
Abstract
The Ncf1 gene, encoding the P47PHOX protein that regulates production of reactive oxygen species (ROS) by the phagocyte NADPH oxidase (NOX2) complex, is associated with autoimmunity and arthritis severity in rats. We have now identified that the single-nucleotide polymorphism (SNP) resulting in an M153T amino acid substitution mediates arthritis resistance and thus explains the molecular polymorphism underlying the earlier identified Ncf1 gene effect. We identified the SNP in position 153 to regulate ROS production using COSPHOX cells transfected with mutated Ncf1. To determine the role of this SNP for control of arthritis, we used the Wistar strain, identified to carry only the postulated arthritis resistant SNP in position 153. When this Ncf1 allele was backcrossed to the arthritis susceptible DA strain, both granulocyte ROS production and arthritis resistance were restored. Position 153 is located in the hinge region between the PX and SH3 domains of P47PHOX. Mutational analysis of this position revealed a need for an −OH group in the side chain but we found no evidence for phosphorylation. The polymorphism did not affect assembly of the P47PHOX/P67PHOX complex in the cytosol or membrane localization, but is likely to operate downstream of assembly, affecting activity of the membrane NOX2 complex. Antioxid. Redox Signal. 14, 2373–2383.
Introduction
Genetic segregation experiments, using strains with variable susceptibility to complex diseases, have shown high efficiency in linkage analysis where the major loci have been indicated (4, 26). Due to the lack of dense recombinations in inbred strain crosses, it has, however, been difficult to position the genes underlying these loci. By using a congenic strategy, in which the linked fragment has been minimized through recombinations and isolated through backcrossing, a limited number of genes have been identified, including the major histocompatibility complex haplotype and the complement factor C5 (8, 13, 34, 44, 49, 66). Another example is the Ncf1 gene, identified in a cross between the pristane-induced arthritis (PIA) susceptible DA and the resistant E3 rat strains (49, 63). The recombinant fragment was reduced to 300 kb containing only two genes, Ncf1 and Gtf2i. Neither of the genes showed differential expression and only Ncf1 contained nonsynonymous SNPs (49). The congenic rat, with a resistant E3-derived fragment on susceptible DA background, did not only show reduced arthritis severity but also increased oxidative burst response. This is compatible with known functions of the P47PHOX protein (also called NCF1), a subunit of the phagocyte NADPH oxidase (NOX2) complex (21), which is responsible for production of reactive oxygen species (ROS) in response to invading pathogens (6, 16). The complex consists of five subunits of which the membrane-integrated catalytic core protein GP91PHOX (CYBB) forms a stable heterodimer with P22PHOX (CYBA), constituting the enzymatic flavocytochrome b588 (CYT b558 ) (7). The other subunits [P40PHOX (NCF4), P47PHOX, and P67PHOX (NCF2)] are in the resting state residing in the cytosol where an auto-inhibitory conformation of the P47PHOX protein prevents the cytosolic complex to bind to the membrane subunits (18, 28). This auto-inhibitory conformation is released upon activation and consequent phosphorylation of P47PHOX, which transport the cytosolic subunits to the membrane to form the functional complex (1, 57).
The finding that low ROS production-mediated arthritis (49) was surprising, as it required a mechanism in which ROS, produced by the NOX2 complex, protected against, rather than exaggerated inflammation, as the general dogma proposes [for an overview see (37, 41)]. These findings were strengthened by the discovery that a spontaneous mutation in the mouse Ncf1 gene had similar effects on arthritis (36).
The three SNPs identified in Ncf1 were all highly polymorphic both in the natural rat population and between the DA and E3 strains (49, 50). To understand the molecular mechanisms underlying the effect on arthritis, we needed to identify the arthritis causative SNP. To do so, we first investigated which of the three SNPs controlled ROS production in vitro by mutating recombinant Ncf1. We found that the amino acid shift from methionine to threonine at position 153 led to a restored ROS production capacity. Next, we selected recombinant haplotypes from inbred strains and made a new congenic strain that had an Ncf1 variant differing only at position 153. Lastly, we investigated the functional difference between threonine and methionine at this position and found that threonine regulated the capacity of the already membrane bound NOX2 complex to produce ROS.
Materials and Methods
Cells and reagents
Calyculin A was from Cell Signaling Technology (Danvers, MA). All other reagents were purchased from Sigma-Aldrich if nothing else is stated. COS-7 cells reconstituted with GP91PHOX, P22PHOX, and P67PHOX (hereafter referred to as COSphox-Ncf1), and COS-7 cells stably expressing P47PHOX in addition to GP91PHOX, P22PHOX, and P67PHOX (referred to as COSphox) (52) kindly provided by Prof. Mary C. Dinauer (Indiana University) were cultured in Dulbecco's complete medium, 10% fetal calf serum, and penicillin–streptomycin (Gibco, Invitrogen). The murine Ra2 microglia cell line (licensed by the Japan Science and Technology Agency, Patent ID US6.673,6,5; JP3410738; EP10/602,234) was kindly provided by Dr. Makoto Sawada (Department of Brain Function, Research Institute of Environmental Medicine, Nagoya University, Nagoya, Japan).
Vector constructs
DA (AF547392) and E3 (AF547393) Ncf1 alleles were PCR amplified and inserted in the pcDNA3.1/Hygro (+) mammalian expression vector (Invitrogen). Mutations in the Ncf1 cDNA were generated using QuickChange II Site-Directed M mutagenesis Kit (Stratagene) according to the manufacturer's recommendations. In all transfection experiments pcDNA3.1/Hygro (+) mammalian expression vector (Invitrogen) was used as control vector. Primer sequences are provided in Supplementary Fig. S1 (Supplementary Data are available online at
Cell transfection and ROS assay
Adherent COS-7 cells were harvested by incubation with trypsin/EDTA for 5 min at 37°C. The cells were resuspended, washed in PBS, and cultured in 96-well plates (NUNC) at a density of 20,000 cells/well overnight. The cells were transiently transfected with Ncf1 with lipofectamine plus, optimum, and DNA PLUS system according to procedures recommended by the manufacturer (Invitrogen) using 100 ng DNA per well. Transfected cells were grown in a complete medium for another 48 h until assayed. Empty pcDNA3 vector was used as a negative control.
Functionality of the NOX2 complex and thus the function of the studied Ncf1 allele was assayed directly in the culture plate using an isoluminol-enhanced chemiluminescence assay (17). Briefly, the cells were gently washed in Hanks balanced salt solution (HBSS) and 100 μl of isoluminol reagent buffer [isoluminol 10–50 μg/ml, HRP type II 2, 5–4 μ/ml, and phorbol 12-myristate 13-acetate (PMA) 200–400 ng/ml, final concentration] was added. Samples were gently mixed and data collection was initiated immediately. The ROS production from the NOX2 complex is initiated within minutes. Extracellular ROS production was followed at 37°C as produced luminescence signal (FluoStar Optima; BMG Labtechnologies) and presented as maximal relative signal during a measurement period of 30 min.
Western blot
Western blot was performed using standard protocols. P47PHOX was detected using a mouse monoclonal anti-P47PHOX (D-10) (sc-17845) from Santa Cruz Biotechnology followed by secondary antibody Rabbit anti-mouse IgG HRP-conjugated (P0260) from DAKO A/S (Glostrup). The sc-17845 antibody is generated against amino acids 196–390 of P47PHOX of human origin, therefore not interfering with the polymorphic area under the study. P67PHOX was detected using goat polyclonal anti-P67PHOX (N-19, sc-7663; Santa Cruz) followed by secondary antibody ImmunoPure® mouse anti-goat IgG HRP-conjugated (31400; Thermo Fisher Scientific).
Animals
Rats, DA and Wistar (originating from Harlan Europe), were kept in a climate-controlled environment with 12 h light/dark cycles and fed standard rodent chow and water ad libitum in the animal facility of Medical Inflammation Research, Lund University, Lund, Sweden, or Karolinska Institute, Solna, Sweden. The Wistar Ncf1 (Ncf1W ) allele was backcrossed to the DA background for 10 generations. The rats were found to be free from common pathogens, including Sendai virus, Hantaan virus, coronavirus, reovirus, cytomegalovirus, and Mycoplasma pulmonalis. The experiments were approved by the local ethical committee license M70/04 and M107/07 (Malmö/Lund, Sweden) and N67-10 (Stockholm, Sweden).
Genotyping
DNA samples were prepared from toe biopsies and assayed on a MegaBACE 1000 (GE Healthcare). Identification of SNPs in the Ncf1 region has earlier been described (49). The SNPs were analyzed using a Pyrosequenser PSQ 96 (Quiagen) according to manufacturer's protocol.
Induction and evaluation of disease
Arthritis was induced at the age of 6–12 weeks. Rats were sex- and age-matched within all experiments. PIA was induced by an s.c. injection at the base of the tail with 200 μl of pristane (Acros Organics). Arthritis development was monitored by inspection evaluating the number of joints affected by arthritis according to a macroscopic scoring system of the four limbs ranging from 0 to 15 (1 point for each swollen or red toe, 1 point for a swollen or red midfoot digit or knuckle and 5 points for a swollen ankle), resulting in a maximum total score of 60 for each rat (33). All scoring was performed in a blinded manner to avoid biased results.
Determination of ROS production ex vivo
The level of intra-cellular oxidative burst ex vivo was measured by preparing single-cell suspensions, hemolyzed with ammonium chloride (0.84% pH 7.4), from blood. Oxidative burst in granulocytes was determined by incubation of cells for 30 min (4°C) with biotin-labeled antibody HIS-48 (anti-granulocytes) (BD Biosciences Pharmingen). According to manufacturer's information the HIS-48 antibody reacts with an antigen expressed on all granulocytes. After washing with PBS, cells were incubated with allophycocyanin-conjugated streptavidin (BD Pharmingen) for 20 min at 4°C. To determine the level of NOX2 activity we used a modified version of the oxidative burst activity flow cytometry assay previously described (64). Briefly, cells were resuspended in Dulbecco's complete medium without FCS after staining, and incubated for 10 min at 37°C with 3 μM dihydrorhodamine-123 (Molecular Probes), which after oxidization by hydrogen peroxide (H2O2), peroxynitrite (ONOO−), and hydroxyl radicals (OH•) to rhodamine-123 emits a bright fluorescent signal upon excitation by blue light. The cells were then stimulated for 20 min at 37°C with PMA (200 ng/ml). Cells were washed with PBS and then acquired on a FACSort (BD Biosciences) and gated on HIS-48-positive cells (granulocytes), R-123 fluorescence intensity measured on FL-1, and results expressed in relative fluorescence units.
Extracellular ROS production was detected in thioglycolate-recruited peritoneal neutrophils using an isoluminol-enhanced chemiluminescence (17). Neutrophils were attracted to the peritoneum with an injection of autoclaved thioglycolate medium (2.4%). About 24 h after injection, peritoneum was washed with ice-cold HBSS and cell concentration was determined. Samples were also analyzed for frequency of granulocytes (HIS-48-positive cells) among white blood cells [Leukocyte common antigen, CD45-positive cells (Mouse anti-rat CD45 clone OX-1; BD Biosciences)] using antibody staining and flow cytometry detection as described above. Cells were washed in HBSS and diluted to a concentration of 2×106 cells/ml and 50 μl of the cell suspension was added to the wells of a white 96-well plate (LumiNUNC; NUNC) and mixed with 50 μl of isoluminol reagent buffer (isoluminol 350 μg/ml, horse radish peroxidase-type II 3.5 μ/ml, and PMA 60 ng/ml or formyl-Met-Leu-Phe (fMLP) 400 nM. Samples were gently mixed and data collection was initiated immediately. Extracellular ROS production was followed at 37°C as produced luminescence signal (Synergy 2 ELISA reader (BioTek Instruments).
Immunoprecipitation
COSphox-Ncf1 cells were seeded on 10-cm dishes (3×10^6 cells/dish), and grown on and transfected when 90%–95% confluence was reached. The cells were transiently transfected with plasmids coding for rat P47PHOX protein (DA or E3) using Lipofectamine 2000 (Invitrogen) according to manufacturer's recommendations. After 48 h incubation, cells were washed twice with ice-cold PBS, and lysed with 1 ml of ice-cold RIPA Buffer (Sigma) containing phenylmethylsulfonylfluoride, protease inhibitor cocktail (Roche), and sodium orthovanadate for 5 min. These whole cell lysates (kept at +4°C throughout the protocol) were collected by centrifugation (8000 g 10 min) and the supernatants were precleared with 5 μg of rabbit IgG and 15 μl of Protein A/G PLUS-Agarose (Santa Cruz) for 1 h. Beads were pelleted and an aliquot (0.5 ml) of the precleared supernatant was incubated with 4 μl of the primary antibody (P67PHOX (N-19), sc-7663 or P47PHOX (D-10), sc-17845; Santa Cruz) that had already been complexed with 20 μl of the Protein A/G PLUS-Agarose by incubating them at +4°C for 1 h with gentle agitation overnight. After 4 h, immunoprecipitates were collected, and the pellets washed 2 times with 1 ml of PBS. Proteins were released into 40 μl of Laemmli sample buffer by boiling the samples for 5 min. Equal aliquots of immunoprecipitates were analyzed by Western blot.
Lentiviral transduction
Ra2 microglia were transduced with separate pLOX TW lentiviral vectors (65) expressing either the tetracycline-inducible transactivator protein or the cDNAs of P47PHOX/DA and/or P47PHOX/E3, the latter two vectors in different ratios as indicated. Virus production and transduction was performed essentially as described (54). Expression levels of P47PHOX after induction with 0.1 μg/ml doxycycline overnight were determined by Western blotting and flow cytometry with rabbit polyclonal anti-P47PHOX antibody poly-B. Superoxide production of the generated P47PHOX/DA and/or P47PHOX/E3 Ra2 cell lines was measured at 37°C by luminol-enhanced chemiluminescence recorded before and after stimulation with 100 ng/ml PMA or 4–8 μM fMLP delivered through the injector module of the Synergy HT microplate reader (54). Luminol has a similar reactivity as isoluminol but is in contrast to isoluminol membrane permeable and thus reacts with both with ROS released from the cell as well as with ROS produced into phagosomes (17). Two series of separately transduced Ra2 cell populations were used for all figures with essentially the same results.
P47PHOX localization and translocation
Ra2 microglia expressing Ncf1E3 or Ncf1DA were seeded in LabTech tissue culture 8-chamber slides with glass bottom and incubated with nonopsonized zymosan particles in full growth medium at 4°C for 30 min to allow zymosan attachment to the cell surface. Subsequently, cells were washed thoroughly with cold medium and incubated for 10 minutes in medium at 37°C to allow internalization of bound zymosan particles. Cells were then fixed and immunofluorescence performed with anti-GP91PHOX mAb 54.1 (15), rabbit pAb anti-P47PHOX poly-B (generated against the peptide (NH2) CRRNSVRFLQQRRRP (−COOH)), and Alexa633-conjugated phalloidin.
Statistics
Quantitative data are expressed as mean±SEM; significance analysis was performed using Mann-Whitney test. All results were compared to those from the control group if nothing else is stated. Statistical significance is represented by *p≤0.05, **p<0.01, and ***p<0.001 throughout the article.
Results
The M153T mutation restores ROS production capacity in vitro
Three SNPs were previously found to be polymorphic between the arthritis susceptible DA strain and the resistant E3 strain. Only two of these polymorphisms were nonsynonymous: M106V and M153T (49). The M106V alteration is located within the P47PHOX membrane phospholipid binding Phox homology (PX) domain (2, 39), whereas the M153T alteration is located between the PX domain and the P22PHOX binding Src homology 3 (SH3) domains (42, 43, 46, 58, 59) (Fig. 1a). To identify which of the SNPs caused the effect on ROS production, we used COS cells expressing the subunits of the functional human NOX2 complex, except for P47PHOX (COSphox-Ncf1 cells) (52). These cells were subsequently reconstituted with the Ncf1 wild-type DA or E3 allele, or with the DA allele mutated at the different polymorphic positions. ROS producing capacity of the cells was assayed using an isoluminol-enhanced chemiluminescence assay reacting with the superoxide anion (O2 .−), produced by the NOX2 complex, exclusively in the extracellular compartment (45). Cells expressing P47PHOX/DA presented with an impaired ROS production compared to cells expressing P47PHOX/E3 (Fig. 1b). When the effect of the two polymorphisms was investigated separately, we found that a substitution of methionine to threonine at position 153 (M153T) restored the ROS producing efficiency of the Ncf1DA allele. The methionine to valine amino acid substitution in position 106 of the DA Ncf1 allele did not significantly alter ROS production (Fig. 1b). After concluding that the SNP resulting in amino acid replacement M153T controlled the induced ROS production, we asked whether the same SNP would also control arthritis.
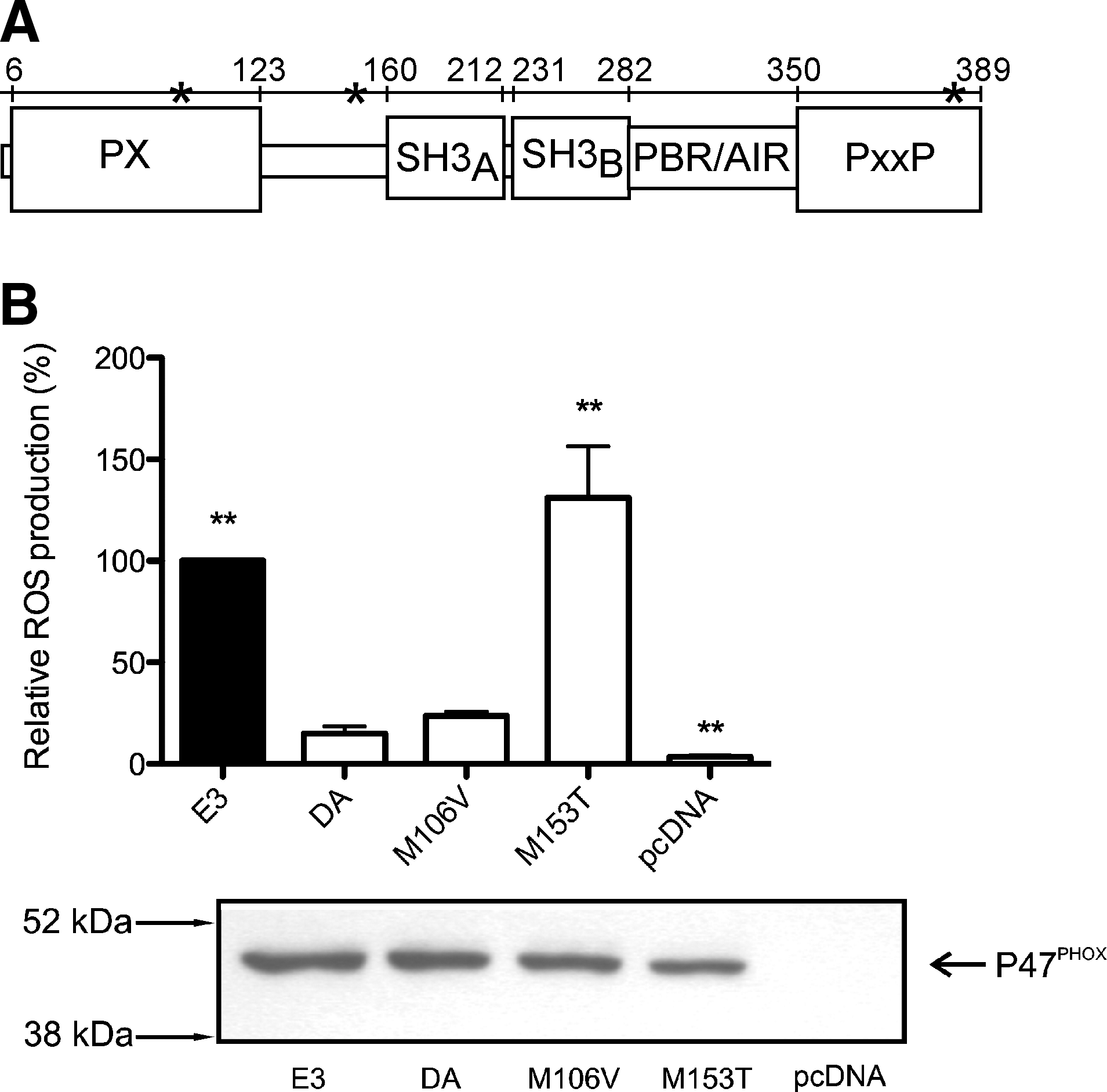
M153T alteration restores ROS production in vivo and protection from severe arthritis
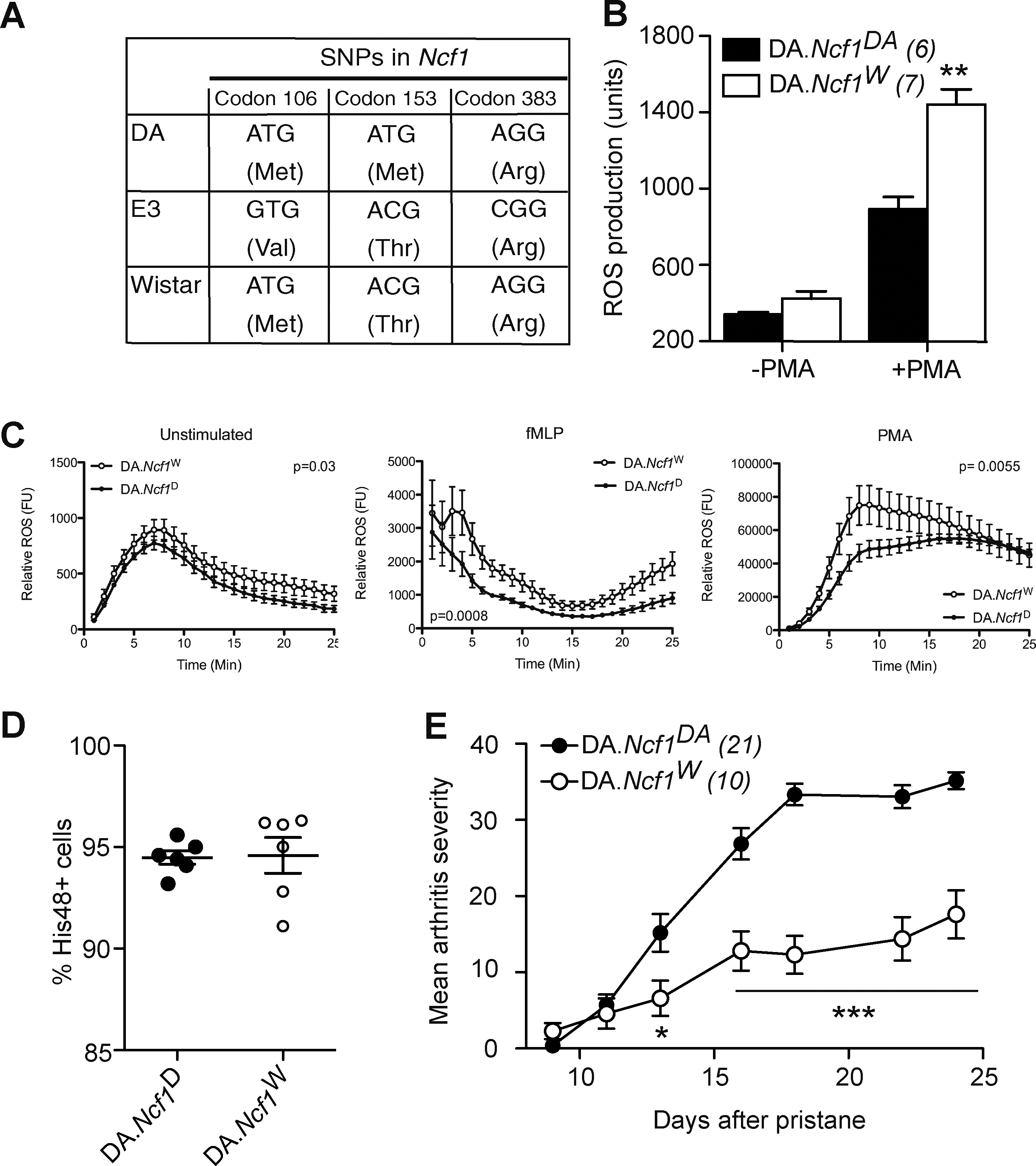
To conclusively determine that the M153T alteration is responsible for the genetic effect of the Pia4 locus (63), we screened a large number of inbred rat strains and found that a substrain of the Wistar strain had Ncf1 DA alleles at two of the three SNPs found to differ between DA and E3. Position 153 was found to be occupied by threonine, which is found in E3 P47PHOX and not DA (Fig. 2a). The Wistar Ncf1 allele (Ncf1W ) was backcrossed to the DA genetic background for 10 generations and the resulting congenic strain (DA.Ncf1W ) (Supplementary Fig. S2) was investigated for ROS production and susceptibility to PIA. Introduction of the Ncf1W allele on the DA genetic background resulted in enhanced ROS production capacity in blood granulocytes, analyzed by a flow cytometry-based assay where intracellular oxidation of rhodamin-123 is measured (55, 64) (Fig. 2b). We could also see an enhanced extracellular ROS production in peritoneal thioglycolate recruited granulocytes in response to PMA and fMLP when using the isoluminol-enhanced chemiluminescence assay in cells that originated from DA.Ncf1W congenic rats (Fig. 2c). This effect was not due to a larger capacity of granulocytes to infiltrate the peritoneum since neither cell concentration in the cell samples (not shown) nor granulocyte frequency differed between the two strains (Fig. 2d). In addition to increasing the capacity of granulocytes to produce ROS, the Ncf1W allele also mediated resistance to severe PIA (Fig. 2e), verifying that the M153T alteration is crucial for the function of the P47PHOX protein, activation of the NOX2 complex, and protection against autoimmunity and arthritis severity.
Only threonine and serine at position 153 can activate the NOX2 complex
To analyze the structural consequences controlled by amino acid 153 of the P47PHOX protein, we created P47PHOX/DA constructs and altered the amino acids at this position. The constructs were used to reconstitute COSphox-Ncf1 cells and ROS production in PMA-stimulated cells was measured using the isoluminol-based chemiluminescence assay (Fig. 3). Our results confirmed earlier published in vitro and in vivo results (24, 35, 49, 60) that cells expressing P47PHOX/DA are not completely deficient of ROS production since cells reconstituted with the negative control, pcDNA, had significantly lower ROS production (Fig. 3a).
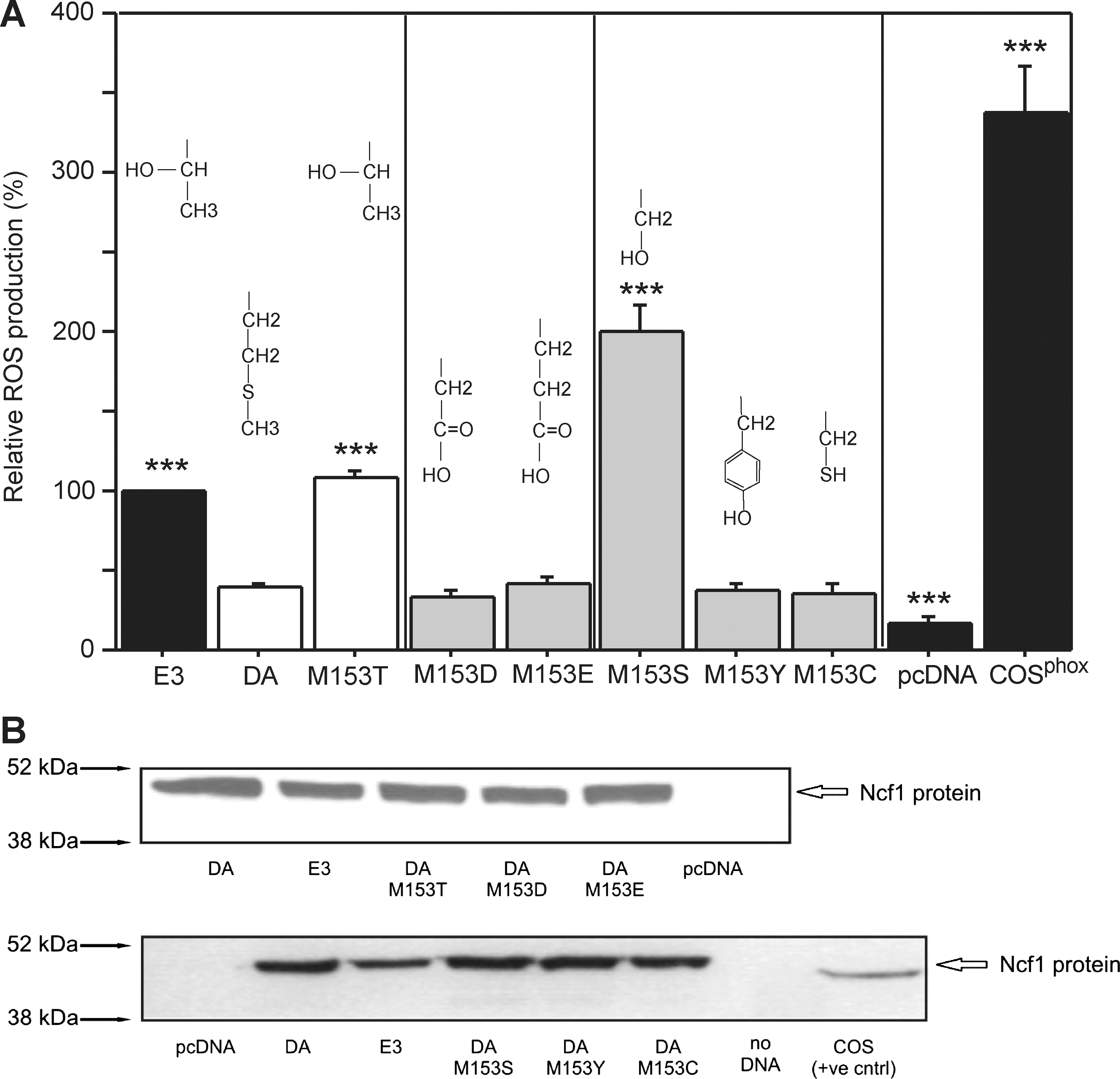
We reconstituted the cells with P47PHOX/DA having serine, tyrosine, or cysteine at position 153 to introduce polar, uncharged amino acids (similar to threonine). Serine and tyrosine have a hydroxyl group and could theoretically be phosphorylated. Cysteine is similar in size with serine, whereas tyrosine has a bulky side chain. However, cysteine also introduces a reactive thiol into the protein that may alter functional outcome. These were also used to assess the importance of the size of the side chain on ROS production. We found that serine, but not tyrosine or cysteine, restored the ROS production, suggesting that activation by a threonine/serine-specific protein kinase(s), such as MAP kinases and protein kinase C, is of importance for a fully functional NOX2 complex. Since both serine and threonine can be phosphorylated, we inserted aspartate and glutamate that are also negative in charge. These alterations did not affect ROS production arguing that phosphorylation is not mediating the effect (Fig. 3a). However, to directly test whether the threonine residue could be phosphorylated, we utilized matrix-assisted laser desorption/ionization-TOF/TOF mass spectrometry to analyze purified P47PHOX/E3 protein from PMA stimulated cells. Phosphopeptides were enriched using TiO2 micro columns in an effort to enrich potential phosphopeptides followed by analysis using dihydroxy benzoic acid, spiked with 1% phosphoric acid as matrix: no peptide with a phosphoryl group was detected. To further enrich the possible phosphorylation, the PMA-stimulated cells were treated with Calyculin A, a potent serine/threonine phosphatase inhibitor (38), to preserve phosphorylated status. Phosphopeptides were this time enriched with TiO2/ZrO2-coated tips and analyzed by mass spectrometry using the dihydroxy benzoic acid matrix as above (see Supplementary Data for further details). Despite these extensive efforts to enrich the phosphorylated peptides, we could not identify phosphorylation at position 153 (data not shown). In addition, analysis of the amino acid sequence of P47PHOX/E3 protein (NetPhos, Center for Biological Sequence Analysis, Denmark) predicted that Threonine 153 is not likely to be phosphorylated (Supplementary Fig. S3) by any kinases, including PKC, p38MAPK, GSK3, PKA, and casein kinase II (NetPhosK).
The M153T mutation does not alter localization or recruitment to phagosomes
The exact effect of mutations in the hinge region between the PX and the SH3 domains is still unknown, but altered localization of the protein has been suggested (56). In resting cells, three of the subunits of the NOX2 complex are residing in the cytosol (P47PHOX, P67PHOX, and P40PHOX), where an auto-inhibited state of P47PHOX prevents binding to the integral membrane subunits (P91PHOX and P22PHOX) (7, 18, 28). Upon activation, phosphorylation of the P47PHOX protein releases the auto-inhibition and permits translocation of the cytosolic subunits to the membrane where they together with CYT b558 and the small GTPase RAC (1/2) (29) form the fully assembled and activated complex (1, 57). Phosphorylation of the auto-inhibitory domain, the tandem SH3 domain, is exposed and allows P47PHOX to translocate to the plasma membrane and bind P22PHOX (11). We wanted to investigate if altered localization of the protein was the reason for the effect of the M153T alteration. First, we investigated if the binding of P47PHOX to P67PHOX was affected by the mutation. However, anti- P67PHOX co-immunoprecipitates analyzed by Western blotting with anti-P47PHOX antibodies showed no major differences in binding capacity between the two genotypes and the same was observed when P47PHOX was pulled down and the co-immunoprecipitated P67PHOX detected (Fig. 4a, b).
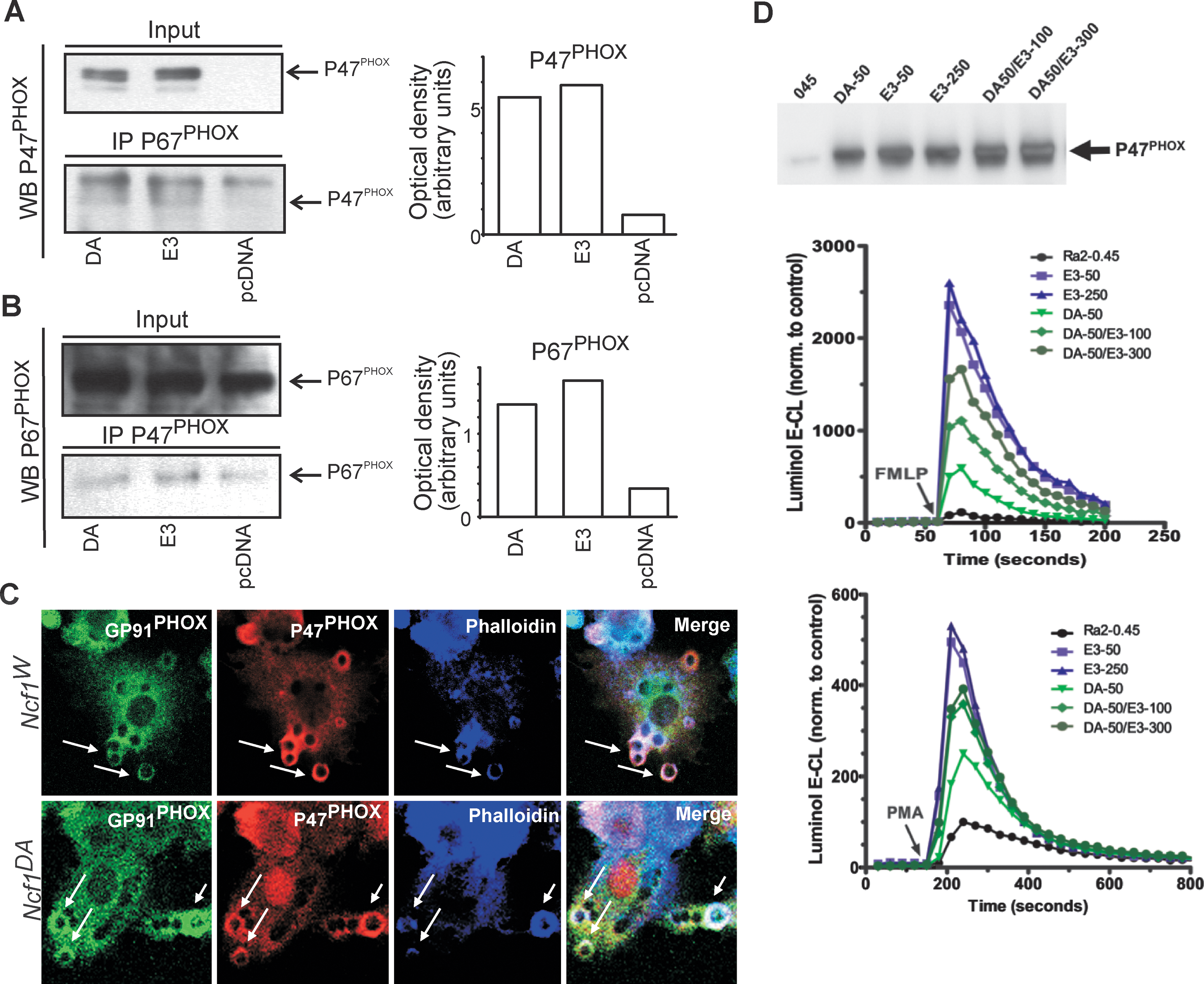
To investigate if the M153T alteration affected the capacity of P47PHOX to translocate to the membrane after activation, Ra2 microglia expressing either P47PHOX/E3 or P47PHOX/DA were allowed to phagocytose surface-bound zymosan particles (heat killed yeast particles) for up to 10 min at 37°C. After cell fixation, immunofluorescence was performed with anti- GP91PHOX (15) and anti-P47PHOX antibodies in combination with Alexa633-conjugated phalloidin (Fig. 4c). Phalloidin specifically binds F-actin and is used here to indicate recently formed phagosomes, as associated F-actin is shed rapidly (within minutes) postinternalization. However, P47PHOX/DA and P47PHOX/E3 were recruited to newly formed phagosomes (co-localizing with CYT b588), with similar kinetics and efficiency, suggesting that the negative effect of the M153T mutation on superoxide production is not due to altered capacity of P47PHOX for stimulus-induced redistribution and binding to the membrane. We found no differences in uptake of particles per cell between P47PHOX/DA and P47PHOX/E3-expressing cells (data not shown).
It is believed that continuous cycling of cytosolic NADPH oxidase subunits is necessary to sustain the oxidative burst (20, 61). It could therefore be speculated that P47PHOX/DA inhibits ROS production in the assembled NOX2 complex due to greatly reduced off-kinetics from the CYT b558 binding sites, effectively preventing new cytosolic subunits to exchange. A similar scenario was also proposed by Shen et al. where P47PHOX protein with glycines in positions 151–158 showed increased binding to liposomes (56). If so, expression of the P47PHOX/DA variant should be dominant negative as the CYT b558 sites available becomes saturated with this P47PHOX species. To test this hypothesis, P47PHOX/DA and P47PHOX/E3 were co-expressed in Ra2 microglia cells at different ratios and the effect on fMLP and PMA-induced superoxide release analyzed. The results, however, do not support a dominant negative role of P47PHOX/DA as P47PHOX/E3 over-expression returned the P47PHOX/DA response to near P47PHOX/E3 levels alone (Fig. 4d).
These observations support the idea that the decreased ROS production associated with P47PHOX/DA does not relate to compromised translocation to the membrane, but to an inherently diminished ability to support the catalytic mechanism of superoxide production in the assembled NOX2 complex.
Discussion
For the first time, we have identified a natural polymorphic quantitative trait nucleotide (QTN) in an earlier identified quantitative trait locus in a disease model for a complex disorder. The identified QTN replaces a threonine at position 153 in the P47PHOX protein with methionine, which leads to a reduced ROS response and increased arthritis severity.
The effect of a polymorphism at position 153 has been unclear as the hinge region between the PX and the SH3 domains for a long time was considered not to be of importance for the function of P47PHOX. Upon activation, P47PHOX is phosphorylated on several serine residues resulting in release of the auto-inhibitory conformation (1, 57) that prevents the cytosolic NOX2 complex subunits to bind CYT b558 and phosphoinositides in the membrane. As a consequence of phosphorylation, the subunits translocate to the membrane where the active NOX2 complex is formed together with the small GTPase RAC (1 or 2) (29). P47PHOX can move by itself to the membrane to interact with the cytochrome and/or phosphoinositides, but P67PHOX is recruited only in association with P47PHOX (31). P47PHOX is, thus, normally regarded to be an organizer of the complex and has been shown to be necessary for ROS production in cellular systems, even though ROS can be produced without involvement of P47PHOX in cell-free systems if P67PHOX and RAC are in excess (22, 40).
There are only a few studies investigating the linker region of P47PHOX that contains the Thr153. A snap-lock function of this region has been proposed, based on the fact that in a coupling model between SH2 and SH3 domains in the Src kinase, the linker region forms a snap-lock that locks the orientation of these two domains together (67). Lately, amino acids 151–158 were shown to be of importance for binding of the PX domain to membrane phosphoinositides and functional ROS production in a cell-free system, whereas region 143–150 did not affect either ROS production or binding to the membrane (56). The fact that neither association with P67PHOX nor redistribution of the P47PHOX protein is altered by the M153T amino acid substitution suggests that this region is not of importance for translocation to the enzymatic core at the membrane sites. The QTN might also alter binding of P47PHOX to the P22PHOX subunit in the membrane. However, the amino acid in position 153 has not been suggested to bind directly to P22PHOX (47).
The importance of this region is also strengthened by the fact that substitution of alanine to Ile152 is not affecting binding to P67PHOX or P22PHOX but inhibits ROS production from the NOX2 complex (60). This is interesting since P47PHOX is often proposed to function just as an adaptor protein to tether P67PHOX to P22PHOX. Our work, together with the work of Taura et al. (60), proposes a direct effect on the activation of the complex. As both serine and threonine, without detectable phosphorylation on threonine, enabled ROS production, a hydroxyl group in this position might be needed for full activity of NOX2 complex. Even though we could not detect phosphorylation on threonine, we cannot completely exclude the possibility that the moiety is phosphorylated upon activation and thus controls the activity of the NOX2 complex. Another possible post-translational modification is glycosylation, but Thr153 in P47PHOX/E3 is not predicted to be glycolysated (NetOGlyc 3.1, Center for Biological Sequence Analysis, Denmark). Our finding that also serine 153 is functional is supported by the fact that serine is found in this position in a P47PHOX ortholog (Supplementary Fig. S4).
Taken together, we have now conclusively identified a naturally occurring polymorphism in rats, resulting in low ROS production and lack of resistance to arthritis development. This polymorphism results in a T153M alteration in the hinge region, located between the PX and SH3 domains that has lately been suggested to be crucial for activation of the NOX2 complex (56, 60). This effect is not mediated by an altered translocation of the P47PHOX protein, but rather depend on the nature of the amino acid and possible conformational effect. Importantly, the identified QTN is highly polymorphic in the wild rat population and is located within the strongest locus controlling development of both models for RA (PIA and collagen-induced arthritis) (48) as well as for multiple sclerosis (experimental autoimmune encephalomyelitis) (10, 12). The increased risk of severe arthritis due to a reduced ROS production is certainly against previous postulations concerning the role of ROS on inflammation, but is now well validated (37). The most likely explanation involves a regulatory effect on autoreactive T cell activation. We hypothesize that ROS production during antigen presentation regulates the reactivity of the responding T cell toward that antigen by altering the T cells redox status (24, 25). A regulatory role of ROS is most presumably an evolutionary conserved mechanism, likely to operate also in other species, such as mice and humans. A corresponding polymorphism is not present in the mouse Ncf1 gene although the pathway has been shown to be functionally similar to rats. In humans, the NCF1 gene is in a region of high instability, with numerous deletions and duplications (5, 9). The human NCF1 gene has several functional and nonfunctional copies, which exists in varying frequencies in different populations (14, 32). Due to this complexity any large-scale genetic analysis, such as a genome-wide association scan, of NCF1 has been impossible. However, this large degree of genetic variability in the NCF1 gene and its duplicates could in numerous ways reduce the functionality of P47PHOX in much the same way as is seen in the rat (14, 19, 32). Two other genes of the NADPH oxidase complex have been found to be genetically associated with RA or other autoimmune conditions. We and others have reported associations of the NCF4 gene (encoding P40PHOX) with RA (51) and Crohn disease (53), and in a large-scale replication analysis, NCF2 (encoding P67PHOX) was found to be strongly associated with Systemic Lupus Erythematosus (23). This study, identifying the underlying QTN causing the Ncf1 effect, now opens a detailed analysis of the role of this important new pathway regulating autoimmune-mediated chronic inflammation.
Footnotes
Acknowledgments
We thank Carlos Palestro, Sandy Liedholm, Rebecka Ljunqvist, and Isabell Bohlin for taking excellent care of the rats. Supported by grants from the Swedish Association against Rheumatism, the Swedish Medical Research Council, the Swedish Foundation for Strategic Research, the Academy of Finland, Sigrid Juselius Foundation, Gigtforeningen, and King Gustav V's 80 year foundation as well as from the European Union grants MASTERSWITCH (HEALTH-F2-2008–223404) and EURATRANS (HEALTH-F4-2010–241504), and by the 6th Framework Programs of the European Union NeuroproMiSe (LSHM-CT-2005–01863) and AUTOCURE (LSHM-CT-2005–018661), the Academy of Finland, Sigrid Juselius Foundation, and King Gustav V's 80-year foundation.
Author Disclosure Statement
This publication reflects only the authors' views. The European Community is not liable for any use that may be made of the information herein. The founders had no role in study design, data collection, and analysis; decision to publish; or preparation of the article. M.H., P.O., and R.H. declare competing interests. No competing financial interests exist for the other authors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.