
Abstract
The forkhead box O (FOXO) family of transcription factors regulates a variety of cellular programs, including cell cycle arrest, reactive oxygen species (ROS) scavenging, and apoptosis, and are of key importance in the decision over cell fate. In animal model systems it has been shown that FOXO is involved in the regulation of long lifespan. FOXO activity is tightly controlled by the insulin signaling pathway and by a multitude of ROS-induced posttranslational modifications. In the cell, ROS levels can be sensed by virtue of stimulatory and inhibitory oxidative modification of cysteine residues within proteins that control various signaling cascades. Recently, it was shown that cysteines in FOXO can also act as sensors of the local redox state. In this review we have outlined the cysteine-dependent redox switches that regulate both the insulin and ROS signaling pathways upstream of FOXO. Further, we describe how FOXO controls ROS levels by transcriptional regulation of a multilayered antioxidant system. Finally, we will discuss how cysteine-based redox signaling to FOXO could play a role in fine-tuning the optimal cellular response to ROS to control organismal lifespan. Antioxid. Redox Signal. 14, 1093–1106.
Introduction

In the literature, regulation of FOXO activity is roughly divided into two main routes: insulin signaling and reactive oxygen species (ROS) signaling. However, in this review we will explain how these two modes of signaling to FOXO are not rather mutually exclusive, but that also insulin signaling is being modulated by ROS and vice versa. Besides the upregulation of antioxidant genes, FOXO also plays a role in the regulation of metabolism, which may affect respiration derived ROS. When we write ROS signaling in this review, we mean signaling induced or mediated by the actual ROS molecules.
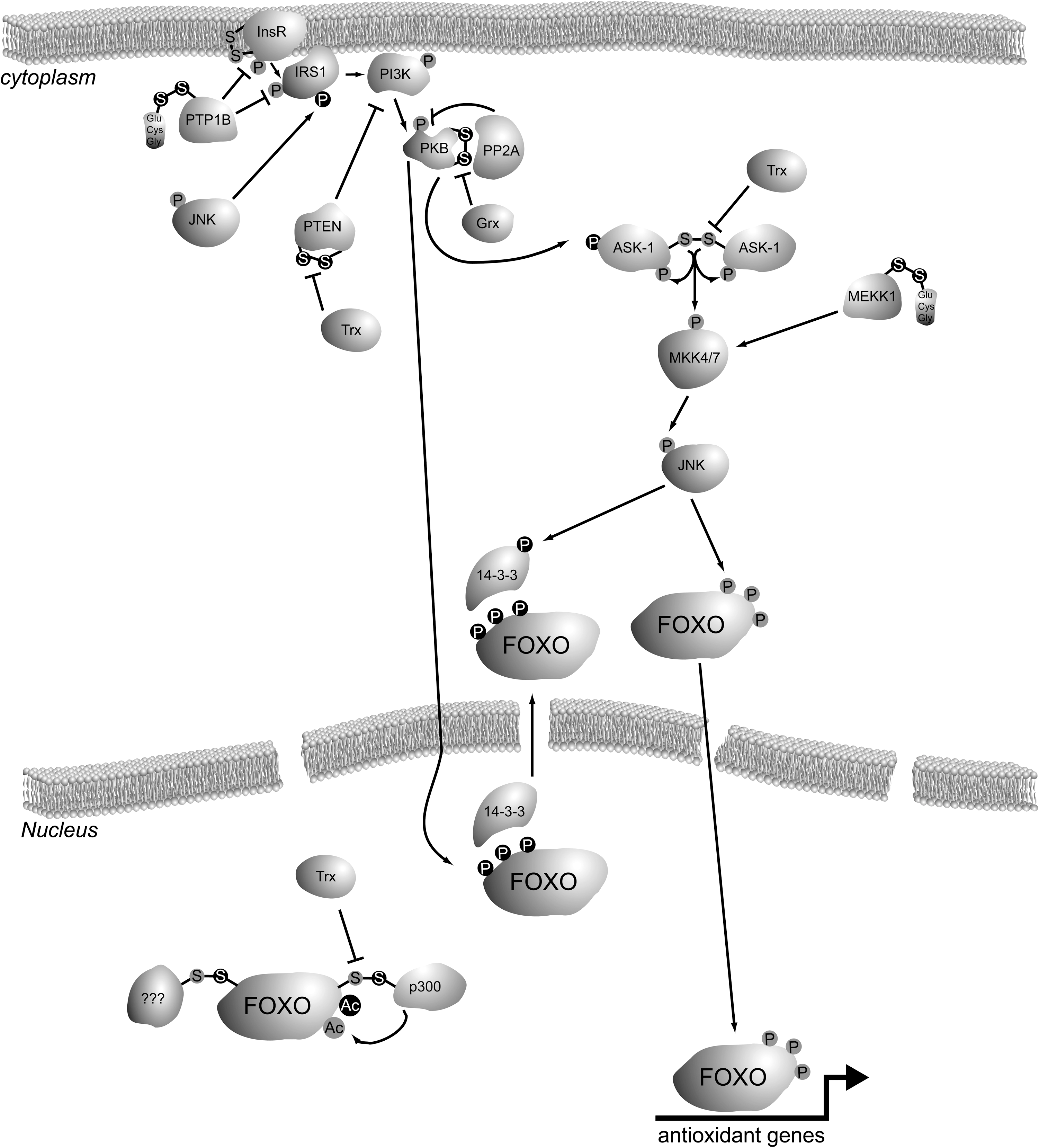
The insulin signaling pathway inhibits FOXO activity: stimulation of cells with insulin (or insulin-like growth factor [IGF]) leads to activation of PI3K and subsequently PKB (Akt) (11), and this action is reversed by the tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN) (49). PKB-mediated phosphorylation of FOXO on three conserved residues creates a binding site for the 14-3-3 adaptor protein, which shuttles FOXO out of the nucleus and thereby prohibits it to function as a transcription factor (8, 38). ROS signaling to FOXO is more complex, as ROS induce the interaction of FOXO with a scala of proteins, including transcriptional coactivators, repressors, and modulators. Many laboratories, including ours, have elucidated that ROS regulate FOXO activity by a multitude of posttranslational modifications (PTMs), including phosphorylation by mitogen-activated protein kinases (MAPKs) like c-Jun N-terminal kinase (JNK) and extracellular regulated kinase (23, 78), acetylation by p300/cAMP responsive element binding (CREB) binding protein (CBP), and deacetylation by Sirt1 (9, 21) and ubiquitinylation by MDM2/Skp2 (31, 78). Whereas insulin/PI3K/PKB signaling shuts down FOXO function, the outcome of ROS signaling is far more complex and quasi contradictory, since some ROS-induced PTMs promote FOXO activity, some act inhibitory, and others seem to dictate a shift in transcriptional targets (13). Why would the regulation of FOXO activity by ROS signaling be so complex? FOXOs can activate programs that lead to damage repair, cell cycle arrest, or apoptosis; hence, FOXO activity judges over a cell's life or death. To be able to make a proper judgment in light of FOXO's evolutionary role in extended lifespan, FOXO activity probably needs to be fine-tuned to meet the appropriate cellular response to certain ROS levels. It is not hard to imagine that if FOXO would induce apoptosis every time a few ROS molecules are present in a cell, tissue renewal sources would become rapidly depleted, working against a prolonged lifespan. On the other hand, if FOXO's response to severely damaging ROS levels would be to frantically attempt to repair all damage and save the cell, potentially hazardous mutations could be passed on to daughter cells, causing, for instance, cancer and thereby limiting organismal lifespan. Hence, one could speculate that FOXO's function as a longevity protein lies not in FOXO being simply active, but in adjusting its activity to adapt to the fittest response to certain stresses cells encounter to ultimately benefit organismal lifespan. In the Free Radical Theory of Aging, it is thought that aging is caused by the build-up of oxidative damage over time (28). It could well be of equal importance how cells in the organism can adequately fine-tune their response to various levels of damage. The multitude of ROS-induced PTMs that have been found to activate, inhibit, or modulate FOXO activity could well reflect such a fine-tuning mechanism.
The above reasoning for a fine-tuning mechanism for FOXO activity in response to certain levels of ROS implies the existence of a cellular system to actually determine how much ROS are present within the cell: the posttranslational modifiers of FOXO in response to ROS are not necessarily the direct sensors of ROS levels but may be the downstream transducers hereof. It was long thought that the intracellular environment was too reducing for cysteines within cytoplasmic or nuclear proteins to be in an oxidized state (33). True, the cell is equipped with systems (e.g., the Glutaredoxin and Thioredoxin systems) to reduce oxidized cysteines, but the fact that such an evolutionary highly conserved system is present in cells argues that apparently cysteine oxidation occurs within cells from time to time; otherwise, there would be no need for a system to reduce these cysteines. It is becoming more and more clear that these reversibly oxidized cysteines in many systems can function as redox sensors and that oxidized cysteines are critical in activating or inactivating enzymes in response to ROS. An illustrative example are the yeast AP-1-like transcription factors Yap1 and Pap1, the activity of which is differentially regulated dependent on ROS levels [for a review, see ref. (51)]. An example in mammalian cells is the regulation of the Nrf2 transcription factor. This transcription factor is continuously being degraded by the proteasome system, because of its cytoplasmic anchor Keap1, which recruits the E3 Ubiquitin ligase Cullin-3 to Nrf2. Upon oxidation of cysteines within Keap1, Nrf2 is released, thereby stabilized and can travel to the nucleus to activate its downstream targets, among which are several antioxidant genes (45). Hence, cysteine oxidation in this case acts as an ROS sensor to boost Nrf2-mediated cellular antioxidant defense. For a review on cysteine redox-dependent processes throughout the cell cycle, see ref. (12).
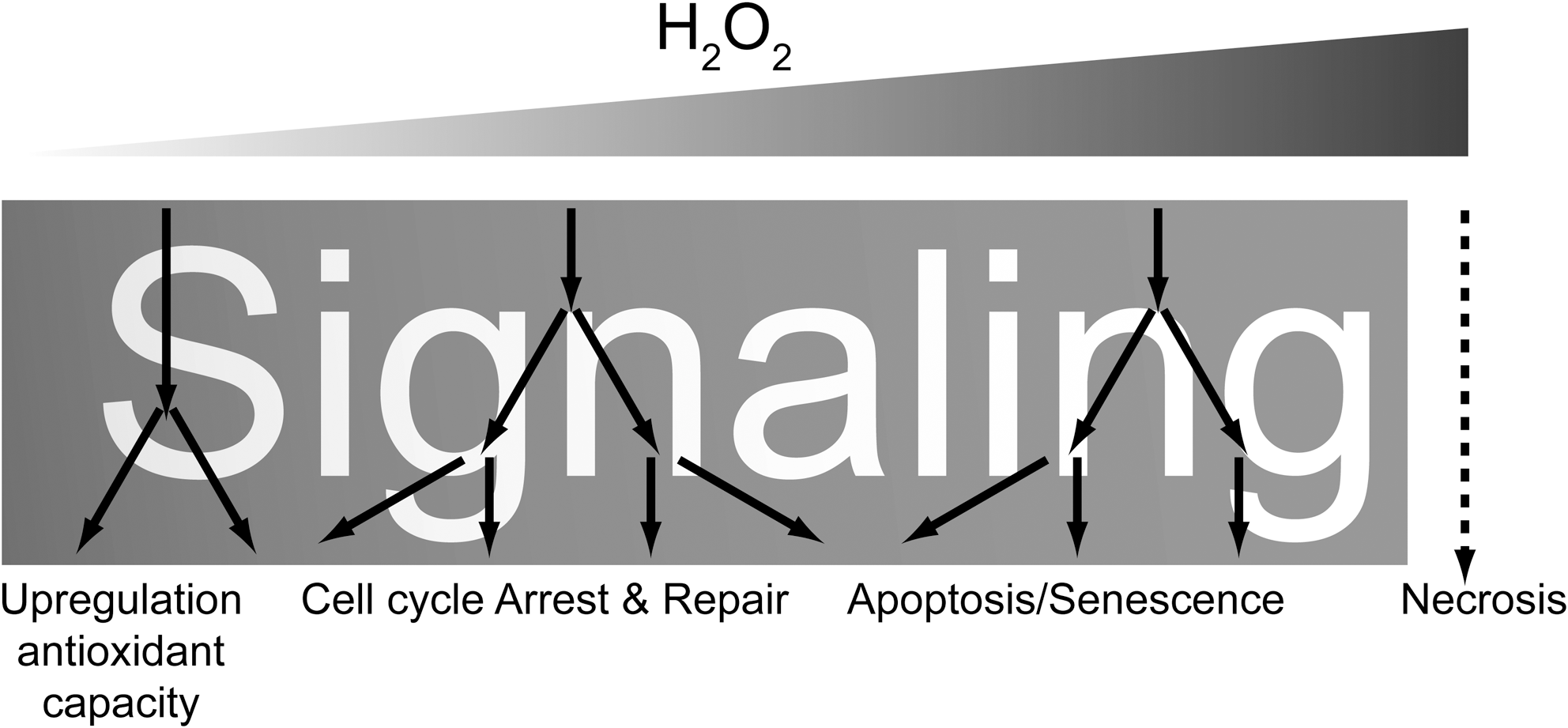
The observation that cellular processes are being regulated through cysteine oxidation has led to the notion that ROS, including hydrogen peroxide, are signaling molecules [see, for instance, ref. (73)]. This is undoubtedly the case, but signaling by ROS is quite different from classical signaling molecules like, for instance, insulin. Insulin binds to the insulin receptor and thereby activates a signaling cascade through the PI3K/PKB pathway. Higher concentrations of insulin will do so too, but in a stronger fashion. Not all cysteines are equally sensitive to ROS (63), unlike, for instance, insulin signaling that is largely proportional to the insulin concentration at the cell surface, higher local concentrations of ROS will oxidize not only more but also different cysteines in different proteins. The latter could enable differential signaling to gradients of local perturbances of the cysteine redox state (Fig. 2).
We have recently shown that also cysteines within FOXO can be oxidized, and that this regulates p300/CBP-mediated acetylation and a subsequent shift in FOXO's transcriptional program from cell cycle arrest to apoptosis (21), but besides FOXO, some of its upstream regulators in both the insulin signaling pathway and the ROS signaling pathway have been described to be subject to regulatory cysteine oxidation. In this review we will delineate how ROS sensing through cysteine oxidation can serve to fine-tune FOXO activity to match an appropriate response to certain ROS levels. Further, we will discuss how FOXO itself modulates ROS levels by regulating the transcription of antioxidant genes. Many excellent reviews have been written recently both on cysteine redox signaling and on FOXO transcription factors. As we attempt to combine the two topics here, we will not be able to cover them in full due to space limitations and we apologize upforehand to our colleagues for omitting important aspects of cysteine redox or FOXO signaling.
Cellular Sources of ROS
During respiration, molecular oxygen is reduced to water in the electron transport chain. When electrons, predominantly from complex I and III, leak prematurely to molecular oxygen, superoxide anions are generated (72). This oxygen radical is highly reactive and has a short half-life within cells due to its spontaneous and enzymatic conversion to hydrogen peroxide. It has been estimated that superoxide anion generation occurs at rates of ∼1 nmol/min/mg mitochondrial protein, thus capable of generating a substantial amount of ROS (67). Another source of superoxide anions and, subsequently, hydrogen peroxide is the Nox family of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. A well-known example of these is found in phagocytic cells, where this system generates the hydrogen peroxide used to neutralize engulfed bacteria. Nox stimulation and subsequent hydrogen peroxide production was shown to occur upon stimulation of cells with several signaling molecules, including epidermal growth factor, platelet-derived growth factor, insulin, and tumor necrosis factor alpha. For a review on Nox, see ref. (25).
Cysteine Redox Chemistry
The thiol side-chain of cysteine makes it reactive to several ROS, including hydrogen peroxide. However, not all cysteines are equally susceptible to oxidation. This generates a degree of specificity in ROS signaling through cysteine residues. The relative reactivity of a cysteine thiol side chain is determined by the direct environment of that cysteine: cysteines that are buried within a protein are, for instance, less susceptible than surface exposed cysteines. Additionally, the local pKa is of importance. Cysteines are most reactive when they are in the thiolate anion form (R-S−). This state is more likely to occur when the local pKa is alkaline, for instance, when the cysteine is flanked by positively charged or basic residues (63). When cysteines partake in the coordination of metals, for example, in Zn-fingers, the Zn atom can also lower the pKa of the cysteine, making it more prone to oxidation. At the same time, oxidation of cysteines that form Zn-fingers is known to lead to the release of Zn, thereby altering the tertiary structure of proteins (39).
Although cysteines can, in principle, react with various ROS, the most important species in redox signaling is hydrogen peroxide. The superoxide anion and the hydroxyl radical are very instable, and it is therefore thought that their steady-state levels are too low to play a role in vivo in cysteine oxidation-based signaling. Hydrogen peroxide has a sufficient long half-life and chemical make-up to diffuse through the cell including membranes, and a sufficient low reactivity to maintain selectivity. Steady-state levels of hydrogen peroxide can reach concentrations in the micromolar range (7). Figure 3 shows some reactions in cysteine oxidation by hydrogen peroxide [adapted from ref. (73)]. The sulfur atom enables the thiol group to undergo multiple rounds of oxidation. The first step of oxidation yields a sulfenic acid (R-S-OH). This reversible, highly reactive intermediate can readily react with another cysteine to form an intramolecular or a mixed disulfide with another protein or with glutathione (glutathionylation). Alternatively, sulfenic acid has been shown to form a, covalent sulfenamide with the backbone nitrogen of a nearby amino acid. Importantly, sulfenic acids can also further oxidize to a sulfinic acid (R-S-O2H) or even sulfonic acid (R-S-O3H), although there are examples of a more stable sulfenic acid intermediate [for a review, see ref. (63)]. Unlike sulfenic acids, sulfenamides and disulfides, cysteine thiols oxidized to sulfinic, and sulfonic acid are considered to be irreversibly modified, although examples, for instance, oxidation of Peroxiredoxin, may exist where sulfinic acid forms are being enzymatically reduced (see also below).
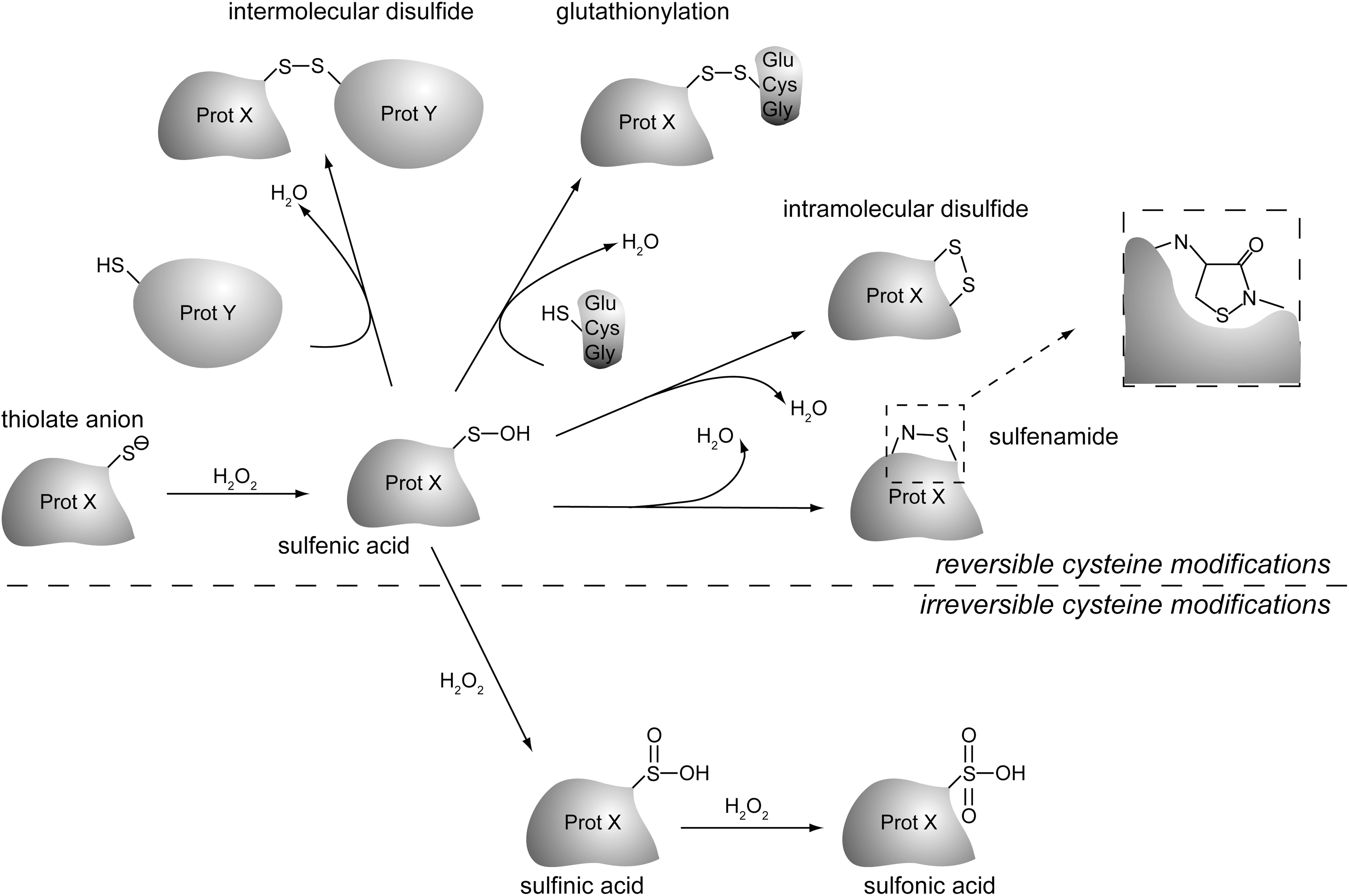
Cysteines can also be S-nitrosylated in a redox-dependent fashion through reaction with reactive nitrogen species. Also S-nitrosylation is reversible and modification of proteins by S-nitrosylation has been shown to regulate several protein–protein interactions in signaling. Like cysteines oxidized to sulfenic acid, nitrosylated cysteines can react to unmodified cysteines to form a protein–protein disulfide or protein glutathionylation (63). The effects of oxidation and nitrosylation on protein cysteine thiols in terms of signaling is quite similar, but we will focus in this review on cysteine oxidation by ROS rather than reactive nitrogen species.
Connecting Cysteine Redox Reactions and Insulin Signaling
Already in the 1970s of the 20th century, it was appreciated that hydrogen peroxide, be it at relatively high levels, had insulin mimetic effects that were mediated by cysteine redox reactions (19). Later it was found that hydrogen peroxide itself can induce the tyrosine autophosphorylation activity of the insulin receptor in the absence of insulin and that oxidation of cysteines within the kinase domain of the insulin receptor prevents inhibitory binding of ADP (65). Although these studies suggest a direct role for cysteine oxidation in activation of the insulin receptor, enhancement of the insulin signaling cascade can be achieved both by enhanced kinase activity and inhibited phosphatase activity, as explained below. This makes it extremely difficult to distinguish what process is most important in altering insulin signaling upon treatment of cells with ROS. Insulin stimulation itself was also shown to induce hydrogen peroxide production via Nox4 at the plasma membrane, although it is not entirely clear how this is regulated (50). In this review we will discuss the main players downstream of both insulin and ROS signaling toward FOXO that are being modulated by redox-sensitive cysteines. For a recent and thorough review on the effects of ROS on insulin signaling, we refer to the work of ref. (3).
Redox Regulation of Insulin Signaling: Protein Tyrosine Phosphatase 1b
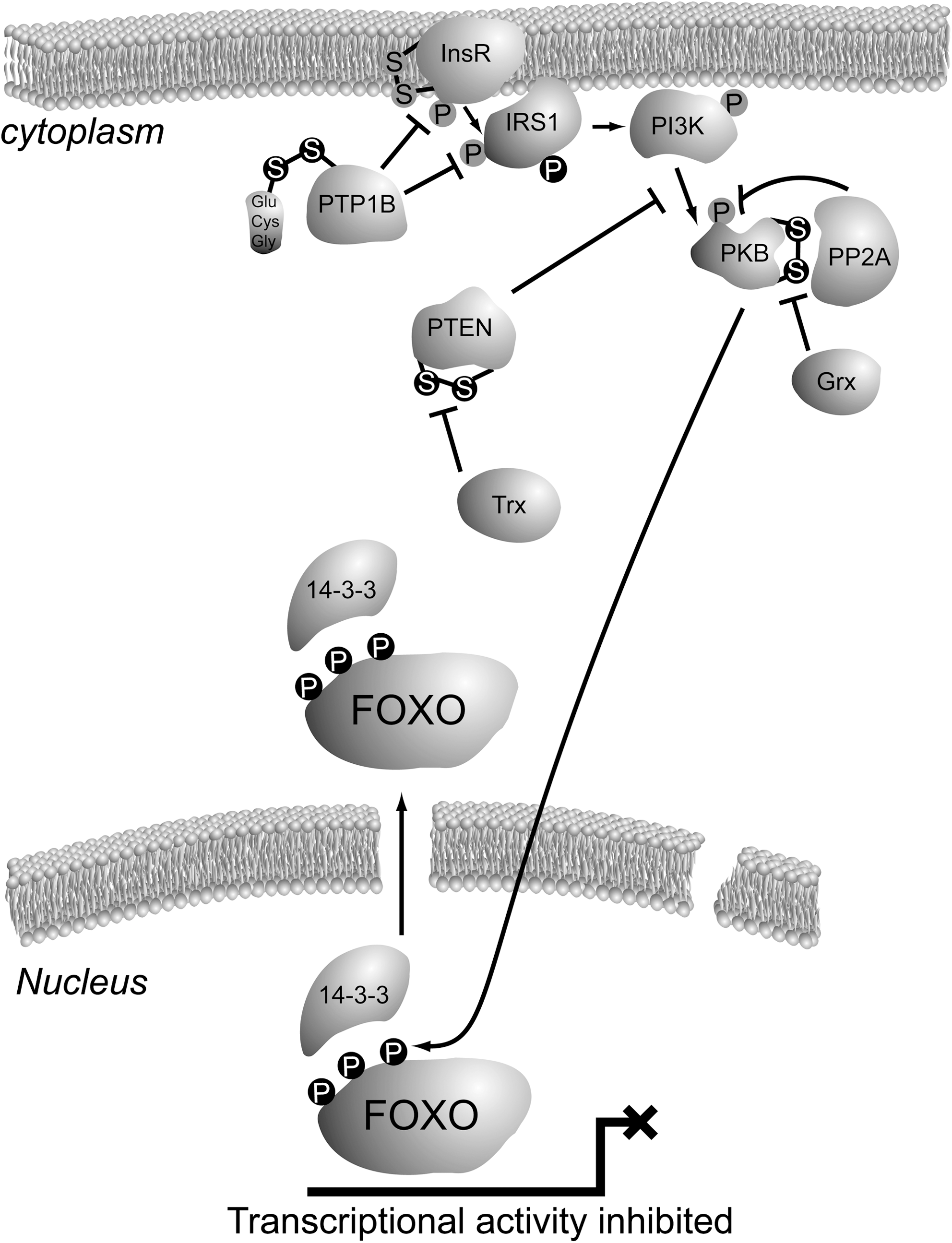
Once the insulin or IGF receptor becomes activated through auto-phosphorylation on tyrosine residues in its intracellular domain, a signaling cascade is set off starting with the phosphorylation of insulin receptor substrate 1 (IRS1) and IRS2 on tyrosines. Tyrosine-phosphorylated IRS forms a docking site for the SH2 domain of the p85 regulatory subunit of PI3K, upon which the p110 catalytic subunit becomes activated due to a conformational change in the complex of these two subunits (66). Both the insulin receptor and IRS1 have been shown to be direct substrates of the protein tyrosine phosphatase 1b (PTP1b) [reviewed in ref. (80)]. In fact, PTP1b is a major regulator of insulin signaling, as loss of PTP1b in genetically engineered mice leads to increased insulin sensitivity (22). The protein tyrosin phosphatases are members of the much larger family of cysteine-dependent phosphatases, characterized by a catalytic cysteine within their active site. For some of the cysteine-dependent phosphatases, including PTP1b (as well as PTEN; see below), it has been shown that their catalytic activity is indeed impaired upon the reaction of ROS with the catalytic cysteine thiol. In PTP1b the initial oxidation leads to a sulfenic acid, which could be easily converted to the irreversible sulfinic or sulfonic acids. However, further reaction to this irreversible modification is prevented by either glutathionylation or formation of a sulfenamide with the main chain nitrogen of an adjacent residue [reviewed in ref. (70)]. In conclusion, ROS alters the insulin signaling route to FOXO by cysteine oxidation of PTP1b; oxidation and therefore inhibition of PTP1b would lead in principle to increased insulin signaling and thus reduced FOXO activity. Hence, ROS negatively regulates FOXO through PTP1b in a cysteine redox-dependent manner.
Redox Regulation of Insulin Signaling to FOXO: PTEN
The PTEN is a lipid phosphatase that dephosphorylates phosphatidylinositol-3-phosphates. Hence, PTEN reverts the action of the PI3K, which is activated downstream of the insulin receptor (49). PTEN is a major gatekeeper in insulin signaling and its function is lost in many cancers, in keeping with the mitogenic effects of PI3K activation. PTEN belongs to the family of protein tyrosine phosphatases and indeed, besides its primary enzymatic activity toward PIP3 lipids, it has been shown to dephosphorylate, for instance, the Focal Adhesion Kinase at tyrosine residues. Like PTP1b, PTEN bears a catalytic cysteine within its active site that can be oxidized in response to exposure to ROS. Oxidation of the PTEN catalytic cysteine leads to formation of a cysteine disulfide between the catalytic Cys124 and Cys71, thereby inactivating the protein (42). The activity of oxidized PTEN can be restored by reduction of the disulfide, which occurs through the action of thioredoxin. Similar to the effect of ROS on PTP1b, cysteine oxidation of PTEN also leads to enhanced insulin signaling and thus inactivation of FOXO.
Redox Regulation of Insulin Signaling to FOXO: PKB
Also, the next player in the insulin signaling cascade, PKB, is directly regulated by oxidation of its cysteines. It has been shown that an intramolecular disulfide bond is formed between Cys297 and Cys311 in PKB upon exposure of cells to ROS (32). These two cysteines are conserved in other members of the AGC kinase family, and were shown to partake in intramolecular disulfides too (27). Oxidation of PKB does not affect its in vitro activity, but in vivo the formation of this bond apparently increases the affinity of PKB for the protein phospatase PP2A, thereby enhancing PKB dephosphorylation and thus inactivation (52). Interestingly, the crystal structure of inactive PKB also showed this disulfide within the activation loop, and when it was reduced with dithiotreitol the structure of the activation loop becomes more distorted. Taken together, these observations could suggest that disulfide formation upon exposure to ROS inactivates PKB by creating a binding surface for PP2A, but that at the same time the disulfide protects PKB from further unfolding when inactive, leaving it readily reactivatable when ROS levels drop and the disulfide is reduced again. The disulfide in the PKB activation loop can be reduced by the glutaredoxin system, consisting of glutaredoxin, glutathione, NADPH, and glutathione disulfide reductase. It is not clear whether, for instance, thioredoxin (Trx) could substitute for the action of glutaredoxin in reduction of the Cys297–Cys311 disulfide within PKB. Since ROS-induced intramolecular disulfide formation within PKB inactivates this kinase, and since PKB-mediated phosphorylation inactivates FOXO, in this case cysteine redox signaling activates FOXO, in contrast to cysteine redox signaling described above for PTP1b and PTEN. An overview of cysteine redox signaling in the insulin/PI3K/PKB pathway to FOXO can be found in Figure 4.
Redox Regulation of JNK/MAPK Signaling to FOXO
The stress-activated protein kinase JNK is a key player in transducing redox signals. When activated upon exposure of cells to ROS, JNK phosphorylates its targets on serine and threonine residues that are followed by proline. FOXOs are also direct targets for JNK (23). This phosphorylation counteracts the PKB-mediated export of FOXO4 from the nucleus, thereby keeping it active upon ROS treatment. The role of JNK signaling to FOXO3 is somewhat controversial. As far as we know, direct JNK-mediated phosphorylation of FOXO3 has not been studied, but effects of JNK activity on FOXO3 localization have been published, although with seemingly contradicting effects. It was shown that JNK activation-mediated export FOXO3a via Crm1 in smooth muscle cells (16). However, other studies showed that JNK activity-mediated nuclear translocation of FOXO3, which was suggested to involve JNK-mediated phosphorylation of 14-3-3 followed by release of FOXO3a from this adaptor protein, which is required for the PKB-mediated nuclear export of FOXO3a (69). FOXO1 is phosphorylated by the p38MAPK and Erk, but not by JNK (2). However, it has been shown that JNK is required for ROS-induced FOXO1 nuclear translocation, suggesting that the mechanism involving 14-3-3 like shown for FOXO3a could work also for other FOXOs. Of note, in C. elegans both JNK and p38MAPK induce nuclear translocation of Daf-16(FOXO) and this is coupled to synergistic lifespan extension (36, 58); for JNK this is conserved in flies (74). Although JNK exerts the effects of ROS on FOXO through phosphorylation of FOXO or factors involved in its nuclear retention, S-Nitrosylation-induced inactivation of JNK has also been shown (61). However, multiple JNK-activating pathways have been shown to be subject to regulatory cysteine oxidation. MAPK (mitogen-activated protein kinase)/ERK (extracellular signal-regulated kinase) kinase kinase 1 (MEKK1), an upstream kinase that activates JNK upon cellular ROS via activation of mitogen-activated protein kinase kinase (MKK)4/7, was shown to be inactivated upon ROS treatment through glutathionylation of Cys1238, which resides in the vicinity of the ATP binding pocket. The cysteine residue itself is not required for ATP binding (a Cys1238Val mutant is active), but redox-dependent glutathionylation sterically hinders ATP binding and thus inactivates MEKK1 activity, which would lead to inhibition of MKK4/7 and subsequent JNK phosphorylation (17). In contrast to the inhibitory effect of ROS signaling on MEKK1, its related kinase apoptosis signal-regulating kinase 1 (ASK-1), which also activates JNK by phosphorylation, is activated in a cysteine redox-dependent manner. It was also shown that the activation is reverted by the action of thioredoxin 1. Several models for the redox-dependent activation of ASK-1 have been proposed. Originally, it was thought that ASK-1 multimers resided in an activating complex, which was kept inactive by binding of Trx-1. Oxidation of the Trx-1 active site would release Trx-1 from the activation complex, and ASK-1 would become active (47). More recently, and in line with the role of Trx-1 as a thiol reductase, it was shown that the multimeric complex of ASK-1 needs to be oxidized to form multiple intermolecular disulfides to be activated and signal via MKK4/7 to JNK (55). Further, the turnover of the intermolecular disulfides between ASK-1 monomers by Trx-1 seemed to be dependent on the covalent association of Trx-1 with Cys250 in ASK-1 (54). Although other models have been suggested too for the ROS (especially hydrogen peroxide)-induced activation of ASK-1 and its regulation by Trx-1, all these models conclude that cysteine redox-mediated interactions activate ASK-1 and subsequently MKK4/7 and JNK. Hence, both MEKK1 and ASK-1 signal to JNK via MKK4/7, but MEKK1 is inhibited, whereas ASK-1 is activated by ROS.
JNK-mediated phosphorylation of FOXO4 has also been shown to be induced upon exposure of cells to hydrogen peroxide via the small GTPase Ral (23). It is unclear, however, how exactly Ral signals to JNK, and whether redox-sensitive cysteines play a role in this regulation.
Finally, JNK can also phosphorylate IRS1 on Ser307, thereby inhibiting its tyrosine phosphorylation by the insulin/IGF receptor. It has also been suggested that Ser307 phosphorylation results in targeting of IRS1 for proteasomal degradation (44). Vice versa, PKB also phosphorylates and thereby inactivates ASK-1. Hence, JNK-mediated phosphorylation of IRS1 prevents insulin-induced negative regulation of FOXO activity, and PKB-mediated phosphorylation of ASK-1 blocks JNK-mediated positive regulation of FOXO (35). An overview of ROS signaling via JNK to FOXO is presented in Figure 5.

FOXO as a Direct Redox Sensor
The above-mentioned examples of cysteine redox regulation upstream of FOXO can be seen as sensors for the local redox state, that subsequently results in a positive or negative signal to FOXO to mediate a certain transcriptional program. Irrespectively, it has now become clear that FOXO itself also acts as a cysteine-redox sensor. Our laboratory has recently shown that cysteines within FOXO4 are subject to oxidation and disulfide formation when ROS levels are increased either by hydrogen peroxide treatment or glucose deprivation-induced mitochondrial ROS production (21). FOXO4 does not dimerize upon ROS treatment, excluding formation of disulfide-dependent multimeric FOXO4 complexes as was shown for ASK-1. Apart from ROS-induced phosphorylation by JNK, FOXO is also acetylated upon treatment of cells with ROS. This acetylation is mediated by the CBP and p300 acetyl transferases. The action of CBP/p300 has a dual effect on FOXO. Whereas acetylation within the DNA binding domain inhibits FOXO-DNA interactions, association of FOXO with CBP/p300 can serve to acetylate histones and thereby open-up the chromtain and expose binding sites for target genes. In C. elegans it was found that the deacetylase sir2 has a daf-16 (FOXO)-dependent effect on lifespan extension, suggesting that acetylation of FOXO has a negative effect on FOXO-mediated lifespan. It was shown that the binding of FOXO to CBP and p300 and the subsequent acetylation of FOXO4 is completely dependent on the presence of redox-sensitive cysteines in FOXO4. An intermolecular disulfide was found between FOXO4 Cys477 and CBP/p300. Redox-dependent disulfide formation between CBP/p300 and its substrates is not a general mechanism, since ROS was not required to induce acetylation of p53 by CBP/p300. Of note, Cys477 in FOXO4 is flanked by two acidic residues, making it unlikely that Cys477 exists in a thiolate anion form that is most prone to oxidation. It could be speculated that the intermolecular disulfide is formed by the reaction of a cysteine in CBP/p300 that is first oxidized to sulfenic acid, with Cys477 in FOXO4. It is not clear which or even whether a specific cysteine in CBP/p300 partakes in the disulfide. It would be interesting to elucidate whether this is the case, and also whether CBP/p300 regulates other transcription factors in a redox-dependent manner. The large number of cysteines in these proteins and the contradicting literature on which domains in CBP/p300 are required for the FOXO interaction would make this a challenging project. In the FOXO4/p300 study it has become clear that FOXO4 interacts with other proteins than CBP/p300 in a disulfide-dependent manner, and each of the five cysteines in FOXO4 seems to have its own favorite binding partners. Investigations into the nature of these interactions are ongoing in our laboratory.
As mentioned, the four human FOXOs have largely overlapping functions, but some specialized functions have been suggested. One possibility to explain differential regulation could be through differential cysteine oxidation. In humans, FOXO3a and FOXO4 have 5 cysteines, FOXO1 has 7, and FOXO6 has 10. Two cysteines in the four FOXOs are conserved: one is situated next to the threonine that is phosphorylated by PKB close to the N-terminus, and the other one is within a small conserved region in the transactivation domain near the C-terminus. The latter cysteine is Cys477 in FOXO4 that was described above as the main site for disulfide formation with CBP/p300. It is not known whether the former conserved cysteine is involved in regulation of PKB-mediated phosphorylation of FOXO, but since it is part of the PKB consensus site this seems plausible. None of the other cysteines in the FOXOs are conserved between any of two family members, posing possibilities for differential regulation through cysteine oxidation. Of note, only FOXO3a and FOXO6 bear cysteines within the DNA binding domain, leaving the possibility that DNA binding affinity of these two FOXOs could be regulated through cysteine oxidation.
Although the CBP/p300 interaction and the novel interactions under investigation represent intermolecular disulfide-mediated interactions, we can at present not exclude that FOXO4 (or other FOXO family members) is also regulated by other types of redox-dependent cysteine modifications like intramolecular disulfides, sulfenic acid formation, or S-nitrosylation.
The Role of Cysteine Reducing Systems
Intracellular oxidized cysteines as well as intracellular hydrogen peroxide are being reduced by an elaborate system of highly conserved cysteine thiol-disulfide exchange enzymes. These enzymes share a common mechanism for reducing oxidized cysteines in target proteins or hydrogen peroxide: reactive cysteines within these enzymes become oxidized themselves in the process, forming either an intramolecular disulfide (Trx, glutaredoxin [Grx], and peroxiredoxin V) or an intermolecular disulfide-dependent homodimer (Peroxiredoxin I-IV [PrxI-IV]). Subsequently, these proteins are enzymatically recycled at the expense of the conversion of NADPH to NADP + either directly or indirectly via a step involving the oxidation and subsequent reduction of glutathione. The recycling enzymes are glutathione reductase and thioredoxin reductase (Fig. 6). Whereas Trx and Grx are involved in the reduction of intramolecular, intermolecular, and mixed disulfides as in glutathionylation, cytoplasmic hydrogen peroxide is scavenged by glutathione peroxidase I (GpxI) and peroxiredoxin I and II (Fig. 7). Peroxiredoxins are recycled by thioredoxin, which is subsequently recycled by thioredoxin reductase [for a review, see ref. (5)]. Because these enzymes use their active site cysteines to neutralize ROS, they are themselves subject to oxidation by hydrogen peroxide, which disables them temporarily. Thus, when hydrogen peroxide induces oxidative modifications that play a role in signaling to FOXO as described above, it will at the same time temporarily inactivate the local cysteine thiol-disulfide exchange enzymes and prolong the signaling effect of cysteine oxidation. Oxidized thioredoxin and other thiol disulfide exchange enzymes are rapidly reduced again. However, when hydrogen peroxide increases further, a Peroxiredoxin active site cysteine can become overoxidized to form a sulfinic acid, which renders the protein inactive (26). It was then shown that Peroxiredoxin in the sulfinic acid form can be reduced by Sulfiredoxin to a disulfide, which is then further reduced by thioredoxin (76). Sestrin [a FOXO transcriptional target (57), see below] was suggested to perform a similar role as sulfiredoxin (10), although this is disputed (75). This mechanism was suggested to serve as a threshold for hydrogen peroxide signaling [known as the floodgate model (77), Fig. 7]. The overoxidation of Peroxiredoxin only takes place at sufficiently high levels of hydrogen peroxide, and because the reduction of the sulfinic acid form of Peroxiredoxin is a relatively slow process, local hydrogen peroxide can build up to fulfill its signaling role, but only when the threshold for inactivation of Peroxiredoxin through sulfinic acid formation has been reached. Although the floodgate model would present an elegant redox switch, some experimental studies contradict this model [reviewed in ref. (51)]. Besides scavenging hydrogen peroxide, another role in redox signaling has been suggested for Prx: in yeast it has been shown that oxidized Prx can transfer its disulfide to a target protein, making it thus positively involved in cysteine oxidation. For a review discussing this activity of Prx, see ref. (24).
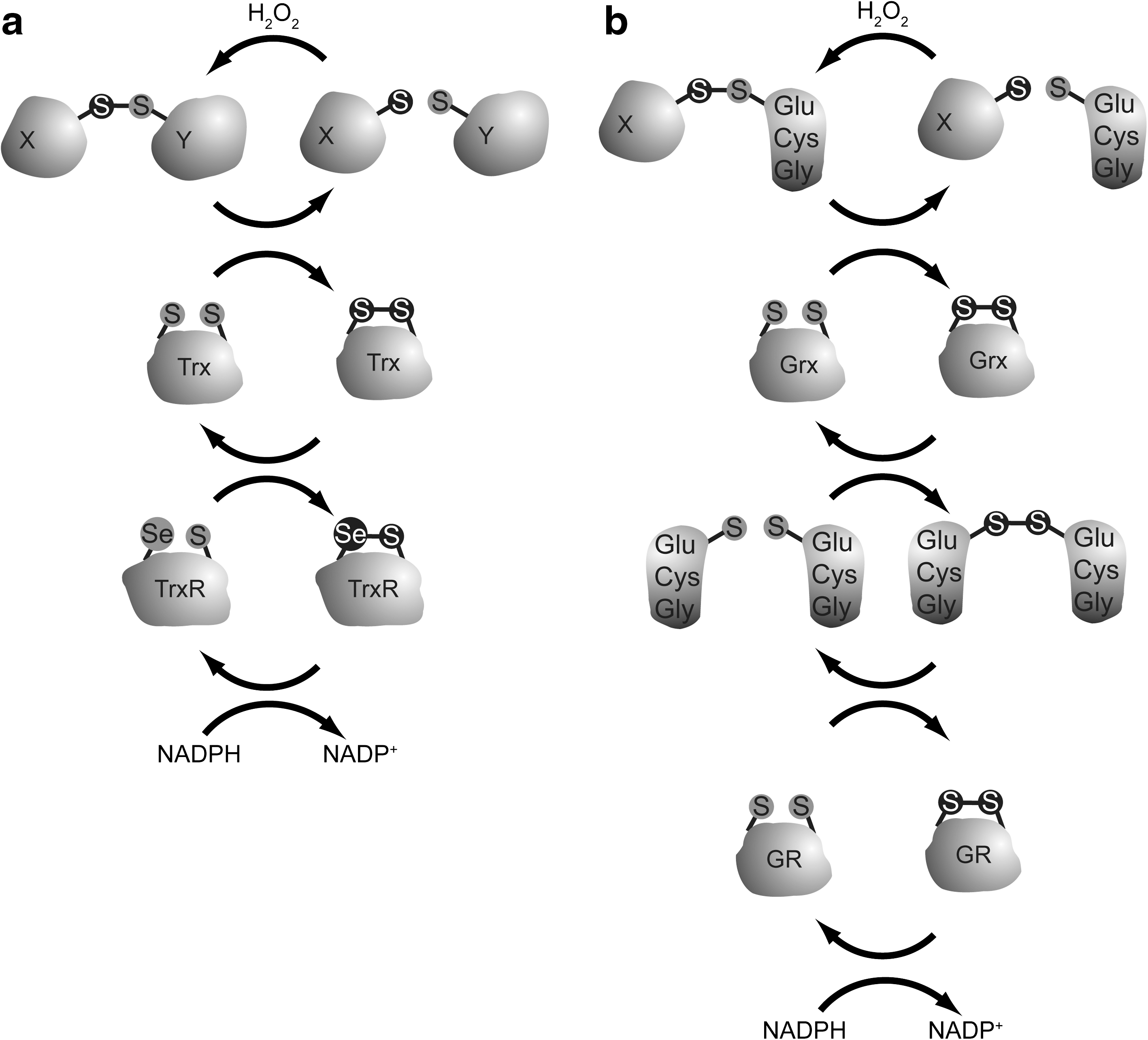

Since the different cysteine thiol disulfide exchange enzymes use overlapping components (Figs. 6 and 7), it is not trivial to determine in vivo which system has the most influence on oxidized cysteines in specific proteins. Nonetheless, there is some difference in substrate specificity, and examples can be found in the literature that seem to show preferential regulation of proteins oxidized at cysteines by one of these enzymes. Redox signaling to FOXO, as we explained, has both positive and negative regulatory effects. This makes it even more difficult to predict what the functional effect will be on FOXO activity when a certain thiol-specific antioxidant enzyme is lost or hyperactive. It was suggested that the oxidation of PTEN is reversed by the action of thioredoxin (42), because thioredoxin coimmunoprecipitates with PTEN, and an inhibitor of Thioredoxin Reductase delays PTEN reduction. Loss of hydrogen peroxide scavenging also influences PTEN, as another study showed that peroxiredoxin I is required to keep PTEN in the reduced state, and that this had a major negative influence on the ability of Ras and ErbB2-induced cellular transformation (14). Also in GpxI knockout mice PTEN was more oxidized, leading to increased insulin sensitivity (48). The inactivating disulfide in PKB was shown to be reduced by glutaredoxin, but it is not clear whether, for instance, thioredoxin could do the same (52). Thioredoxin does regulate ASK-1 disulfide-dependent autophosphorylation, and also the turnover of the FOXO4/p300 heterodimer was shown to be likely regulated by thioredoxin. Taken together, cysteine redox signaling can be reversed by reductive enzymes like Trx and Grx, or prevented by scavenging hydrogen peroxide through Prx and Gpx. When oxidized, Prx can transfer its disulfide to target proteins to modulate redox signaling.
FOXO as a Transcriptional Regulator of ROS Scavenging Enzymes
As mentioned above, nematodes mutant for Daf-2 (insulin/IGF receptor) display a 3–4-fold increase in both median and maximum lifespan. Daf-2 was shown not only to regulate the Age (lifespan extension) and the Daf-c (dauer constitutive) phenotype, but also the so-called oxidative stress-resistant (Oxr) phenotype (29). Like the Age phenotype, the Oxr phenotype requires Daf-16 (FOXO), and it was appreciated that a key antioxidant gene, sod-3, was regulated through the Daf-2/Daf-16 network. Sod-3, like sod-2, encodes a C. elegans homolog of manganese superoxide dismutase (MnSOD), which scavenges superoxide anion radicals by converting them to hydrogen peroxide. MnSOD was also shown to be directly transcriptionally regulated by FOXO in human cells (37). As mentioned, superoxide anion radicals are highly reactive and they randomly damage proteins, lipids, and DNA in their vicinity. The dismutation to hydrogen peroxide leads not only to a less reactive ROS, but in contrast to superoxide anion radicals, hydrogen peroxide formed could play a role in redox signaling. However, hydrogen peroxide can also be damaging, especially when converted to hydroxyl radicals and hydroxyl anions under influence of transition metals in the Fenton reaction. Hydroxyl radicals are even more reactive than superoxide anions and therefore have too little specificity to have a role in signaling. In this respect, it is important that excess hydrogen peroxide is being neutralized. This can be done by glutathione peroxidases, peroxiredoxins, and catalase. Large-scale RNAi screening for genes that confer the daf-16-dependent Age and Oxr phenotype confirmed that indeed a number of antioxidant genes are involved in these phenotypes, including the previously found sod-3 as well as ctl-1 and ctl-2, which encode a cytosolic and a peroxisomal catalase, respectively (43, 53). Mammals only have a peroxisomal catalase, and also this protein is regulated by FOXO (56). FOXO also transcriptionally regulate Peroxiredoxins (15), which could serve to scavenge hydrogen peroxide derived from the action of superoxide dismutases. Notably, in flies it was shown that a homolog of PrxII (Jafrac1) mediates resistance to paraquat, a superoxide anion radical-generating compound (41). As explained in the floodgate model, overoxidation renders peroxiredoxins (PRXs) temporarily inactive; hence, increased PRXs production is one-way FOXOs may aid in ensuring continued peroxide scavenging. Alternatively, oxidation of PRXs can be reversed by sulfiredoxin and the small redox family of sestrins, comprising Sesn1-3, respectively, ensuring further scavenging of cellular peroxides (10). Sesn1 and 2 are under direct regulation by p53, whereas Sesn3 is a transcriptional target for FOXO. Indeed, Sesn3 expression is significantly downregulated in foxo1,3,4−/− mouse embryonic fibroblasts resulting in elevated cellular ROS levels (57). Thus, FOXOs not only ensure peroxide scavenging through increased transcription of Catalase and Prxs, but also enable recycling of the catalytic cysteines in PRXs that are overoxidized to sulfinic acid through upregulation of Sesn3, but it is to be noted that the sulfiredoxin-like activity of Sestrins is debated (75).
The FOXO-dependent regulation of MnSOD, catalase, and PRX and Sestrin indicates that FOXO controls the antioxidant system in a triple layered manner. ROS is eliminated at three oxidation states: from superoxide reduction, via hydrogen peroxide scavenging, to sulfinic acid reduction. This triple-layered control of ROS levels might be in fact the only way to successfully protect cells from ROS, as scavenging of only superoxide would lead to higher hydrogen peroxide levels and reducing only sulfinic acid would not protect cells from the highly reactive superoxide anion.
FOXO, ROS Scavenging and Lifespan Regulation
The free radical theory of aging predicts that boosting the antioxidant system will prevent aging and thereby extend lifespan. The observation that in C. elegans daf-16/FOXO regulates both lifespan extension and oxidative stress resistance was therefore seen as supportive of the free radical theory of aging. However, the link seems to be more complex. A recent study shows that although sod-2 and sod-3 (MnSOD) single and double mutants were highly sensitive to oxidative stress, no effect was observed on lifespan. Further, the same study showed that when sod2::sod3 double mutants were crossed with daf-2 (InsR) mutants, the Oxr phenotype of these worms was lost, but not the Age phenotype (30). Lifespan was affected variably in the daf-2 mutant crossed with sod-2, sod-3, or double mutant, suggesting that oxidative stress resistance per se is not directly correlated to longevity. Surprisingly, dauer formation in the Daf-2 mutant was also variably affected by sod gene expression, with the sod-2 mutation having a suppressive and sod-3 mutation a stimulating effect. Atmospheric oxygen tension influenced the observed effects on dauer, suggesting that oxidative stress plays a role in the this. Hence, loss of MnSOD in C. elegans affects both lifespan and dauer in the daf-2 mutant. It was proposed that in C. elegans MnSOD fine-tunes insulin like signaling pathway to control longevity not only by acting as antioxidants but also by acting as controllers of redox signaling (30).
In flies the effect of overexpression of MnSOD on lifespan is unclear, as studies have been published that show lifespan extension (68), little effect on lifespan (59), and lifespan reduction when combined with catalase overexpression (4). Also in mice oxidative stress levels and lifespan do not seem to correlate. Functional MnSOD is clearly important for normal lifespan, as mice that lack MnSOD die shortly after birth with severe oxidative stress and damage in metabolically active tissues (40, 46). However, another study shows that transgenic mice overexpressing MnSOD, copper/zinc SOD (CuZnSOD), or combinations of both MnSOD and CuZnSOD or Catalase and CuZnSOD are protected against oxidative stress but do not have any increase in lifespan (62). Similarly, a reduced gene dosage of Sod2 (encoding MnSOD) either alone or in combination with GpxI ablation had no effect on lifespan in a study comparing wild-type mice with Sod2+/−, GpxI+/−, Sod2+/−::GpxI+/−, and Sod2+/−::GpxI−/− mice (remember that the Sod2−/− genotype is lethal before or shortly after birth) (79).
These examples illustrate that lifespan and antioxidant capacity are not always simply correlated. In line with this, it was shown that despite the popularity of high-dose antioxidant food supplements that are being sold to boost vitality and youthfulness, these substances may actually reduce lifespan (6). Upregulation of the cellular antioxidant system will indeed protect cells from oxidative damage, but it will also alter the cellular redox state and thus redox signaling. It maybe, as suggested in the aforementioned C. elegans study, that fine-tuning redox signaling to meet an optimal cellular response to certain ROS levels is of equal importance for organismal lifespan than ROS scavenging per se (30). In this reasoning the role of FOXO may lie in keeping the redox sensing and signaling system fit to rapidly adapt. Hormesis is the lifespan extension observed when worms and flies exposed to low levels of (oxidative) stress, and may be an example of training cellular redox signaling pathways. FOXO/daf-16 has been shown to be required for this hormetic response (18).
In this review we describe an intricate cysteine redox signaling network that could function to balance insulin signaling and ROS signaling to FOXO. An overview of this network can be found in Figure 8. As we have outlined, some cysteine redox-dependent effects within the same pathway promote FOXO activity, whereas others inhibit FOXO activity. Why would two opposing signals impinge on the same signaling cascade? If we take the opposing effects of cysteine modifications on ASK-1 and MEKK1 signaling to JNK as an example, JNK is not the only downstream effector of both ASK-1 and MEKK1. It could well be that the combination of JNK activation and other MEKK1 downstream targets leads to, for instance, cellular survival, whereas activation of JNK in combination with other ASK-1 downstream targets leads to apoptosis. FOXO can induce multiple cellular programs, including apoptosis and protection against oxidative stress. Switching between these programs might depend on binding to differential cofactors or on combinations of PTMs, which could in theory be downstream of either MEKK1 or ASK-1. In such a model, it would be the combination of signaling of JNK to FOXO and another ASK-1 or MEKK-1 signal that dictates what phenotypic outcome is mediated by FOXO's transcriptional activity. PTP1b, PTEN, and PKB are all negatively regulated by cysteine modification, but since PTEN and PTP1b oppose PKB activation, the outcome of oxidation of these proteins have reciprocal effects on the activity of FOXO. The relative reactivity of the cysteines in regulators within and upstream of FOXO, as well as the local redox state around these proteins, will determine to what extent insulin signaling and ROS signaling will influence FOXO's transcriptional response. Cysteine redox signaling networks like described in this review might prove to be key in making an optimal cellular decision in terms of organismal lifespan regulation.
Footnotes
Acknowledgment
P.L.J.d.K., T.B.D., and B.M.T.B. are being supported by grants from the Dutch Cancer Society/KWF Kankerbestrijding.