
Abstract
Drosophila melanogaster is one of the most widely used model organisms. About 77% of known human disease genes have an ortholog in Drosophila, and many of the cellular signaling pathways are common between fruit flies and mammals. For example, a key signaling pathway in the regulation of growth and metabolism, the insulin/insulin-like growth factor 1 signaling pathway, is well conserved between flies and humans. Downstream effectors of this pathway are the Forkhead box class O (FOXO) family of transcription factors, with four members in mammals and a single FOXO protein in Drosophila, dFOXO. Research in Drosophila has been critical to elucidate the molecular mechanisms by which FOXO transcription factors regulate insulin signaling. In this review, we summarize the studies leading to dFOXO identification and its characterization as a central regulator of metabolism, life span, cell cycle, growth, and stress resistance. Antioxid. Redox Signal. 14, 635–647.
Introduction
Identification and Characterization of dFOXO
The fly ortholog of mammalian FOXO transcription factors, dFOXO, was independently identified in three laboratories (50, 56, 85). dFOXO is a protein of 613 amino acids encoded by a gene containing 11 exons. One of the reports (56) identified a shorter protein (463 amino acids), perhaps derived from alternative splicing, that lacks part of the glutamine-rich activation domain characteristic of Drosophila transcription factors, and thus has different transcriptional properties. The biological relevance for the existence of at least two dFOXO isoforms is unknown.
dFOXO regulation by posttranslational mechanisms
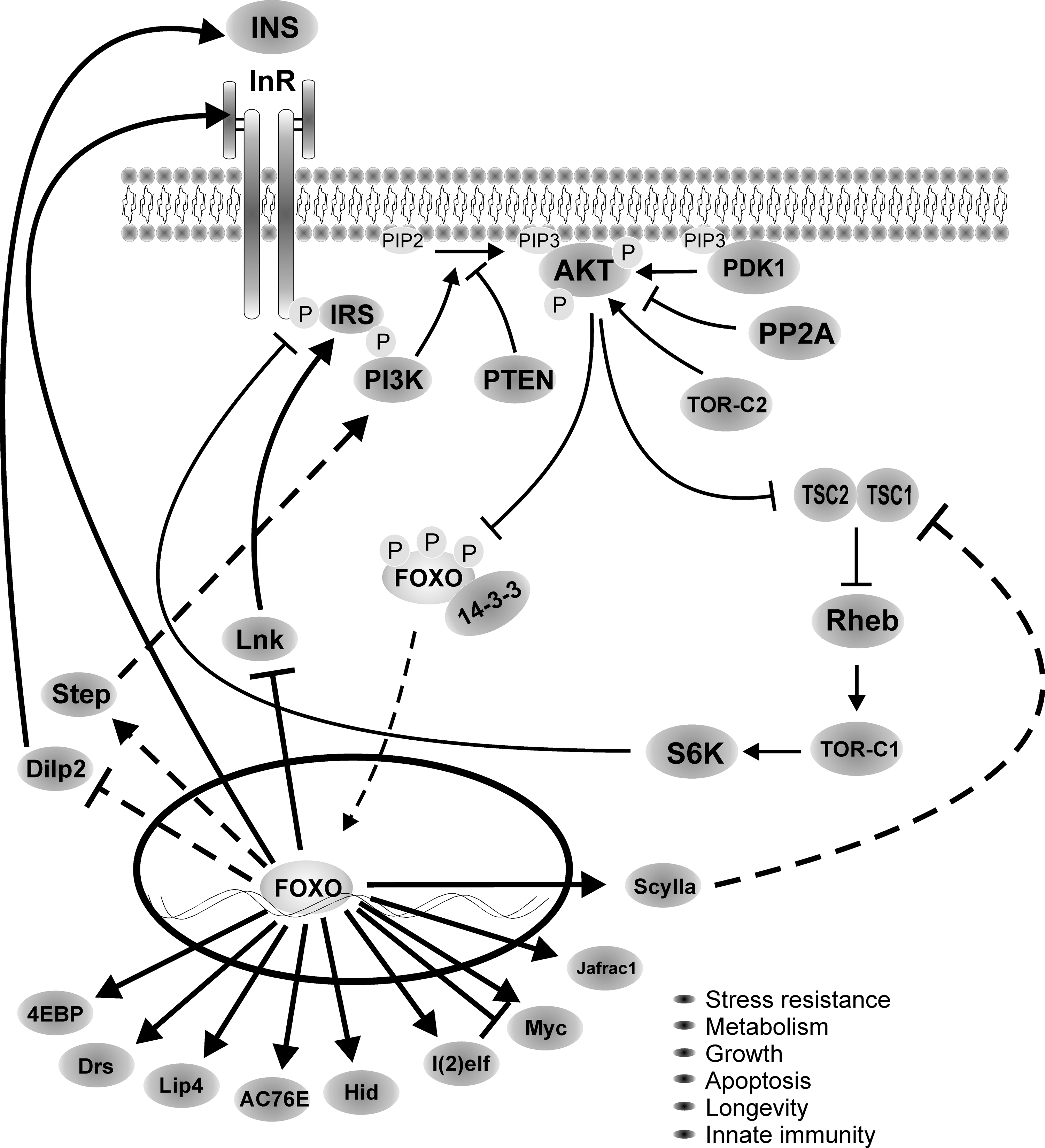
Little is known on the transcriptional regulation of dFOXO, and most of the focus has been on understanding how posttranslational modifications affect its activity (Fig. 2). At the time dFOXO was identified, it was known that mammalian FOXOs are inhibited by insulin/IGF signaling through phosphorylation by Akt (6, 12, 55, 89, 97). Akt phosphorylation in three conserved residues (T28, S193, and S258 in FOXO4) leads to nuclear exclusion and sequestration in the cytoplasm by binding to 14-3-3 proteins, which inhibits FOXO activity (12). The three amino acids modified by Akt in mammalian FOXOs are conserved in dFOXO (T44, S190, and S259); thus, an obvious step in the characterization of dFOXO function was to mutate these residues and determine whether Drosophila Akt inhibits dFOXO. These experiments showed that a mutant dFOXO lacking dAkt phosphorylation sites no longer responds to insulin inhibition and remains in the nucleus, and it is constitutively active (85), confirming that the molecular mechanism of dFOXO regulation by dAkt is conserved in flies. Similarly to its mammalian orthologs, inhibition of dFOXO by dAkt leads to binding to 14-3-3 proteins (74). As we will describe below, dAkt phosphorylation of dFOXO in Drosophila has a key role in the regulation of growth, as well as in the adaptation to nutrient availability.
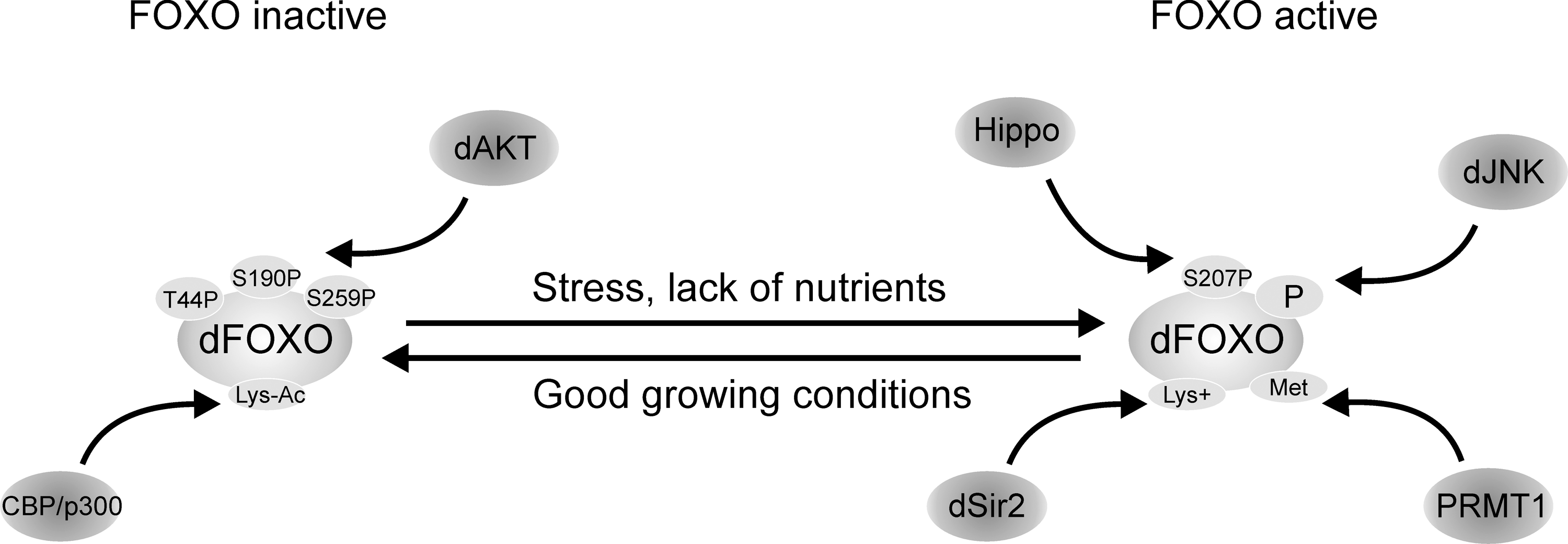
Mammalian FOXO3a is phosphorylated on Ser207 by mammalian Ste20-like kinase 1 (MST1). This modification disrupts its interaction with 14-3-3 proteins, promotes FOXO nuclear translocation, and thereby induces cell death (60). In C. elegans Cst-1, the MST1 ortholog, also phosphorylates daf-16. In flies Drosophila MST1, Hippo was found among the kinases that regulate dFOXO activity (67), and hippo mutants result in increased tissue growth and impaired apoptosis (40), both phenotypes consistent with reduced dFOXO activity (increased dFOXO activity causes growth inhibition and increased apoptosis (50, 63, 85)). Therefore, although in flies direct phosphorylation of dFOXO by Hippo has not been demonstrated, these results suggest that FOXO phosphorylation by MST1 is a general conserved mechanism in mammals, flies, and worms by which organisms respond to oxidative stress and regulate apoptosis.
In addition to Akt and MST1, several kinases phosphorylate and modulate FOXO activity. Mammalian FOXO is phosphorylated in vivo by Jun-N-terminal kinase (JNK) (27), casein kinase 1 (90), 5′ adenosine monophosphate-activated protein (36), inhibitor of nuclear factor kappa B kinase beta subunit (43), cyclic guanosine monophosphate–dependent kinase I (8), cyclin-dependent kinases 1 and 2 (44, 62, 111), and serum- and glucocorticoid-inducible kinase (13). In flies, in addition to Hippo and dAkt, dFOXO's function is modulated by JNK (108) and Drosophila target of rapamycin (dTOR) (39, 58, 64), and its activity and localization is regulated by several other kinases (67), but direct phosphorylation has not been demonstrated in either case.
A second way to modulate FOXO activity is through acetylation. Mammalian FOXOs are acetylated by CBP/p300 (29, 81, 105), and deacetylated by Sirt1 (14, 22, 29, 70, 105, 107). Acetylation occurs upon oxidative stress (105, 107) and inhibits FOXO activity. Sirt1 deacetylation increases FOXO activity, induces cell cycle arrest, and increases resistance to oxidative stress, inhibiting at the same time FOXO-induced apoptosis (14). In flies, direct deacetylation of dFOXO by dSir2 (fly ortholog of mammalian Sirt1) has not been demonstrated; however, overexpression of dSir2 by an eye-specific driver causes a rough eye phenotype due to increased apoptosis (37). This phenotype is partially rescued by a loss of function of foxo, demonstrating a genetic interaction between dSir2 and dFOXO. This result suggests that dFOXO activation by dSir2 has a role in the regulation of cell survival, at least in the context of eye development.
In addition to phosphorylation and acetylation, mammalian FOXOs are regulated by ubiquitination and methylation. FOXO ubiquitination and subsequent proteasomal degradation is induced by Akt phosophorylation as a mean to inhibit FOXO activity and enhance cell growth and survival (2, 83, 104). FOXO methylation counteracts Akt phosphorylation and it has an important role in the regulation of apoptosis induced by oxidative stress (110). To our knowledge, ubiquitination and methylation have not been explored in Drosophila.
Environmental inputs affecting dFOXO activity
FOXO transcription factors are downstream effectors of insulin signaling, but their activity is also modulated, directly or indirectly, by other external inputs, like the level of nutrients, a variety of cellular stresses (oxidative stress, UV light, etc.), and developmental signals. However, insulin/IGF remains the main signaling input converging in FOXO. Insulin receptor (InR) is highly conserved between mammals and flies, and dInR responds well to mammalian insulin (28). dFOXO activity is repressed by mammalian insulin in vitro (85). In Drosophila there are seven insulin-like peptides (Dilps) (10), and overexpression of any of them leads to enhanced growth (46). Dilp2 is the closest in similarity to human insulin/IGF, and it is the source of insulin during larval growth (10). Dilp2, 3, and 5 are expressed in small cell clusters in the central region of the brain and genetic ablation of these cells produces developmental delay, growth retardation, and elevated carbohydrate levels in larval hemolymph (46, 91). These insulin-producing cells therefore are functionally analogous to pancreatic islet β cells.
Nutrient abundance regulates dFOXO activity indirectly by affecting insulin levels and dInR signaling. For example, dFOXO activity is repressed by nutrients (sugar and amino acids), and activated by starvation (85, 86), as a consequence on the effect that nutrients have in insulin levels and dInR signaling.
Stress signals, like UV light, heat, or reactive oxygen species (ROS), activate dFOXO through the JNK signaling pathway (27, 63, 108). Importantly, activation of dFOXO by stress represses insulin signaling and thus growth, so FOXO is a central integrator that modulates cell functions as a response to different environmental cues (nutrients, stress). We describe below in detail the different dFOXO cellular functions and how this fascinating system is tightly regulated.
dFOXO Cellular Functions
Regulation of metabolic response by dFOXO
dFOXO has a central role modulating the cellular response to nutrients (Fig. 3). Flies lacking dFOXO show no developmental phenotype, and are undistinguishable from wild-type flies in normal conditions. The only apparent phenotype observed for foxo mutants grown in nutrient-rich medium is that under conditions of oxidative stress, foxo null flies are more sensitive than wild-type flies and live shorter (50). These results can be interpreted as dFOXO has a minor role in fly biology; however, genetic epistasis experiments performed with mutants in the insulin signaling pathway ruled out this idea. Flies mutant for chico, the fly InR substrate, are smaller because they have fewer and smaller cells. Loss of one foxo copy dominantly suppresses the cell number reduction in chico mutant flies without affecting cell size. The suppression is more pronounced when both copies of foxo are removed in a chico mutant background (50). The observation that growth deficiency in insulin signaling mutants can be fully rescued by foxo loss of function indicates that dFOXO is kept inactive when insulin signaling is sufficiently high. Further confirmation of the role of dFOXO in metabolism came from transcriptional analysis: dFOXO has a major role in regulating the transcriptional response to changes in nutrient conditions. Gershman and collaborators (31) studied the transcriptional response of flies upon feeding/fasting. Twenty percent of the Drosophila genes monitored (3519 genes) are regulated in response to changes in nutrient conditions, and approximately one-third of those genes (995 genes, 28%) are regulated by dFOXO. Upon feeding, transcript changes reflected rapid downregulation of the nutrient-sensing insulin and target of rapamycin pathways, shifting of fuel metabolism from lipid to glucose oxidation, and increased purine synthesis, tricarboxylic acid cycle-biosynthetic functions, and mitochondria biogenesis. In a second study, Teleman and collaborators (100) fasted larvae from wild-type and foxo null mutant flies. They collected adipose and muscle, and studied transcriptional changes. The vast majority of the genes differentially expressed in these tissues depended on the presence of dFOXO, since these changes were not observed upon fasting in foxo null flies. Gene families affected upon fasting are amino acid, lipid, carbohydrate, and energy metabolism. These results demonstrate that dFOXO has a major role in regulating the transcriptional response to nutrient availability. Why did the initial experiments with foxo null flies (50) show no apparent metabolic phenotype? This likely reflects the fact that these mutants were reared under abundant nutrient conditions, supporting the idea that dFOXO is required for successful adaptation in periods of reduced nutrient availability (see below).
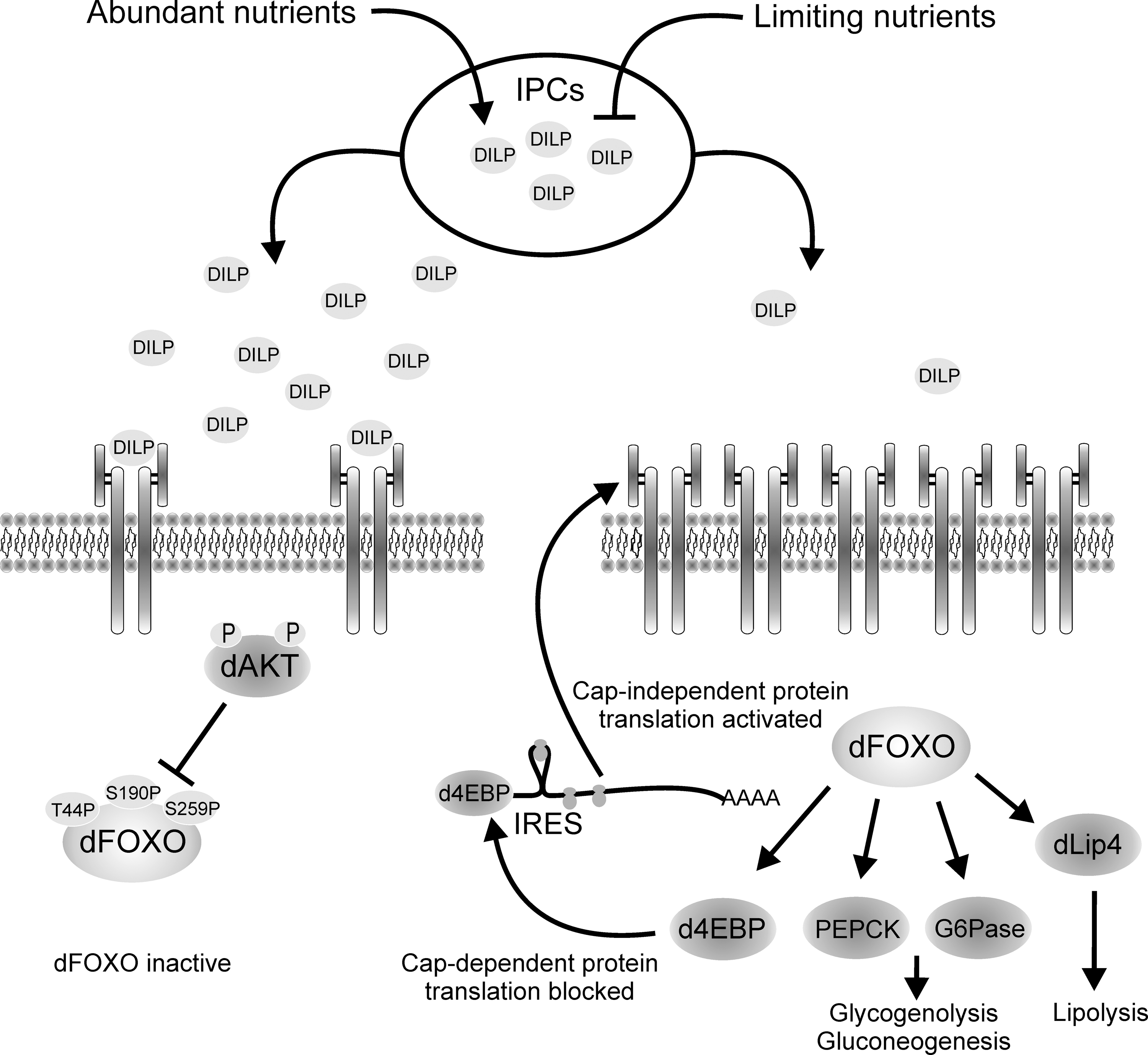
The molecular mechanism by which insulin signaling regulates the transcriptional response to nutrients was unraveled by studying dFOXO function in Drosophila. The insulin/IGF receptor pathway has evolved to deal with changes in fasting and feeding conditions, and to adjust cell growth accordingly. In situations of nutrient abundance, insulin/IGF receptor signaling is turned on by direct binding of insulin/IGF to its receptor. This response leads to cell growth and division, which are coordinately promoted in the different cell types within the organism. By contrast, when nutrients are scarce, insulin/IGF signaling is turned off and growth is inhibited. Research first in Drosophila (85), and subsequently in mammals (86), demonstrated that the InR itself is directly activated by dFOXO/FOXO1. This provides a feedback mechanism to regulate rapidly and sensitively the organism response to nutrients (Fig. 3). When nutrient conditions are limiting, insulin/IGF levels are low, insulin/IGF signaling is turned off, and dFOXO/FOXO1 is dephosphorylated, nuclear, and active. In these conditions dFOXO/FOXO1 targets are activated, among them InR. This accumulates receptor molecules in the cell surface, which makes cells more sensitive to small variations in insulin levels. When nutrient availability is high, insulin/IGF signaling is activated and FOXO is inhibited by phosphorylation through Akt and transcriptionally inactive. In flies, Dilps are expressed in response to nutrient load and act in a similar way to mammalian insulin/IGF (46, 91). The phenotype of flies lacking the Dilp-producing cells includes certain features of diabetes mellitus (91).
One-way growth and division synchronized to nutrient availability is through the regulation of protein translation. In situations of nutrient deprivation, the survival response is initiated by dramatically downregulating metabolism to save on energy. This is achieved by repressing protein translation, and only proteins with essential function are translated. Therefore, cells developed mechanisms to integrate response to nutrients and protein translation, and once again dFOXO plays a central role in this process. In addition to modulating the sensitivity of cells to nutrient changes, dFOXO also regulates growth through its direct effect in the translational inhibitor Drosophila translational initiator eI4E binding protein (d4EBP), a direct dFOXO transcriptional target (50, 85). d4EBP binds and inhibits the translational initiation factor eIF-4E, a factor required for productive protein translation that depends on the presence of an m7Gppp cap structure in the 5′ end of the mRNA. Upon high nutrient availability, dFOXO is turned off, and translation and cell growth are activated. Conversely, upon lack of nutrients, dFOXO is active and activates expression of d4EBP, which inhibits translation and growth, saving valuable energy that it is redirected to survival functions. This way the organism can adapt growth and metabolism to the level of nutrients in the environment (85).
Upon dFOXO activation, d4EBP is expressed and general translation is inhibited, yet some mRNAs continue to be translated, for example, the mRNA for dInR. How do cells achieve selective translation of certain mRNAs? The answer came once again from studying the Drosophila system. dInR has three promoters that produce three mRNAs with long 5′ untranslated regions with multiple AUG codons upstream of the authentic translation start site (16). All three dInR mRNAs have functional internal ribosome entry sites (IRES), small hairpins in the mRNA structure that allow ribosome binding, and initiation of translation in a 5′ cap-independent manner (65). The 5′ untranslated region of foxo mRNA also has a functional IRES, suggesting that dFOXO translation is regulated by a similar system (Eugenia Villa-Cuesta and Marc Tatar, personal communication). Turning off cap-dependent translation by dFOXO-dependent d4EBP activation makes ribosomes available for cap-independent translation, which leads to enhanced dInR translation and ultimately enhances insulin sensitivity (65). Therefore, when faced with limited nutrient availability, flies exploit a sophisticated system to optimize the production of dInR. The transcript for human InR remains associated with polysomes when cap-dependent translation is inhibited, suggesting that amplification of insulin signaling by IRES-dependent translation is also conserved in mammals (47). This mechanism provides a fascinating example of coordination between transcription and translation in eukaryotes, evolved to respond in a fast and sensitive way to changing nutrient conditions.
FOXO also acts as a key modulator of energy metabolism by integrating insulin responses to glucose and lipid homeostasis. FOXO1 activation increases glycogenolysis and gluconeogenesis by activating transcription of glucose-6-phosphatase and phosphoenolpyruvate carboxykinase mRNAs (38, 93). In addition, work in Drosophila showed that dFOXO regulates expression of lipase 4 (dlip4), a Drosophila ortholog of human acid lipases (106). Thus, when nutrients are limited, insulin levels drop, and FOXO increases glycogenolysis, gluconeogenesis, and lipolysis by activating glucose-6-phosphatase, phosphoenolpyruvate carboxykinase, and dLip4, which would release glucose and fatty acids as a source of energy. When nutrients are abundant, insulin is secreted, FOXO is repressed, and glucose and fatty acids are deposited as energy stores. Insulin resistance is a major feature of pathological states such as obesity and diabetes. A negative consequence of insulin resistance is enhanced lipolysis and hepatic glucose production, which causes excessive release of free fatty acids and glucose, and deregulates glucose and fatty acid homeostasis. Thus, accurate FOXO regulation of these metabolic processes is critical to keep a healthy energy homeostasis.
FOXO function and life span
The lifespan of an organism is determined by the ability to maintain cellular and physiological processes such as genetic stability, stress resistance, and metabolic control sustaining proper tissue function and decreasing the probability of death. Caloric restriction and changes in insulin/IGF signaling are well-known interventions affecting aging in various species (11, 19). Drosophila has turned out to be a valuable model to study these questions. Initial observations in C. elegans demonstrated an association between insulin/IGF signaling, longevity, and DAF16 (worm FOXO) activity (61, 76). In flies, dFOXO regulation of life span was demonstrated by overexpression of dFOXO in the adult fat body (33, 45). The Drosophila fat body is the equivalent of mammalian liver and white adipose tissue, the latter being previously shown in mouse to be important for life span extension mediated by insulin/IGF signaling (7). Hwangbo and coworkers employed a driver solely active in the head fat body and observed an average increase of 15.5% and 19.4% in male and female lifespan, respectively. Giannakou and coworkers used a different driver specifically overexpressing dFOXO in abdomen and head fat body and reported an increase of 20%–50% in female lifespan only. Hwangbo et al. observed a concomitant reduction in Drosophila dilp2 expression, suggesting that dFOXO might affect lifespan indirectly by decreasing the overall insulin/IGF signaling of the organism. Further studies demonstrated that dFOXO overexpression reduces the mortality rate equally at all ages but does not affect the age-enhanced increase in mortality (32). These results suggest that overexpression of dFOXO in the fat body does not protect from the cumulative damage associated to aging but rather decreases the probability of death equally at all ages, perhaps by protecting tissue function. This is further supported by the finding that dFOXO overexpression in the heart abolishes an age-associated increase in cardiac failure rate with no effect in life span (109).
One plausible explanation for the above observations is that dFOXO overexpression in the fly mimics caloric restriction, which has been shown to prolong lifespan by affecting the baseline mortality but not the increase in mortality with age (84). A generally considered view is that lifespan extension by caloric restriction is mediated by signals from the nutrient sensing TOR and insulin/IGF pathways. However, the experimental evidence supporting this hypothesis is still inadequate and partly conflicting [for discussion see ref. (82)]. The question whether dFOXO, as a target of insulin/IGF signaling, is involved in lifespan extension mediated by caloric restriction was directly addressed by Giannakou et al. (32) and Min et al. (69), who challenged foxo null flies on varying food dilutions. The results indicate that foxo null flies in general are short lived but respond to caloric restriction similarly to the control flies, by increasing their median lifespan at intermediate food concentrations. Similar experiments with flies overexpressing dFOXO in their fat body showed that median life span was increased, but only when the flies were not dietary restricted. At the range of food dilutions where caloric restriction causes lifespan extension, or upon complete starvation, overexpressed dFOXO gives no survival advantage. These results imply that caloric restriction and dFOXO mediate their lifespan effects through separate signaling routes (32, 69). However, the underlying molecular mechanisms might overlap. This perspective is consistent with the findings in C. elegans, where another forkhead box transcription factor, FOXA, has been shown to mediate lifespan extension induced by caloric restriction. FOXA and FOXO are known to have overlapping consensus DNA binding sites (80). The redundancy between dFOXO and other forkhead box transcription factors in fly is yet to be explored.
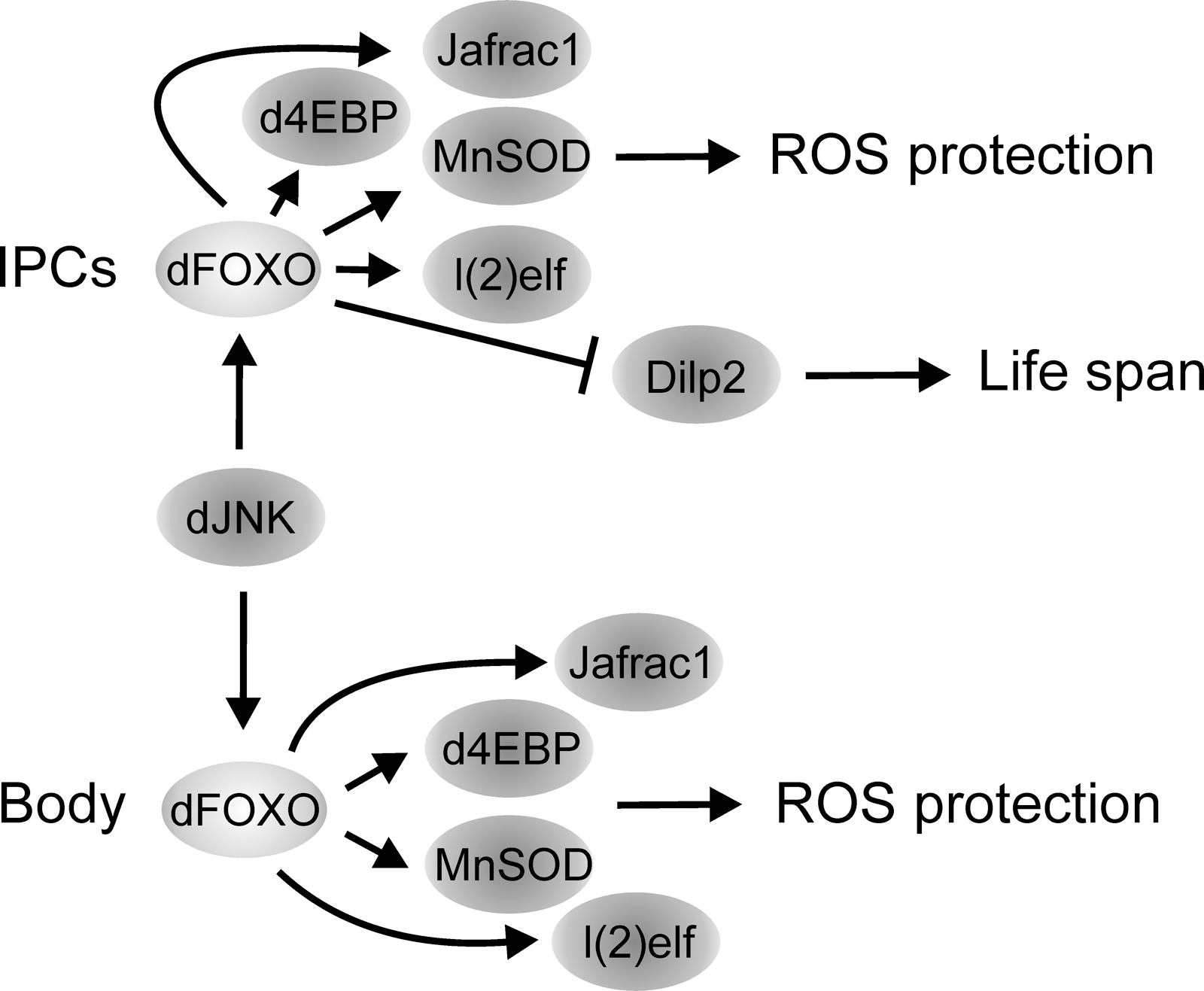
The role of dFOXO in regulating lifespan is not restricted to its function in the fat body. In fact, dFOXO also mediates lifespan extension through activation by JNK in the insulin-producing cells (108). Elevated JNK signaling in these secretory cells extends lifespan by reducing systemic insulin signaling through dFOXO-dependent repression of dilp2 expression (45, 52). Accordingly, upon adverse environmental conditions, such as elevated oxidative stress, the JNK-dFOXO signaling results in reduced Dilp2 production and protects the organism by reallocating resources from growth and metabolism to somatic maintenance. The mechanism might be conserved in other species, since in C. elegans several insulin-like peptides are under transcriptional control of DAF16 (71). In addition, JNK-dFOXO signaling holds also cell-autonomous function in regulating lifespan: JNK signaling regulates, through dFOXO, several genes involved in stress response and lifespan regulation. These include d4ebp, l(2)efl, and jafrac1 (59, 108). Jafrac1 (a Drosophila Peroxiredoxin II that acts as a neuronal protective agent against oxidative stress) and d4EBP mediate lifespan extension through mitochondrial function, a cellular organ intimately linked to longevity and aging. Overexpression of Jafrac1 in neurons extends lifespan and restores ROS-induced loss of ATP in mitochondria (59). Indeed, neurons are a critical tissue, along with fat body, in regulating longevity (11). In addition, d4EBP has an important role in mediating changes in mitochondrial activity and lifespan extension linked to caloric restriction (114). Finally, the ubiquitous overexpression of another dFOXO target, the antioxidant enzyme manganese superoxide dismutase, was found to significantly increase the adult lifespan (96).
On the basis of evidence presented above, it is clear that dFOXO is an important factor in regulating organism survival. Two separate modes of action are suggested: (a) dFOXO is involved in the systemic regulation of insulin/IGF signaling by reducing the level of circulating Dilp2, resulting in global reduction of insulin/IGF signaling, a mechanism shown to increase lifespan in fly (18, 98), worm (53) and possibly in mouse (7), and (b) dFOXO extends lifespan cell autonomously by inducing several genes involved in somatic maintenance and stress protection (Fig. 4). It is tempting to speculate that enhanced dFOXO activity in a tissue-specific manner could prolong the healthy function of such tissue, perhaps by sustaining mitochondrial function. The conservation of this process among species is unclear, since mice overexpressing FOXO1 in their skeletal muscle were not long lived (17), and it may also vary among tissues since inhibiting insulin signaling in the Drosophila heart protects it from age-dependent changes, but does not extend lifespan (109). More experimental evidence investigating the role of FOXO in other tissues, such as the liver, adipose tissue, and neurons is needed to resolve this question.
dFOXO function in tissue growth
FOXO has a well-defined role as a mediator of nutrient-regulated growth, and much of our knowledge of this process is derived from studies in Drosophila (Fig. 5) (24, 30, 39, 45, 50, 85, 100). Overexpression of a constitutively nuclear dFOXO in cell culture results in proliferation arrest (85). Correspondingly, tissue-specific overexpression of dFOXO during development reduces organ size by loss of cell number (50, 85) and/or an increase in apoptosis (63). This phenotype is exacerbated by genetically inhibiting insulin/IGF signaling or by lowering nutrient levels, demonstrating that these effectors act in the same pathway (50). Further evidence was provided by the finding that the growth phenotype of impaired insulin/IGF signaling, that is, small tissue size, was suppressed in foxo null flies. However, foxo null flies are normal in size, suggesting that dFOXO is dispensable under adequate growing conditions (50). These findings further support the role of dFOXO in regulation of response to nutrients discussed in the previous section, and link nutrient availability, dFOXO activity, and growth regulation by inhibiting cell division. The mechanism by which dFOXO exerts its function on cell division has been demonstrated in more detail in mammalian cells where FOXO induces expression of cyclin-dependent kinase inhibitors p27kip1 and p21cip1 and the downregulation of Cyclin D resulting in G1 arrest (68, 92, 94). Another situation when FOXO regulates growth is in conditions of hyperactive TOR signaling. For example, dFOXO restricts overgrowth of tsc1 (tuberous sclerosis complex 1, a TOR regulator) null tissue by regulating scylla expression and possibly other genes that inhibit insulin and TOR pathway activity (39, 50). Scylla is a negative regulator of growth acting through inhibition of TOR signaling (87). The mechanism is conserved since the mammalian Scylla ortholog, Redd1, is also upregulated by elevated TOR signaling through FOXO (39). In addition, dFOXO induces expression of Sestrin, another inhibitor of TOR signaling (58). Sestrin acts as a feedback inhibitor of TOR, thereby preventing pathologies caused by chronic TOR activation (58). Taken together, these findings demonstrate that FOXO restricts tissue growth under adverse nutritional conditions. On the other hand, at instances of elevated or prolonged TOR signaling, FOXO is mediating a negative feedback loop on insulin signaling, which protects organism from hypertrophy. Both mechanisms ensure proper growth regulation and maintenance of tissue homeostasis under varying nutrient conditions.
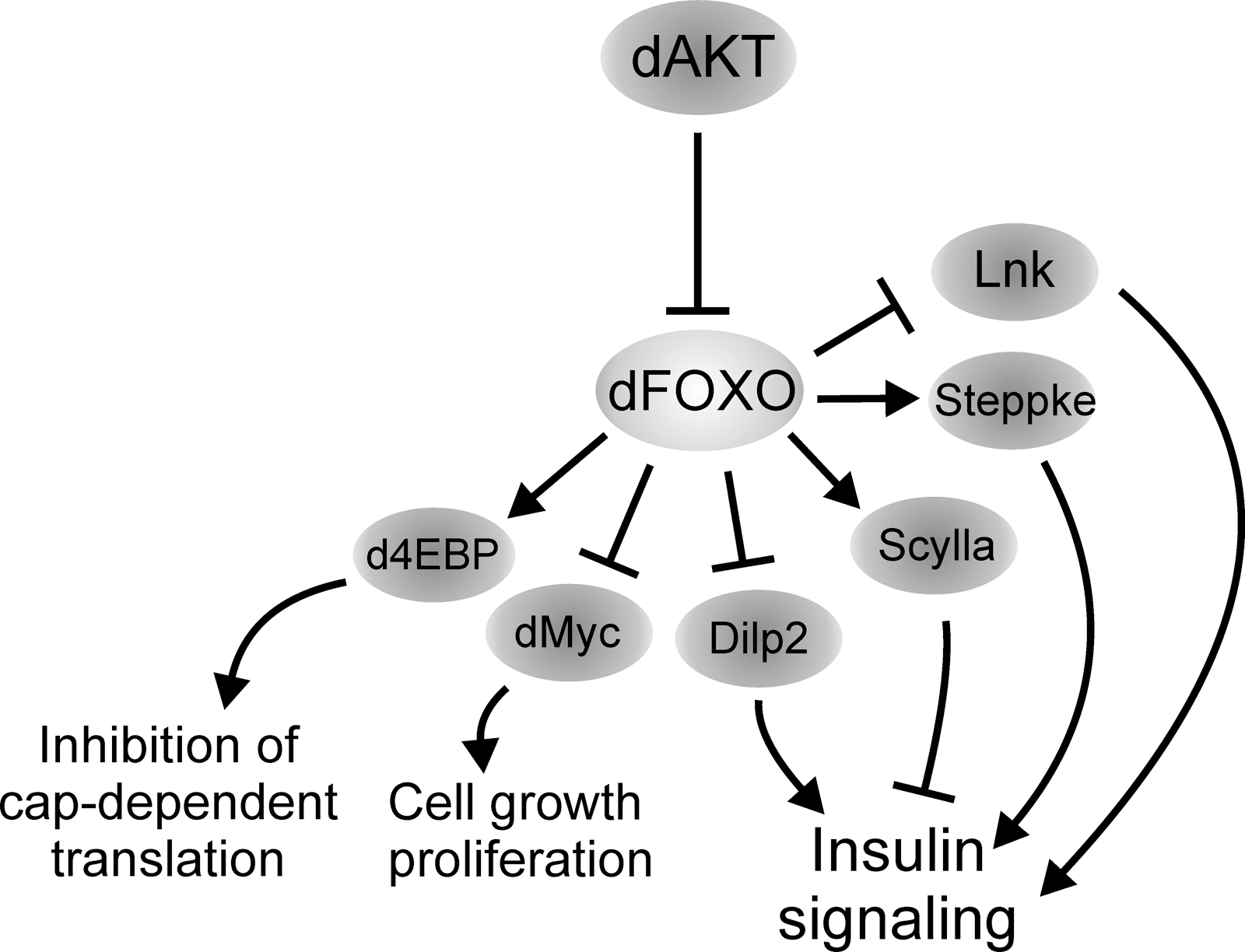
The size of an organism and tissues within are determined by changes in cell number and cell size, and these parameters are regulated through separate mechanisms (48). The involvement of dFOXO in cell size regulation is not as clear as in cell division. Gain-of-function studies in Drosophila tissue provided conflicting results possibly due to differences in timing and strength of overexpression, and possibly to the fact that, as discussed above, the overexpressed dFOXO proteins were not identical (50, 56). Impaired insulin/IGF signaling during development affects both cell size and number, but a foxo null background suppresses only the latter (50). However, the results from microarray profiling experiments of Drosophila tissue suggest an important role for dFOXO in cell growth during nutritional deprivation (100). When larvae are reared at low food concentration, development is delayed and the size of the emerging adults is reduced. Teleman and coworkers investigated the starvation-induced gene expression program in the larval fat body and in the muscle tissue, and observed that it was highly dependent on dFOXO (100). Among genes activated by dFOXO in these conditions they found components of the signal recognition particle and ribosomal proteins. Hence, dFOXO seems to contribute to the nutrient-regulated control of the cellular biosynthesis of macromolecules. Further supporting the role of dFOXO in the regulation of cell size, a key translational regulator, d4EBP, and dMyc, a transcription factor with a known role in ribosomal biogenesis [reviewed by ref. (21)], are regulated by dFOXO (50, 85, 100). The regulation of dMyc by dFOXO upon starvation takes place in the fat body and in the muscle tissue, yet in opposite fashion. In the fat body dFOXO activates dMyc expression, which allows dMyc to sustain the fat tissue metabolically active ensuring sufficient mobilization of energy reserves for survival and growth. Inability to activate this mechanism results in reduced growth in conditions of low food supply. In muscles, however, dFOXO inhibits dMyc expression and growth (24, 100). Elevated dFOXO activity in muscle tissue has also been found to significantly reduce larval feeding, yielding systemically reduced developmental growth (24). The mechanism behind this regulation is unknown.
In addition to protein translation and cell division, dFOXO may affect organism growth by regulating transcription of genes that control upstream insulin signaling, like dInR, Steppke, and Lnk (30, 85, 95). Flies mutant for inr, steppke, and lnk are small (30, 95, 98), due to reductions in cell count and size. Therefore, dFOXO, through a tight regulation of insulin signaling, cell division, and protein translation, plays a critical role in the regulation of organ size.
The studies discussed above demonstrate the role of dFOXO as a cell-autonomous regulator of tissue growth. However, dFOXO is also involved in organism-wide systemic growth regulation. As mentioned before, activated JNK signaling results in reduced brain dilp2 mRNA levels in a dFOXO-dependent fashion and a concomitant reduction in body size (108). Given the role of JNK as a stress-induced pathway, this suggests that upon elevated stress, such as increased ROS, JNK activates dFOXO in the insulin-producing cells, leading to downregulation of dilp2 expression and correspondingly to inhibition of insulin/IGF signaling in the peripheral tissues. This system would slow down growth rate and allocate resources to cellular maintenance and stress protection. How dFOXO activity is balanced and how its activity modulates dilp2 mRNA levels in the insulin-producing cells are not yet completely clear. However, the data suggest that nutrient availability and stress mutually modulate dFOXO activity in the insulin-producing cells, which in turn regulates growth in the peripheral tissues.
The final size of a Drosophila adult is determined by the rate and length of the growth period at larval stages. The rate of growth is determined by tissue intrinsic cues such as locally produced morphogens and extrinsic cues such as nutrition and temperature (48). The latter are translated to growth regulation in the peripheral tissues mainly through the action of Dilps secreted from the insulin-producing cells (46, 91) and the actions of insulin and TOR signaling, which serve as mediators between nutrition and growth [reviewed by ref. (41)]. The length of the larval growing period is regulated by ecdysone, a steroid hormone produced by the prothoracic gland, which initiates the larval molting cycle and terminates growth either temporarily (larval molt) or permanently (pupae formation). The secretion of ecdysone is regulated by the prothoracicotropic hormone, which itself is regulated by the larval volume and, consequently, by the concentration of the juvenile hormone (JH) [reviewed by ref. (75)]. The combined action of these hormones balances growth and molting in a developing larva [reviewed by ref. (23)]. There is emerging evidence that dFOXO plays a role in adjusting the functions of this endocrine system and hence the timing and rate of developmental growth. Disruption of insulin/IGF signaling in the corpora allatum, an endocrine tissue responsible for JH production, results into a small fly phenotype (5). Overexpression of a dFOXO target gene, adenylate cyclase 76e, in the corpora allatum recapitulates this phenotype (66). In addition, flies carrying a hypomorphic allele of the inr and a null mutation of the InR substrate chico have lowered JH titer (98, 103). These data imply that dFOXO, as a target of the insulin/IGF signaling, is participating in the JH biosynthesis or its release to the haemolymph, which in turn would modulate the length of the larval growing period. However, direct evidence is still missing. On the other hand, modulation of the insulin/IGF signaling levels in the prothoracic gland by overexpression of a dominant negative form of PI3K results in decreased ecdysone titer and an increased growth rate (20). Although not directly shown, it is possible that dFOXO has a role in the ecdysone production or its release from prothoracic gland. Ecdysone was observed to exert its growth regulation through modulation of the insulin/IGF signaling and dFOXO activity in the fat body (20). These data imply that dFOXO has an important role in mediating the endocrine signaling responsible for the rate and timing of larval growth.
dFOXO function in stress resistance
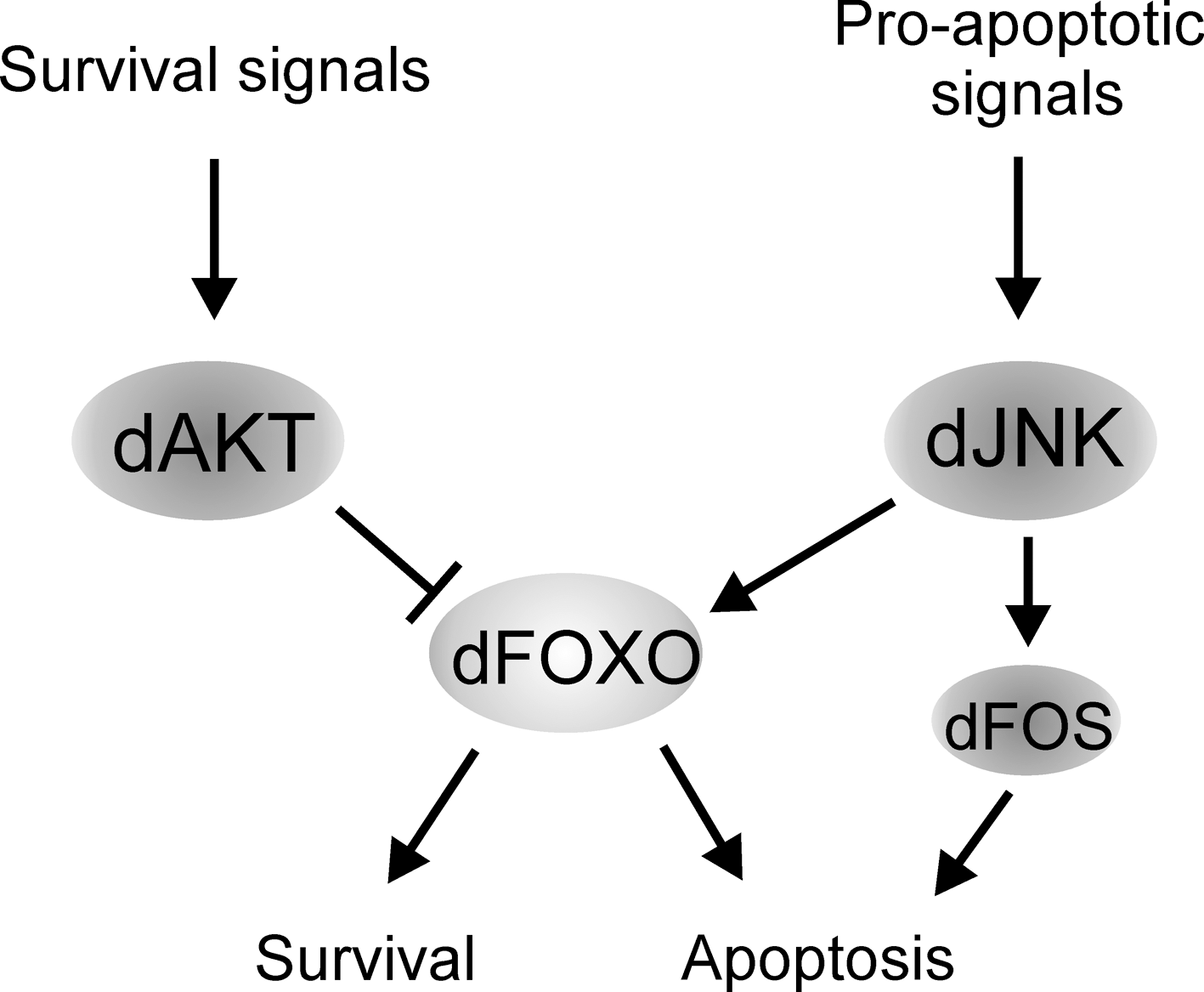
One of the most prominent roles of FOXO proteins is their function in cellular and organism-wide protection from stress (Fig. 6). These include oxidative stress (59, 77), genotoxic stress (44, 63), hypoxia (3), inflammation (4), and nutritional deprivation (35, 57, 86). For example, in mammalian cells, elevated oxidative stress induces FOXO nuclear localization through Sirt1 and JNK-dependent deacetylation and phosphorylation, respectively. Once in the nucleus, FOXO activates a pattern of gene expression promoting stress resistance and survival (14, 27). However, depending on the type of the cell, quality and strength of the stress stimuli, FOXO activity can shift the cellular response from survival to apoptosis. The decision between these alternatives depends on the balance of the activation of pro-apoptotic, for example, bim and fas ligand (12, 25, 34), and pro-survival, for example, manganese superoxide dismutase, growth arrest and DNA damage 45, and catalase (14, 54, 73, 102) genes. It is not completely understood how the distinction between activation of the different gene expression programs is achieved. However, it is likely that it involves specificity in FOXO posttranslational modifications, specific FOXO cofactor binding partners, and perhaps interaction with other transcription factors. Variations in these parameters can lead to deviations in the strength and duration of FOXO activity and, correspondingly, to qualitative and quantitative differences in target gene activation. Studies of how Drosophila tissue respond to genotoxic stress induced by UV light have provided an example of this process. Genotoxic stress in the developing retina induces JNK-mediated apoptosis, which is achieved through activation of the dFOXO and Fos transcription factors (63). These proteins combinatorially activate expression of the pro-apoptotic gene head involution defective (hid). It was found that the severity of the tissue loss through JNK-dFOXO/Fos-mediated apoptosis was enhanced by reducing the level of signaling through the EGF and InRs. This finding suggests that the presence of survival signals modulate the cellular response to dFOXO/Fos activation. As a result, survival and stress resistance is promoted. Hence, dFOXO is working as a signal integrator, in concert with other transcription factors, balancing the decision between survival and apoptosis in an appropriate manner (Fig. 7). In this setting dFOXO is not acting alone but in concert with another transcription factor, dFos. Similar cooperational functions might exist in other FOXO-regulated processes. Indeed, how FOXO is networked with other transcription factors and transcriptional cofactors remains largely unknown.
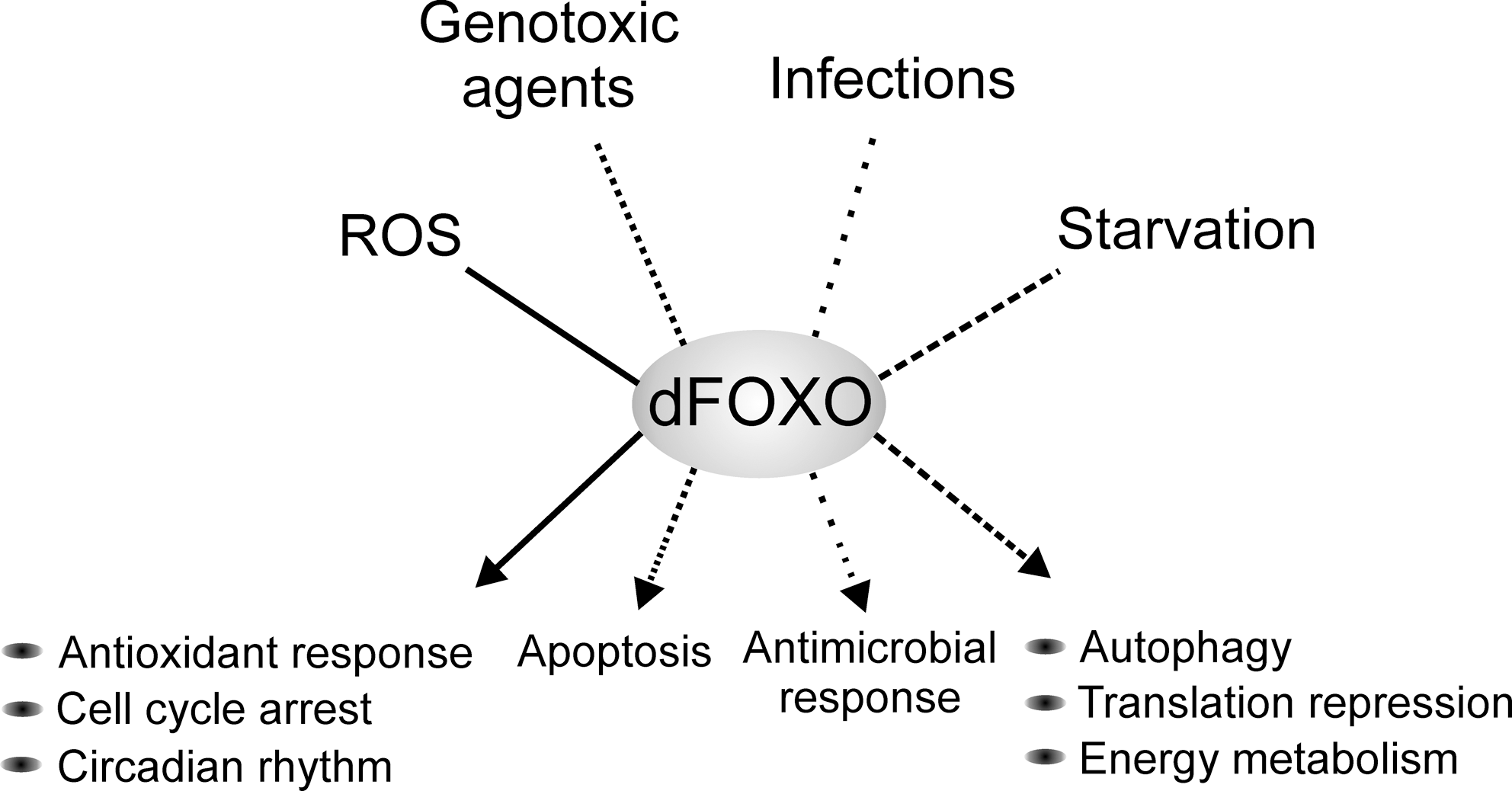
Oxidative stress is the result of accumulation of ROS, which are highly reactive byproducts of cellular respiration. The main source of intracellular ROS is the mitochondrial respiratory chain and an increase in ROS concentration poses a threat to the integrity of cellular processes. Consistent with its known role as a protector of oxidative stress in mammalian cells, dFOXO induces expression of genes such as l(2)efl, hsp68, fer1HCH, sestrin, and jafrac1, which have known antioxidant or chaperone functions (58, 59, 108). Accordingly, foxo null flies are sensitized to oxidative stress and die rapidly under paraquat or H2O2 treatment (50). Ubiquitous overexpression of another dFOXO target, d4EBP, fully rescues the oxidative stress sensitivity of the foxo null flies (101). This finding suggests that repression of cap-dependent translation is crucial for survival under oxidative stress. Taken together, these results propose that in Drosophila increased oxidative stress results in an antioxidant response orchestrated by dFOXO in parallel with a global reduction of protein synthesis, reallocating resources from cell growth to protection and cellular maintenance.
The inability to properly sense and respond to physiological ROS levels leads to defects affecting processes like cell division, differentiation, and circadian rhythm. Given that dFOXO has a central role in the modulation of the cellular ROS levels, it is not surprising that dFOXO has been linked to the effect of ROS in these processes. foxo null flies are incapable of adjusting their circadian behavioral rhythms under low doses of paraquat (113). A simultaneous decrease in the central molecular clock gene expression period was observed in lateral neurons, the tissue responsible for the systemic circadian rhythm regulation. dFOXO acts in the lateral neurons from the fat body in a non-cell-autonomous manner, although the underlying mechanism is unknown. In addition, foxo null flies were also found to display an enhanced age-dependent decline in their ability to maintain the circadian behavioral rhythms. However, the causal relationship between aging and the disruption of the circadian rhythms is not clear. It would be interesting to learn whether the lifespan-promoting function of dFOXO is partly attributed to its role in maintaining the circadian rhythm.
Oxidative stress is also known to modulate cell cycle progression and differentiation. In Drosophila, knockdown of a respiratory chain complex I component, Pdsw, in the developing eye, leads to an increased concentration of cellular ROS, upregulation of the p27kip1 ortholog Dacapo, and a subsequent G1 arrest (79). This cell cycle block is mediated by ROS-induced activation of the JNK-dFOXO signaling. Although the in vivo significance of the finding is not known, it is possibly a mechanism ensuring sufficient time for DNA repair before the cell can proceed to the next cell cycle. A similar mechanism is employed in the differentiation of Drosophila hematopoietic progenitors. An increase in ROS by either modulation of the antioxidant enzyme levels or by disrupting the respiratory complex I function in the hematopoietic stem cell-like compartment triggers premature blood cell differentiation. The differentiation is suppressed by inhibition of the JNK signaling and enhanced by overexpression of dFOXO (78). Although the exact downstream targets responsible for the blood cell differentiation are unknown, these findings demonstrate that the role of dFOXO is not merely protective. Physiological concentrations of ROS are important regulators of cellular processes and dFOXO is an important mediator in the cascade triggering the downstream events.
Drosophila has been used extensively to study organismal response to nutritional deprivation. Flies starved for nutrients undergo rapid changes in cell growth, metabolism, and locomotor activity. The breakdown of energy reserves (fat and carbohydrates) is needed to maintain cellular homeostasis and survival, so the resistance to starvation is strongly correlated with the amount of reserves (26). However, adult dFOXO null flies exhibit no difference in fat reserves and are not less resistant to complete starvation as the wild-type flies (50). These results are in sharp contrast to the findings that d4EBP, a direct target of dFOXO, plays a key role in adjusting the energy metabolism and/or consumption under starvation. The inability to hinder cap-dependent translational activity, due to lack of d4EBP, leads to rapid loss of energy reserves and, consequently, early lethality (99). Hence, d4EBP has been considered to serve as a metabolic brake under starvation. In addition, starvation-responsive gene expression is highly dependent on dFOXO (31) and the inability to activate dMyc expression by dFOXO upon starvation results in reduced starvation resistance (100). Hence, dFOXO has a critical role in the regulation of organism responses to starvation. The fact that foxo null flies are equally resistant to starvation as wild-type flies remains enigmatic and it suggests the existence of unknown factors that contribute to this phenotype. Another function by which dFOXO regulates stress response to starvation is through autophagy, an important survival mechanism under nutritional deprivation. Autophagy is activated by nitrogen or amino acid starvation, and promotes the survival of the cells by recycling dispensable cellular constituents for reuse in synthetic processes. Fasting-induced autophagy in the fat body is abolished in foxo null tissue, and overexpression of dFOXO induces autophagy (49). Thus, dFOXO-dependent increase in autophagy is an additional way to redirect metabolic reserves upon starvation.
Another source of stress is infections caused by external organisms. In Drosophila the innate immune system protects against invading bacteria and fungi by increasing expression of antimicrobial peptides, like drosomycin, diptericin, and metchnikowin. Under pathogen stimulation these peptides are activated by toll-like receptor and immune deficiency signaling. Recently, Becker and coworkers showed that an alternative signaling pathway to activate expression of these peptides is through dFOXO (4). They showed that under normal physiological conditions, and not as response to pathogens, dFOXO activates antimicrobial peptides in response to the oscillating energy status of cells and tissues. The authors suggest that this mechanism would modulate the defense reaction when animals are suffering from energy shortage or stress. This is another elegant example of the key role of dFOXO in the regulation of stress response.
Concluding Remarks
Multiple questions still remain open in the field of Drosophila FOXO: Why there are such large differences among life span experiments? What is the effect of gender in life span? What promoters are bound by this transcription factor? What is the functional role of the two (and possibly more) dFOXO variants? Is dFOXO directly phosophorylated by dTOR or JNK, is it directly deacetylated by dSir2? Does dFOXO directly regulate Dilp2 (and perhaps other Dilps) expression? How is dFOXO regulated transcriptionally? In vivo chromatin immunoprecipitation experiments with dFOXO antibodies, life span experiments in parallel with multiple tissue-specific drivers, coimmunoprecipitation experiments, etc., could address some of these questions.
Research in Drosophila has been critical to explain several of the molecular mechanisms by which FOXO transcription factors regulate insulin signaling. After the identification of dFOXO in 2003, several labs got interested in this transcription factor and subsequent findings explained the central role of dFOXO in the regulation of insulin signaling. dFOXO remains an attractive topic due to its central role affecting processes as diverse as metabolism, life span, stress resistance, cell cycle, and growth. FOXO transcription factors are a good example of how exquisitely cells evolved mechanisms to deal with changes in environmental cues, and to respond rapidly and efficiently to these changes. What surprises will dFOXO deliver in the future? We will wait impatiently for new developments in the field.
Footnotes
Acknowledgments
The authors thank Tobias Dansen for his invitation to write this article. We especially thank Kieran Harvey, Ville Hietakangas, Henri Jasper, Linda Partridge, Marc Tatar, Ingo Zinke, and the two anonymous reviewers for critically reading this article and for their invaluable suggestions. The authors thank Johanna Seppäläinen for style improvements and proofreading.