
Abstract
Mitochondria are at the center of cellular energy metabolism and regulate cell life and death. The cell biological aspect of mitochondria, especially mitochondrial dynamics, has drawn much attention through implications in human pathology, including neurological disorders and metabolic diseases. Mitochondrial fission and fusion are the main processes governing the morphological plasticity and are controlled by multiple factors, including mechanochemical enzymes and accessory proteins. Emerging evidence suggests that mitochondrial dynamics plays an important role in metabolism–secretion coupling in pancreatic β-cells as well as complications of diabetes. This review describes an overview of mechanistic and functional aspects of mitochondrial fission and fusion, and comments on the recent advances connecting mitochondrial dynamics with diabetes and diabetic complications. Antioxid. Redox Signal. 14, 439–457.
Introduction
Biology textbooks frequently depict mitochondria as granular oval-shaped organelles, likely stemming from modern electron micrographic images. However, when these organelles were first observed in cells, they must have appeared to be a mixture of threads and grains and thus named “mitochondrion” from “mitos-khondros” for “thread-grain” in Greek. More recent microscopic observations of cells have re-established the original view of mitochondria as filamentous tubular organelles, and additionally observed that mitochondria are not static, but highly dynamic, constantly changing shape and location in cells (11, 12). The morphological plasticity of mitochondria includes changes in size, tubule branching, extension/retraction, looping, rolling/unrolling, and so on (11, 12, 183). The molecular mechanisms mediating these shape changes are largely undefined. However, in the past decades, the molecular machineries mediating fission and fusion of the mitochondrial tubule have been identified, allowing mechanistic studies of some of these processes. One enticing question regarding mitochondrial dynamics is, why cells change mitochondrial shapes? To rephrase, what is the form–function relationship of mitochondria? Do different shapes merely reflect varying functional states of mitochondria, or do they have an active role in modulating bioenergetic activities of mitochondria?
Mitochondrial morphology change is associated with many pathological conditions, notably neurodegeneration and aging. In addition, apoptosis is almost inevitably accompanied by fragmentation/granulation of mitochondria. A growing number of studies have begun to investigate mitochondrial morphology as an important parameter for many disease-related disorders. Metabolic diseases are not an exception and recently attention has been drawn to the role of mitochondrial fission/fusion in pancreatic β-cell function, as well as hyperglycemic complications. In this review, we will first describe mitochondrial fission and fusion machineries and their presumed mechanisms mediating mitochondrial membrane remodeling. In the second part, the functional/physiological significance of mitochondrial fission and fusion will be discussed and finally highlighting where mitochondrial dynamics stands in the context of diabetic and hyperglycemic complications.
Mitochondrial Shape Change by Fission and Fusion
In typical cultured mammalian cells, mitochondria form reticular networks composed of elemental tubules. While the bulk of the mitochondrial networks crowd the perinuclear region, various sized mitochondria ranging from small spheres to rods to elongated tubules are readily found in peripheral cytoplasmic regions (Fig. 1A). Time-lapse imaging of mitochondria reveals fission and fusion, changing the sizes of mitochondrial tubules (Fig. 1B and C). Studies of yeast mitochondria indicated that mitochondrial fission and fusion occurred at a relatively balanced frequency (123, 151). We quantified mitochondrial fission and fusion, and confirmed the balanced fission and fusion in mammalian cells as well (Fig. 2A). Interestingly, against the notion that cytoskeleton may provide a scaffold for pulling mitochondria apart or bringing them together for fission and fusion (184), disruption of microtubule networks did not affect the frequency of fission and fusion (Fig. 2A).
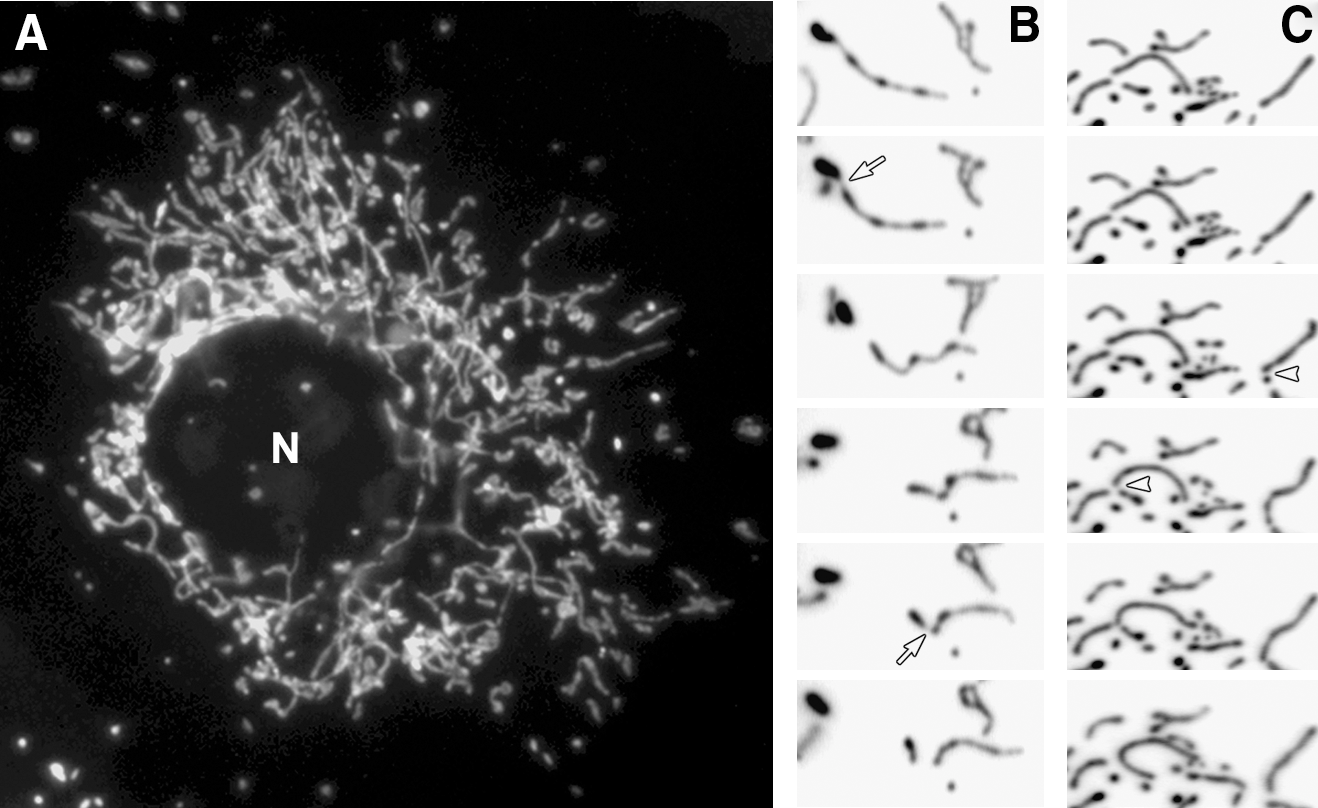
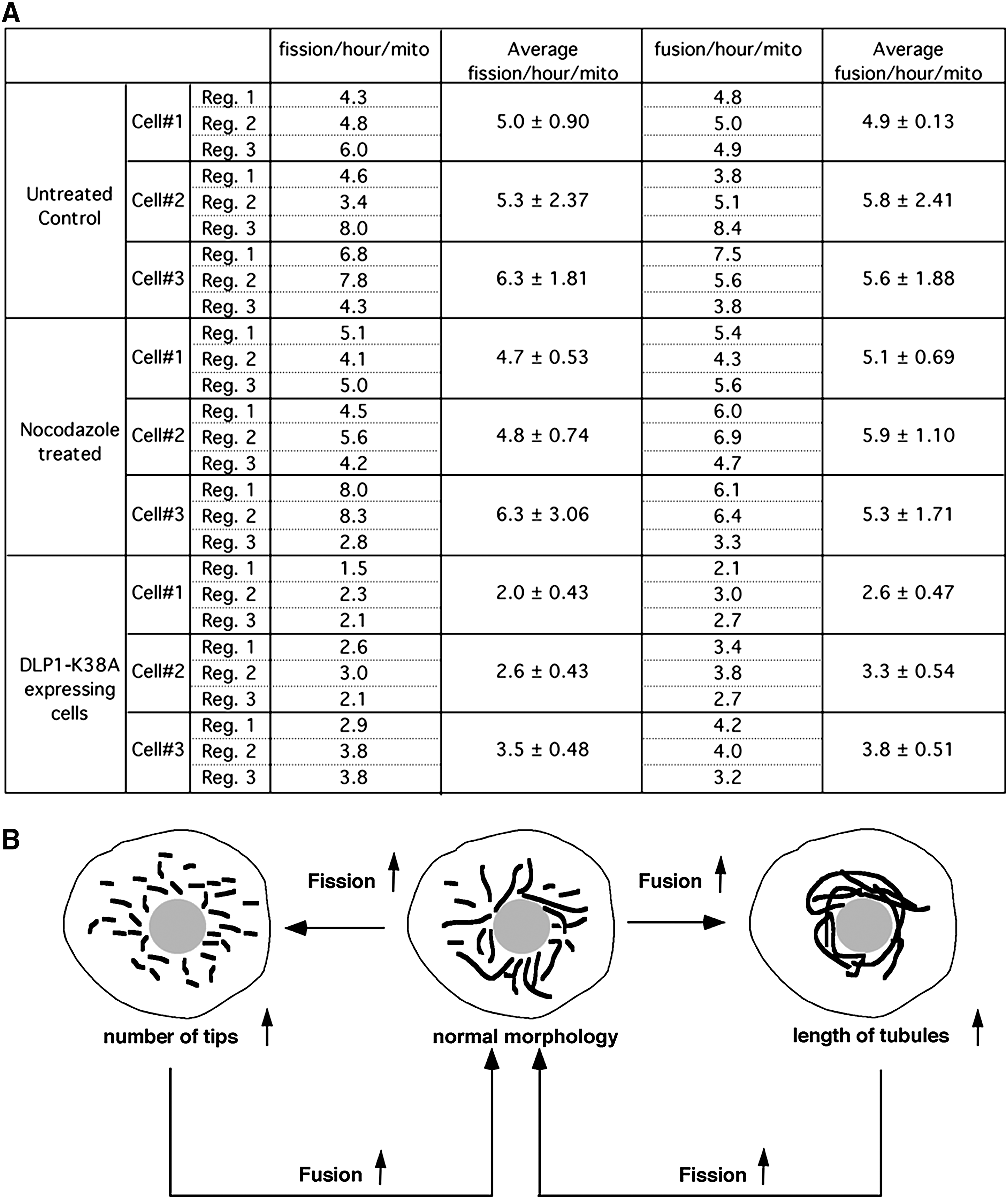
A shift of the fission–fusion balance alters mitochondrial morphology: more fission leads to fragmented mitochondria, whereas excess fusion results in elongated mitochondrial tubules (Fig. 2B). The quantification of fission and fusion frequency in cells containing excessively fused mitochondria via reduced fission (by a dominant-negative [DN] fission mutant: dynamin-like protein 1 [DLP1]-K38A) showed decreases in both fission and fusion (Fig. 2A). The extent of the fusion decrease is similar to that of fission, indicating that a new fission–fusion balance is established in these cells after the initial excess fusion that has produced elongated mitochondria. This observation brings up an interesting notion that normal mitochondrial morphology is maintained by a simple feedback control governing the fission–fusion balance. This notion is based on the predictions that the probability of mitochondrial fission is proportional to the length of mitochondrial tubules. Additionally, we observed in live cell imaging that the majority of fusion events occurred from one tip of a mitochondrial tubule to either another tip or side of other mitochondria with few fusion events occurring between the two sides of mitochondrial tubules. This suggests that mitochondrial fusion requires at least one mitochondrial tip and predicts that the mitochondrial fusion probability is proportional to the number of tips of mitochondria. As illustrated in Figure 2B, upon the increase of mitochondrial fusion, mitochondrial tubules become longer, which would increase the fission frequency and subsequently the normal morphology is regained. In the same manner, increased fission generates more mitochondrial tips, allowing the restoration of normal morphology through the increased fusion probability. Mechanistically, this feedback control predicts a close connection between fission and fusion, perhaps through direct and indirect communications among machineries mediating mitochondrial fission and fusion.
Abnormal mitochondrial morphology via the faulty feedback control is often associated with pathological conditions, suggesting that dysregulation of fission and fusion can cause disease states or vice versa. While this functional aspect of mitochondrial morphology will be discussed later, we will first describe the proteins and mechanisms that mediate mitochondrial fission and fusion. Although fission and fusion machineries have been identified in phylogenetically diverse organisms, this review will not cover beyond those in the mammalian system unless necessary.
Mitochondrial fission machinery in mammals
The DLP1/dynamin-related protein 1 (Drp1) mediates mitochondrial fission in mammals (133, 156) (Fig. 3A). DLP1 was identified as an immunologically related protein to dynamin (186). DLP1 is expressed in multiple different spliced variants, ranging 705–761 amino acids (81, 186). While these splicing variants are expressed ubiquitously in all tissues, their relative levels among tissues vary, most prominently in brain (186). DLP1 is a member of dynamin family proteins that are known to mechanically squeeze cellular membranes in a guanosine triphosphate (GTP)-dependent manner. Like conventional dynamin, DLP1 assembles into a ring-like structure (156, 187) and has a capacity to constrict synthetic liposomes into tubules in vitro (187) (Fig. 3B). DN mutants of DLP1 cause excessive elongation of mitochondria in cells, supporting the role of DLP1 in mitochondrial fission (133, 156). Subcellular fractionation experiments showed that most of the cellular DLP1 is in the cytosol, whereas only a small amount is associated with the mitochondrial fraction (153, 156, 186). Immunolocalization of DLP1 in cells revealed DLP1 forming distinct spots on the mitochondrial tubules (133, 156). These observations suggest that DLP1 shuttles between the cytosol and specific sites of mitochondria, but the period that DLP1 resides on the mitochondrial surface is relatively short. The current model for mitochondrial fission is that DLP1 binds to the mitochondrial surface where it assembles into a ring-like structure that squeezes and severs the mitochondrial tubule via its GTPase activity (Fig. 3C). In addition to the fact that DLP1 is abundant in the cytosol, increasing the level of DLP1 in cells does not enhance mitochondrial fission (133), suggesting the presence of limiting factors or regulatory mechanisms in mitochondrial fission. However, one report indicated that extreme overexpression could induce mitochondrial fragmentation (164). Given that DLP1 can directly bind to the membrane and tubulate them in vitro (187), it is possible that excess DLP1 in cells indiscreetly assembles onto and pinches mitochondrial tubules.
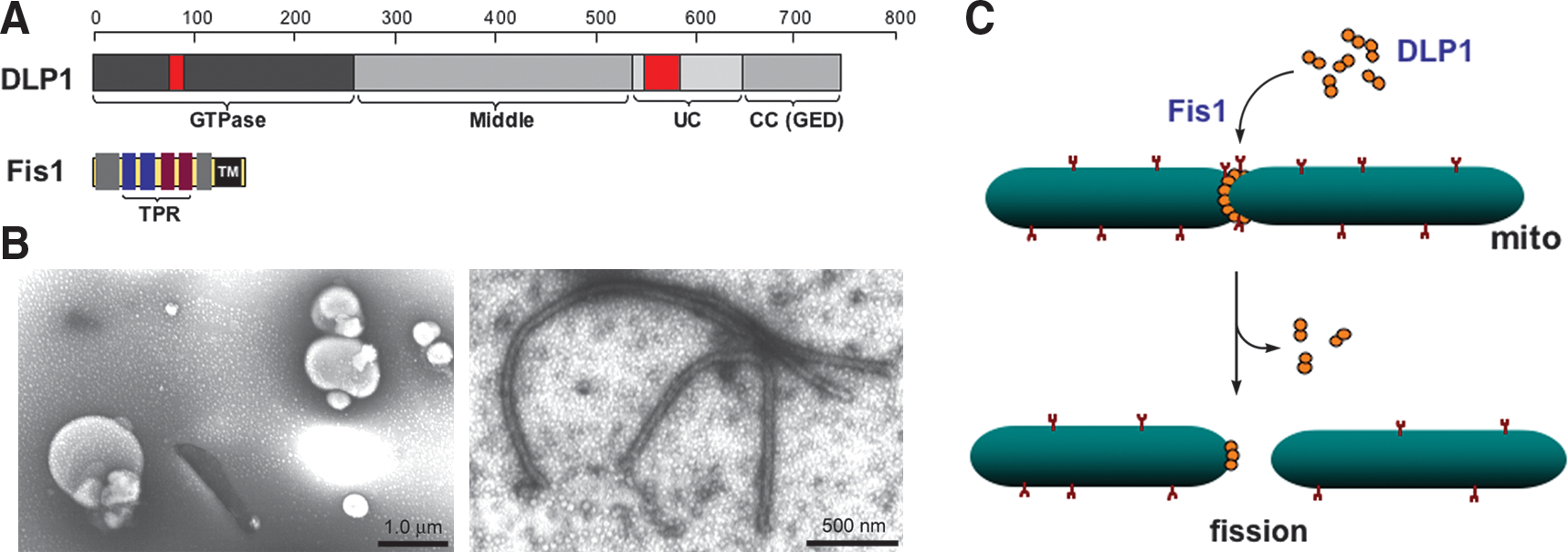
Fission protein 1 (Fis1), a mitochondrial outer membrane protein, is another protein participating in the mitochondrial fission process (Fig. 3A). Overexpression of the human protein hFis1 alone fragmented mitochondria by increasing fission, and this mitochondrial fragmentation required intact DLP1 function (160, 185), indicating that hFis1 is a limiting factor in the DLP1-mediated mitochondrial fission. The protein structure and membrane topology of hFis1 imply that it may function as a receptor recruiting DLP1 onto the mitochondrial surface. Fis1 is a small helix-rich protein (17 kDa) anchored in the mitochondrial outer membrane through a C-terminal single transmembrane domain, allowing the bulk of the protein to be exposed to the cytosol (160, 185). The cytosolic domain of Fis1 contains six α-helices of which the core four helices form two tandem tetratricopeptide repeat (TPR) motifs that are known to participate in protein–protein interactions (50, 163) (Fig. 3A). DLP1 was shown to interact transiently with the hFis1 TPR, which is regulated by the N-terminal first α1-helix of hFis1 (188). Mutations of the hFis1 TPR not only disrupted the DLP1 interaction but also exerted a DN effect on mitochondrial fission (188). Subsequent studies indicated that hFis1 forms homo-oligomers on the mitochondrial surface that are likely the functional configuration of this protein for the DLP1 interaction during mitochondrial fission (150). Whether Fis1 is a bona fide DLP1 receptor in mitochondrial fission is unclear. Fis1 has been shown to localize evenly throughout the mitochondrial surface (78, 163) and changes of Fis1 levels did not affect the DLP1 distribution/level in mitochondria (96, 163). On the contrary, knockdown of Fis1 prevented the formation of DLP1 puncta on mitochondria in metabolically noxious conditions that lead to mitochondrial fragmentation (115). In addition, phosphorylation of DLP1 has been shown to increase the Fis1-DLP1 interaction in vitro (69) (see below). It is likely that other factors, including signal-mediated modifications in participating proteins, and local lipid molecules, play a role in regulating DLP1-mediated mitochondrial fission.
Several studies showed that DLP1 is phosphorylated by different protein kinases to modulate mitochondrial fission. Cyclic adenosine monophosphate-dependent protein kinase (PKA) was demonstrated to phosphorylate DLP1 at a C-terminal serine residue (28, 37), which was dephosphorylated by the calcium-dependent protein phosphatase calcineurin (37). PKA-mediated DLP1 phosphorylation decreases the GTPase activity, thus attenuating mitochondrial fission (28). The same serine residue was reported to be phosphorylated by the calmodulin-dependent kinase Iα (CaMKIα) in neurons (69). However, in opposition to the PKA-mediated phosphorylation, the CaMKIα-mediated DLP1 phosphorylation increased mitochondrial fission. DLP1 phosphorylated by CaMKIα showed an increase of both mitochondrial translocation of DLP1 in cells and binding to Fis1 in vitro (69). Cyclin-dependent kinase 1 also phosphorylates DLP1 at a different serine residue of DLP1 to induce mitochondrial fragmentation in mitosis (165). In addition to phosphorylation, DLP1 undergoes sumoylation and ubiquitylation that may affect the DLP1 association to mitochondria and protein stability (18, 57, 70, 117, 177, 181, 194). Recently, it has been shown that increased nitric oxide induces mitochondrial fission through S-nitrosylation (SNO) of DLP1 (SNO-Drp1) (33). The SNO occurred at the cysteine residue within the C-terminal GTPase effector domain, and is necessary for the DLP1 dimer formation and the GTPase activity. An increase of SNO-Drp1 was observed in Alzheimer's brain, but not in Parkinson's, and β-amyloid protein can induce the DLP1 SNO in cultured neurons, suggesting the involvement of the SNO-Drp1-mediated mitochondrial fission in the pathogenesis of Alzheimer's disease (33).
Mitochondrial fusion machinery in mammals
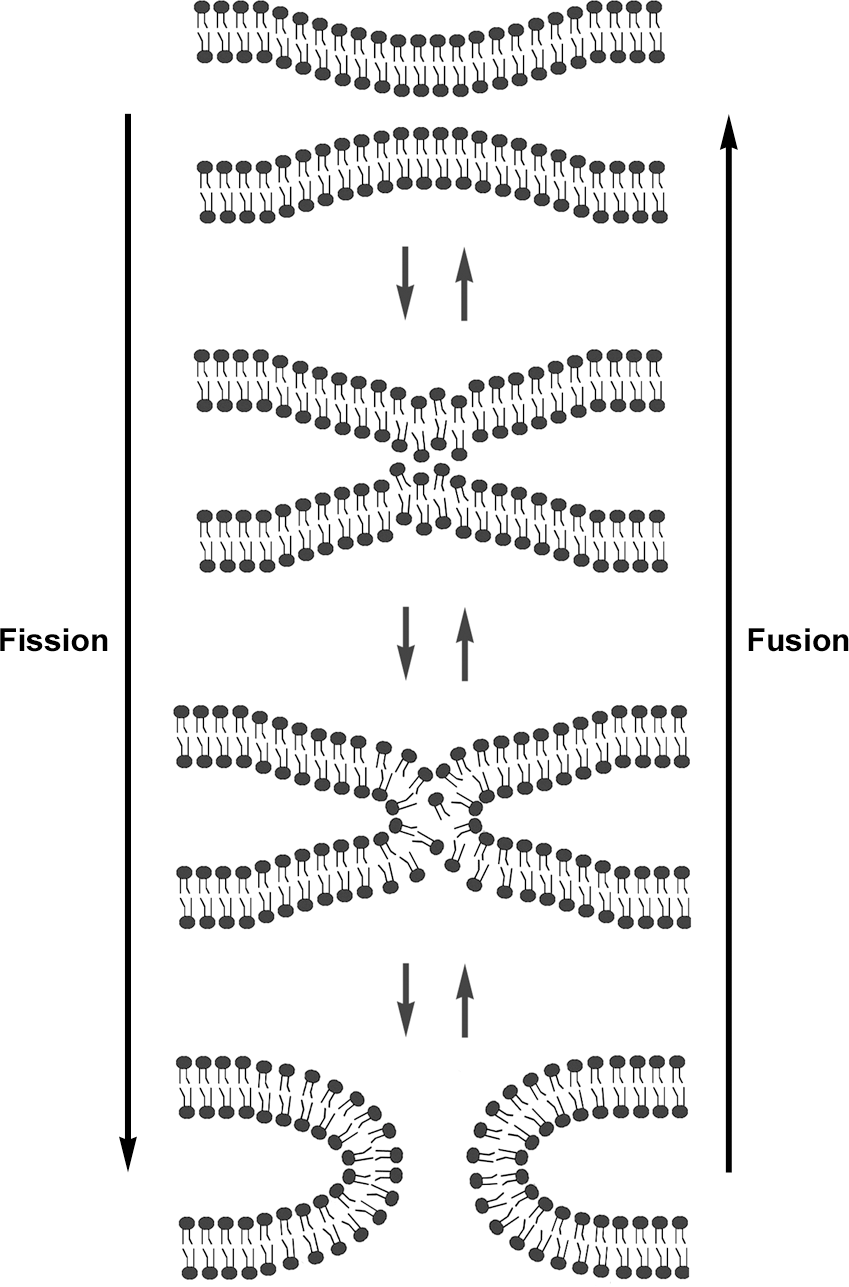
Mitochondria are double-membrane-bound organelles. Therefore, unlike the fission process that can conceivably be achieved by simple pinching from the outer surface of mitochondria, the completion of mitochondrial fusion is predicted to occur sequentially: fusion of the outer membrane followed by that of the inner membrane. Indeed, two different proteins, mitofusin (Mfn) and optic atrophy 1 (OPA1), associated with the outer and inner membranes, respectively, mediate mitochondrial fusion. Interestingly, like the fission protein DLP1/Drp1, both fusion proteins, Mfn and OPA1, also belong to the dynamin GTPase family (Fig. 4A). Although fusion of two separate membranes is considered a topologically opposite process to the membrane fission, the final steps of fission and fusion are thermodynamically identical processes in which lipid molecules of two contacting membranes mix and rearrange to form new bilayers (Fig. 5). Therefore, regardless of fission or fusion, a main task of dynamin proteins is likely to generate mechanical force that brings two membranes in close contact to allow lipid molecule mixing. Consistent with this idea, mitochondrial fusion is initiated by tethering of two opposing mitochondria mediated by the Mfn protein (91). Mfn is an 85-kD protein anchored at the outer mitochondrial membrane. Mammalian cells have two Mfn isoforms, Mfn1 and Mfn2. Both isoforms share a conserved structure containing the N-terminal GTPase domain, two heptad-repeat regions (HR1 and HR2), and two transmembrane domains near the C-terminus (146) (Fig. 4A). HR1 and HR2 lie at opposite sides of the transmembrane region that spans the outer membrane twice, exposing the GTPase, HR1, and HR2 domains to the cytosol (91, 142). Studies with Mfn1 showed that the C-terminal HR2 region interacts with the HR2 of another Mfn molecule in the adjacent mitochondrion through an antiparallel interaction, which allows tethering of the two membranes (Fig. 4B). The distance between the two outer membranes upon the HR2-mediated tethering was estimated to be ∼160 Å (91). Subsequently, the GTP-mediated conformational change of Mfn proteins would further draw two membranes closer for the outer membrane fusion.

The inner membrane-associated OPA1 is presumed to mediate the fusion of the inner membranes. The human OPA1 gene expresses eight alternatively spliced variants (43). The OPA1 protein is further processed by differential proteolytic cleavages, producing long and short forms of OPA1 (74). Long forms are integrated in the mitochondrial inner membrane through the N-terminal transmembrane domain (Fig. 4). The OPA1 cleavage occurs within the membrane, producing short forms that remain associated with the inner membrane at the inter-membrane space. Different proteases, including presenilin-associated rhomboid-like, i-AAA (Yme1L), m-AAA (paraplegin and AFG3L1 and 2) proteases, and the inner membrane metalloprotease OMA1, were found to participate in the cleavage of OPA1 (36, 53, 54, 64, 71, 74, 157). It has been shown that the successful fusion requires the presence of both long and short forms, whereas either long forms or short forms alone do not support mitochondrial fusion (157). Although partial outer membrane fusion was observed in the absence of the inner membrane fusion in OPA1-null cells (158), OPA1 knockout or knockdown generally caused fragmentation of mitochondria even with intact Mfn proteins in the outer membrane. This suggests that functional coordination of Mfn and OPA1 is necessary for successful fusion of mitochondria (35, 41, 65, 124). In yeast, the physical connection between Fzo1p (yeast Mfn orthologue) and Mgm1p (yeast OPA1 ortholog), mediated by the transmembrane linker Ugo1p, may coordinate outer and inner membrane fusion. To date, however, no such linker protein has been identified in mammals. In mammalian cells, OPA1 appears to function with the Mfn1 isoform for mitochondrial fusion, suggesting a functional distinction between the two Mfn isoforms (35). Interestingly, Mfn2 has an additional function in tethering the mitochondria to the endoplasmic reticulum (42). OPA1 also has an additional function in regulating cristae junction opening for cytochrome c release during apoptosis (59) (Fig. 4B).
Other proteins in mitochondrial morphogenesis
While DLP1, Fis1, Mfn, and OPA1 are the most studied proteins for mitochondrial fission and fusion, additional proteins have been identified and implicated in mitochondrial morphogenesis. MTP18 is an 18-kDa mitochondrial inter-membrane space protein whose expression is regulated by phosphatidylinositol 3-kinase signaling (170). Knockdown of MTP18 induced interconnected mitochondrial tubules and it was suggested to act downstream of Fis1 (168, 170). Two outer membrane proteins, ganglioside-induced differentiation associated protein 1 (120) and mitochondrial fission factor, were also implicated in mitochondrial fission (60). A Bin-Amphiphysin-Rvs domain-containing protein, endophilin B1, has been shown to change mitochondrial morphology upon its downregulation, presumably by changing membrane curvature (82). Although these proteins affect mitochondrial morphology in the DLP1/Fis1-dependent fashion one way or another, no physical interactions between these proteins and DLP1 or Fis1 has been identified. More directly involved in the DLP1/Fis1-mediated process, a mitochondrial outer membrane-associated ubiquitin E3 ligase, MARCH-V, also referred to as MITOL, has been shown to bind to DLP1 and Fis1 as well as the fusion protein Mfn2 subsequently ubiquitinating DLP1 and Fis1 for degradation (117, 181). Accordingly, knockdown of MARCH-V led to mitochondrial fragmentation through decreasing the degradation of fission proteins (117, 181). However, a contradictory report indicated that MARCH-V inhibition results in mitochondrial elongation and net formation, possibly by inhibiting DLP1 assembly or postfission turnover at the fission site (83).
Mitochondrial phospholipase D was implicated in mitochondrial fusion, possibly downstream of Mfn-mediated tethering (34). Mitochondrial phospholipase D binds to the outer surface of mitochondria, where it hydrolyzes cadiolipin of an adjacent mitochondrial outer membrane to generate a fusogenic lipid phosphatidic acid, which would promote transmitochondrial membrane adherence (34). Several Mfn-binding proteins have been identified that regulate mitochondrial morphology. MIB, Mfn-binding protein, is a member of the medium-chain dehydrogenase/reductase superfamily, localized in the cytosol (56). Knockdown of MIB elongated mitochondria, indicating that it is a negative regulator of the Mfn function (56). Stoml2 or stomatin-like protein 2 interacts with Mfn2 and prohibitin 1 and 2 (39, 67), and is associated with the mitochondrial inner membrane and faces the intermembrane space (67). A recent study showed that stomatin-like protein 2 participates in the stress-induced mitochondrial hyperfusion through the OPA1 and Mfn1 functions but independently of Mfn2 and prohibitins (169). In addition, pro- and antiapoptotic proteins Bax, Bak, Bcl-2, and Bcl-xL have been shown to interact with the Mfn protein and affect mitochondrial morphology (see below) (22, 45). Locations and functions of proteins implicated in mitochondrial fission and fusion along with their associated mitochondrial phenotypes are summarized in Tables 1 and 2.
DLP1, dynamin-like protein 1; Drp1, dynamin-related protein 1; Fis1, fission protein 1; GDAP1, ganglioside-induced differentiation associated protein 1; MARCH-V, membrane-associated RING-CH-V; Mfn, mitofusin; MIB, mitofusin-binding protein; MitoPLD, mitochondrial phospholipase D; MTP18, mitochondrial protein 18 kDa; OPA1, optic atrophy 1; SLP-2, stomatin-like protein 2.
Functional Significance of Mitochondrial Fission and Fusion
One obvious question regarding mitochondrial dynamics is why cells bother changing mitochondrial morphology as it consumes a great deal of cellular energy. Clearly, mutations in mitochondrial fission and fusion genes cause human disease or death, indicating their roles in pathophysiology. Abnormal regulation of apoptosis is destined for developing disease conditions. Mitochondria are the central regulator of apoptosis, and mitochondrial morphology undergoes a drastic change during apoptosis, implying mitochondrial fission/fusion in apoptotic regulation.
Mitochondrial fission and fusion determine the continuity or separation of mitochondrial membranes and matrix in which bioenergetic/biosynthetic activities take place. Aberrations in the regulation of mitochondrial continuity are pathologic as mentioned above. Despite the clear link between fission/fusion and activity/function of mitochondria, knowledge regarding the mechanistic correlation of these two components is only rudimentary. In this section, the apoptotic role of mitochondrial fission/fusion and recent efforts to identify mechanistic links between mitochondrial morphology and function will be discussed.
Mitochondrial fission and fusion in apoptosis
Mitochondrial morphology changes during apoptosis. More specifically, tubular mitochondria disintegrate and become fragmented when the mitochondrial outer membrane is permeabilized for cytochrome c release. The time frame for mitochondrial fragmentation and the mitochondrial outer membrane permeabilization (MOMP) is almost simultaneous, suggesting a close correlation between the two processes. The fission proteins DLP1 and Fis1 mediate apoptosis-associated mitochondrial fragmentation, as the inhibition of DLP1 or Fis1 by knockdown or DN approach maintained tubular mitochondria upon apoptotic stimulation (58, 96). Further, cytochrome c release, caspase activation, and cell death are delayed upon inhibition of mitochondrial fission (3, 21, 22, 58, 96). Overexpression of the fusion proteins Mfn1 and Mfn2, which increases connected mitochondria, also delayed cytochrome c release and Bax/Bak activation upon apoptotic stimulation (77, 119, 162). In addition, Fis1 overexpression-induced mitochondrial fragmentation caused cytochrome c release and apoptosis (78, 188). Knockdown of the fusion proteins Mfn1, Mfn2, or OPA1 fragmented mitochondria and increased the sensitivity to apoptotic stimuli (96, 124, 162). These results suggest that mitochondrial fragmentation actively participates in promoting the MOMP (22, 58, 78, 96, 188). However, others reported contradicting results in which overexpression of Mfn1, Mfn2, or the DN DLP1-K38A had little effect on apoptosis (152). Additionally, it has been shown that the exodus of mitochondrial factors such as Smac/DIABLO, Omi, Tim8a, and adenylate kinase 2 upon MOMP still occurs with normal kinetics when mitochondrial fission is inhibited (55, 129). This indicates that MOMP and mitochondrial fragmentation are separate events and that DLP1-mediated fission is specific only for the release of cytochrome c. Moreover, an apoptotic induction leads to cell death in DLP1-knockout cells that maintain tubular mitochondria through the cytochrome c release phase, albeit with delayed apoptotic progression, demonstrating that mitochondrial fission is dispensable for apoptosis (76). Therefore, the significance of mitochondrial fission in apoptosis is unclear based on the nonexisting or delaying effect rather than a strong blocking effect presented by the absence of DLP1 function. Interestingly, in DLP1 knockout cells, DLP1-independent fragmentation of mitochondria was observed in the later stage of apoptosis after cytochrome c release (76).
Whether mitochondrial fragmentation is an active process or a coincidental phenomenon in apoptosis is still a matter of debate. Mitochondrial fragmentation appears neither sufficient nor necessary for apoptosis. However, given that almost all apoptosis is accompanied by mitochondrial fragmentation, there ought to be a reason for this phenomenon. A clue may be found in the recent finding that the MOMP-directing pro- and antiapoptotic Bcl-2 proteins interact with the fission and fusion machineries influencing mitochondrial morphology. The pro-apoptotic protein Bak has been shown to be responsible for mitochondrial fragmentation during apoptosis, potentially by differential interactions with the two Mfn isoforms, Mfn1 and Mfn2 (22). Mfn proteins were also shown to interact with Bax in the same study (22). Karbowski et al. reported that Bax altered Mfn2 activity and localization to increase mitochondrial fusion in healthy, nonapoptotic conditions (84). Mfn2 was also reported to interact with antiapoptotic Bcl-xL and Bcl-2 (45). Bcl-xL was found to interact with the Fis1 under overexpressing conditions. This interaction prevented the Fis1-induced cytochrome c release and apoptosis without inhibiting the Fis1-induced mitochondrial fragmentation (78). Fis1 was also shown to interact with Bcl-2, protecting cells from apoptosis by increasing mitochondrial calcium retention capacity (90). In addition, Bcl-xL has been shown to increase DLP1-mediated mitochondrial fission in neurons (14, 100). Although these studies indicate that there are physical interactions between fission/fusion proteins and Bcl-2 family proteins, morphological manifestations resulting from these interactions are not consistent with the fragmentation-MOMP axis, again bringing up skepticism regarding the active role of mitochondrial fragmentation in apoptosis. An emerging theory that reconciles this inconsistent effect is that mitochondrial fragmentation is the result of a regulatory function of Bcl-2 proteins in mitochondrial morphogenesis rather than having a direct role in MOMP/apoptosis (4). Bcl-2 proteins are critical regulators of cell survival and have a broad role in regulating cellular processes throughout the cell including calcium metabolism, autophagy, insulin secretion, and cell cycle control (40, 131, 149, 191). Because proper control of mitochondrial morphology is important for cell survival, cells may have developed a surveillance mechanism for mitochondrial morphology by employing Bcl-2 proteins in mitochondrial morphogenesis. Whether morphological change of mitochondria has a direct role in apoptosis or not, it is evident that there are physical and functional associations of Bcl-2 proteins that alter mitochondrial morphology. Further studies will define how these interactions differentially regulate mitochondrial fission and fusion in normal and apoptotic conditions.
Mitochondrial morphology, diseases, and bioenergetics
The functional importance of mitochondrial fission/fusion is reflected in the human diseases caused by mutations in mitochondrial fission and fusion genes. The mitochondrial fusion protein OPA1 is named after a disease, optic atrophy type 1, the most prevalent hereditary optic neuropathy that results in progressive loss of visual acuity (1, 44). Mutations in another fusion protein Mfn2 cause Charcot-Marie-Tooth type 2A, a peripheral neuropathy (193). One report identified a mutation in the DLP1 gene as a direct cause of the premature death of a newborn (178). In addition, Charcot-Marie-Tooth type 4A was reported to be associated with mutations in ganglioside-induced differentiation associated protein 1, a protein implicated in mitochondrial fission (8, 38, 118, 120). Prominent neurodegenerative diseases, Alzheimer's, Huntington's, and Parkinson's are also associated with dysregulation of mitochondrial morphology. Readers can refer to recent review articles dealing with this subject (30, 88, 89, 103, 161, 174, 175). In addition, genetic knockout of Mfn1, Mfn2, OPA1, or DLP1 causes embryonic lethality in mice, demonstrating that mitochondrial fission and fusion are required for embryonic development (32, 41, 76).
These disease states are the physiological consequences of dysregulation of mitochondrial morphogenesis, presenting circumstantial evidence for the significance of the mitochondrial morphology control. Plausibly, failure to maintain normal mitochondrial morphology would be detrimental for the mitochondrial bioenergetic activity. Several studies have shown that this is the case by manipulating mitochondrial morphology through overexpression or knockdown/knockout of fission and fusion proteins. Embryonic fibroblasts from Mfn1 and 2 double-knockout mice contained completely fragmented mitochondria. These morphologically altered mitochondria resulted in slower cell growth, loss and heterogeneity of the mitochondrial inner membrane potential, and decreased respiration, indicating the reduced mitochondrial function upon mitochondrial fragmentation (31). OPA1 knockdown also induced mitochondrial fragmentation and resulted in similar functional defects of mitochondria. In the same study, mitochondrial fragmentation was also observed upon overexpression of OPA1; however, these fragmented mitochondria were fusion competent and showed no defects in mitochondrial function (31). This suggests that mitochondrial fusion, not the fragmentation per se, is necessary to maintain the normal function of mitochondria. Conversely, mitochondrial fusion has been shown to require the mitochondrial inner membrane potential (75, 97, 112). Disruption of the membrane potential by p-trifluoromethoxyphenylhydrazone/carbonylcyanide m-chlorophenylhydrazone resulted in fragmented mitochondria by excess fission. The inner membrane fusion protein OPA1 seems to be a key mediator in this process, as the loss of mitochondrial membrane potential increases OPA1 cleavage and prevents fusion (52, 74). This observation raises an intriguing possibility that the OPA1 processing is a bioenergetic sensor, linking the functional state of mitochondria to their morphology. The membrane potential-dependent cleavage of OPA1 would determine whether given mitochondria undergo fusion or remain separated depending on their energetic capacity.
Inhibiting mitochondrial fission was also reported to impair mitochondrial function. Downregulation of DLP1 by RNA interference induced highly fused and interconnected mitochondria. This morphological deformation was accompanied by a multitude of anomalies in mitochondrial function, showing decreases in mitochondrial membrane potential, respiration, adenosine triphosphate (ATP) content, and cell proliferation as well as increases in ROS levels and oxidative damage, along with mitochondrial DNA loss and autophagic activation (9, 128). In contrast, embryonic fibroblasts from DLP1 knockout mice appeared normal in these aspects (76). Mitochondrial membrane potential, respiration, ATP levels, mitochondrial DNA levels, and autophagy were unaffected in DLP1-knockout fibroblasts, suggesting that DLP1 is dispensable for maintaining mitochondrial function in embryonic fibroblasts. In the DLP1 knockdown study, the functional defects of mitochondria were observed in HeLa cells derived from malignant tumor. Cancer cells are known to have different mitochondrial properties (143, 176), which might have contributed to the discrepancy between the two different experimental systems.
The experimental results described thus far clearly indicate that there is an intricate form–function relationship of mitochondria. One intriguing question is how the large-scale morphological change of mitochondria through fission and fusion influences mitochondrial bioenergetic activity. Mitochondria become completely fragmented when they are isolated away from the context of cells or tissues. However, these isolated mitochondria retain all the necessary bioenergetic components and maintain normal respiratory capacity. Interestingly, isolated mitochondria from DLP1-depleted cells revealed decreased respiration along with marked uncoupling of oxidative phosphorylation (OXPHOS) compared to those from normal cells (128), despite their identically fragmented morphology resulting from the isolation process. This observation suggests that the functional defect of mitochondria in DLP1-depleted cells is due to an intrinsic change in respiratory capacity. Related to this notion, Benard et al. proposed that an increase of mitochondrial membrane fluidity upon DLP1 depletion would decrease the constraint exerted on the OXPHOS supercomplexes within the inner membrane, causing functional defects through alterations in complex organization and substrate channeling (9, 10).
Mitochondrial Dynamics in Diabetes and Hyperglycemic Complications
Mitochondria play an important role in insulin secretion, and mitochondrial DNA mutation is a known etiology of a subset of diabetes. As a source of ROS and as a regulator of apoptosis, mitochondria also participate in the development of diabetic complications. Given the presence of the intricate form–function relationship of mitochondria, mitochondrial dynamics is likely an important contributing factor to pathology of diabetes.
Mitochondrial dysfunction in diabetes
Insulin is synthesized in the pancreatic β-cells and stored in the secretory vesicles. The secretion of insulin is controlled by blood glucose levels, amino acids, and other circulating hormones. An increase of glucose uptake by β-cells following an elevation of blood glucose concentration augments OXPHOS by β-cell mitochondria. The rise in ATP/ADP ratio inhibits the ATP-sensitive potassium channels on the cell membrane, which depolarizes the plasma membrane, leading to calcium influx into cells. Elevated cytosolic calcium stimulates the exocytosis of insulin-containing vesicles for insulin secretion into the circulation (13, 72, 105 –108). This sequence of events leading to the insulin secretion requires normal bioenergetic activity of mitochondria. A critical role of mitochondria in insulin secretion is evident from the mitochondrial diabetes, for maternally inherited diabetes and deafness (MIDD). MIDD is a hereditary disorder accounting for ∼1% of total diabetes cases. The etiology of MIDD is the impairment of β-cell function caused mostly by A3243G mutation in the tRNALeu gene of the mitochondrial DNA. Patients carrying this mutation have almost 100% chance to gradually develop diabetes upon aging, which is associated with pancreatic β-cell dysfunction and a marked reduction in both first- and second-phase insulin secretion (104). Knockout of the nuclear gene encoding mitochondrial transcription factor A in pancreatic β-cells resulted in diabetes in mice (154). Islets from the mitochondrial transcription factor A knockout mice had mitochondrial dysfunction, including mitochondrial DNA depletion and deficient OXPHOS. The mutant islets displayed impairments in calcium signaling and insulin release in response to glucose stimulation, demonstrating a critical role of mitochondrial function in insulin secretion.
In addition to mitochondrial diabetes, the most common type of diabetes (type 2 diabetes) is also associated with mitochondrial dysfunction (102). Defects in insulin secretion are well documented in type 2 diabetes (80, 95, 141) and mitochondrial dysfunction impairs the insulin secretion as just described. The most notable characteristic of type 2 diabetes is insulin resistance, the decreased ability of target tissues to respond properly to normal circulating concentrations of insulin. Decreases in mitochondrial oxidative enzyme activity and lipid oxidation were observed in skeletal muscles of insulin-resistant and obese patients (85 –87, 155). Type 2 diabetics also displayed downregulation of genes for mitochondrial biogenesis and OXPHOS (116, 130). The development of insulin resistance has been shown to occur concomitantly with mitochondrial dysfunction in humans and rodents (16, 19, 73, 139, 159). Several intervention studies also showed that both mitochondrial function and insulin sensitivity are improved upon exercise training, calorie restriction, and resveratrol treatment (92, 93, 125, 140, 167). However, some studies indicated that the improvement of insulin sensitivity occurs without enhancement of mitochondrial function (148, 166). In addition, genetic disruption of mitochondrial function in mice did not affect insulin sensitivity in skeletal muscle (135, 180). Although mitochondrial dysfunction is widely recognized as a causal factor for insulin resistance in type 2 diabetes, more mechanistic studies are necessary to further delineate the cause-and-effect relationship between these two pathological phenomena.
Mitochondrial dynamics in pancreatic β-cells and insulin secretion
Normal mitochondrial function is required for proper regulation of insulin secretion in pancreatic β-cells. Considering the intricate relationship between mitochondrial function and morphology, it is likely that mitochondrial dynamics plays a role in insulin secretion in β-cells. Type 2 diabetic patients exhibit both functional and morphological alterations of mitochondria in the pancreatic islets, including the reduced mitochondrial membrane potential, decreased ATP production, and high density of swollen mitochondria, along with impaired insulin secretion (2, 46). Shorter and swollen mitochondria were also reported in the β-cells from Zucker diabetic fatty rat model (15). These observations suggest potential disruption of mitochondrial dynamics in diabetic individuals and animals.
While the dysfunction of mitochondria with respect to their bioenergetic activity has been studied extensively in the context of diabetes, it is only recently that β-cell mitochondrial dynamics and its role in insulin secretion are being investigated. Using a mitochondrial matrix-targeted photoactivatable green fluorescent protein, Molina et al. examined mitochondrial morphology and dynamics in pancreatic islets and cultured β-cells (115). Photoactivation image analyses for mitochondrial connectivity indicated that densely organized mitochondria in primary mouse β-cells consist of individual tubules mostly at the length shorter than 2.5 μm, suggesting that β-cell mitochondria are shorter than those in other cell types such as fibroblasts. This result is somewhat in contrast to other reports in which mitochondria in human and rat primary islet cells formed tubular networks throughout the cytoplasm (15, 127). Elegant electron tomograms of intact islet revealed tubular and branched mitochondria in β-cells with the length ranging 0.5–5 μm (122). Recently, 4Pi microscopy, which has higher axial resolution, was employed to examine the mitochondrial morphology of pancreatic β-cells. Image analyses revealed that mitochondria of rat islet β-cells form highly interconnected reticulum similar to those observed in the insulinoma β-cell line (49, 134). In contrast, disintegration of tubular mitochondrial network was observed in islet β-cells from diabetic Goto Kakizaki rats (49). Individual mitochondria in healthy β-cells are dynamic, actively communicating with one another and with the rest of the network through frequent fusion and fission events, as evidenced by the observation that locally photoactivated mitochondrial matrix fluorescence rapidly becomes dispersed throughout the entire cell (115).
Decreased islet size and β-cell mass caused by β-cell apoptosis are also established features of type 2 diabetes (25, 51, 137, 145, 182), contributing to reduced circulating insulin levels. In a cultured β-cell model, high glucose concentrations induced apoptosis (114). Inhibiting mitochondrial fission prevented the high-glucose-induced β-cell apoptosis (114), suggesting that mitochondrial fission may regulate the circulating insulin levels by participating in pancreatic β-cell apoptosis in diabetes. Under apoptosis-inducing glucolipotoxic conditions, namely, high glucose and high fatty acid concentrations mimicking the type 2 diabetic milieu, β-cell mitochondria became fragmented by decreased fusion, and the glucose-stimulated insulin secretion was significantly reduced (115). Downregulation of Fis1 under the glucolipotoxic conditions rescued mitochondrial morphology and protected these cells from apoptosis, but was not able to restore the insulin secretion capacity (115). These results suggest that, despite the restoration of normal mitochondrial appearance by inhibiting fission under the fusion-limiting conditions, mitochondrial bioenergetic dysfunction is sustained in these cells. These observations support the notion that the dynamic nature of mitochondrial morphology by continuous fission and fusion, but not the static normal shape, is necessary to maintain proper mitochondrial function and activity.
The initial indication for the involvement of mitochondrial dynamics in insulin secretion was that downregulation of the mitochondrial Fis1 in β-cells significantly reduced glucose-induced insulin secretion presumably through decreased respiration (171). To directly study the role of mitochondrial dynamics in insulin secretion, mitochondrial morphology was manipulated in pancreatic β-cells, and the effect on the metabolism–secretion coupling was examined (127). Fis1 overexpression in the β-cell line induced extensive mitochondrial fragmentation and impaired the glucose-stimulated insulin secretion via decreased cellular ATP levels. A similar perturbation of metabolism–secretion coupling was observed upon overexpression of the fusion protein Mfn1, despite the apparently opposite mitochondrial morphology induced by this manipulation. However, overexpression of the DN-Mfn1 that caused mitochondrial fragmentation affected neither the cellular ATP level nor the insulin secretion. Because Fis1 and DN-Mfn1 cause almost identical mitochondrial fragmentation upon overexpression but have different effects on the metabolism–secretion coupling, it is unlikely that mitochondrial fragmentation itself is the determining factor for mitochondrial energy metabolism and insulin secretion in pancreatic β-cells (127). Further, overexpression of wild-type Mfn1 adversely affected the metabolism–secretion coupling similar to Fis1 overexpression despite forming the seemingly opposite mitochondrial morphology (127). These observations seem contradictory to the general notion that mitochondrial fusion is a requisite for maintaining normal mitochondrial function (29, 31, 32, 147, 179, 184); hence, it would be predicted that DN-Mfn1 inhibits mitochondrial fusion and resulting fragmentation would be associated with mitochondrial dysfunction and disruption of insulin secretion in β-cells. Instead, it was found that the metabolism–secretion coupling in β-cells is closely associated with cytoplasmic mitochondrial volume rather than the mitochondrial morphology (127). Overexpression of hFis1 and Mfn1 greatly decreased mitochondrial volume, whereas DN-Mfn1 overexpression had no effect on it. Mitochondrial flux through biogenesis and autophagic removal determines cellular mitochondrial contents. Mitochondrial fission and fusion are believed to be closely linked to autophagy (7, 171, 172). In this regard, inhibiting mitochondrial fission decreases glucose-stimulated insulin secretion in β-cells by accumulating dysfunctional mitochondria resulting from dysregulated autophagy (171). It has been reported that autophagy is associated with diabetes (62, 68, 110, 111, 113). It is evident that mitochondrial fission and fusion play an important role in metabolism–secretion coupling of pancreatic β-cells by regulating bioenergetics and homeostasis of mitochondria. In addition, mitochondrial dynamics is also likely a contributing factor for circulating insulin levels in diabetes by participating in β-cell apoptosis.
Mitochondrial dynamics in hyperglycemia, ROS production, and diabetic complications
Aside from pancreatic defects, deformation of mitochondria is frequently observed in other organs of hyperglycemic patients and diabetic animal models. Mitochondria in hepatocytes from hyperglycemic patients are swollen, and show disarrayed cristae and reduced electron density of matrix (173). In skeletal muscles from obese and type 2 diabetic subjects, mitochondria are smaller and have less defined internal structure with some vacuolization (85). Reduction of Mfn2 in both mRNA and protein levels were observed in skeletal muscles of obese Zucker rats and obese human subjects (5, 6). The reduction in Mfn2 levels was correlated with a smaller mitochondrial size and higher density, indicative of a fragmented mitochondrial network in obese Zucker rat skeletal muscle (6). In cultured cells, high glucose concentrations induce mitochondrial fragmentation in cell lines derived from liver, heart, endothelium, pancreatic islets, as well as in primary cell cultures of neuronal and the cardiovascular system (98, 114, 126, 189, 190). Mitochondrial fragmentation in high-glucose or high-glucose/high-fat conditions requires mitochondrial fission machinery (114, 115, 189, 190). Treatment of cultured dorsal root ganglia (DRG) neurons with high glucose concentrations caused mitochondrial fragmentation and increased expression and mitochondrial localization of the fission protein DLP1, indicating the activation of mitochondrial fission (98). Increases of expression, activation, and mitochondrial localization of pro-apoptotic proteins Bim and Bax were also observed in high-glucose treatment, indicating that high-glucose-induced mitochondrial fragmentation in DRG is associated with apoptosis. An increased DLP1 expression was also observed in DRG from streptozotocin-induced diabetic rats, pancreatic β-cells incubated in high glucose concentrations, and 3T3-L1 adipocytes in high free fatty acids (61, 98, 114).
ROS are a group of small molecules including superoxide anion, hydroxyl radical, and hydrogen peroxide. While a low level of ROS is an important cellular signaling component, high levels of ROS cause protein oxidation, lipid peroxidation, and DNA damage referred to altogether as oxidative stress that contributes to the development of the multitude of pathological conditions, including diabetic complications. Of multiple ROS sources in cells, nicotinamide adenine dinucleotide phosphate oxidase and mitochondria have been implicated in diabetic vascular injury in hyperglycemic conditions (23, 27, 48, 63, 121, 136). Mitochondria utilize the electrochemical gradient generated by the electron transport chain (ETC) to produce ATP. During the electron transfer, the ETC also produces ROS as a byproduct that, under normal conditions, are kept below harmful levels by antioxidant mechanisms. In hyperglycemic conditions, increased metabolic input overwhelms the ETC and causes electron backup within the ETC, allowing increased slippage of electrons resulting in excess formation of superoxide anions (Fig. 6). Brownlee postulated that superoxide anions produced from the mitochondria under hyperglycemic conditions inhibit the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase, diverting glycolytic intermediates into other harmful pathways implicated in the development of diabetic complications (23, 24, 121). These pathways include increased polyol pathway flux, increased formation of advanced glycation end products, activation of protein kinase C isoforms, and increased hexosamine pathway flux (23, 24) (Fig. 6). Several reports also indicate that increased ROS in hyperglycemia directly induce apoptosis and oxidative stress, causing organ damage in diabetes (26, 47, 144) (Fig. 6).
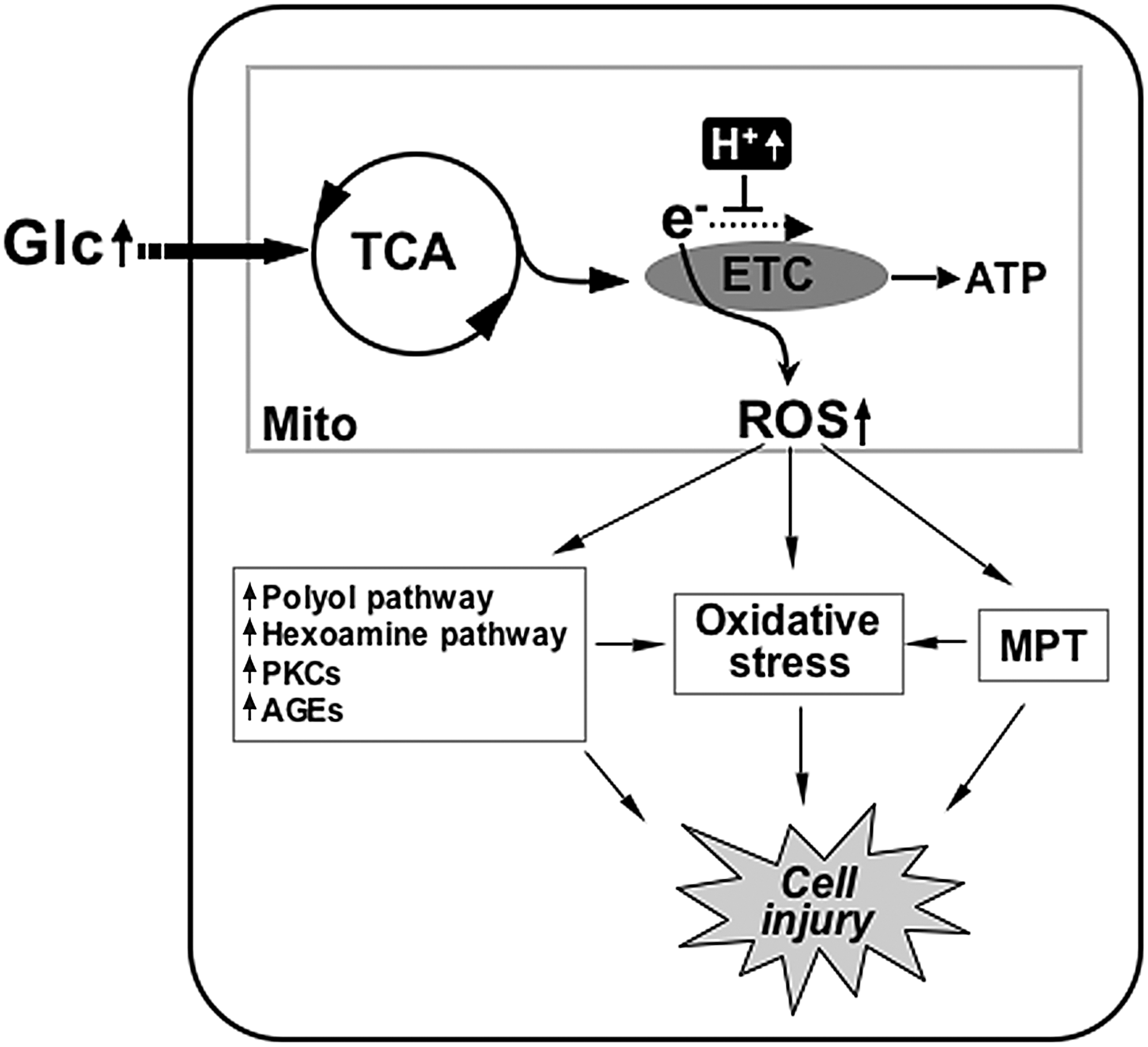
Our studies have identified the involvement of mitochondrial dynamics in high-glucose-induced ROS production (189). Acute exposure of cells to high glucose concentrations induced rapid fragmentation and recovery of mitochondria that occur concomitantly with an increase and decrease of ROS levels (189). Inhibiting mitochondrial fission or increasing fusion normalized ROS levels in the high-glucose incubation (189). An important contribution of this study is that the morphological change of mitochondria is an upstream causal factor for the ROS production under acute hyperglycemic stimulation, defining mitochondrial dynamics as a controlling factor regulating mitochondrial activity (189). Another report also showed that downregulation of Fis1 decreased the ROS level in acute high-glucose stimulation (171). As briefly discussed in the previous section, the underlying mechanisms of how alterations in mitochondrial morphology perturb mitochondrial bioenergetic activity to overproduce ROS remain unknown. Hackenbrock's classical observations indicate that active mitochondria are more condensed and have an electron dense matrix (66). Possibly, small short mitochondria formed upon high-glucose stimulation represent contracted and condensed mitochondria. The reversibility of mitochondrial fragmentation in the early high-glucose incubation suggests that the high-glucose-induced mitochondrial fragmentation is a physiological event, as opposed to the pathological fragmentation sustained in apoptosis. It is possible that morphological change of mitochondria in high-glucose conditions is a cellular response to increased metabolic substrate to facilitate metabolic input into mitochondria by increasing total surface area. In addition, recent electron tomographic analyses indicate that the dynamic change of the mitochondrial internal structure is closely associated with functional states of mitochondria (109). Ultrastructural change of the mitochondrial inner membrane was observed in nitric-oxide-induced mitochondrial fragmentation (7). Manipulations of the levels of Mfn2 have been shown to alter expression of mitochondrial respiratory chain complexes, leading to different glucose oxidation activities and conversion between orthodox and condensed mitochondria (132). Components of the mitochondrial ETC are arranged in an orderly fashion within the mitochondrial membrane for efficient transfer of electrons. Mitochondrial shape change in hyperglycemic exposure may alter structural organization and arrangement of respiratory chain complexes within the membrane, leading to perturbation of ETC activity or its coupling to ATP synthesis conducive to ROS overproduction. Changes in metabolic channeling, ETC complex organization, OXPHOS coupling efficiency, and membrane fluidity are plausible mechanisms to alter the mitochondrial activity upon hyperglycemia-induced mitochondrial fragmentation to increase ROS production.
Pathologic complications of hyperglycemia are associated with apoptosis and oxidative stress arising from increased levels of ROS, and accompanied by fragmentation of mitochondria (114, 173, 190). Under the pathological conditions, it is still debatable whether the elevated ROS cause mitochondrial fragmentation or vice versa. Inhibition of mitochondrial complex II in neuronal cells induced an ROS increase and mitochondrial fragmentation. The antioxidant N-acetyl-L-cysteine significantly decreased mitochondrial fragmentation under this condition, indicating that ROS is responsible for mitochondrial fragmentation (101). Treatment of cells with hydrogen peroxide results in an increase of ROS inside the cells that causes transient changes in the mitochondrial morphology (79). In a model of nitric-oxide-mediated neuronal injury, Barsoum et al. reported that nitrosative and oxidative stress induced DLP1-mediated mitochondrial fragmentation, which preceded neuronal cell death (7). In contrast to these observations, it has been reported that blocking mitochondrial fragmentation in hyperglycemic incubation decreases ROS levels, mitochondrial permeability transition (MPT), cytochrome c release, and apoptotic cell death, indicating that mitochondrial fragmentation in hyperglycemic insult is causal to the ROS increase (114, 190). High levels of ROS can induce the MPT, followed by MOMP and apoptosis (17, 94, 99, 192). The MPT allows leakage of mitochondrial glutathione resulting in accumulation of more ROS due to the diminished glutathione level (20). Dislocation of cytochrome c upon the MOMP would inhibit the ETC and further enhance ROS generation (20). During apoptosis, activated caspase cleaves the p75 subunit of mitochondrial complex I, causing ETC dysfunction and ROS increase (138). It is likely that ROS, MPT, MOMP, and caspase activation constitute a cyclic event that amplifies ROS accumulation, accelerating apoptotic cell death (Fig. 7). Mitochondrial fragmentation in hyperglycemic insult could be either the cause or consequence of the ROS increase, as discussed. However, due to the cyclic nature of the hyperglycemia-induced mitochondrial fragmentation, ROS increase, and apoptosis, blocking mitochondrial fission would be effective in preventing the ROS amplification and cell injury during hyperglycemic complications, regardless of where the hyperglycemia-induced mitochondrial fragmentation acts (Fig. 7). In this regard, mitochondrial dynamics could be a novel therapeutic target to decrease the pathological effects of increased ROS production in hyperglycemia-associated disorders.

Perspectives
Abnormal mitochondrial morphology is frequently seen in many pathological conditions. These include prominent neurological disorders and the focus of this review, diabetes and its associated complications. Defining the form–function relationship of mitochondria will be crucial in our understanding of pathologies of the disease states associated with their dysfunction. Given the long-standing ascribed function of mitochondria as metabolic regulators of the cell, it is not surprising that they play an intricate role in diabetes and hyperglycemia, ailments triggered by metabolic excess. More surprisingly perhaps is the intimate role that mitochondrial dynamics appears to play in the progression of this disease state, through insult to β-cells, and observed pathologies in other tissue systems. Observations in multiple cell and tissue types correlate the fragmentation of mitochondria with hyperglycemia and/or hyperlipidemia but the nature of this, causative or resulting, remains an open question.
Mitochondrial fission itself, which we have described as a result of hyperglycemic insult, can be detrimental to the cell through the production of ROS molecules. ROS damage to mitochondria has a compounding effect on ROS production and damage to cellular contents, and, as such, damaged mitochondria are degraded through autophagy, also termed “mitophagy.” This completes what can be viewed as the mitochondrial life cycle, in which mitochondrial dynamics represent a crossroads for maintenance required to adjust to perturbations in the cellular environment (Fig. 8). Studies of morphological disintegration of mitochondria in apoptosis have also brought Bcl-2 family proteins to the mitochondrial dynamics. Through this, mitochondrial dynamics controls the proper maintenance of tissues and organ systems, which, when misregulated, progresses to pathologies associated with disease.
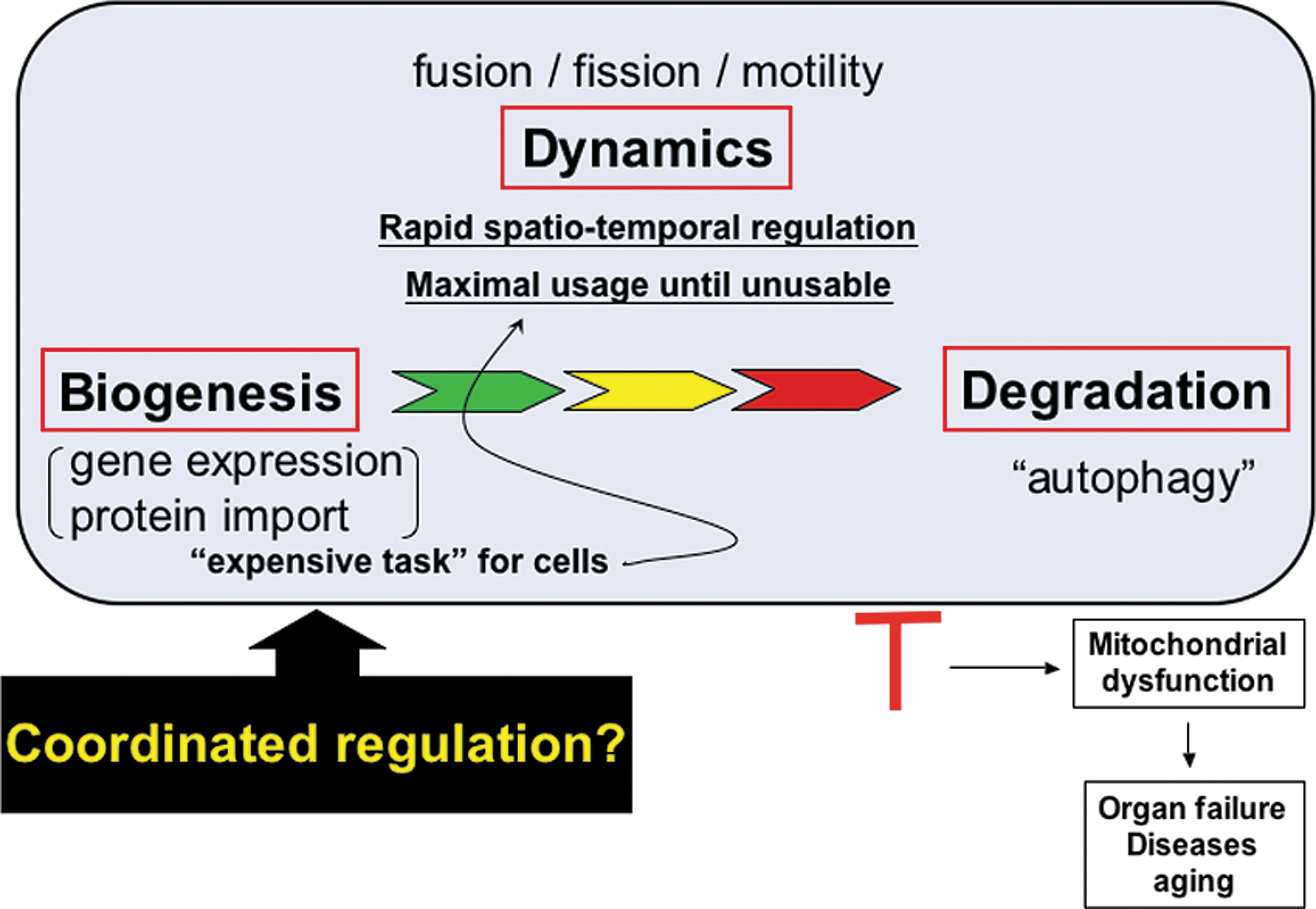
Mitochondrial dynamics which started with a simple microscopic observation has come a long way to find an important place in the human pathophysiology. The field of mitochondrial dynamics is still young and many questions remain to be answered. As discussed, mounting evidence suggests that proper control of mitochondrial morphology has an immense impact on the diabetic pathophysiology. Elucidating the functional interactions between the machineries participating in mitochondrial dynamics, and differential signaling pathways operating in normal and pathologic conditions will assist in delineating the pathophysiological role of mitochondrial dynamics in diabetes and lead to novel therapeutic interventions in combating this disease.
Footnotes
Acknowledgment
This work was supported by National Institutes of Health grants DK 078618 and DK 061991.