
Abstract
Fluid shear stress plays a critical role in the regulation of vascular biology and its pathology, such as atherosclerosis, via modulation of redox balance. Both pro-atherogenic (either oscillatory or turbulent, nonunidirectional) shear stress and anti-atherogenic (either steady or pulsatile, unidirectional laminar) shear stress stimulate production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) that are involved in signal transduction of gene expression. Nonunidirectional shear stress induces pro-atherogenic genes encoding adhesion molecules and chemokines in a manner dependent on production of both superoxide and nitric oxide. Steady or pulsatile laminar shear stress induces expression of genes encoding cytoprotective enzymes for glutathione biosynthesis and detoxification, which are regulated by the transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf2). We show that pulsatile laminar shear stress (PLSS)-induced expression of adhesion molecules and chemokines was enhanced in human umbilical vein endothelial cells (HUVEC) treated with Nrf2 siRNA and arterial endothelial cells isolated from Nrf2 knockout mice. Hence, we propose the hypothesis that PLSS maintains the endothelium in an anti-atherogenic state via intracellular antioxidant levels increased as a result of Nrf2 activation, thereby preventing excess ROS/RNS production required for pro-atherogenic gene expression. Antioxid. Redox Signal. 15, 1415–1426.
Introduction
To investigate the response of endothelial cells upon exposure to shear stress, many studies have been performed under a variety of experimental conditions with various flow-exposing apparatuses (86a). These experiments show that both oscillatory and either steady or pulsatile laminar shear stress evoke generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in vascular cells (33, 39, 55, 76). The mechanisms by which shear stress induces either pro-atherogenic or anti-atherogenic responses in endothelial cells have been the subject of intense studies over the past 2 decades. In this article, we will present the ROS/RNS-regulated mechanisms underlying the anti-atherogenic response of endothelial cells to pulsatile laminar shear stress (PLSS).
Materials and Methods
The details of materials and methods are shown in the Supplementary Data (available online at
ROS and/or RNS Production by Fluid Shear Stress
Fluid shear stress caused by the dragging force generated by blood flow on endothelial cells plays a critical role in production of ROS and RNS in the vasculature. Exposure of endothelial cells to fluid shear stress activates NADPH oxidase, resulting in production of superoxide (O2 •−) (22, 29, 42, 111). Xanthine oxidoreductase also contributes to O2 •− production in response to oscillatory shear stress (73). In addition to O2 •−, nitric oxide (NO) is generated via activation of endothelial nitric oxide synthase (eNOS) in vascular endothelial cells (12, 74, 108, 112) and inducible nitric oxide synthase (iNOS) in smooth muscle cells by either steady or pulsatile laminar shear stress (31, 91). NO plays an important role in vasodilation (36, 75, 86) and anti-inflammation (15, 27, 44). For example, inhibition of nuclear factor kappa B (NF-κB) by NO has been linked to downregulation of vascular cell adhesion molecule-1 (VCAM-1) gene expression, leading to decrease in monocyte binding to the endothelium (21, 68, 103). However, NO may also react with O2 •−, forming peroxynitrite (ONOO−), one of the highly reactive species at a rapid diffusion-limited rate (k = ∼1 × 1010 M −1 s−1) (6), which in turn modifies proteins and lipids (4, 5, 84, 85). ONOO− also induces oxidative damage and enhances adhesion molecules expression in the vasculature (77, 92). When endothelial cells were sheared in the presence of LDL, oscillatory flow caused higher levels of LDL 3-nitrotyrosine, a footprint of ONOO- formation, compared to pulsatile flow (39). The importance of ONOO− in development of atherosclerosis is also implicated by detection of 3-nitrotyrosine in human atherosclerotic lesions (3, 7, 17, 82, 96).
Several lines of evidence show that both oscillatory and either steady or pulsatile laminar shear stress produce O2 •− and NO; however, NO production in endothelial cells by steady or pulsatile laminar shear stress is significantly higher than that by oscillatory shear stress (39, 72). On the other hand, oscillatory flow induces O2 •− production much more than steady or pulsatile laminar flow (13, 22, 43, 73), but induces eNOS upregulation to a much lesser extent compared to steady or pulsatile laminar flow (8, 39, 93). In addition, the high level of O2 •− generated by oscillatory shear stress reacts with NO to form ONOO−, resulting in less bioavailable NO under oscillatory flow conditions. In contrast, either steady or pulsatile flow upregulated the expression of eNOS, CuZn superoxide dismutase (CuZnSOD), and MnSOD (1, 18). It is well known that reduced NO availability can lead to vascular dysfunction, including intimal hyperplasia and expression of adhesion molecules (70, 102). Based on the above, it is reasonable to propose that ROS/RNS and their reaction products can cause fluid shear stress to be either pro- or anti-atherogenic.
Activation of Transcription Factor Nrf2 by Laminar Shear Stress
The DNA microarray is a powerful tool used to reveal gene expression profiles of cells exposed to different types of shear stress. Brooks et al. compared gene expression of endothelial cells in response to disturbed flow and steady laminar flow (9) and showed that expression of adhesion molecules such as VCAM-1 and intercellular adhesion molecule-1 (ICAM-1) and inflammatory molecules such as monocyte chemotactic protein 1 (MCP-1) and the receptors for interleukins was selectively induced by disturbed shear stress. The recent study by Conway et al. (16) reported that the reversing component of disturbed flow was primarily responsible for the upregulation of endothelial receptors and monocyte adhesion. The expression of VCAM-1 and ICAM-1 at sites of the predisposed to lesion formation in rabbit and mouse was also shown (46). NF-κB and activator protein 1 (AP-1) are known as major transcription factors regulating these inflammatory genes (8, 24, 41, 48, 71, 104, 114). Our studies using DNA microarrays showed that PLSS (2 dyn/cm2) induced antioxidant enzymes such as heme oxygenase 1 (HO-1), glutamate-cysteine ligase modifier (GCLM), glutamate-cysteine ligase catalysis (GCLC), and NADPH quinone oxidoreductase 1 (NQO1) in human umbilical vein endothelial cells (HUVEC) (106, 107; Table 1), which were regulated by stabilization of Nrf2. Similar results were shown by Chen et al. (14).
, Nrf2-regulated genes; PLSS, pulsatile laminar shear stress.
Microarray analysis was performed by using gene chip U133 (Affymetrix Inc., Santa Clara, CA) and calculation was performed as described in a previous article (98).
Nrf2 is a well-characterized transcription factor that plays an important role in the antioxidant response element (ARE)-mediated expression of a group of genes encoding phase II detoxification enzymes and antioxidant proteins, such as glutathione-S-transferase, HO-1, peroxiredoxin 1, NQO1, GCLM, and GCLC (47, 49). These enzymes are crucial for protecting cells from electrophile toxicity and oxidative stress. Under basal conditions, Nrf2 is negatively regulated by Kelch-like ECH-associated protein 1 (Keap1), which facilitates the degradation of Nrf2 through the proteasome (53). A variety of environmental stresses such as ultraviolet irradiation and exposure to cigarette smoke or heavy metals are known to induce ARE-mediated antioxidant proteins via Nrf2 activation (30, 37, 61, 62). In response to these stimuli, oxidative stress occurs with electrophile generation in cells. Electrophilic compounds are believed to attack the reactive cysteine residues in Keap1 intervening region (IVR), leading to a conformational change in the Keap1-Nrf2 association motif. The dissociation of Nrf2 from Keap1 and phosphorylation of Nrf2 prevent its proteasomal degradation, leading to accumulation of newly synthesized Nrf2 and its translocation to the nucleus (52, 65, 100, 101). Multiple sets of reactive cysteine residues in Keap1 have been identified, and other signaling molecules are also reported to be involved in Nrf2 activation (11, 51, 64, 69, 78, 83, 87 –90, 105, 109), suggesting that the molecular mechanisms for Nrf2 activation by various stimuli are different.
We found that in response to PLSS, Nrf2 was markedly accumulated and translocated into the nucleus (Fig. 1A) (106) and Nrf2-regulated cytoprotective genes were induced in HUVEC (Table 1) (107). Figure 1B showed increasing expression of GCLM measured by quantitative real-time PCR (qRT-PCR) which is almost completely abolished in HUVEC transfected with Nrf2 siRNA. Even more, these cytoprotective genes (Table 1) were not induced in endothelial cells isolated from Nrf2-deficient mice (Table 2). These results suggested that Nrf2 was essential for upregulation of cytoprotective genes under PLSS. We have previously revealed that steady laminar shear stress but not oscillatory shear stress enhances binding of Nrf2 to the regulatory region of NQO1 (38). Although the molecular mechanisms are not clear yet, we have presented one plausible explanation by presuming the presence of factor (X) which is induced by oscillatory flow or inhibited by laminar flow. The differential responses of Nrf2 target genes to laminar flow and oscillatory flow were shown in vivo by Zakker et al. (113). Both the accumulation of Nrf2 and the induction of GCLM mRNA by PLSS were significantly suppressed by N-acetylcysteine (NAC) (Fig. 2) (106). Besides our findings (106,107), several groups have reported that steady or pulsatile laminar shear stress activates Nrf2 for endothelium protection, which is blocked by ROS scavengers (14, 20, 34, 38, 40, 56). These results suggest that ROS produced by steady or pulsatile laminar shear stress plays important roles in activation of Nrf2, leading to a protective response by inducing antioxidant enzyme genes in endothelial cells. It is worthwhile to identify which ROS and/or RNS are responsible for Nrf2 activation, thereby causing laminar shear stress to be anti-atherogenic.
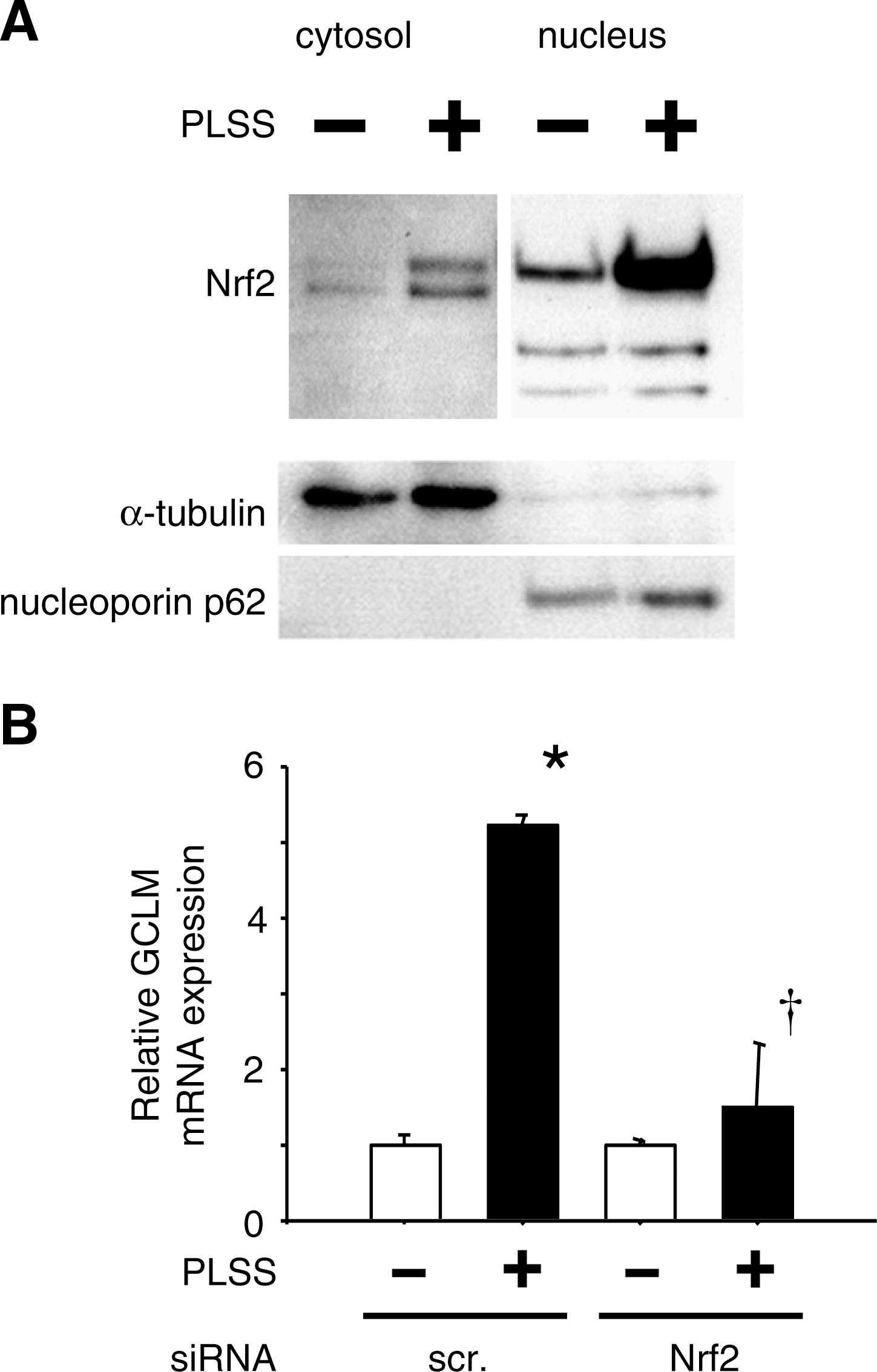

PLSS, pulsatile laminar shear stress.
Microarray analysis was performed by using gene chip MOE 430A (Affymetrix Inc.) and calculation was performed as described in a previous article (98).
Another laminar shear stress-induced transcription factor, Kruppel-like factor 2 (Klf2) has been reported to act as a key role in anti-atherosclerosis (8, 25). Nrf2 is one of target molecules of Klf2 and acts in synergy with Klf2 to control approximately 70% of the genes induced by laminar shear stress (25). More details about the function of Klf2 in response to shear stress are reviewed by Nayak et al. (77a) and Nigro et al. (77b) in this Forum.
ROS/RNS Responsible for Activation of Nrf2
Several publications have shown that O2 •− is produced by NADPH oxidase activation in response to oscillatory shear stress (42, 95). Our recent article (106) has shown that involvement of O2 •− produced by PLSS in GCLM expression was implicated by using the XO inhibitor oxypurinol and NADPH oxidase inhibitor diphenyleneiodonium (DPI; Fig. 3A). Furthermore, knockdown of one of the components of NADPH oxidase, p22, by using siRNA in HUVEC showed the same results (Figs. 3B–3D). McNally et al. has reported that NADPH oxidase maintains endothelial cell XO levels in endothelial cells and that XO is responsible for increased reactive oxygen species production in response to oscillatory shear stress (73). These results suggest that PLSS induces O2 •− production via activation of NADPH oxidase and XO, resulting in expression of Nrf2-regulated genes.
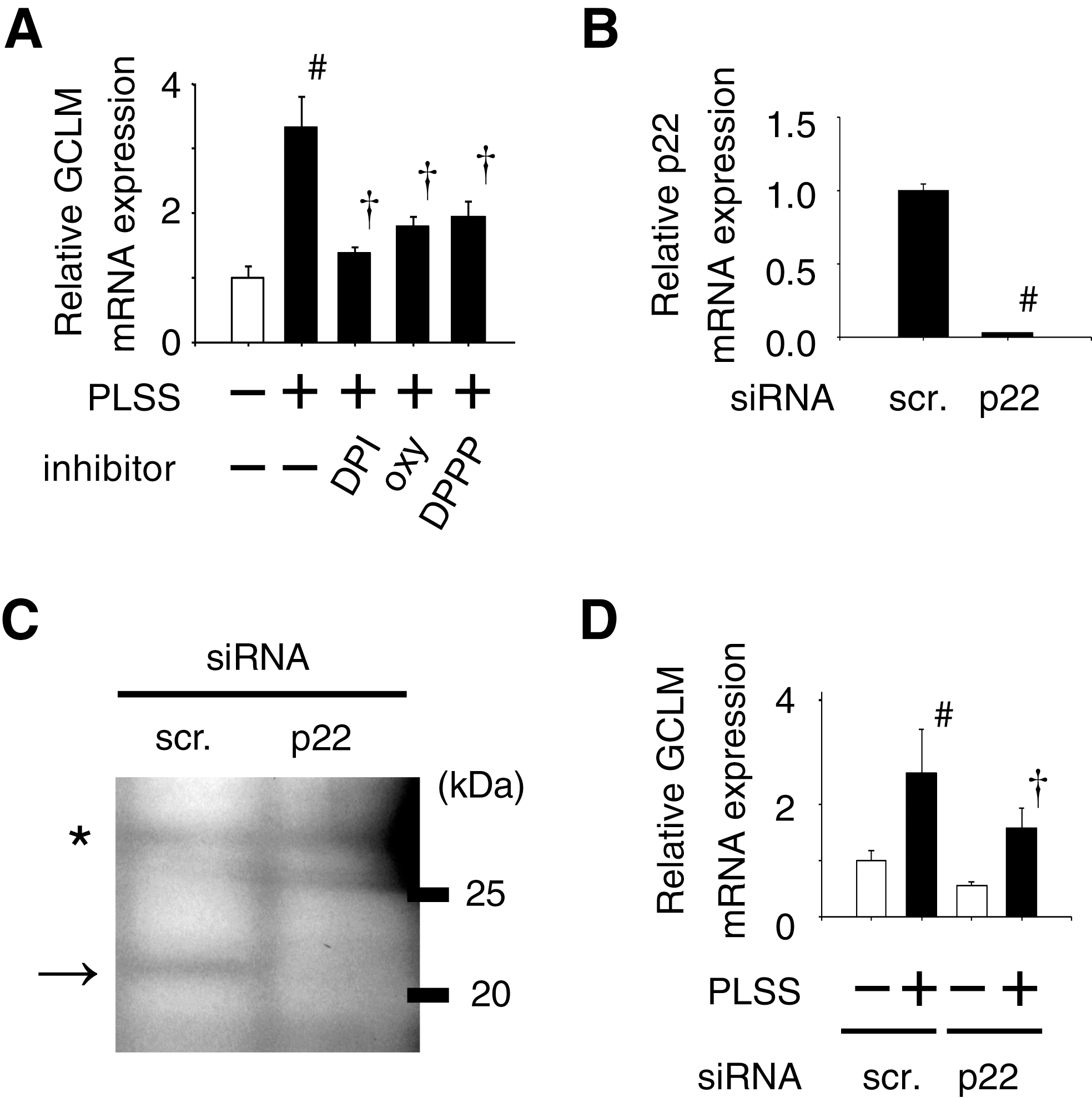
ROS/RNS can initiate lipid peroxidation in which lipid hydroperoxides are formed as primary products converting to electrophilic compounds such as aldehydes (54, 67). Keap1 reacts with electrophiles and dissociates Nrf2, resulting in its nuclear translocation. The hypothesis that lipid peroxidation products play a role in Nrf2-regulated gene expression is supported by using a reducing reagent of lipid hydroperoxide, diphenylpyrenylphosphine (DPPP) (79, 80). DPPP significantly attenuates PLSS-induced expression of GCLM in HUVEC (Fig. 3A) (106).
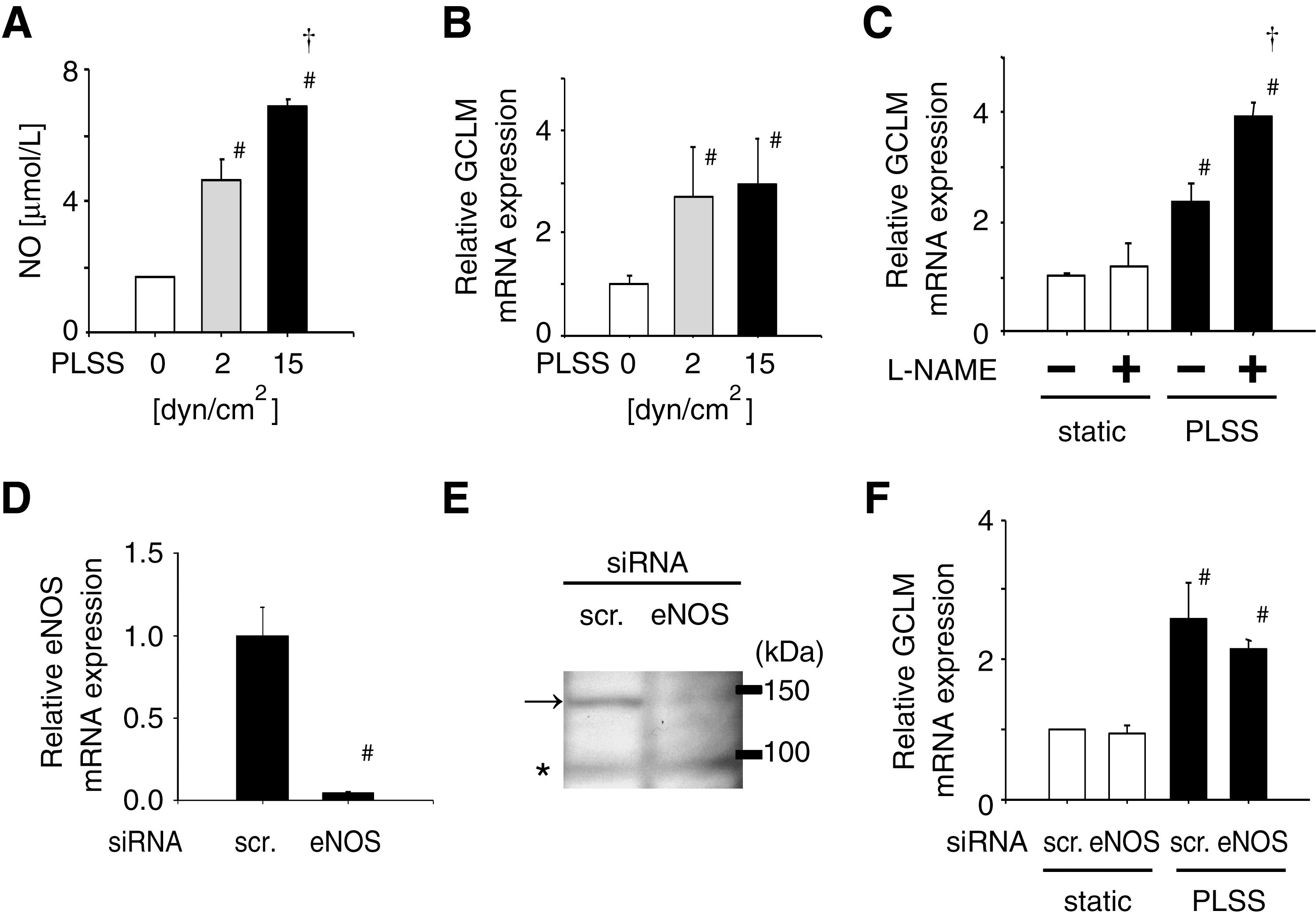
As a number of reports have shown, NO production increases with PLSS (Fig. 4A) (106); however, expression of GCLM was not affected by the extent of shear stress and NO production (Fig. 4B). Even more, NOS inhibitor L-NAME enhanced the expression of GCLM significantly (Fig. 4C). Furthermore, knockdown of eNOS using siRNA (Figs. 4D and 4E) did not change the expression of GCLM induced by PLSS (Fig. 4F). These results suggest that NO produced in HUVEC in response to PLSS is not involved in the expression of GCLM. There are some reports showing NO-dependent, HO-1 upregulation via Nrf2 activation in aortic endothelial cells exposed to steady laminar shear stress (34) or treated with NO donor (10, 11, 34). It is assumed that NO produced in HUVEC in response to PLSS does not work as NO produced in aortic endothelial cells exposed to steady laminar shear stress or derived from NO donor.
Regulation of Oxidative Stress by Nrf2 Activation
The ROS production by PLSS was detected by using a fluorescence dye, 2’7’-dichlorodihydrofluorescindiacetate (DCFH-DA), which is enhanced by transfecting cells with Nrf2 siRNA (Fig. 5), implicating that Nrf2 suppresses the extent of oxidative damage caused by PLSS via upregulation of antioxidant enzymes. For example, biosynthesis of glutathione (GSH), one of key molecules protecting cells from oxidative damage (19, 60, 99) is dependent on activity of glutamate-cysteine ligase (GCL), one of Nrf2-regulated antioxidant enzymes.

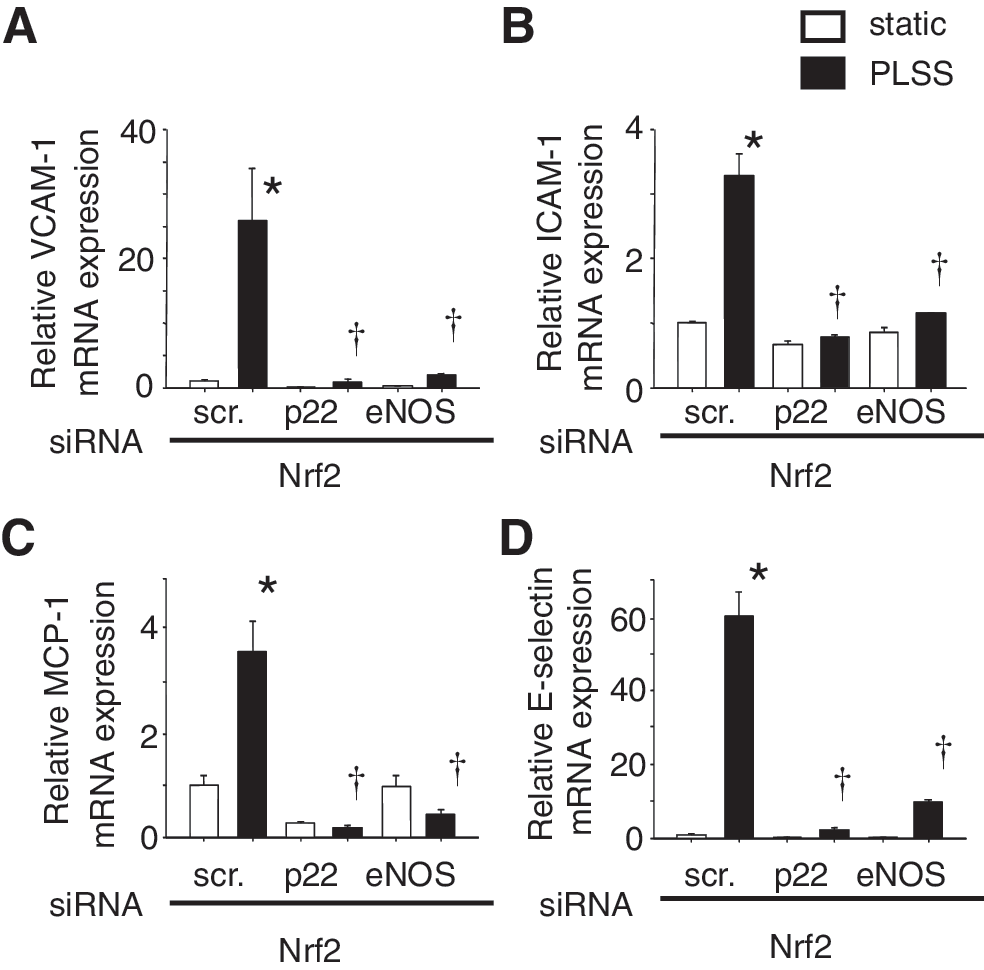
The expression of adhesion molecules such as VCAM-1, ICAM-1, E-selectin, and monocyte chemoattractant protein-1 (MCP-1) was induced in HUVEC in response to PLSS (Fig. 6). Moreover, the expression of these genes was increased in Nrf2 knocked-down cells. It is assumed that the difference in the extent of adhesion molecule expression sustained in vivo may determine the fate of endothelium as either pro-atherogenic or anti-atherogenic.
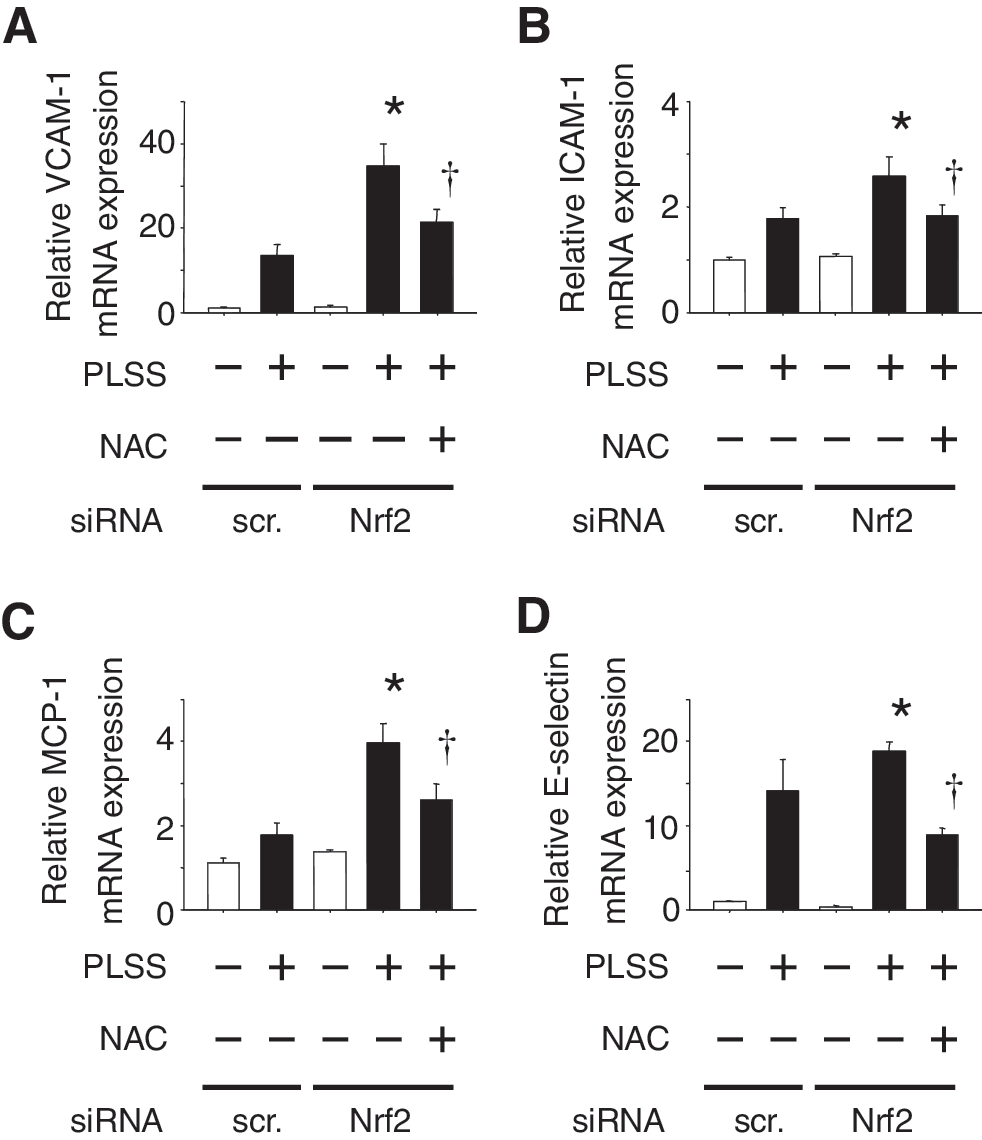
The experiments using siRNA against p22 or eNOS revealed that both O2 •− and NO were required for induction of adhesion molecules and chemokines by PLSS in Nrf2 knocked down HUVEC (Fig. 7). These results suggest that ONOO− may play an important role in signal transduction for expression of these pro-atherogenic genes. Evidence supports the contribution of ONOO− in induction of adhesion molecules and chemokines via activation of NF-κB and AP-1 (5, 35, 45, 50, 57 –59, 94, 110, 115). ONOO− formation is linked to shear stress-mediated activation of a member of the mitogen-activated protein kinase family (MAPK), c-Jun N-terminal kinase (JNK) (28) and ERK (116). ONOO− easily penetrates cell membranes and modulates target lipids, proteins, and DNA via generation of highly reactive radical, nitric dioxide (NO2) by reacting with carbon dioxide. Also peroxynitrous acid (ONOOH) derived from a reaction between ONOO− and hydrogen leads to generation of hydroxy radical (•OH) and NO2 (97). Thus, ONOO− causes oxidative stress in vascular cells and contributes to development of atherosclerosis. Dickhout et al. have reported the involvement of ONOO− in atherogenesis by causing ER stress (23). It would be interesting to test whether ONOO− scavengers, such as uric acid, can attenuate the expression of pro-atherogenic genes induced by oscillatory shear stress.
The molecular mechanisms underlying the protective effect of PLSS remain to be defined; however, inhibition of O2 •− production via induction of Nrf2-regulated antioxidant enzymes, which in turn limits the formation of ONOO−, may be involved. It is proposed that these antioxidant enzymes may keep oxidative stress under the threshold necessary to induce expression of pro-atherogenic genes.
Anti-Atherogenic Roles of Nrf2 In Vivo and Ex Vivo
Zakkar et al. have reported that VCAM-1 is highly induced in the aortic arch of Nrf2-deficient mice (113). We have established a primary culture system of mouse arterial endothelial cells (MAEC) (63). MAEC isolated from wild-type and Nrf2-deficient mice were exposed to PLSS for 8 h, and gene expression was analyzed using DNA microarrays. As shown in Table 2, the upregulation of antioxidant genes by PLSS (Table 1) (107) was strongly suppressed in MAEC isolated from Nrf2-deficient mice, as well as their basal levels. Interestingly, the expression levels of adhesion molecules and cytokines including VCAM-1, ICAM-1, E-selectin, and MCP-1 were markedly increased in Nrf2-deficient MAEC exposed to PLSS (Table 3). These results agree with data obtained from Nrf2 knocked down HUVEC (Fig. 6).
PLSS, pulsatile laminar shear stress.
Microarray analysis was performed by using gene chip MOE 430A (Affymetrix Inc.), and calculation was performed as described in a previous article (98).
The global genomic analysis of adult pig inner aortic arch by Passerini et al. revealed the coexistence of pro- and anti-atherosclerotic transcript profiles in susceptible regions where endothelial cells are exposed to disturbed flow (81). They suggested the introduction of additional risk factors might shift this balance to favor lesion development. Hajra et al. also suggested that NF-κB signal transduction was primed for activation in high probability regions in mouse proximal aorta on encountering an activation stimulus such as hypercholesterolemia (32). These in vivo data suggest that endothelial cells possess an anti-atherosclerotic phenotype even under disturbed flow without additional pro-atherogenic factors present and support our interpretation of in vitro and ex vivo experimental data showing that Nrf2 activation by steady or pulsatile, laminar flow can induce an anti-atherogenic environment.
Conclusions
The modulation of redox balance in the vasculature by fluid shear stress regulates in the development of atherosclerotic lesions (Fig. 8). Both unidirectional and nonunidirectional shear stress produce ROS/RNS that have potential to activate signal transduction pathways leading to pro-atherogenic gene expression. The expression of pro-atherogenic genes such as adhesion molecules and chemokines requires both O2 •− and NO, implying ONOO- is one of the key molecules affecting induction of pro-atherogenic genes. On the other hand, pulsatile laminar shear stress induces expression of Nrf2-regulated genes via O2 •− production in HUVEC, which is not affected by NO production. The enhancement of GSH biosynthesis by induction of GCL due to Nrf2 activation can prevent generation of excess amount of ROS/RNS that normally led to pro-atherogenic gene expression. The precise molecular mechanisms underlying pulsatile laminar shear stress specifically activating Nrf2 are unclear at this time and are interesting issues to be elucidated by further investigations.

Footnotes
Acknowledgments
This study was supported by the Program of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO), by Focus 21 Project of New Energy and Industrial Technology Development Organization (NEDO), and by Special Coordination Fund for Science and Technology and the Academic Frontier Research Project on "New Frontier of Biomedical Engineering Research" of the Ministry of Education, Culture, Sports, Science and Technology.
We thank Dr. Ken Itoh, Hirosaki University School of Medicine, for advice based on his animal studies.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.