
Abstract
Modification of α-conotoxin frameworks through cyclization via an oligopeptide linker has previously been shown as an effective strategy for improving in vivo stability. We have extended this strategy by investigating cyclic analogs of α-conotoxin AuIB, a selective α3β4 nicotinic acetylcholine receptor (nAChR) antagonist, to examine a range of oligopeptide linker lengths on the oxidative formation of disulfide bonds, activity at nAChRs, and stability to degradation by chymotrypsin. Upon nondirected random oxidation, the ribbon isomer formed preferentially with the globular isomer occurring as a minor by-product. Therefore, a regioselective disulfide bond forming strategy was used to prepare the cAuIB-2 globular isomer in high yield and purity. The cAuIB-2 globular isomer exhibited a threefold decrease in activity for the α3β4 nAChR compared to wild-type-AuIB, although it was selective for α3β4 over α7 and α4β2 subtypes. On the other hand, the cAuIB-2 ribbon isomer was shown to be inactive at all three nAChR subtypes. Nonetheless, all of the cyclic analogs were found to be significantly more stable to degradation by chymotrypsin than wild-type AuIB. As such, the cAuIB-2 globular isomer could constitute a useful probe for studying the role of the α3β4 nAChR in a range of in vivo experimental paradigms. Antioxid. Redox Signal. 14, 65–76.
Introduction
The α-conotoxins are a class of conotoxins that demonstrate unique selectivities for different subtypes of nicotinic acetylcholine receptors (nAChRs) (3). nAChRs are ligand-gated pentameric ion channels constructed from five subunits. To date, 17 nAChR subunits have been cloned and are defined as α1-α10, β1-β4, δ-, γ-, and ɛ-subunits (36). Acetylcholine-mediated activations of receptors assembled by these subunits produces fast synaptic neurotransmission and modulates the release of additional neurotransmitters. Muscle-type nAChRs are composed of α1, β1, δ-, γ-, and ɛ-subunits, whereas neuronal nAChRs are composed of any homo- or heteromeric combination of five α2-α10 and β2-β4 subunits (25). The α4β2, α3β4, and α7 nAChRs are the predominant subtypes expressed in mammalian brain; however, a large number of other subunit combinations and stoichiometric ratios for neuronal nAChRs are available, thus underlining the therapeutic importance of developing subtype specific ligands for these receptors (44).
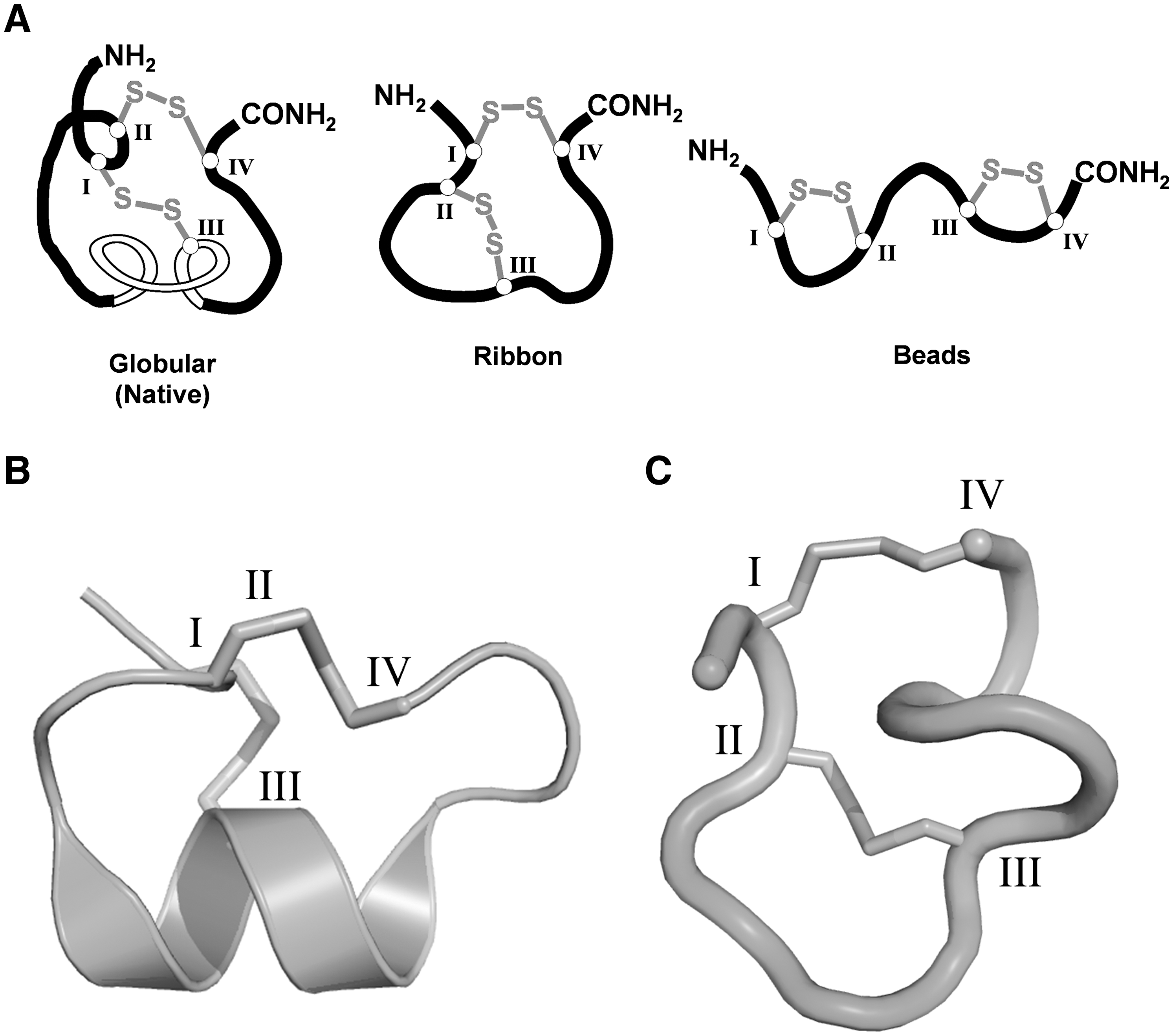
Although small in size (12–20 amino acids), α-conotoxins exhibit well-defined three-dimensional structures characterized by a 310-helical barrel that projects residues into the orthosteric binding site of the nAChR (38). Their highly rigid structural frameworks can be attributed to the presence of two highly conserved disulfide bonds that exist in a CysI-CysIII and CysII-CysIV (globular) arrangement (Fig. 1A). This gives rise to a framework of two intervening loops of hypervariable amino acids denoted m and n, respectively (19). The majority of α-conotoxins characterized to date that target neuronal nAChRs possess a 4/7 loop framework, although others continue to be characterized including 4/6 (AuIB), 4/4 (BuIA), and 4/3 (ImI, ImII, and RgIA) (8) (Fig. 2). In addition to the globular disulfide bond isomer, there are two other possible disulfide bond arrangements that can form, defined as the CysI-CysIV, CysII-CysIII (ribbon), and CysI-CysII, CysIII-CysIV (beads) isomers (Fig. 1A). Engineering α-conotoxins with these non-native arrangements results in analogs with very different structural and pharmacological properties (18, 21, 41).

α-Conotoxin AuIB is the only α-conotoxin identified to date that possesses the 4/6 loop framework. Structurally, AuIB resembles other α-conotoxins that possess the 4/7 loop framework (Fig. 2) (10). AuIB is a specific antagonist of the neuronal α3β4 nAChR, making it a useful tool for probing the physiological functions maintained by this nAChR subtype (37). Antagonists of α3β4 nAChRs have been shown to decrease nicotine self-administration in rats, suggesting that this receptor is also a potential target for treating nicotine dependence (23, 43). As such, the discovery of new α3β4 nAChR antagonists could have profound implications as in vivo research tools in the development of novel smoking cessation treatments.
AuIB blocks nicotine-stimulated hippocampal norepinephrine release but not striatal dopamine release, demonstrating that AuIB is able to discriminate among different native nAChR subtypes (37). Further, the ribbon isomer of AuIB has been found to be 10-fold more active for the α3β4 nAChR than the native globular isomer (18), although it has also been reported that significant differences in activity between the two isomers exist between native and oocyte-expressed α3β4 receptors (41). The ribbon isomer exhibited reduced structural definition when compared with the globular isomer (Fig. 1B, C). As such, observed differences in pharmacological activity can be attributed to the increased flexibility of the ribbon isomer, which would be more readily able to adopt the biologically active conformation upon receptor binding (18).
Despite their potential as drug candidates and their application as important pharmacological tools, α-conotoxins like many other classes of peptides exhibit poor in vivo stability and short half-lives as a result of their inherent vulnerability to enzymatic degradation, limiting their potential for use in in vivo experiments and as drug candidates (31). Moreover, while conotoxins contain multiple disulfide bonds that restrict their conformation, the disulfide bonds themselves are not entirely stable in the presence of physiological reducing agents such as glutathione and serum albumin (5). N-to-C cyclization of peptides is one method commonly used to stabilize the conformation of many classes of peptides in solution to enhance their in vivo stability (1, 22, 28). Further, there is a growing interest in a class of peptides known as cyclotides, which possess multiple disulfide bonds in addition to their circular backbone that exhibit phenomenal stability under a variety of stress conditions (13, 14).
In view of the superior stability of cyclic peptides over linear peptides, N-to-C cyclization of α-conotoxins is a feasible approach to improve their in vivo stability, a principle that has been demonstrated previously with the synthesis of cyclic α-conotoxin MII and χ-conotoxin MrIA analogs (11, 34). In these studies, the cyclic conotoxin analogs were found to possess greater resistance to proteolysis, as well as enhanced stability in human plasma, yet the analogs retained the biological activity and the three dimensional conformations when compared to the native conotoxin. It was recently reported that N-to-C cyclization of α-conotoxin ImI through short oligopeptide linker units (1–3 amino acids) showed a clear preference for the formation of the non-native ribbon disulfide bond isomer (6). Although each of these studies was successful in producing α-conotoxin analogs with enhanced stability, a limited number of linker sizes were investigated (6, 11, 34).
In this report, we describe the synthesis, pharmacological characterization, and stability of a series of N-to-C cyclic analogs of α4/6-conotoxin AuIB. Given that the ribbon isomer of α-conotoxin AuIB displays a higher antagonistic potency for α3β4 nAChRs (18), we reasoned that N-to-C cyclization through short oligopeptide linker units would result in the spontaneous formation of the ribbon isomer and could possibly lead to α3β4 nAChR antagonists with enhanced in vivo stability. In comparison to previous studies concerning cyclized conotoxin analogs (6, 11, 34), our study investigates a wider range of linker lengths of between one and seven amino acid residues, and assesses their influence on the formation of disulfide bond isomers, in vitro stability and biological activity. In a complementary study, Lovelace et al. also synthesized a series of cAuIB derivatives, which focused on linkers of 4–7 amino acid residues, with additional structural characterization by nuclear magnetic resonance (NMR) spectroscopy (35). The findings of their study are broadly consistent with those reported in this article.
Materials and Methods
Chemistry
Materials
Reagents and materials were purchased from commercial suppliers and used without further purification. Protected amino acid derivatives and 4-methylbenzhydrylamine (MBHA) resin were purchased from ChemImpex (Wood Dale, IL) and S-tritylmercaptopropyl MBHA resin from Peptides International (Louisville, KY). 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate was purchased from ChemPep (Miami, FL).
Liquid chromatography mass spectrometry analysis
Liquid chromatography mass spectrometry (LC-MS) spectra were recorded using a Shimadzu 2010EV liquid chromatography mass spectrometry system (Kyoto, Japan) equipped with a photo diode array detector. A Phenomenex Jupiter column (C18, 5 × 0.46 cmID, 5 μm) (Torrance, CA) was used to achieve analytical separations. Gradients of 10% aqueous MeCN + 0.05% formic acid (buffer A) and 90% aqueous MeCN + 0.046% formic acid (buffer B) were employed. Preparative reversed phase high performance liquid chromatography (RP-HPLC) was performed using a Shimadzu LC8A preparative system. A Phenomenex Luna preparative RP-HPLC column (C18, 15 × 2.5 cmID, 10 μm) was used to achieve chromatographic separations. Gradients of 10% aqueous MeCN + 0.5% trifluoroacetic acid (TFA) (buffer A) and 90% aqueous MeCN + 0.5% TFA (buffer B) were employed.
LC-MS/MS spectra were recorded using Agilent 1200 series solvent delivery system equipped with an auto injector coupled to an Agilent 6410 triple quadrupole mass spectrometer equipped with an electrospray ionization source. Gradients of 10% aqueous acetonitrile + 0.05% formic acid (buffer A) and 90% aqueous acetonitrile + 0.046% formic acid (buffer B) were employed with a flow rate of 0.75 ml/min. A Zorbax column (C18 column, 2.1 × 0.50 cmID, 2 μm) was used.
Peptide synthesis
All peptides were assembled in parallel using the tea bag methodology as previously described (24), employing 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate activation of N α-tert-butyloxycarbonyl protected amino acids with in situ neutralization chemistry (42). The following amino acid protecting groups were used: Cys, 4-methylbenzyl; Asp, O-cyclohexyl; Asn, xanthyl; Thr, benzyl; Tyr, 2-bromobenzyloxycarbonyl; Ser, benzyl. The directed disulfide bond synthesis of cAuIB-2 utilized acetomidomethyl (Acm) protection at positions Cys2 and Cys7 for the globular isomer, or Cys2 and Cys14 for the ribbon isomer in the linear thioester precursor peptides. Native α-conotoxin AuIB and Ac-AuIB were assembled on MBHA resin, and thioester peptide precursors were assembled on S-tritylmercaptopropyl MBHA resin. The N-terminal of Ac-AuIB was acetylated using a mixture of acetic anhydride/diisopropylethylamine/dimethylformamide (1:1:8), (2 × 30 min) before cleavage. Cleavages were performed by treating each tea bag containing the peptide-resin with 5 ml of HF/p-cresol (9:1) (v/v) for 2 h at 0°C. After evaporation of the HF under a stream of nitrogen, the crude peptide thioesters were precipitated and washed with cold methyl-tert-butylether ether (2 × 10 ml), filtered, and lyophilized from 95% aqueous acetic acid and again from 50% aqueous acetonitrile + 0.1% TFA.
The crude linear reduced thioester peptides (30 mg) were shaken in aqueous 0.1 M phosphate buffer pH 8.2 (50 ml) in an open flask and the oxidations monitored by LC-MS analysis to ensure reaction completion. The mixtures were acidified to pH 2 with TFA and each peak was isolated to >95% purity by preparative RP-HPLC. The second disulfide bond in cAuIB-2 was formed by treating the partially protected/oxidized cyclic peptide (10 mg) in 10 mM HCl containing 80% aqueous methanol (25 ml) with ∼10 equivalents of I2 per Acm group. The mixture was stirred for 5 min under an inert N2 atmosphere and the reaction quenched by drop-wise addition of 0.1M Na2O3S2 until colorless. The fully oxidized peptide was loaded onto a preparative RP-HPLC column by direct infusion and eluted and fractionated accordingly using a linear gradient to >95% purity.
Circular dichroism (CD) spectra were recorded on a Jasco J-720 spectropolarimeter (Easton, MD) at ambient temperature in the low UV range (190–260 nm) using a quartz cell with a 1-mm path length. Samples were dissolved in 50 mM phosphate buffer, pH 7.2 at a concentration of 30 μM. Each spectrum represents an average of 5 scans (scan speed 50 nm/min).
Disulfide bond determination
A modified reduction/alkylation strategy based on the method of Clark et al. was used to determine the disulfide bond connectivity of cAuIB-1 (11). Partial reduction was achieved by incubating the peptide (50 μg in 0.2M citrate buffer (pH 3, 50 μl) with 50 equivalents of tris-(2-carboxyethyl)phosphine hydrochloride (TCEP) at 25°C for 5 min, after which it was injected onto RP-HPLC and fractionated using a linear gradient. Fractions were analyzed by LC-MS and those containing the partially reduced conotoxin (+2 amu) were pooled, and an equal volume of 50 mM N-ethylmaleimide in n-propanol was added and incubated at 37°C for 1 h to alkylate the free thiol groups on cysteine residues. The partially reduced/alkylated conotoxin was then isolated by RP-HPLC and remaining disulfide bonds were reduced by treatment with 50 equivalents of TCEP in 0.2 M citrate overnight at 37°C, isolated by RP-HPLC, and lyophilized.
The reduced/alkylated/cyclic peptide was dissolved in 50 mM tris buffered to pH 7.4 (50 μl) and 1 μl of a solution of bovine pancreatic α-chymotrypsin (Sigma-Aldrich, St. Louis, MO) (1 mg/ml) dissolved in 50 mM tris buffered to pH 7.4 was added. The peptide was incubated overnight at 37°C and the reduced/alkylated/linear peptide was analyzed by LC-MS/MS to identify location of the two alkylated cysteine residues in the amino acid sequence.
Chymotrypsin stability assay
A solution containing the α-conotoxin AuIB analog in 50 mM tris buffer, pH 7.4 (0.5 μM), was prepared, together with a stock solution of chymotrypsin in 50 mM tris, pH 7.4 (1 mg/ml). Both solutions were preincubated to 37°C, before 5 μl of the chymotrypsin stock solution was added to 200 μl of each conotoxin analog stock solutions. About 20 μl aliquots were removed and quenched with 10 μl of 5% TFA/acetonitrile at successive time intervals. Stability experiments were performed in triplicate and each sample was analyzed by LC-MS. Percentage degradation was determined by measuring the ratio of the peak height of the proteolytically cleaved conotoxin and the residual conotoxin peak.
Pharmacology
Materials
Culture media, serum, antibiotics, and buffers for cell culture were obtained from Invitrogen (Paisley, United Kingdom). ACh was purchased from Sigma (St. Louis, MO) and epibatidine from Tocris (Bristol, United Kingdom). The rα3β4-HEK293 (46), mα4β2-HEK293T (27), and hα7-GH3 cell lines (20) were generous gifts from Drs. Y. Xiao and K. Kellar (Georgetown University School of Medicine, Washington, DC), Dr. J.A. Stitzel (University of Colorado, Boulder, CO), and Dr. D. Feuerbach (Novartis Institutes of Biomedicinal Research, Basel, Switzerland), respectively.
Cell culture
The cell lines used in this study were cultured at 37°C in a humidified 5% CO2 incubator. The cells were maintained in the culture medium (Dulbecco's modified Eagle's medium supplemented with penicillin [100 U/ml], streptomycin [100 μg/ml], and 10% fetal bovine serum). The rα3β4-HEK293 cell line was maintained in the culture medium supplemented with 1 mg/ml G-418, the hα7-GH3 cell line in the culture medium supplemented with 0.1 mg/ml G-418, and the mα4β2-HEK293T cell line in the culture medium supplemented with 0.1 mg/ml zeozin and 0.5 mg/ml hygromycin.
Functional assays
The functional properties of the AuIB analogs as nAChR antagonists were characterized by using the Ca2+/Fluo-4 assay for the hα7-GH3 and rα3β4-HEK293 cell lines and by using the FLIPR™ Membrane Potential (FMP) Blue assay for the mα4β2-HEK293T cell line. The cells were split into poly-
In the Ca2+/Fluo-4 assay, the medium was aspirated and the cells were incubated in 50 μl loading buffer (Hank's buffered saline solution containing 20 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, and 2.5 mM probenecid, pH 7.4, supplemented with 6 mM Fluo–4/AM [Molecular Probes, Eugene, OR]) at 37°C for 1 h. The loading buffer was aspirated, the cells were washed once with 100 μl assay buffer (Hank's buffered saline solution containing 20 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, and 2.5 mM probenecid, pH 7.4), and then 100 μl assay buffer containing various concentrations of the AuIB analogs was added to the wells. For the experiments with α7-GH3 cell line, the assay buffer was supplemented with 100 μM genistein. After a 30 min incubation at 37°C in a humidified 5% CO2 incubator, the 96-well plate was assayed in a NOVOstar™ microplate reader (BMG Labtechnologies, Offenburg, Germany) measuring emission (in fluorescence units) at 520 nm caused by excitation at 485 nm before and up to 60 s after addition of 33 μl agonist solution (the agonists were dissolved in assay buffer).
In the FMP assay, the medium was aspirated, and the cells were washed with 100 μl Krebs buffer (140 mM NaCl/4.7 mM KCl/2.5 mM CaCl2/1.2 mM MgCl2/11 mM HEPES/10 mM
Results
Design and synthesis of analogs
A series of seven cyclic analogs of AuIB were synthesized by incorporating variable length peptide linker units consisting of one to seven amino acid residues to link the N- and C-termini to assess their impact on the random formation of disulfide bond isomers under oxidative condition, as well as their in vitro stability and nAChR activity (Fig. 3). Analogs described in this study are named with respect to their linker length; for example, cAuIB-2 refers to the cyclic AuIB analog with a two residue linker. Linkers were designed using functionally inert Ala and Gly residues, since these residues were not expected to interfere with receptor binding. Moreover, the linker has the potential to serve as an anchor point in future studies to attach various chemical moieties that could potentially enhance the pharmacokinetic properties of these molecules (17). In addition to the cyclic analogs, an N-terminal acetylated AuIB analog (Ac-AuIB) was also prepared to assess the requirement for an N-terminal charge.

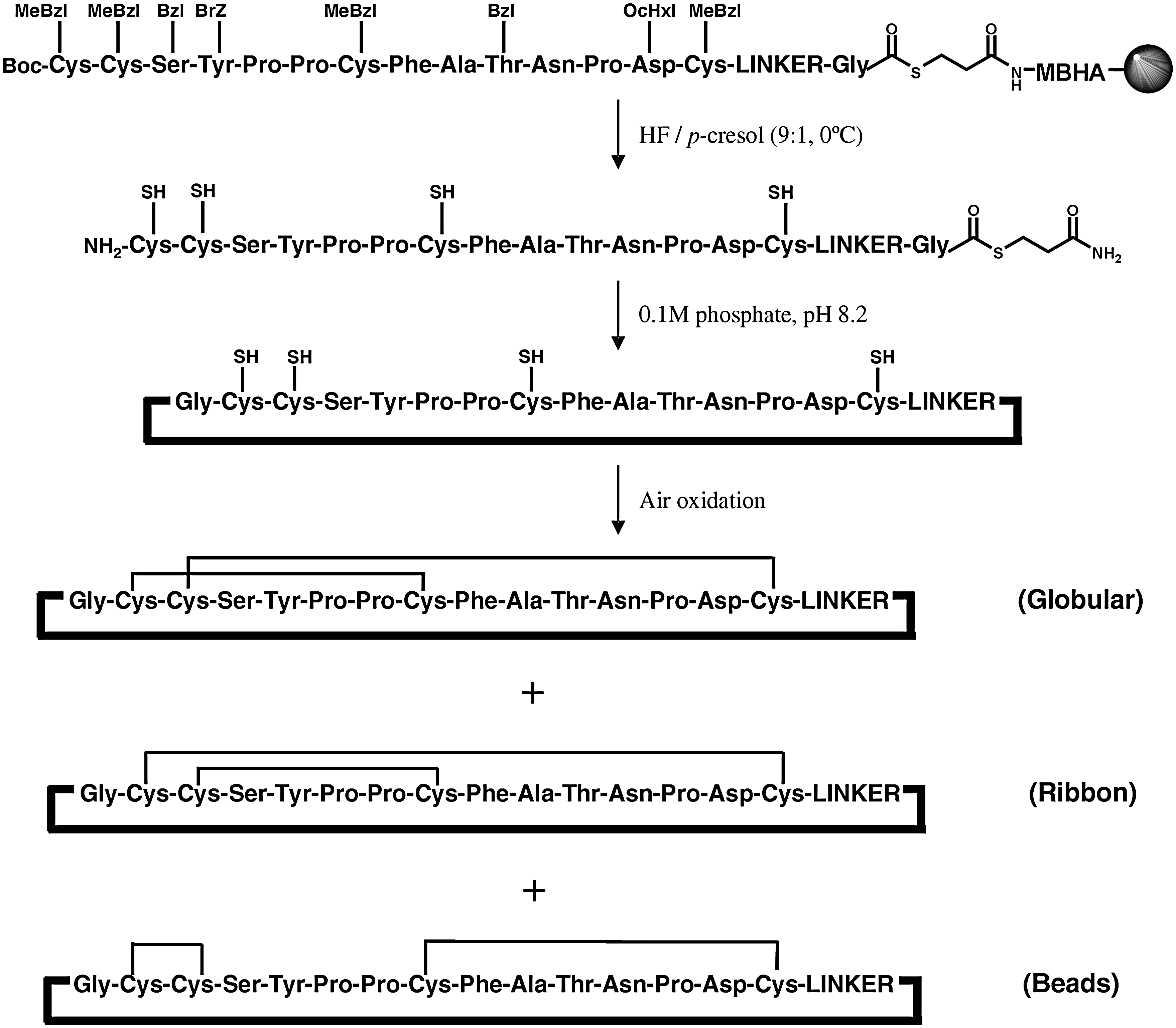
An intramolecular native chemical ligation reaction was used to perform the N-to-C cyclization (9, 16). Conveniently, α-conotoxins contain multiple cysteine residues that give rise to four ligation sites. The least sterically hindered Gly-Cys site was selected and the linear thioester precursors were assembled accordingly (Fig. 4). An S-mercaptopropionamide thioester was used at the C-terminal and the linear thioester peptide precursors were conveniently assembled in parallel using a modified procedure with polypropylene tea bags (7), employing tert-butyloxycarbonyl in situ neutralization chemistry (42). After chain assembly, the peptides were cleaved from the resin using HF/p-cresol (9:1) to afford the crude linear thioester precursors. The identities and purity of the synthetic thioesters were confirmed by LC-MS analysis (Table 1), and the peptides were used in the subsequent cyclization step without further purification.
Values are given for the linear reduced and linear oxidized conotoxins, respectively.
Ac, acetyl; WT, wild-type.
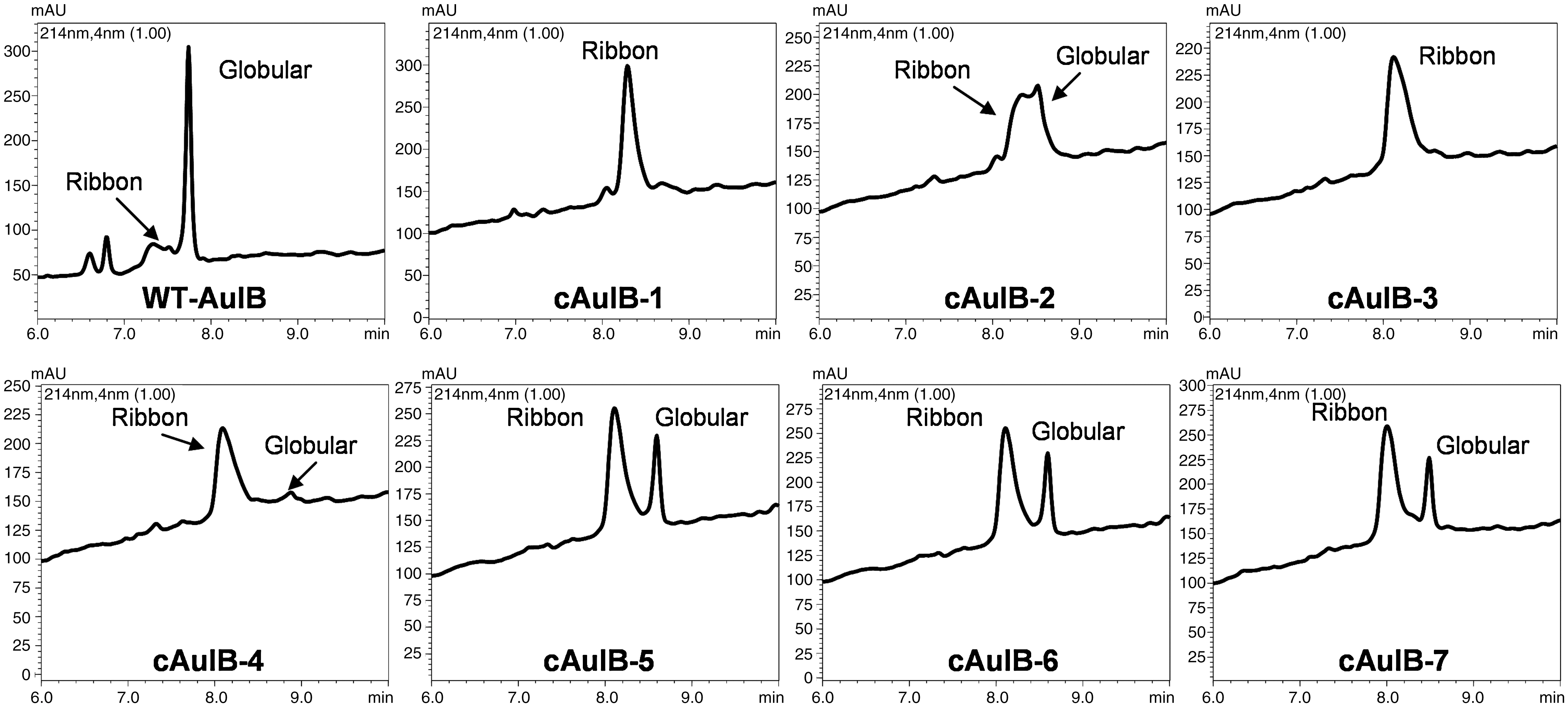
Cyclization and random oxidation of the seven peptide thioesters was performed by stirring the crude linear thioester precursors in aqueous 0.1 M phosphate buffer, pH 8.2. Although reducing reagents such as thiophenol or 2-mercaptoethanesulfonic acid are often included in native chemical ligation reactions to catalyze the formation of more reactive thioesters (15), they were excluded in our synthesis to generate an oxidizing environment, which allows the in situ formation of the disulfide bonds. This one pot approach was advantageous, since isolation of the intermediates was not required. The cyclization was monitored by LC-MS (Table 1), and the N-to-C cyclization step occurred very rapidly (ca. <1 min) for all analogs. On the other hand, complete oxidation of the cysteine residues is much slower, ranging from 2 to 3 days with cAuIB-5, -6, and -7 requiring longer reaction times. Although cAuIB-1, -3, and -4 predominantly yielded one major disulfide bond isomer, cAuIB-2, -5, -6, and -7 formed one additional disulfide bond isomer (Fig. 5), which in the case of cAuIB-5, -6, and -7 were isolated to >95% purity by preparative RP-HPLC. However, the minor peaks of cAuIB-5, -6, and -7 were isolated in very small yields. Further, the two disulfide isomers of cAuIB-2 were not readily separated by RP-HPLC; thus, a directed disulfide bond forming strategy utilizing orthogonal cysteine protecting groups was required to obtain both isomers separately (Fig. 6). In this strategy, an additional iodine-mediated oxidation/deprotection of the Acm protected cysteine residues was used to form the second disulfide bond after the initial one pot cyclization/oxidation step. Subsequent purification by preparative HPLC yielded both isomers in high yield and purity, with each isomer exhibiting distinctly different analytical LC-MS retention times (Fig. 7). Co-injection of both globular and ribbon isomers of cAuIB-2 confirmed that both products were indeed different, allowing for the clear characterization of both isomers obtained using random oxidation strategy.
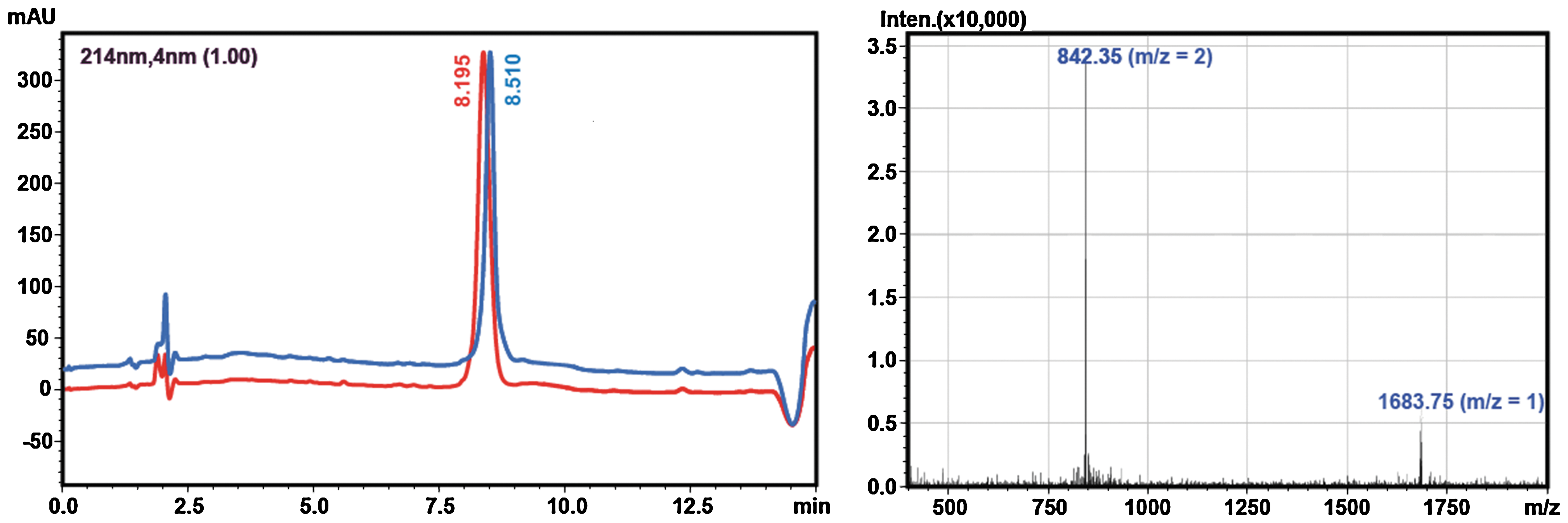
Determination of disulfide bond connectivity
To unambiguously confirm the disulfide bond connectivity of cAuIB-1, a modified reduction/alkylation strategy was used as previously described (11). The cyclic oxidized conotoxin was partially reduced with TCEP in acidic buffer to avoid scrambling of the disulfide bonds, followed by alkylation of the partially reduced peptide with N-ethylmaleimide. The cyclic peptide was then treated with α-chymotrypsin to cleave the cyclic backbone, followed by full reduction of the second disulfide bond using TCEP. The resulting linear peptide was sequenced using LC-MS/MS, which confirmed the location of the N-ethylmaleimide groups as being Cys3 and Cys8. Hence, cAuIB-1 was designated as the ribbon isomer.
We have previously used CD spectroscopy to characterize the secondary structure and assign the disulfide connectivity of α-conotoxins (Fig. 8) (2). Analogs that exhibit overlapping CD spectra can be assumed to share the same folding characteristics and disulfide bonding. As the cAuIB-2 globular and ribbon isomers were obtained separately using an orthogonal disulfide bond synthesis strategy, these were used to assist in identifying the disulfide connectivity of other isomers. Wild-type (WT)-AuIB exhibited a CD spectrum typical of a peptide with helical elements, with characteristic minima occurring at 222 nm (26). The cAuIB-2 globular isomer exhibited increased helical content compared to WT-AuIB, whereas the cAuIB-2 ribbon isomer displayed significantly less helical content, suggesting a more random structure. A similar trend was observed for each of the two isolated isomers of cAuIB-5, -6, and -7, allowing identification of each isolated product as either the globular or ribbon isomer (Fig. 8).
Pharmacological characterization
The functional properties of WT-AuIB and the AuIB analogs were characterized at three major neuronal nAChR subtypes, the α3β4, α7 and α4β2, using two fluorescence-based screening assays (Ca2+/Fluo-4 and FMP Blue assays) (Fig. 9). We and others have previously found that these fluorescence-based functional assays are in agreement with reported electrophysiological recordings (20, 46). WT-AuIB inhibited the α3β4 nAChR signaling elicited by 100 nM epibatidine with an IC50 value of 9.1 μM, whereas it did not display any significant inhibition of α4β2 and α7 nAChR at concentrations up to 300 μM. The antagonist activity displayed by WT-AuIB at the α3β4 nAChR in the assay was somewhat lower (∼10-fold) than previously reported for the α-conotoxin at the nAChR expressed in Xenopus oocytes (37, 41). This difference is likely to arise from the different expression systems and assays used. Ac-AuIB exhibited slightly lower antagonist potency (17 μM) than WT-AuIB (Fig. 9). The cAuIB-2 globular isomer was the only cyclic analog that displayed activity at the α3β4 nAChR, exhibiting an IC50 of 24 μM (Fig. 9), which represents a 2.6-fold decrease in inhibitory activity compared to WT-AuIB. In contrast, all cyclic ribbon isomer AuIB analogs displayed an IC50 > 300 μM at the α3β4 nAChR subtype and therefore were defined as inactive. Further, none of the cyclic analogs displayed significant inhibitory activities at the α7 or α4β2 nAChR subtypes. As such, the cAuIB-2 globular isomer displays significant selectivity for the α3β4 nAChR over the α4β2 and α7 subtypes.

Chymotrypsin stability
Enzymatic degradation was used to assess the stability of the ribbon isomer of each analog. AuIB contains a chymotrypsin cleavage site, specifically the C-terminal of Phe9; thus, its resistance to proteolysis can be used as a marker for peptide stability. Each analog was incubated with chymotrypsin at 37°C in 50 mM tris buffer at pH 7.4 and their hydrolysis monitored at various time intervals by LC-MS (Fig. 10). All of the cyclic analogs demonstrated an improved stability compared to WT-AuIB. However, the degree of stability correlates with linker length in the ribbon isomers, with cAuIB-3, -4, -5, -6, and -7 possessing significantly greater proteolytic resistance. Although cAuIB-1 (92% degradation after 48 h) exhibited no significant increase in stability compared to WT-AuIB (86% degradation after 48 h), the globular and ribbon isomers of cAuIB-2 both exhibited significantly increased stability compared to WT-AuIB; however, the globular isomer was found to be slightly less stable than the ribbon isomer (60% and 46% degradation after 48 h, respectively).

Discussion
The approval of Prialt by the U.S. Food and Drug Administration in 2004 as an intrathecal analgesic (39), together with the development of other conotoxin drug leads that are currently undergoing clinical trials for the treatment of neuropathic pain, has aroused much interest in the therapeutic potential of conotoxins for treating a wide range of neuropathological conditions (32). As such, structural templates that improve the pharmacokinetic properties of conotoxin drug leads are required to enhance their chemical and biological stability for use as in vivo probes and as drugs leads. Cyclization of conotoxins through their N- and C-termini has previously been shown to improve their stability under a range of biological conditions (6, 11, 34). Previous studies have shown that a shorter linker length linking the N- and C-termini can promote the formation of the ribbon disulfide bond isomer of α-conotoxins (6). Given that the ribbon isomer of α4/6-conotoxin AuIB has been shown to possess a 10-fold greater potency as an antagonist at native α3β4 nAChRs than the globular isomer (18), we investigated the relationship between linker length on disulfide bond formation, conformation, stability, and biological activity of N-to-C cyclic analogs of AuIB at nAChRs.
Seven analogs that incorporated between one to seven amino acid linker units to link the N- and C-termini of AuIB were used in this study to investigate the impact of linker length on the formation of disulfide bond isomers, stability, and pharmacological activity. Parallel assembly of the linear thioester precursors using polypropylene tea bags allowed rapid access to multiple cyclic conotoxin analogs. A one-pot cyclization/oxidation utilizing an intermolecular native chemical ligation reaction was used to prepare the cyclic conotoxins as reported previously (6, 11), and the formation of disulfide bond isomers was monitored by LC-MS. Linker length was found to have a major influence on the formation of disulfide bond isomers, with a clear preference for the formation of the ribbon isomer occurring in analogs with shorter linker lengths. However, the globular isomer was formed as a minor isomer in cAuIB-5, -6, and -7. The beads isomer was not observed for any of the analogs in this study. The globular isomer of cAuIB-2 formed along with the ribbon isomer in a 1:1 ratio, although these proved difficult to isolate by RP-HPLC. As such, both isomers of cAuIB-2 were synthesized separately using an orthogonal cysteine protecting group strategy, allowing sufficient quantities to be readily obtained in high yield and purity for pharmacological characterization.
CD spectroscopy of the ribbon and globular isomers of cAuIB-2, synthesized using regioselective control of disulfide bonds, allowed a convenient means for identifying different disulfide bond isomers obtained synthetically in the random oxidation strategy. Although the ribbon isomer of WT-AuIB has been found to possess greater activity at the native α3β4 nAChRs (18), none of the cyclic ribbon isomers in this study were shown to possess any significant activity for the α3β4 nAChR. This may be due to the restricted conformational mobility of the cyclic AuIB ribbon isomer analogs compared to the WT-AuIB ribbon isomer, which was previously found to possess a flexible backbone solution structure and would readily adopt the active conformation upon receptor binding (18). On the other hand, the cAuIB-2 globular isomer exhibited a moderate decrease in antagonist potency for the α3β4 nAChR compared to WT-AuIB. This observation can be rationalized by absence of an N-terminal charge group, where the activity of Ac-AuIB was also lower for the α3β4 nAChR than WT-AuIB. In this regard, the cAuIB-2 globular isomer exhibits comparable activity to Ac-AuIB. From a drug design perspective, the modest decrease in activity at the α3β4 nAChR displayed by cAuIB-2 would most likely be offset by the significantly improved in vivo stability. Moreover, cAuIB-2 globular isomer was shown to be selective for this receptor subtype over the α7 and α4β2 subtypes, thus may find potential use as a stable probe for studying the role of α3β4 nAChR in in vivo animal models.
Although cAuIB-2 was the only analog possessing the globular disulfide connectivity that was isolated in sufficient yield for pharmacological characterization, cAuIB-5, -6, and -7 globular isomers may exhibit similar pharmacological properties. In the case of cyclic analogs of α4/7-conotoxin MII, a linker of at least six residues was required to maintain structure and activity for the α3β2 nAChR (11). We have found that in AuIB, a two-residue linker can yield analogs that possess a moderate decrease in activity compared to the native conotoxin. We speculate that this is the result of the restricted flexibility of the smaller n loop in AuIB, which has one less residue and may result in a tighter backbone fold compared to the 4/7 α-conotoxins. As such, precise fine-tuning of the linker unit is required for each different α-conotoxin loop spacing to minimize perturbing the three-dimensional structure (10). However, full characterization of cyclic AuIB analogs by NMR spectroscopy is required, which has been the subject of a separate investigation by Lovelace et al. (35).
Resistance to proteolytic cleavage by α-chymotrypsin was used as a model to determine the in vitro stability of the analogs. Although each of the cyclic analog ribbon isomers exhibited improved resistance to proteolytic cleavage by chymotrypsin, longer linker lengths (i.e., cAuIB-3, -4, -5, -6, and -7) clearly demonstrate greater stability. In contrast, cAuIB-1 exhibited no significant increase in stability when compared to WT-AuIB (11). Although the ribbon isomer of WT-AuIB was not tested in the chymotrypsin stability assay, a comparison of the stability of the globular and ribbon isomers of cAuIB-2 shows that the globular isomer is moderately less stable to chymotrypsin degradation than its corresponding ribbon isomer. This result is somewhat surprising considering that the globular isomers of cyclic ImI analogs were previously shown to be more stable than the corresponding cyclic ribbon isomers (6). Nonetheless, when compared to WT-AuIB and cAuIB-1 (ribbon), the cAuIB-2 globular isomer exhibited significantly improved stability under the same conditions, with ∼60% degradation occurring over 48 h.
In summary, we have demonstrated that N-to-C cyclization of α-conotoxin AuIB is a valuable means for achieving improved stability while maintaining biological activity and promises to be a useful strategy in the application of designing drug leads based on other novel α-conotoxin frameworks. Given the improved stability of the cAuIB-2 globular isomer compared to WT-AuIB, this analog could find use as a probe for studying the role of α3β4 nAChRs in a number of in vivo experimental paradigms.
Footnotes
Acknowledgments
C.J.A. acknowledges a postdoctoral grant from the Drug Research Academy, The Faculty of Pharmaceutical Sciences, University of Copenhagen, and research funding from the State of Florida. A.A.J. was supported by the Lundbeck Foundation and the Danish Medical Research Council. Drs. Xiao, Kellar, Stitzel, and Feuerbach are thanked for their generous gifts of the nAChR-expressing cell lines.
Author Disclosure Statement
No competing financial interests exist.