apoptotic proteins and suppressing Akt-dependent pathways in the prostate of TRAMP mice. Increasing evidence points out that the Akt-dependent signaling regulatory proteins are critical regulators of prostate tumorigenesis in vivo (21, 37, 151). Our latest data present the first experimental evidence that inhibition of Akt signaling pathway might be an important cellular mechanism for prevention of prostate tumor growth in TRAMP mice by broccoli sprout. The data also suggest that both the activation of Nrf2 pathway and the inhibition of Akt are required for effective prevention of PCa growth in TRAMP mice (78). Our current understanding of the interplays between Nrf2 and other signaling pathways is incomplete; research is undergoing to shed more light on this aspect.
TRAMP model has been used extensively to evaluate agents against PCa by various chemopreventive compounds, including difluoromethylornithine (43), R-flurbiprofen (163), toremifene (130), green tea (3, 44), genistein (158), and non-steroidal antiinflammatory drugs, celecoxib and exisulind (42, 114), silibinin (131), phenethyl isothiocyanate and curcumin (11), and methyl selenium (159). Most recently, we found that gamma tocopherol-enriched mixed tocopherols suppressed PIN and tumor development in the TRAMP mice, upregulated expression of Nrf2, and induced phase II detoxification and antioxidant enzymes (12). Again, these findings substantiate the important role of Nrf2 pathway in cancer prevention, at least, in TRAMP mice, by chemopreventive agents (Fig. 7B).
Potential epigenomic regulation of Nrf2 in TRAMP mice
The currently accepted paradigm of the regulation of Nrf2 signaling appears to be mainly achieved via posttranslational mechanism, as discussed previously. Briefly, Nrf2 is functionally suppressed by Keap1 protein, which binds to and sequesters Nrf2 in the cytoplasm leading to the degradation of Nrf2, and thus prevents Nrf2 to translocate into nucleus to activate its targeted genes (102). When stimulated by oxidative stress or upstream kinases, the ubiquitination and the proteosomal degradation of Nrf2 are impeded and Nrf2 is being released from Keap1, translocates into the nucleus, can dimerize with Maf proteins, binds to ARE, and transcriptionally activates the Nrf2-targeted genes. To date, it is not clear as how expression of Nrf2 in human PCa or in TRAMP mouse tumor is suppressed. However, our most recent studies indicate that expression of Nrf2 is epigenetically silenced by DNA hypermethylation in TRAMP prostate tumor (173), which warrants further studies.
Interplay Between Inflammation and Nrf2 Pathways
Dysfunctional Nrf2 signaling/suppression of Nrf2 in aging TRAMP mice (Fig. 7B) appears to be similar to the Nrf2 KO mice (Fig. 5) and causes a persisting chronic inflammation (Fig. 6B) with increased oxidative stress, and eventually could contribute to and driving carcinogenesis if other factors are present. Interplay between Nrf2 signaling pathway and inflammatory pathway has recently be reviewed (82, 101). Increasing evidence has shown that Nrf2 could play an important role in defense against oxidative stress possibly by activation of cellular antioxidant machinery as well as suppression of proinflammatory signaling pathways. In addition, in vivo and in vitro data suggest that many dietary chemopreventive compounds can differentially regulate Nrf2-mediated antioxidant/antiinflammatory signaling pathways as the first line defense or induce apoptosis once the cells have been damaged (101), and many other pathways can also regulate Nrf2, including a wide variety of kinase signaling pathways such as MAPKs, PI3K, PI3K, and PERK as discussed earlier in regulation of Nrf2 signaling by other pathways section (65, 138). Many signaling pathways are involved in the cytokines and inflammatory response, such as nuclear factor-kappa-B (NF-κB) (30, 127) and MAPKs (155) pathways. Many chemopreventive compounds that act through Nrf2 pathway are also having antiinflammatory activities (82, 83, 101). As an initial study, we explored the relationship between Nrf2 and NF-κB, a key transcriptional factor involved in inflammatory response (111). Our bioinformatic analysis of the promoter regions of Nrf2 and NF-κB genes reveals that 75% of the members in the regulatory network are MAPKs, which appears to be consistent with the current role of MAPKs in modulation of both Keap1-Nrf2-ARE (63, 137, 144, 170, 175) and NF-κB signaling pathways (27, 110). Collectively, we are presenting a simplified model for the potential concerted modulation of Nrf2 and NF-κB in inflammation and carcinogenesis via upstream MAPKs pathway (Fig. 8).
Conclusion and Future Perspective
Many oncogenic transcription factors can be targeted for preventing cancer from forming (22), and Nrf2 is one of the very promising target as evidenced from the findings of increased Nrf2-dependent susceptibility in Nrf2 KO mice to various chemical-carcinogen-induced cancers and the effectiveness of chemopreventive compounds to inhibit carcinogenesis in the counterparts Nrf2 WT mice (with intact Nrf2 function) (Table 1). In addition, the role of Nrf2 in carcinogenesis of PCa in TRAMP mice appears to be implicated with the downregulation of expression of Nrf2 and its target genes (12), which can be explained in part via epigenetic mechanism (173). Epigenetic is also modulating the CRC carcinogenesis (104). A variety of antioxidants and antiinflammatory drugs, which are likely to be capable of attenuating procarcinogenic genomic damage from ROS and reactive nitrogen species, are also under current development for PCa prevention (10, 115).
On the other hand, Nrf2-regulated high expression of phase II/antioxidant enzymes during chemotherapy might be one of the major causes of ineffectiveness of chemotherapeutic agents, and such dark side of Nrf2 has been reviewed recently by Zhang's group (97). As we progress with future studies, optimization of the timing of when to give chemopreventive compounds to patients in lieu of the potential dark side of Nrf2 in cancer chemoresistance would need to be considered, in particular with many advanced tumors that are resistance to radiation and chemotherapeutic drugs. Nevertheless, the studies using Nrf2 KO models have clearly shown the protective role of Nrf2 in cancer prevention particularly during the earlier phases of initiation of carcinogenesis. The dark side of Nrf2 would need further investigation, and may depend on the animal models and the compounds tested. It is believed that basal Nrf2 is indeed a carcinogenesis protective transcriptional factor under normal physiological conditions when it is functioning properly. However, the high levels of Nrf2 as well as many other drug resistance genes, tumor oncogenes, and growth factors found in malignant tumors will need further integration and investigation in terms of translating to patients.
Footnotes
Acknowledgments
This work was supported in part by Grant no.30801410 from the Natural Science Foundation of China (R.H.), NIH R01-CA094828 (A.N.T.K.), and NIH R01-AG023497 (R.Y.). The authors thank all the members of Dr. Tony Kong's lab for their help in the discussion and preparation of this article.