
Abstract
Nitroxyl (HNO), the one electron reduced and protonated form of nitric oxide (NO•), is rapidly emerging as a novel nitrogen oxide with distinct pharmacology and therapeutic advantages over its redox sibling. Whilst the cardioprotective effects of HNO in heart failure have been established, it is apparent that HNO may also confer a number of vasoprotective properties. Like NO•, HNO induces vasodilatation, inhibits platelet aggregation, and limits vascular smooth muscle cell proliferation. In addition, HNO can be putatively generated within the vasculature, and recent evidence suggests it also serves as an endothelium-derived relaxing factor (EDRF). Significantly, HNO targets signaling pathways distinct from NO• with an ability to activate KV and KATP channels in resistance arteries, cause coronary vasodilatation in part via release of calcitonin-gene related peptide (CGRP), and exhibits resistance to scavenging by superoxide and vascular tolerance development. As such, HNO synthesis and bioavailability may be preserved and/or enhanced during disease states, in particular those associated with oxidative stress. Moreover, it may compensate, in part, for a loss of NO• signaling. Here we explore the vasoprotective actions of HNO and discuss the therapeutic potential of HNO donors in the treatment of vascular dysfunction. Antioxid. Redox Signal. 14, 1675–1686.
Introduction
Interestingly, nitroxyl (HNO), the one electron reduced and protonated form of NO• is rapidly emerging as a novel redox sibling of NO• with distinct pharmacological actions and therapeutic advantages over NO• (35) (Table 1). In particular, its cardioprotective actions have received much attention with HNO, unlike NO•, increasing myocardial contractility (via thiol interaction) (8, 63, 79) and conferring protection in the setting of acute experimental heart failure (62). The concomitant ability of HNO to serve as a positive cardiac inotrope and to unload the heart (via vasodilatation) is of benefit in the treatment of heart failure. Thus, together with its myocardial effects, the vascular actions of HNO are also likely to be of importance and warrant further investigation.
Like NO•, HNO may be produced endogenously within the vasculature and has similar vasoprotective properties such as the ability to induce vasorelaxation (3, 19, 20, 33), inhibit platelet aggregation (5, 56), and inhibit vascular smooth muscle cell (VSMC) proliferation (80). In contrast to NO•, HNO targets distinct signaling pathways in the vasculature that include activation of voltage-sensitive K+ channels (Kv) (19, 33) and the release of calcitonin-gene related peptide (CGRP) (20). In addition, HNO is resistant to scavenging by superoxide (.O2 -) (54) and is not susceptible to tolerance development (34, 37). As such, the vascular actions of HNO may be preserved under disease conditions, whereas those of NO• are compromised (i.e., during oxidative stress). Thus, HNO donors may offer a superior alternative to traditional nitrovasodilators for the treatment of vascular dysfunction. This review discusses the vasoprotective actions of HNO in the context of cardiovascular health and disease, exploring the role of HNO as an endogenous vascular signaling molecule, as well as the mechanisms via which it modulates vascular function and the therapeutic potential of HNO donors in the treatment of vascular disease.
Chemical and Biological Properties of HNO
Prior to discussing the vasoprotective actions of HNO, it is important to briefly consider the chemistry and biological targets of this nitrogen oxide. HNO has been shown to be a weak acid with a pKa value of approximately 11.4 (4), suggesting that at physiological pH, HNO rather than the nitroxyl anion (NO-) will predominate. HNO is highly reactive and undergoes rapid dimerization and dehydration to nitrous oxide (N2O) (51). As such, HNO cannot be stored as a stable molecule and is typically studied using HNO donor compounds. Readers are referred to Miranda (51) for a comprehensive review on the chemistry of HNO.
HNO donors are essential tools to elucidate the biological actions of HNO, and in particular Angeli's salt, discovered over 100 years ago, has thus far been the mainstay of the HNO research field (35). Angeli's salt (Na2N2O3) spontaneously decomposes to generate HNO and nitrite (NO2 -) (32), however, its short half-life (∼2.5 minutes) and concomitant release of NO2 - confers limitations on its usefulness. Isopropylamine NONOate (IPA/NO), a primary amine diadiazeniumdiolate, has more recently been used to study HNO-induced effects in the cardiovascular system (52). Yet, whilst IPA/NO spontaneously decomposes at physiological pH to generate HNO and exerts similar physiological effects as Angeli's salt (52), it too has a short half life (∼2.3 minutes), may also generate NO• (at pH < 7), and its nitrosamine byproduct may exert nonspecific effects (35). Ultimately, a pure and longer-lasting HNO donor is required to further our understanding of the biological actions of HNO.
Despite the limitations of currently available HNO donors, they have enabled the pharmacology of this nitrogen oxide to be substantially delineated. Indeed, the biological activity of HNO is governed predominantly by its high reactivity with metallo- and thiol-containing proteins (22, 61). Thus, HNO reduces metals such as iron, copper, and manganese (22, 53, 58) and preferentially targets ferric (Fe3+) versus ferrous (Fe2+) heme groups in a number of proteins (53). In the vasculature, the heme-containing protein, sGC, represents a major cellular target of HNO. Moreover, the direct interaction of HNO with thiols underlies many of the distinct pharmacological actions of HNO versus NO• (35) and may direct the actions of HNO to thiol-containing receptors, ion channels, and enzymes in the blood vessel wall. Studies exploring the vasoprotective properties of HNO have exploited its high reactivity with thiols to distinguish its actions from those of NO• and provide evidence for its endogenous generation. Thus, high concentrations of thiols such as L-cysteine, N-acetyl-L-cysteine (NAC), and dithiothreitol will attenuate the actions of HNO but not those attributable to NO• (20, 33, 34, 63, 64). Conversely, NO• scavengers such as carboxy-PTIO ((2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxy-3-oxide)) and hydroxocobalamin will scavenge NO• but not HNO (18, 33, 34, 42, 83).
Employing HNO donors such as Angeli's salt and IPA/NO and thiol-based HNO scavengers, the capacity for HNO to modulate vascular tone, .O2 - generation, VSMC proliferation and platelet function has been examined.
HNO as an Endothelium-Derived Relaxing Factor
In 1980, Furchgott and Zawadzki identified that a factor was released from the endothelium that evoked VSMC relaxation (24). This endothelium-derived relaxing factor (EDRF) was subsequently identified as NO• (60) and its biosynthetic pathway rapidly elucidated (55). Within the endothelium, nitric oxide synthase (NOS) catalyses NADPH-dependent oxidation of L-arginine to form the unstable intermediate N-hydroxy-L-arginine (NOHA), into which O2 is incorporated to yield NO• and L-citrulline (55). Following synthesis, NO• diffuses rapidly to the VSMC whereby it stimulates sGC to form cGMP that can interact with a number of downstream targets, including cGMP-dependent protein kinases (cGKs), phosphodiesterases (PDEs), and cGMP-modulated cation channels to cause vasorelaxation (40).
The importance of NO• as an endogenous regulator of vascular tone is irrefutable, yet over the last 10 years evidence has emerged that NO• may not be the sole endothelial-derived nitrogen oxide, with HNO likely to also play such a role (3, 23, 45, 83). Thus, like NO•, HNO elevates VSMC cGMP to mediate vasorelaxation (23, 34) and can potentially be generated via a number of biosynthetic pathways in the vasculature (Fig. 1). These include direct production of HNO from NOS itself, whereby HNO serves as an intermediate in the conversion of L-arginine to NO• (29, 72). In particular, superoxide dismutase (SOD) facilitates oxidation of HNO to NO•. Moreover, reduced levels of the NOS cofactor, tetrahydrobiopterin (BH4), results in the partial uncoupling of NADPH oxidation and NO• synthesis to promote the production of HNO over NO• (71). HNO can also be formed after oxidative degradation of the NOS intermediate, NOHA (66). NOHA represents a feasible biosynthetic pathway for the generation of HNO in vivo, given it is detected in plasma (27) and produced by some cells (90). In addition, heme protein-mediated oxidation of hydroxylamine (NH2OH), a product of cellular and NOS metabolism, leads to HNO generation (15). Additionally, HNO can be formed from non-NOS sources. Whilst a direct reduction of NO• to HNO is unlikely to occur spontaneously (4, 61), a number of enzymes catalyze this reaction, including mitochondrial cytochrome c, ubiquinol, hemoglobin, xanthine oxidase, and manganese SOD (35, 61). Finally, S-nitrosothiols have also been known to generate HNO via S-thiolation, a reaction between S-nitrosothiols and other thiol species (87).

Whilst definitive proof for the endogenous generation of HNO is absent due to the lack of direct and sensitive detection methods for HNO in mammalian cells, its role as an EDRF can be inferred from pharmacological studies. Thus, in large conduit arteries, the profile of the EDRF response resembles HNO more closely than NO•. For example, the HNO scavenger L-cysteine reduces HNO- and ACh-mediated vasorelaxation in rat (18) and mouse (83) aortae, yet responses to NO• gas are unchanged or enhanced in the presence of L-cysteine. We have shown evidence for a similar contribution of HNO to ACh-mediated vasorelaxation in resistance-like arteries, particularly following inhibition of endothelium-derived hyperpolarizing factor (EDHF) (3). Together, these findings suggest that HNO contributes, at least in part, to the EDRF response previously attributed to NO• and that these two redox siblings work in concert to mediate endothelium-dependent vasorelaxation.
It is likely that in the blood vessel wall, HNO is derived predominantly from endothelial NOS (eNOS), as the component of endothelium-dependent relaxation attributed to HNO is, for the most part, sensitive to the NOS inhibitor N-nitro-L-arginine methyl ester (L-NAME) (3). Interestingly, neuronal NOS (nNOS) may also serve as a source of HNO in situ, given the attenuation of nitrergic transmission by L-cysteine (11, 42). Whether HNO can also be generated endogenously from non-NOS sources is unclear. Our finding in rat small mesenteric arteries that L-NAME does not abolish the component of endothelium-dependent relaxation attributed to HNO (3) may be indicative of the release of HNO from a non-NOS source such as preformed thiol stores (87) or, alternatively, incomplete inhibition of NOS by L-NAME.
Together with its ability to cause vasorelaxation, exogenous HNO induces VSMC hyperpolarization via opening of Kv channels (33). As such, endogenous HNO may also serve as an EDHF distinct from the classical EDHF, which is NOS-independent and targets calcium-sensitive K+ channels (KCa) channels (17). Evidence in support of this concept comes from our recent findings in rat small mesenteric arteries where, following negation of EDHF (by KCa inhibition), a hyperpolarization response to ACh persisted which, like that of HNO, was sensitive to Kv channel inhibition (3). Similarly, in mouse mesenteric arteries, HNO-induced VSMC hyperpolarization contributes to ACh-mediated vasorelaxation (3). These findings highlight a role for endogenous HNO in local VSMC hyperpolarization and vasodilatation.
We have now advanced this concept further, with recent evidence that exogenous HNO can initiate, and endogenous HNO mediates (in response to ACh) spreading vasodilatation in pressurized rat small mesenteric arteries (88). Spreading vasodilatation is dependent upon VSMC hyperpolarization and is of physiological importance (16). Thus, the local action of a vasodilator is conducted upstream to ensure a significant drop in vascular resistance and thereby a sufficient increase in tissue perfusion. Whilst endothelium-derived HNO appears to initiate spreading vasodilatation, similar observations have not been made with NO• (86).
The identification of HNO as an EDRF and EDHF in the vasculature has heightened interest in this nitrogen oxide (45). Together, NO• and HNO appear to play an integral role in the control of vascular tone and we hypothesize that HNO production and/or bioavailability is preserved during oxidative stress and in disease. Thus, uncoupled NOS preferentially generates HNO over NO• (71), HNO is resistant to scavenging by .O2 - (54), and disease-associated thiol depletion (6) may lead to a reduction in HNO scavenging. As such, HNO may compensate, at least in part, for a loss of endogenous NO• (3) and classical EDHF (17) under pathophysiological conditions. We eagerly await the future development of methods to detect HNO production in biological systems such that the endogenous generation of HNO can be proved conclusively.
Vasodilator Properties of HNO Donors
HNO donors are known to be potent vasodilators both in vitro and in vivo (35). The seminal work of Fukuto et al. (23) demonstrated an ability of the HNO donor, Angeli's salt, to induce relaxation of isolated rabbit aorta and bovine intrapulmonary artery. Such an effect of HNO was distinguished from that of NO• via the sensitivity of HNO-mediated vasorelaxation to scavenging by thiols (64). Subsequently, Angeli's salt has been shown to induce vasorelaxation in other large conduit (18, 34, 83) and small resistance-like arteries (3, 19, 33). Similarly, in the intact circulation, Angeli's salt dilates the rat coronary (20) and cat pulmonary (12) vascular beds and decreases mean arterial blood pressure in anaesthetized rabbits (44), conscious dogs (62, 63), and rats (37). Although HNO appears to be a preferential venodilator in vivo (63), such an effect is lost in the setting of heart failure with equivalent arterial and venous dilation observed (62). Importantly, there is now evidence that HNO is a potent vasodilator in the human vasculature, with Angeli's salt inducing vasorelaxation of human isolated radial arteries (Andrews et al., unpublished, Fig. 2).

sGC/cGMP signaling
HNO signals predominantly via the sGC/cGMP pathway to mediate vasorelaxation (Fig. 3). Thus, Angeli's salt elicits an increase in vascular cGMP levels (23, 34) and its vasorelaxant responses are resistant to the NO• scavengers carboxy-PTIO and hydroxocobalamin (3, 18 –20, 33, 83) and markedly attenuated by the sGC inhibitor 1H-(1,2,4)oxadiazole(4,3,-a)quinoxaline-1-one (ODQ) (3, 19, 20, 33, 34, 83). Interestingly, HNO-mediated vasorelaxation is more susceptible to inhibition by ODQ than vasodilator responses to NO gas or NO• donors such as diethylamine-NONOate (DEA/NO) (3, 19, 33, 34, 83). Together such findings indicate that HNO directly targets sGC, yet this concept remains a matter of contention.

Biochemical studies originally suggested that NO• was the only nitrogen oxide capable of activating sGC (14), with HNO presumably requiring oxidation to NO• prior to sGC stimulation. However, those studies were performed in the presence of high concentrations of thiols, sufficient to scavenge HNO and possibly negate its effect. Recent studies that have re-examined the interaction of HNO with sGC have now yielded contrasting results (50, 89). Thus, in the absence of extracellular thiols and under anaerobic conditions, Miller and colleagues demonstrated an ability of the HNO donors, Angeli's salt and 1-nitrosocyclohexyl trifluroacetate (NCTFA), to directly stimulate purified bovine lung sGC (up to 60-fold), an effect independent of oxidation of HNO to NO• (50). In contrast, utilizing purified bovine lung sGC and cultured endothelial cells, Zeller and colleagues reported that Angeli's salt had no significant effect on sGC activity and cGMP levels in the absence of SOD (89). Given that Cu(II), Zn(II)-SOD (SOD1) has been shown to facilitate the conversion of HNO to NO• in a cell-free assay (58), those investigators proposed that Angeli's salt activates sGC predominantly via SOD-induced oxidation of HNO to NO• (89). Whether the intracellular concentration of SOD is sufficient to facilitate such a conversion in the intact cell remains to be determined. Clearly further work is required to elucidate the precise mechanisms by which HNO activates sGC.
Aside from the mechanism(s) of sGC activation by HNO, a number of other interesting observations have been made regarding its interaction with that heme-containing protein. First, high concentrations of Angeli's salt (0.1 mM) cause inhibition of sGC activity via an apparent oxidative modification of cysteine thiols (50). From a therapeutic standpoint, such an action of HNO may be beneficial in that it would limit excessive vasodilatation by HNO donors. Second, the preference of HNO for Fe3+ versus Fe2+ heme groups (53) suggests that HNO may target the oxidized (Fe3+) rather than reduced (Fe2+) form of sGC. Given the predominance of NO•-insensitive, oxidized sGC in the diseased vasculature (76), such a property of HNO may allow its vasorelaxant activity to be sustained and/or enhanced in the setting of disease whereas traditional NO• donors may be compromised. However, surprisingly recent studies failed to demonstrate an ability of HNO to stimulate oxidized sGC (50, 89) and like NO•, HNO was found to activate the reduced, ferrous heme on sGC. Such findings may be indicative of a kinetically slow interaction of HNO with the ferric form of sGC as compared with its ability to dimerize or react with thiols (50, 89). In addition, the presence of multiple cysteine residues on sGC may contribute to the resistance of ferric sGC to reductive nitrosylation by HNO (50).
cGMP-independent signaling
Whilst the cGMP-dependence of HNO induced vasorelaxation in situ is evident, the HNO donors, Angeli's salt and IPA/NO, do not elevate plasma cGMP following intravenous administration (52, 62, 63). Such discrepancies may be indicative of plasma cGMP levels not reflecting changes at the cellular level, differential sensitivity in the detection of plasma versus cellular cGMP, and/or HNO targeting sGC/cGMP-independent pathways in vivo. Indeed, administration of Angeli's salt and IPA/NO to conscious dogs leads to an elevation in plasma levels of the sensory neuropeptide, CGRP, an effect not observed with NO• donors (52, 62, 63). CGRP serves as a vasodilator, targeting CGRP1 receptors on the endothelium and VSMC to stimulate NOS and adenylate cyclase, respectively (Fig. 3) (7). Interestingly, CGRP appears to contribute, at least in part, to Angeli's salt-mediated vasorelaxation in the rat coronary vasculature (20). The mechanisms underlying HNO-mediated release of CGRP from sensory neurons remain to be elucidated and its role in vivo is unclear given that vasodepressor responses to HNO donors are unchanged in the presence of the CGRP receptor antagonist CGRP8-37 (63). Nevertheless, CGRP remains a valuable biomarker to distinguish between the effects of HNO and NO• in the circulation.
K+ channel activation
Another important pathway involved in HNO-induced vasodilatation is through activation of K+ channels (Fig. 3). Specifically, we have identified an ability of HNO to target Kv (3, 19, 20, 33) and ATP-sensitive K+ channels (KATP) (20) in the resistance vasculature. For instance, Angeli's salt-induced relaxation of small mesenteric (rat and mouse) and coronary (rat) arteries is attenuated by the Kv channel inhibitor, 4-aminopyridine (4-AP) (3, 19, 33) and KATP channel inhibitor, glibenclamide (20), respectively. Importantly, electrophysiological studies have confirmed an ability of HNO to target Kv channels with Angeli's salt-induced VSMC hyperpolarisation abolished by 4-AP (19). Although NO• can also activate Kv channels in vitro and in vivo (75), this is not evident in rat small mesenteric arteries. Rather, in these vessels, NO• targets KCa channels via a cGMP-independent mechanism (65). Moreover, we have shown that HNO is more efficacious in eliciting VSMC hyperpolarization than NO• in resistance arteries (19). Together, these findings serve to further highlight the distinct vascular actions of NO• and HNO and indicate that the nature of HNO signaling (i.e, type of K+ channel activated, role of CGRP) may be dependent on the vessel size and vascular bed.
Given the reactivity of HNO with thiols (35, 61), it is tempting to speculate that HNO directly modifies cysteine residues on Kv and KATP channels to modulate their activity. However, recent findings suggest that HNO primarily activates K+ channels via a cGMP-dependent mechanism. Thus, in rat small mesenteric arteries, sGC inhibition (ODQ) virtually abolishes Angeli's salt-induced VSMC hyperpolarization (19). Furthermore, VSMC hyperpolarization in response to the cGMP elevating agent YC-1 (a NO•-independent stimulator of sGC) is attenuated by 4-AP (19). Together these findings indicate that HNO induces VSMC hyperpolarization via cGMP-dependent activation of Kv channels.
It is clear that the vascular actions of HNO and NO• differ with respect to the K+ channels they target and their dependence upon sGC/cGMP signaling (i.e, HNO > NO•). However, given both HNO and NO• will lead to an elevation in vascular cGMP levels, it is difficult to reconcile the finding that HNO, but not NO•, targets K+ channels via a cGMP-dependent mechanism. It is possible that the distinct chemistry of these two redox siblings confers such biological differences. Given that HNO is resistant to scavenging by .O2 -, we predict that it has a longer intracellular half-life than NO•, potentially leading to alternate cellular targets and modes of activation. In addition, cellular thiol concentrations may compartmentalise the actions of HNO such that it targets membrane-bound molecules where the thiol concentration is low (85).
HNO's distinct pharmacology (Table 1), in conjunction with its vasodilator capacity, may confer therapeutic advantages over traditional NO• donors. A major limitation of currently used nitrovasodilators such as the organic nitrate, GTN, is that they develop tolerance with continued use. The mechanisms underlying tolerance development are likely to be multifactorial, involving reduced biotransformation of GTN, desensitization of sGC, increased activity of cGMP-degrading PDEs, or reduced NO• bioavailability (41). Importantly we have shown that unlike GTN, Angeli's salt does not develop tolerance following administration either acutely in vitro (34) or chronically in vivo (37). Moreover, cross-tolerance is not observed such that the vasodilator efficacy of HNO is sustained in animals rendered tolerant to GTN (34, 37). This is of particular relevance in a clinical setting as HNO donors may be of use in patients resistant to the effects of GTN and administered alone or in conjunction with traditional nitrovasodilators for the treatment of vascular pathologies such as angina and heart failure.
In addition to tolerance development, a loss of potency of NO
Importantly, HNO can induce relaxation of human arteries (Fig. 2), with a similar potency and efficacy as GTN. Coupled with its lack of tolerance development and potential for preserved bioavailability under conditions of oxidative stress, HNO donors may represent a realistic novel therapeutic approach to the treatment of vascular disorders such as angina, hypertension, and heart failure.
Anti-Aggregatory Properties of HNO Donors
NO• plays an important role in the prevention of platelet adhesion and aggregation (82). Like its redox sibling, HNO also modulates platelet function by exerting anti-aggregatory actions. Thus, Angeli's salt has been shown to inhibit aggregation of human platelets induced by adenosine diphosphate (ADP), arachidonic acid, adrenaline, thrombin, and collagen (5, 56). A role for HNO in these actions was confirmed by partial reversal of the anti-aggregatory effects of Angeli's salt by the HNO scavenger, L-cysteine (5). Moreover, HNO decreases markers of platelet activation (5) and has been found to modify cysteine residues in up to 10 platelet proteins, some of which are involved in cytoskeletal changes, metabolic processes, and platelet activation (30).
Currently, the mechanisms by which HNO inhibits platelet aggregation remain to be fully elucidated. Most of the inhibitory effects of NO• in platelets are via the sGC/cGMP pathway (82). Similarly, HNO appears to target platelet sGC, given that Angeli's salt increases platelet cGMP levels and its anti-aggregatory actions are partially reversed by the sGC inhibitor, ODQ (5). The effects of cGMP in the platelet are transduced predominantly via cGMP-PDEs and cGKs (82). Interestingly, despite an ability of Angeli's salt to elevate platelet cGMP, its anti-agreggatory effect in human platelets is resistant to a cGK inhibitor, yet reversed by a cAMP-dependent protein kinase (cAK) inhibitor (5). Similar observations in human platelets have been made with NO• donors (36, 46). Taken together, these findings are indicative of potential cross-talk between the cGMP and cAMP signaling pathways following platelet stimulation with HNO (Fig. 4), and PDE3 may serve as the link. Thus, binding of cGMP to PDE3 inhibits its hydrolysis of cAMP, which can then accumulate (36, 46) subsequently activating cAK and inhibiting platelet aggregation. Clearly, the potential role of cAMP/cAK in the regulation of platelet function by HNO warrants further investigation.
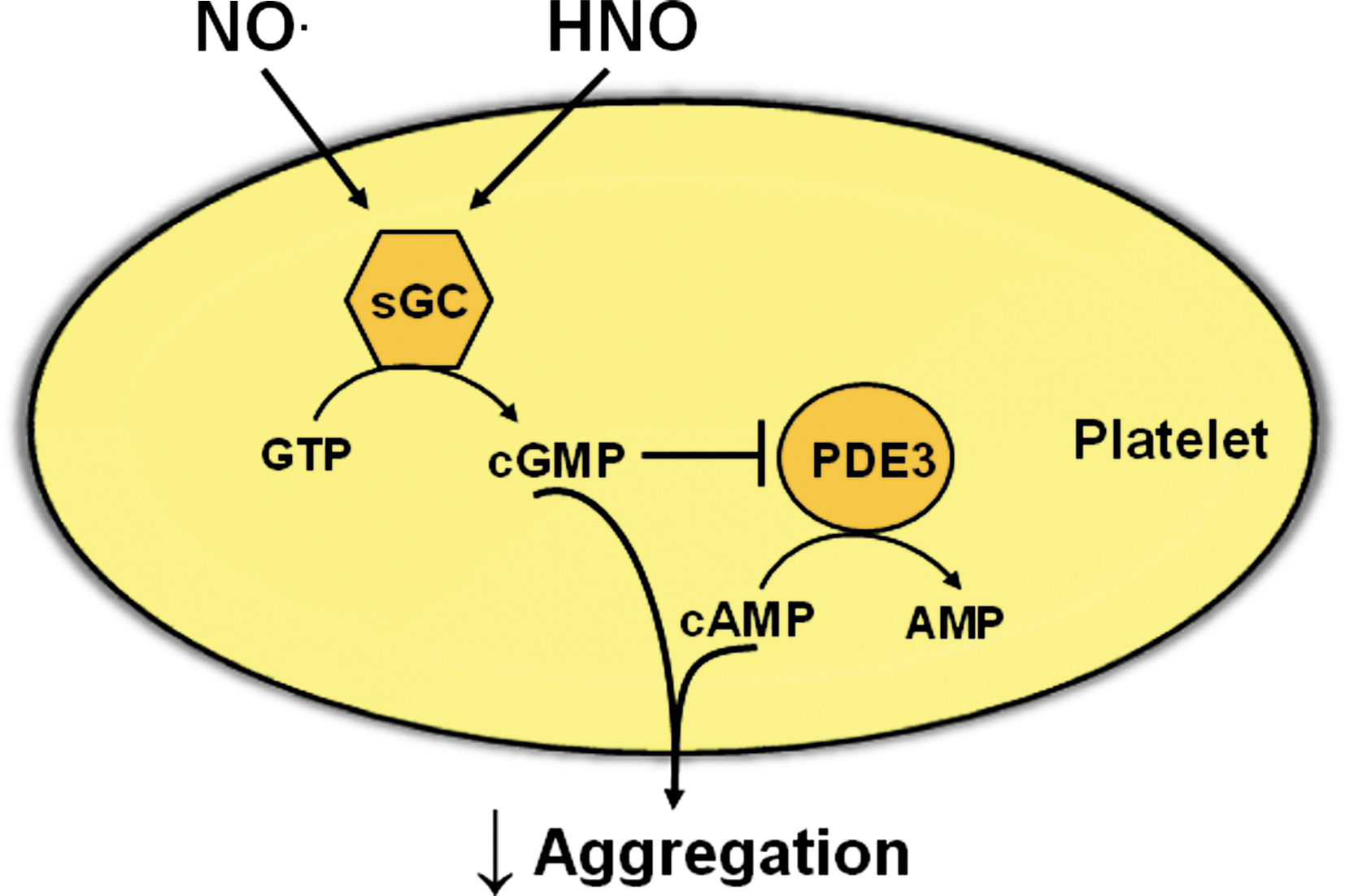
With an ability to inhibit platelet aggregation, HNO donors may be of use in the treatment of atherothrombotic syndromes and offer advantages over traditional nitrovasodilators. Thus, patients with cardiovascular diseases such as angina, ischemic heart disease, and diabetes often display resistance to the anti-aggregatory effects of NO• (9, 84). Such resistance is independent of prior exposure to organic nitrates and may arise as a consequence of decreased NO• efficacy due to scavenging by .O2 - and dysfunction of sGC (9). Given the resistance of HNO to scavenging by .O2 -, it is anticipated that HNO donors will retain their anti-aggregatory properties under conditions of oxidative stress. Indeed, the anti-aggregatory effect of Angeli's salt is sustained in patients suffering from sickle cell disease (56), a condition associated with vascular oxidative stress (25). Furthermore, we have made similar observations in platelets from hypercholesterolemic mice (Bullen et al., unpublished). Together, these results indicate that platelets do not develop resistance to HNO donors during disease and as such these compounds may serve as effective anti-aggregatory agents.
Whilst it is well recognized that NO• also modulates the function of other blood cell types such as leukocytes (2), little is known with respect to the effects of HNO on leukocyte adhesion, rolling, and intravasation. To date, HNO has been shown to stimulate human neutrophil migration (81), and indirect evidence suggests that it may increase neutrophil accumulation during myocardial ischemia (44). Future studies exploring HNO-mediated modulation of leukocyte function will be of importance, particularly in light of recent findings that the innate and acquired immune systems are central to the pathology of vascular diseases such as hypertension (26).
.O2 - Suppressing Properties of HNO Donors
An augmentation of ROS production and/or impairment in ROS metabolism is thought to lead to vascular oxidative stress, which plays a pivotal role in the pathogenesis of numerous cardiovascular pathologies (21, 48). Indeed, increased levels of ROS such as .O2 -, hydrogen peroxide (H2O2), hydroxyl (OH.), and ONOO- cause many of the vascular changes associated with cardiovascular disease, including endothelial dysfunction, vascular remodeling, and inflammation (78). Although there are several sources of pathological ROS, the family of enzymes called the NADPH-oxidases are emerging as strong candidates for the excessive ROS production that leads to oxidative stress (49, 74). As such, an ability to limit ROS production by NADPH-oxidase and/or other sources is a desirable trait of a vasoprotective drug.
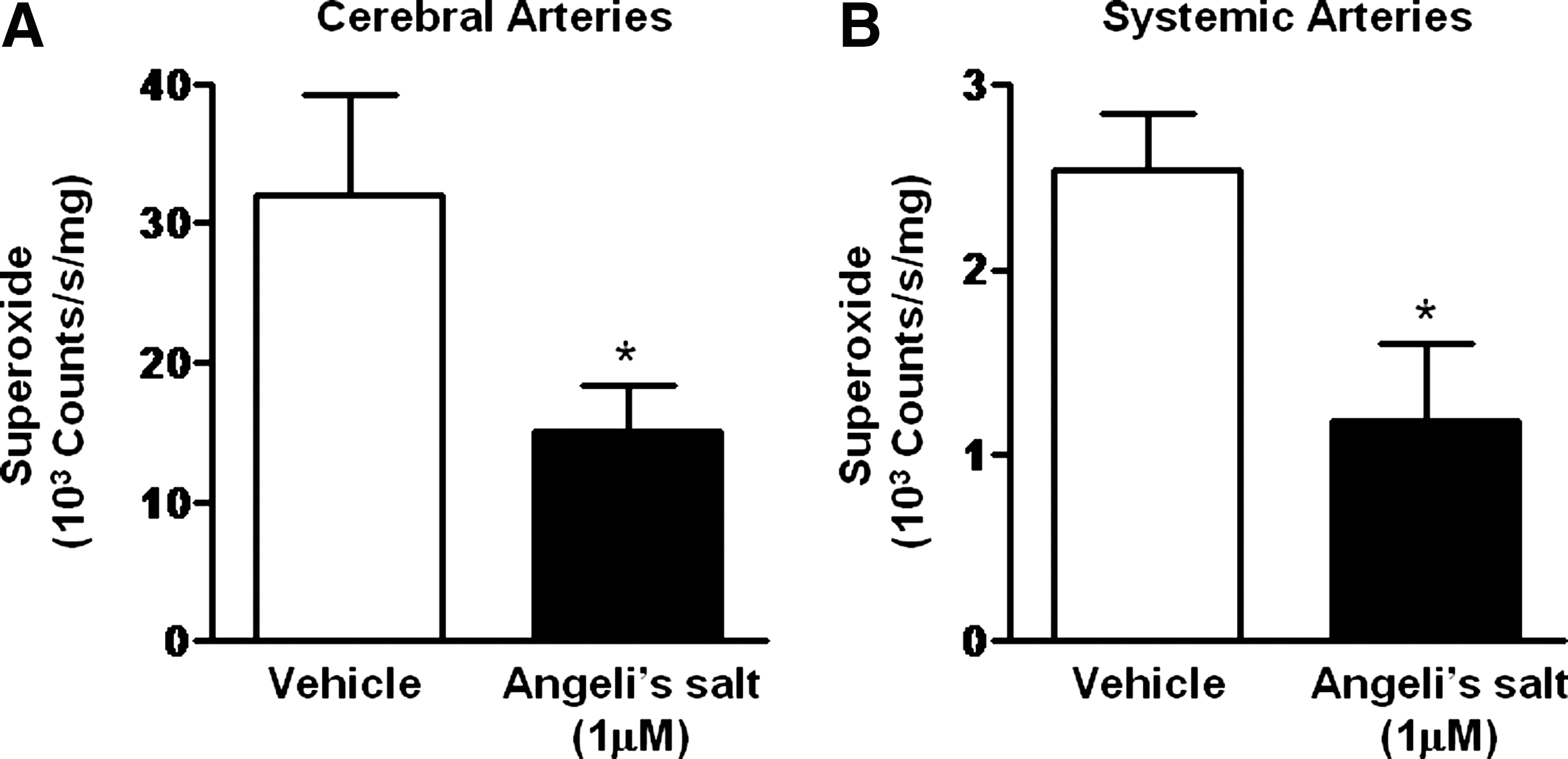
Evidence exists that HNO limits oxidative stress via a number of mechanisms. For example, HNO may serve as a one-electron reductant via donation of its hydrogen atom. In nonvascular cells, HNO has been shown to inhibit membrane lipid peroxidation (43) and stimulate the expression and activity of the antioxidant protein, heme oxygenase-1 (56). Moreover, we have preliminary evidence that HNO suppresses activity of the .O2 - generating enzyme, NADPH oxidase, in neonatal rat cardiomyocytes (68). Based upon these observations, it is anticipated that HNO donors will also limit NADPH oxidase activity in the vasculature. We have previously shown that in human endothelial cells, prolonged treatment with the NO• donor DETA/NONOate leads to inhibition of NADPH oxidase-stimulated .O2 - production, possibly via S-nitrosylation of its regulatory cytosolic subunit p47phox (73). Pilot studies also indicate that the HNO donors, Angeli's salt and IPA/NO, rapidly attenuate .O2 - production by NADPH oxidase, in both the cerebral and peripheral vasculature (Miller et al., unpublished, Fig. 5). Such a property of HNO appears to be sGC/cGMP-independent and may arise as a consequence of post-translational modifications of reactive cysteine thiols within NADPH oxidase. By suppressing .O2 - generation, HNO donors may help to preserve the bioavailability of endogenous NO• and limit oxidation of sGC, thereby maintaining vascular NO•- and HNO-mediated signaling. Given that NADPH oxidase has been identified as a major contributor to oxidative burden in the vasculature (74), an ability of HNO to modulate its activity is of significant potential therapeutic benefit and the mechanisms underlying such actions warrant further investigation.
Regulation of Growth and Proliferation of Vascular Cells by HNO Donors
Amongst its vasoprotective actions, NO• is known to regulate VSMC proliferation and migration with stimulatory and inhibitory effects observed at low and high concentrations, respectively (40). The antiproliferative actions of NO• have the potential to limit VSMC migration and mitogenesis in atherosclerosis (69). Conversely, NO• stimulates endothelial cell proliferation, leading to endothelial regeneration following vascular injury (1). Until recently, little was known with respect to HNO-mediated regulation of endothelial and VSMC proliferation and migration.
However, Tsihilis and colleagues (80) have recently shown that a high concentration of IPA/NO (1 mM) inhibits proliferation, but not migration, of VSMC and endothelial cells in culture. This effect of IPA/NO in VSMC was via S-phase cell cycle arrest, yet NO• induces G0/G1 cell cycle arrest in the same cell type (80). In addition, following topical application of IPA/NO powder (10 mg) to the periadventitial surface of carotid arteries immediately post balloon injury, a modest reduction in neointimal hyperplasia together with reduced VSMC proliferation and macrophage infiltration was observed 14 days later (80).
Whilst IPA/NO limited neointimal hyperplasia following vascular injury, it also appeared to attenuate endothelial regeneration (80). Although it is currently unclear if such effects were due to HNO itself or other components of IPA/NO decomposition (i.e, isopropylamine, isopropanol), such findings are indicative of a potential anti-angiogenic property of HNO. This notion is further supported by the observation that Angeli's salt reduces overall blood vessel density in mouse tumours with an associated trend for decreased serum levels of vascular endothelial growth factor (VEGF) (59). Such an anti-angiogenic activity may indicate potential for the use of HNO donors in the treatment of cancer, yet it also raises the question as to their ability to preserve endothelial integrity post vascular injury.
It is important to note that research on the vascular actions of HNO is in its infancy, and many important questions remain unanswered. Thus, whilst in vivo administration of IPA/NO led to a modest attenuation of neointimal hyperplasia and impaired endothelial regeneration, which was associated with high mortality (80), it remains to be determined if these effects were attributable to HNO itself or to the breakdown products of IPA/NO. Moreover, in the same study the mode of administration of IPA/NO (i.e., topical application) prevents accurate determination of the effective concentration of HNO donor applied and may limit access of HNO across the blood vessel wall, as well as facilitating nonspecific effects such as a chemical interaction with the adjacent vagal nerve. Further investigation into the antiproliferative effects of HNO in the vasculature will be essential to ascertain the clinical feasibility of such an action of HNO donors.
Therapeutic Potential of HNO Donors
The therapeutic application of HNO-donating compounds is tenable given the HNO donor, cyanamide, is currently used in the treatment of chronic alcoholism with minimal adverse effects (13, 61). Attention is now being afforded to the potential use of HNO donors in the treatment of heart failure, given the unique ability of HNO to increase myocardial contractility and unload the heart (via vasodilatation) (62, 67). In fact, a pure, small molecule HNO donor, CXL-1020 developed by Cardioxyl Pharmaceuticals (Chapel Hill, NC), is currently being tested in a Phase I/IIA clinical trial in patients with stable heart failure and we await the outcomes of this trial with interest.
With respect to the vascular actions of HNO, its ability to induce vasodilatation, inhibit platelet aggregation, and suppress .O2 - generation, coupled with its resistance to scavenging by .O2 - and lack of tolerance development indicate that HNO donors may be of use in the treatment of vascular dysfunction associated with angina and atherothrombotic syndromes. The clinical efficacy of traditional nitrovasodilators, such as GTN, in these pathologies is limited by their susceptibility to tolerance development and potential resistance in platelets. As such, HNO donors may represent novel stand-alone or combination therapies (i.e., with GTN), and be of particular use in those patients exhibiting tolerance and/or resistance to NO• treatment.
However, it should be recognized that the apparent therapeutic benefits of HNO may be tempered by possible nonspecific and toxic effects. Thus, toxic actions of HNO, albeit at high concentrations (2–4 mM Angeli's salt), have been reported in cells such as neurons (28) and involve DNA oxidation and thiol loss. Moreover, Angeli's salt has been shown to exacerbate ischemia-reperfusion injury in both the brain (10) and heart (44). It remains to be determined if such deleterious effects of HNO are due to its ability to decrease blood pressure and potentially reduce organ perfusion, stimulate neutrophil migration (77), or directly modulate cellular function. Despite these potential adverse effects of HNO, the anti-alcoholism drug, cyanamide, has few reported side effects in man and the long-term administration of Angeli's salt in animals is well tolerated (LD50 > 130 mg/kg) with no observable carcinogenesis (39).
Clearly more work is required before the therapeutic utility of HNO donors can be fully assessed. Nevertheless the vasoprotective actions of HNO coupled with its distinct pharmacology, as compared with NO•, confer potential for the use of HNO donors in the treatment of vascular disease.
Perspectives
The redox siblings, NO• and HNO, have distinct biological and pharmacological properties which are readily apparent in the cardiovascular system (35) (Table 1). In recent years, considerable attention has been afforded to HNO as it has been demonstrated to increase myocardial contractility and decrease cardiac preload in the setting of heart failure. Likely to be of equal therapeutic importance is the action of HNO in the vasculature, with evidence emerging that endogenous and exogenous HNO target novel signaling pathways to confer a number of vasoprotective properties.
Whilst the role of HNO as an endogenous modulator of vascular function remains to be proven conclusively, we should no longer consider NO• as the sole endothelium-derived nitrogen oxide. Rather, with the potential to be generated from NOS-dependent sources, HNO appears to work in concert with NO• to mediate endothelium-dependent vasodilatation and it may compensate for a disease-associated reduction in NO• bioavailability. The field now awaits the development of new approaches to detect HNO selectively in the intact cell and thus confirm its endogenous generation.
In the vasculature, HNO shares some similar features with NO•, such as an ability to induce vasodilatation, limit VSMC proliferation and .O2 - generation, and inhibit platelet aggregation (Fig. 6). However, often in contrast to traditional nitrovasodilators, HNO donors appear to activate distinct vascular signaling mechanisms (i.e., Kv and KATP channels, CGRP release), are not scavenged by .O2 - nor do they develop vascular tolerance. Intriguingly, these properties may allow the vasoprotective actions of HNO to be preserved under conditions of oxidative stress in which those to NO• are compromised.
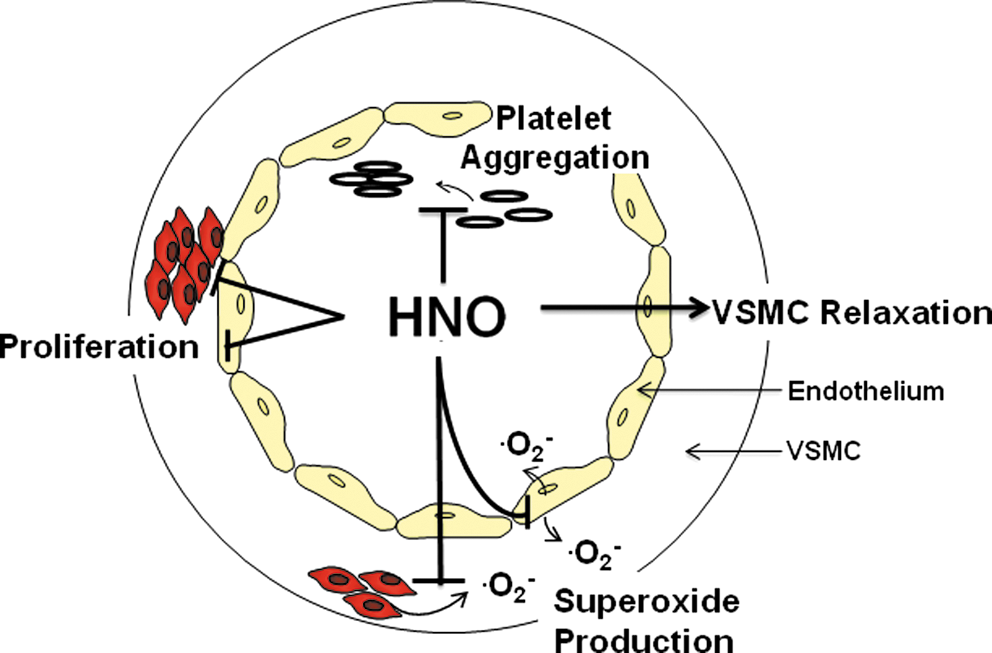
Although it is clear that HNO donors offer considerable advantages over traditional nitrovasodilators, a number of important issues must be addressed before the therapeutic potential of HNO donors can be fully realized. Namely, the potential for HNO to inhibit endothelial regeneration and exert nonspecific effects due to its high thiol reactivity, requires further investigation. A comprehensive evaluation of the efficacy of HNO donors under disease and oxidative stress conditions is also needed. In addition, pure, longer-acting HNO donors are urgently required for experimental evaluation in order to advance the field through the study of long-term vascular effects of HNO.
In summary, the vasoprotective actions of HNO coupled with its lack of tolerance development and potential for preserved bioavailability under conditions of oxidative stress indicate that HNO donors may represent novel strategies for the treatment of vascular dysfunction associated with diseases such as angina, hypertension, and atherosclerosis. Undoubtedly, as research continues in this area, further novel properties and therapeutic applications of HNO will emerge.
Footnotes
Acknowledgments
Michelle Bullen is supported by an Australian Postgraduate Award and Dr. Alyson Miller by a Career Development Award and grants from the National Health & Medical Research Council (NHMRC) of Australia. Dr. Jennifer Irvine is the recipient of a Heart Foundation Postdoctoral Fellowship (Australia) and Dr. Rebecca Ritchie is supported by grants from the NHMRC and is the recipient of a NHMRC Senior Research Fellowship. A/Prof. Chris Sobey is a NHMRC Senior Research Fellow and both himself and Dr. Barbara Kemp-Harper are supported by grants from the NHMRC.