
Abstract
Autophagy is a highly orchestrated cellular process by which proteins and organelles are degraded via an elaborate lysosomal pathway to generate free amino acids and sugars for ATP during metabolic stress. At present, the exact role of autophagy in the heart is highly debated but suggested to play a key role in regulating cell turnover in cardiomyopathies and heart failure. The signaling pathways and molecular effectors that govern autophagy are incomplete, as are the mechanisms that determine whether autophagy promotes or prevents cell death. The mitochondrion has been identified as a key organelle centrally involved in regulating autophagy. Certain members of the Bcl-2 gene family, including Beclin-1, Bcl-2 nineteen kilodaltons interacting protein (Bnip3), and Nix/Bnip3L, provoke mitochondrial perturbations leading to permeability transition pore opening, resulting in apoptosis, autophagy, or both. These and other aspects of autophagy processes have been discussed. Antioxid. Redox Signal. 14, 2245–2250.
Introduction
Autophagy
Autophagy is a cellular catabolic process that involves the breakdown of long-lived proteins and damaged organelles via an elaborate lysosomal-mediated pathway. In this process, the formation of a double-membrane structure called the autophagosome sequesters ubiquitinated protein aggregates or organelles such as mitochondria, endoplasmic reticulum, or other cytoplasmic constituents for degradation. Autophagy occurs in mammalian cells by three main pathways, microautophagy, chaperone-mediated autophagy, and macroautophagy, the latter being the most common form of autophagy and we will refer to it hereafter as autophagy (14). In the context of the heart, autophagy occurs under normal physiological conditions and represents a major pathway by which cells rid themselves of damaged proteins or macromolecular structures. Indeed, the proteasome is crucial for protein degradation and protein elimination, whereas autophagy appears to be more of a homeostatic process by which cells generate sufficient nutrients for ATP production and cell survival. However, defects in its activation or suppression result in cardiomyopathy (20, 36, 37). Notably, autophagy is rapidly induced by cellular stresses such as nutrient deprivation and hypoxia, presumably as a survival mechanism to recycle organelles and proteins as nutrients.
Despite the obvious beneficial effects, there is increasing awareness that defects in regulatory pathways that govern autophagy may be centrally involved and contribute to several human pathologies including cancer and neurodegenerative diseases [reviewed in refs. (19, 38)]. Central to this issue, new emerging evidence including data from our laboratory suggests that autophagy may also play a critical role in cardiovascular disease. Notably, autophagy has been detected in a number of cardiac pathologies including hypoxia (22), ischemia followed by reperfusion, and myocardial infarction and, more recently, in patients with end-stage heart failure (24, 29). This is best exemplified by Danon disease, which results in the accumulation of autophagosomes from a defect in the lysosome-associated membrane protein gene (LAMP-2), a membrane protein important for autophagosome–lyosome fusion reviewed in refs. (7, 33, 35). Despite the occurrence of autophagy in a variety of cardiac pathologies, its significance is undetermined with even less known of the molecular mechanisms that regulate autophagy under normal or disease conditions. Hence, whether autophagy per se is adaptive, maladaptive, or indifferent in the context of normal and failing remains to be elucidated.
Regulation of Autophagosome Formation
The autophaghic process begins with the formation of an intracellular membrane fragment referred to as a phagophore that can be either synthesized de novo or derived from the other membranous structures within the cell (11, 30). Notably, the autophagy (Atg) proteins first identified in yeast, Atg5, Atg12, and Atg16, are essential for the early membrane formation and engulfment of intracellular targets including damaged mitochondria, pathogens, and macromolecules (13, 21, 27, 40). Microtubule-associated protein light chain (MAPLC3) is cleaved by the protease activity of Atg4, with the subsequent LC3-I becoming lipidated with phosphatidylethanolamine to form LC3-II (10, 12). LC3-II is recruited to the preautophagosomal membrane, where it associates with the Atg8 homolog gamma-butyric acid receptor–associated protein to form the mature autophagolysosome. It is within this final structure the degradative lysosomal enzymes catabolize the autophagosome, liberating amino acids and other small molecules that can be used for further anabolic processes (8, 17).
Molecular Regulation of Autophagy
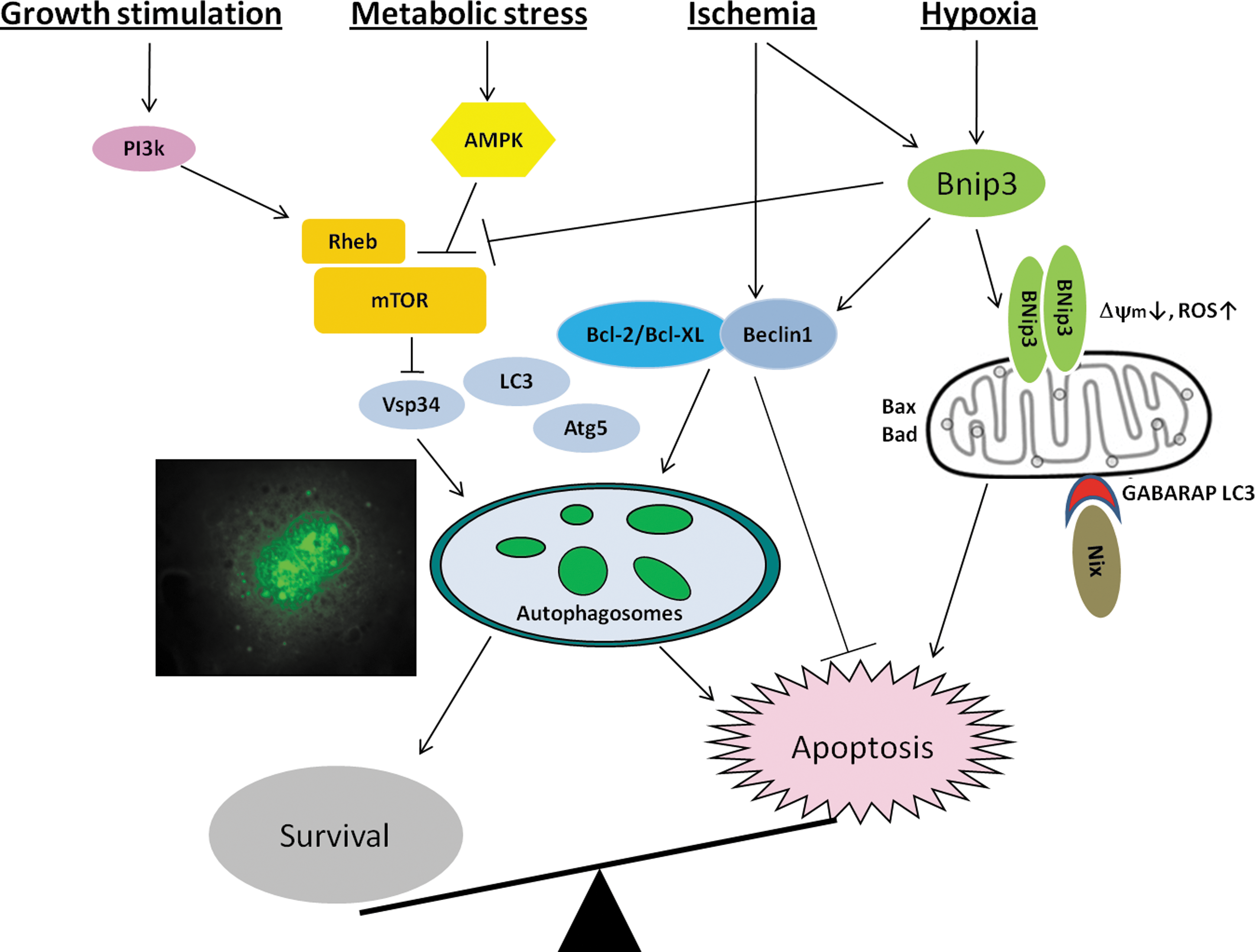
The decision to activate autophagy is complex and likely matched to the cell's energy status. In fact, the mammalian target of rapamycin (mTOR), a highly conserved serine-theonine kinase that regulates protein synthesis and cell growth, has been suggested to be a key regulator of autophagy. Notably, mTOR is activated in cells downstream PI3K/Akt signaling pathway, which relieves the inhibition of the tuberous sclerosis complex proteins (TSC1/TSC2) on the small GTPase Rheb (Ras-homology in brain) essential for mTOR activation. mTOR activation results in the phosphorylation of its downstream targets including p70S6K and 4EBP-1, increasing protein synthesis and cell growth. Interestingly, the class III PI-3K Vps34 essential for autophagosome formation is inhibited by mTOR, thereby linking mTOR activation to inhibition of autophagy (41). However, autophagy can reportedly occur through mechanism independent of mTOR. Another interesting association between energy status and mTOR activity is linked to the ratio of adenosine monophosphate/ATP, because adenosine monophosphate kinase (AMPK), which is activated during metabolic stress such as hypoxia, promotes autophagy by inhibiting mTOR—supporting a survival role for autophagy in cells.
Crosstalk Between Autophagy and Apoptosis
Although the majority of studies particularly in the context of cancer and proliferative disorders support a survival role for autophagy, there is growing evidence that autophagy may also promote cell death. In fact, it has been proposed that basal levels of autophagy are essential for normal cellular homeostasis, whereas defects in autophagy involving either the formation or removal of the autophagosome have been implicated in several human pathologies as described above. Perhaps most compelling is the potential for crosstalk between autophagy and apoptosis for cell killing that may involve the mitochondrion as a common point of convergence of both pathways. However, whether autophagy kills cells via interaction with the apoptotic machinery or whether autophagy induced cell death involves an entirely independent pathway is presently unknown. Hence, the coincident detection and presence of autophagy along with nonapoptotic cell death observed in different cardiac pathologies could easily be misinterpreted as a “cause and effect” relationship; however, another tenable explanation may simply be that autophagy per se was inadequate to suppress cell death and promote survival. In this respect, there has been considerable interest in exploring the relationship of certain members of the Bcl-2 gene family as regulators of both apoptosis and autophagy linked to the mitochondria.
Bcl-2 Family and Autophagy
The link to autophagy by the Bcl-2 gene family is perhaps best exemplified by studies involving the BH3 domain protein Beclin-1. Beclin-1 was first identified as Bcl-2 interacting protein, and it was shown to attenuate apoptosis induced upon viral infection (16). Studies by Shimizu et al. (34) in Bax/Bak−/− double knockout mouse embryonic fibroblasts showed that Bcl-2 and Bcl-XL both augmented nonapoptotic cell death induced by etoposide, which was dependent on Beclin-1 and Atg5, suggesting that Bcl-2 and/or Bcl-XL may promote autophagy. However, subsequent studies using nutrient starvation as a physiological trigger of autophagy have provided contrary evidence indicating that Bcl-2 and Bcl-XL suppress rather than promote autophagy (25). This view was later substantiated by other reports that Bcl-2 inhibits autophagy by its association with Beclin-1 at the ER. In studies in HL-1 cardiac myocytes, autophagy was measured by assessing autophagic “flux,” rather than the “steady-state” amount of GFP-LC3–positive autophagosomes as done by Pattingre et al. (25). Brady et al. provide strong evidence that Bcl-2 may have a proautophagic role because Beclin-1 mutants that could not interact with Bcl-2 suppressed autophagy. The work by Brady et al. (4) suggests that Bcl-2 may be innately proautophagic; however, during conditions when Bcl-2 is localized to ER or SR, it may become antiautophagic.
The Autophagy Protein Atg5 as a Regulator of Apoptosis
Further evidence for crosstalk between autophagy and apoptosis is exemplified by a study demonstrating that calpain-mediated cleavage of Atg5 triggers mitochondrial perturbations leading to cytochrome c release and apoptosis through the intrinsic death pathway (42). Interestingly, Bcl-XL was found to interact with truncated Atg5, suggesting a potential secondary apoptotic mechanism whereby Atg5 may antagonize the cytoprotective actions of Bcl-XL. Moreover, Atg5–FADD complexes have been reported, thereby linking the extrinsic death receptor pathway activation with autophagy proteins (28). Although there are several examples of a relationship between autophagy and apoptosis, they remain independent processes.
Autophagy and Ischemia-Reperfusion
Recent investigations have reported an important protective role for autophagy during ischemia-reperfusion (I/R). Using HL-1 cardiomyocytes, Hamacher-Brady et al. (9) showed a substantial reduction in autophagosome synthesis and degradation during ischemia, and only partial recovery during reperfusion. Beclin-1 overexpression during I/R prevented the disruption of autophagosome formation and inhibited Bax activity and presumably apoptosis. Interestingly, the authors further established that Beclin-1–Bcl-2 interactions were necessary to activate autophagy (4). Inhibition of autophagy by Beclin-1 knockdown, dominant negative inhibition of Atg5, or 3-methyladenine increased the extent of apoptosis after I/R (9). Collectively, these findings support a model in which early activation of autophagy may promote cell survival role by antagonizing, or at least delaying, apoptosis onset (3, 6). Despite these findings highlighting a protective survival role for autophagy during ischemic injury, these findings must be interpreted with caution in view of contrasting studies demonstrating Beclin-1's ability to induce autophagic cell death in the heart (39). This point illustrates that the exact role or nature of autophagy in the heart is not well understood.
As previously stated, a convergence point that may operationally link apoptosis and autophagy is the mitochondrion. Previously, we established the mitochondrion as a crucial component of the intrinsic death pathway during hypoxic injury in vivo and in vitro. Further, we identified Bcl-2 nineteen kilodaltons interacting protein (Bnip3) as a hypoxia-inducible factor crucial for provoking mitochondrial defects and cell death of ventricular myocytes. The closest homolog to Bnip3 is Nix/BnipL. However, unlike Bnip3, Nix is not transcriptionally activated during hypoxia. In fact, Bnip3 and Nix are regulated transcriptionally by distinct biochemical signals. Notably, Nix triggers mitochondrial defects and cell death of ventricular in response to pathological hypertrophy. An underlying assumption of many of the studies is that Bnip3 and Nix kill cardiomyocytes via caspase-dependent apoptosis. However, it was recognized early on that, in certain cells, Bnip3 can trigger programmed cell death via mitochondrial permeability pore transition but independent of caspase activation. Notably, permeability transition pore opening has been postulated to be a central event in the generation of reactive oxygen species presumably through the uncoupling of electron transport and dissipation of electrical gradient across the inner mitochondrial membrane. Given the extraordinary linkage between mitochondrial function and cell survival, there has been considerable interest in the role of mitochondrial perturbations induced by Bnip3 and autophagy. This raises the perplexing question of how can Bnip3, on the one hand, induce apoptosis and, on the other hand, survival via autophagy? One explanation to this apparent conundrum may involve the temporal and spatial expression of Bnip3 during hypoxic or ischemic injury. For example, cells overexpressing cardiac-restricted Bnip3 transgene displayed evidence of apoptosis, yet some myocytes displayed increased evidence of autophagy/mitophagy (Fig. 1) (J.A. Shaw and L.A. Kirshenbaum, unpublished data). Although there is no evidence that Bnip3 directly transactivates beclin-1, or directly regulates its transcription, it implies that Beclin-1 may be important for Bnip3-induced autophagy in response to hypoxic injury. Alternatively, the cell death induced by Bnip3 may instead represent a failed attempt of Beclin-1 to otherwise rescue the apoptosis induced by Bnip3 or other death effector proteins. Whether Beclin-1 alone or in combination with other Atg proteins is necessary for Bnip3-induced autophagy during hypoxia or I/R injury is unknown and an active area of investigation. Nevertheless, Bnip3 has the propensity of provoking apoptosis, autophagy, and nonnecrotic-like cell death.
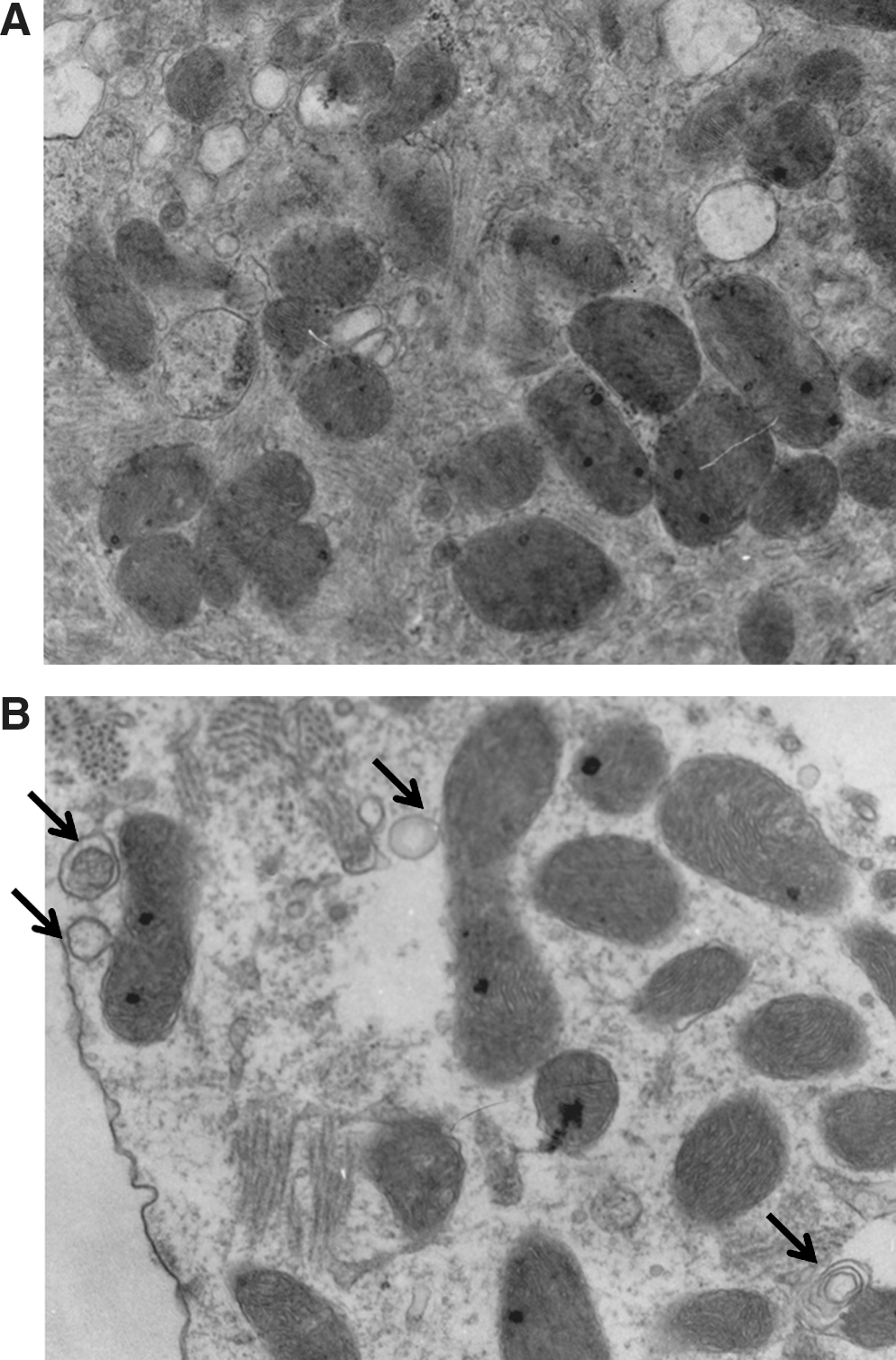
Another explanation purports that the given primary mode by Bnip3-induced cell death involves perturbations to mitochondrial function (31), it is possible that permeability transition pore opening by Bnip3 may also induce autophagy (6). Interestingly, a recent report demonstrated Rheb, a key factor essential for mTOR activation, was inhibited by Bnip3 (15). Hence, inhibition of Rheb by Bnip3 conceptually links Bnip3 to the mTOR pathway and may explain in part how Bnip3 can induce both apoptosis and autophagy.
Although the significance of Bnip3's effects on mitochondrial function beyond the obvious is unknown, it is possible that Bnip3 may serve as a potential mechanism by which damaged or irreversibly injured mitochondria are removed or discarded by the cell via “mitophagy.” In fact, it could be envisioned that the removal of damaged mitochondria that have undergone permeability transition postpones or delays induction of Bnip3-mediated apoptosis. Whether this is the case is highly speculative but strongly supported by a recent report documenting the importance of Nix/Bnip3L for mitochondrial clearance and reticulocyte differentiation (43). In this respect, it has been suggested that the recruitment of Nix to damaged mitochondria serves as a docking site for LC3-gamma-butyric acid receptor–associated protein to remove mitochondria via mitophagy. Whether a similar process is functionally operational during ischemic injury or heart failure remains to be investigated. Nevertheless, these studies provide new important insight into the role and significance of Bnip3 and Nix proteins in autophagy (Fig. 2). Future studies specifically designed to dissect the contribution of Bcl-2 proteins alone or in combination with Atg proteins for removal of damaged mitochondria by mitophagy are required to definitively prove whether autophagy during metabolic crisis permits the dying cell an opportunity to effectively “live twice.”
Footnotes
Acknowledgments
The authors are grateful to Dr. H. Weisman and Dr. D. Baetz for critical comments on the manuscript, Pam Lowe for editorial assistance and manuscript preparation, and Floribeth Aguilar for technical assistance. This work was supported by grants to L.A.K. from the CIHR and St. Boniface Hospital Research Foundation. L.A.K. is a Canada Research Chair in Molecular Cardiology.