
Abstract
Autophagy (macroautophagy), or the degradation of large numbers of cytoplasmic components, is induced by extracellular and intracellular signals, including oxidative stress, ceramide, and endoplasmic reticulum stress. This dynamic process involves membrane formation and fusion, including autophagosome formation, autophagosome–lysosome fusion, and the degradation of intra-autophagosomal contents by lysosomal hydrolases. Autophagy is associated with tumorigenesis, neurodegenerative diseases, cardiomyopathy, Crohn's disease, fatty liver, type II diabetes, defense against intracellular pathogens, antigen presentation, and longevity. Among the proteins and multimolecular complexes that contribute to autophagosome formation are the PI(3)-binding proteins, the PI3-phosphatases, the Rab proteins, the Atg1/ULK1 protein-kinase complex, the Atg9•Atg2-Atg18 complex, the Vps34-Atg6/beclin1 class III PI3-kinase complex, and the Atg12 and Atg8/LC3 conjugation systems. Two ubiquitin-like modifications, the Atg12 and LC3 conjugations, are essential for membrane elongation and autophagosome formation. Recent findings have revealed that processes of selective autophagy, including pexophagy, mitophagy, ERphagy (reticulophagy), and the p62-dependent degradation of ubiquitin-positive aggregates, are physiologically important in various disease states, whereas “classical” autophagy is considered nonselective degradation. Processes of selective autophagy require specific Atg proteins in addition to the “core” Atg complexes. Finally, methods to monitor autophagic activity in mammalian cells are described. Antioxid. Redox Signal. 14, 2201–2214.
Introduction
About 30 years after the first description of autophagy, autophagic bodies, as intermediate compartments with intra-autophagosomal membranes, were first identified in yeast vacuoles under starvation conditions (100). Taking advantage of yeast genetics, autophagy-defective (atg/apg/aut) mutants of Saccharomyces cerevisiae were isolated as yeasts showing little or no accumulation of autophagic bodies under starvation conditions (75, 113). The ATG genes were later isolated and characterized (Table 1) (75, 113). Most ATG genes contribute to autophagosome formation, with many being well conserved from yeast to mammals. In this review, the word “autophagy” denotes macroautophagy unless otherwise indicated.
Cvt, cytoplasm-to-vacuole targeting; PE, phosphatidylethanolamine; PI3-kinase, phosphoinositide 3-kinase; PI(3)P, phosphatidylinositol 3-phosphate.
Autophagy and Diseases
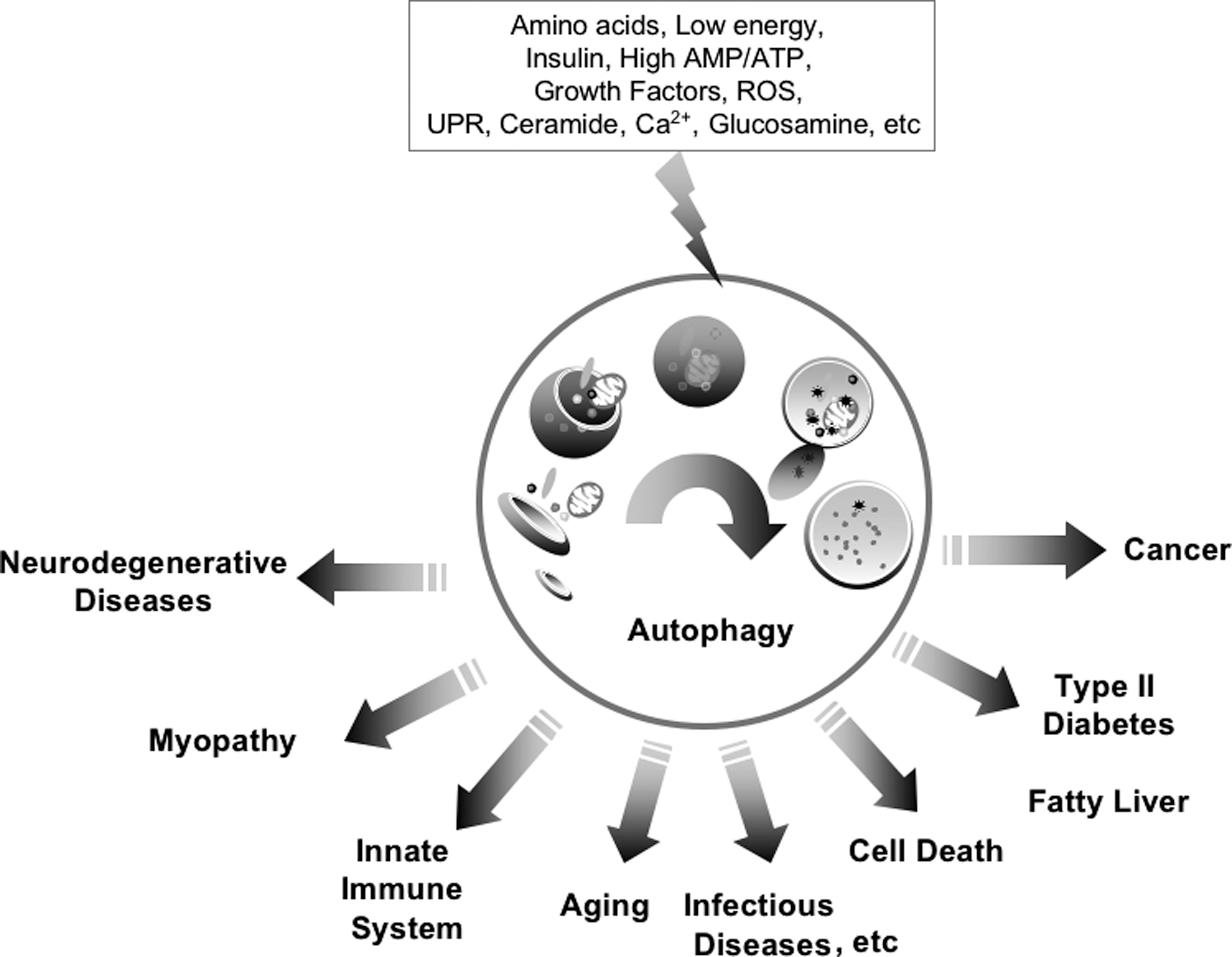
Autophagy is a process during which cytoplasmic components are degraded in bulk and includes the degradation of proteins, lipids, sugars, and organelles (mitochondria, endoplasmic reticulum [ER], and peroxisomes) (12, 59). Autophagy can be induced by extracellular and intracellular signals, including oxidative stress, growth factors, ceramide, ER stress, and glucose, amino acid, and serum starvation (Fig. 2) (12, 59). Autophagy contributes to cellular survival against nutrient deprivation and the turnover of damaged organelles and is related to tumorigenesis, neurodegenerative diseases (including Parkinson's, Alzheimer's, Huntington's, and Creutzfeldt-Jakob diseases), cardiomyopathy (e.g., Danon disease), Crohn's disease, fatty liver, type II diabetes, defenses against intracellular pathogens, antigen presentation, and longevity (12, 49, 63, 73).
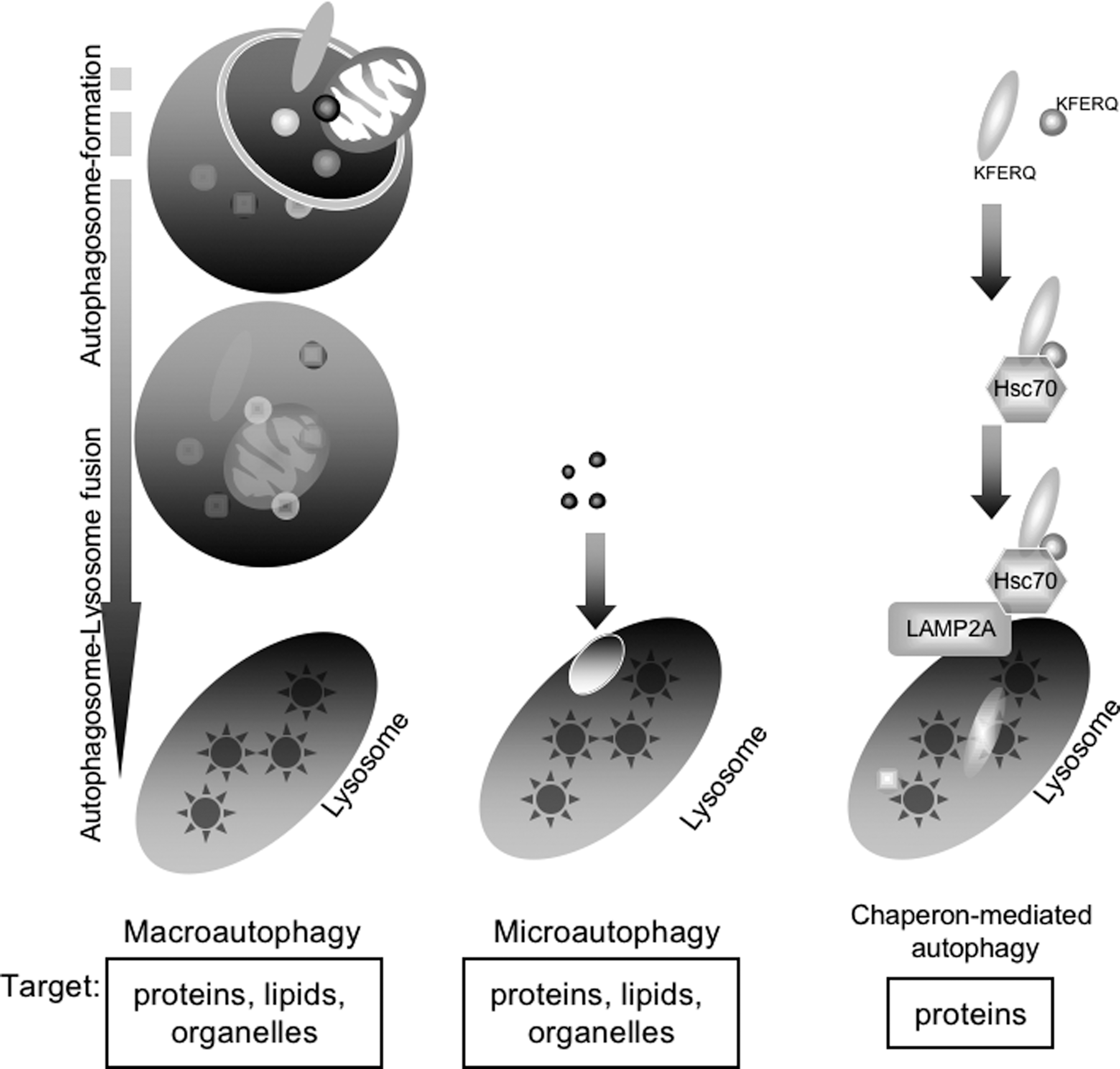
Autophagic Flow in Mammalian Cells: From the Formation of Isolation Membranes to Autolysosomes
Following the induction of autophagy, the omegasome is formed from the ER, resulting in the formation of a cup-shaped structure, the isolation membrane, also called the preautophagosome or phagophore (Fig. 3, Initiation) (62, 67). The isolation membrane subsequently elongates to engulf cytoplasmic components, including organelles (Fig. 3, Elongation), and finally closes to form the autophagosome (initial autophagic vacuole [AVi]), a structure with a double membrane (Fig. 3, Maturation) (67, 95). The outer membrane of the autophagosome fuses with a lysosome to form an autolysosome (degradative autophagic vacuole [AVd]) (Fig. 3, Autophagosome–Lysosome fusion). Finally, lysosomal hydrolytic enzymes, including cathepsins (proteases) and lipases, degrade the intra-autophagosomal components and the inner membrane of the autophagosome (Fig. 3, Degradation) (10). In rat hepatocytes under starvation conditions, the entire process of autophagy occurs within 10–15 min.

“Core” Atg Complexes for Nucleation of Autophagosomes
The “core” Atg proteins are divided into five subgroups: the Atg1/ULK1 protein-kinase complex, the Atg9•Atg2-Atg18 complex, the Vps34-Atg6/beclin1 class III phosphoinositide 3-kinase (PI3-kinase) complex, the Atg12 conjugation system, and the Atg8/LC3 conjugation (lipidation) system (Table 1). In mammals, the Atg1/ULK1 protein-kinase complex is composed of ULK1 (a protein kinase), Atg13, FIP200, and Atg101 (19, 22, 23, 29, 57, 60). ULK1/Atg1 and Atg13 are downstream of the mTorC1 complex. FIP200 is important for the stability and phosphorylation of ULK1. Atg101 is a protein conserved in various eukaryotes (from Schizosaccharomyces pombe to mammals), but not in S. cerevisiae. Atg101 is important for the stability and basal phosphorylation of Atg13 and ULK1 (23, 57). Atg13 is responsible for Atg14 recruitment to the preautophagosomal structure in yeasts (80), and Atg14 is a subunit of the autophagy-specific Vps34 class III PI3-kinase complex in yeasts and mammals (25, 33, 37, 80), suggesting that the ULK1-Atg13-FIP200-Atg101 complex interacts with the Atg14-Vps34 class III PI3-kinase complex. Atg29 and Atg31 are Atg1-interacting proteins in yeasts, but have no mammalian homologs (30, 36).
The Atg9•Atg2-Atg18 complex is composed of Atg9/mAtg9, Atg2, and Atg18/WIPI-1. Atg9 is the only integral Atg membrane protein in yeasts (77, 97); its mammalian homologs are mAtg9/Atg9L1, which is expressed ubiquitously, and Atg9L2, which is specific to the placenta and pituitary gland (120). Under nutrient-rich conditions, mAtg9 localizes to the trans-Golgi network and partial endosomes, whereas, under starvation conditions, mAtg9 localizes to autophagosomes in a process dependent on ULK1 (120). During autophagy, mAtg9 and the phosphatidylinositol 3-phosphate [PI(3)P]–binding protein WIPI-1/Atg18 localize to LC3-positive puncta, making LC3-II/Atg8-PE a promising autophagosomal marker (79, 84). Atg18 constitutively interacts with Atg2 in yeasts, and Atg9 interacts with the Atg2-Atg18 complex under starvation conditions (90). Atg27 is required for autophagy-dependent cycling of Atg9 in yeasts (118). To date, mammalian homologs of Atg2 and Atg27 have not been identified. According to the yeast Atg9 model, mAtg9 would interact with the mammalian Atg18-Atg2 complex during autophagy.
The subunit composition of the Vps34-Atg6/beclin1 class III PI3-kinase complex has been shown to change depending on its function (33, 37, 55, 97, 121). The Vps34-Vps15-beclin1/Atg6 complex is a core complex of class III PI3-kinase (92). In yeasts, the Atg14-Vps34-Vps15-Atg6 complex plays an indispensable role in autophagy, whereas the Vps38-Vps34-Vps15-Atg6 complex is essential for vacuolar protein sorting (37). In mammals, at least three types of class III PI3-kinase complexes contribute to autophagy (25, 50, 51, 55, 99, 121). The Atg14-Vps34-Vps15-beclin1 complex is essential for autophagosome formation, and the UVRAG/Vps38-Vps34-Vps15-beclin1 complex functions positively in autophagosome maturation and endocytic traffic (25, 51). In contrast, the Rubicon-UVRAG-Vps38-Vps34-Vps15-beclin1 complex negatively regulates autophagosome maturation and endocytic traffic (55). Ambra1, a protein containing a WD40 domain that activates beclin1-regulated autophagy, regulates autophagy, and has a crucial role in embryogenesis (14). In sensory neurons, Vps34-independent autophagosome formation and autophagy have been identified as a noncanonical autophagy pathway, requiring Atg7 (122) and suggesting the possibility of a mammalian-specific alternative autophagy pathway independent of Vps34.
Two Ubiquitin-Like Conjugation Systems Are Essential for Autophagosome Formation
Two ubiquitin-like conjugation systems, the Atg12 and Atg8/LC3 (lipidation) conjugation systems, contribute directly to the elongation of isolation membranes and the maturation of autophagosomes (Fig. 4) (11, 81, 110). In the Atg12 conjugation system, Atg12 is activated by Atg7, an E1-like enzyme; transferred to Atg10, an E2-like enzyme, and conjugated to Atg5 to form Atg12-Atg5 conjugates (38, 65, 66, 89, 103). At present, no E3-like enzyme for Atg12 conjugation has been identified. Although the amino acid structures of Atg12 and ubiquitin are dissimilar, Atg12 possesses a ubiquitin-fold (65). As in ubiquitin, the carboxyl terminal Gly of Atg12 is essential for the formation of thio-ester bonds with the active site Cys residues of Atg7 and Atg10 and is also essential for the formation of amide bonds with the Lys residues in Atg5 (65, 70, 76, 89, 103, 107). Atg16 interacts with Atg5, forming a multimeric complex (47, 62, 64).
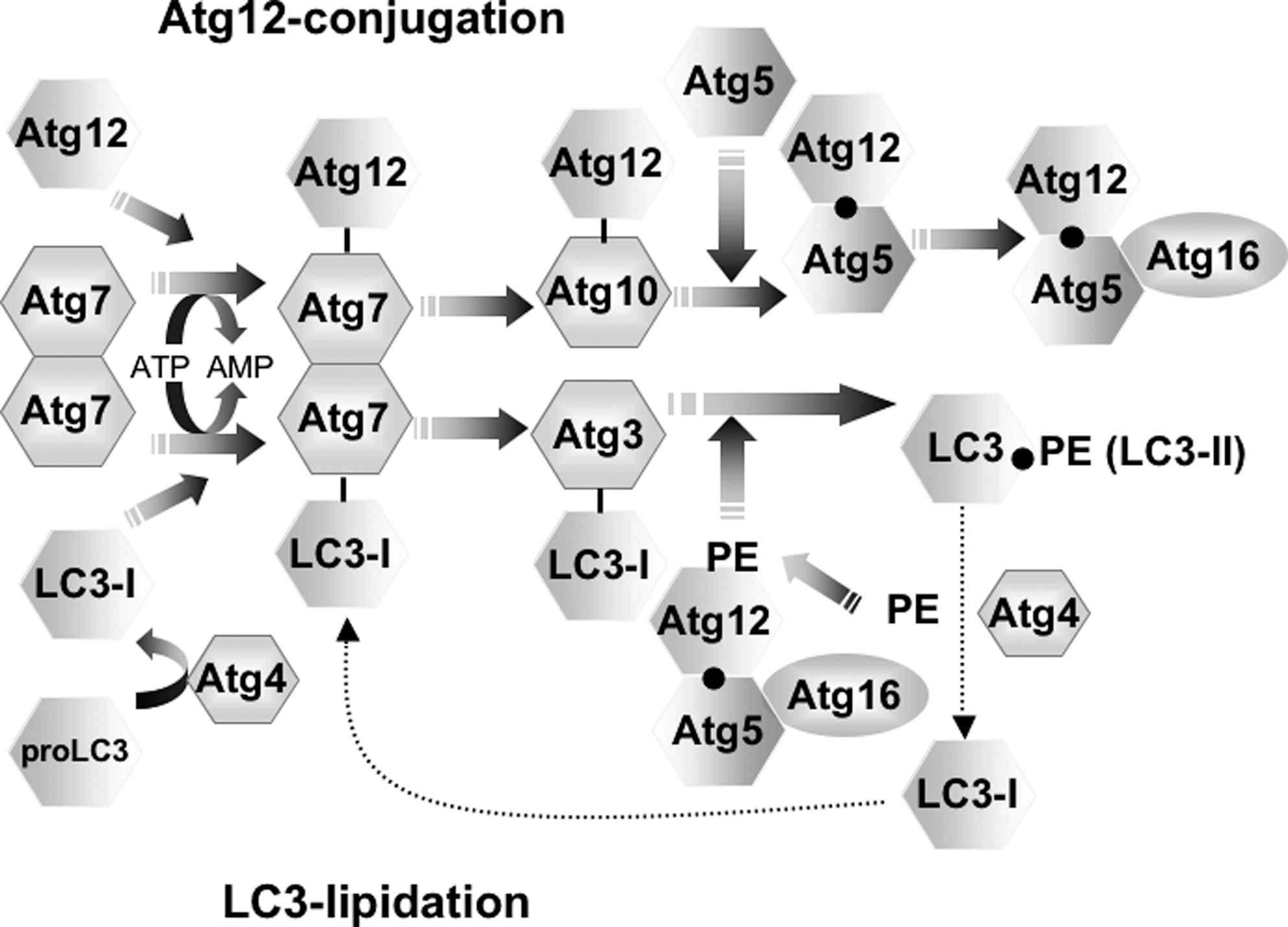
The second ubiquitin-like conjugation system, the Atg8/LC3 conjugation system, is unique, in that its target is a phospholipid, phosphatidylethanolamine (PE) (24, 31). Therefore, the Atg8/LC3 conjugation system has been called Atg8/LC3 “lipidation.” In yeasts, Atg8 is synthesized as proAtg8, which is cleaved by Atg4, a cysteine protease, to expose Gly at its carboxyl terminus. Atg8 is activated by the same E1-like enzyme, Atg7, transferred to Atg3, a second E2-like enzyme, and finally conjugated to PE to form Atg8-PE (106, 107). Atg8-PE localizes to autophagosomes, whereas Atg8 is present in the cytosol. Atg8-PE on the cytosolic surface of autophagosomes is cleaved to Atg8 by Atg4 to recycle Atg8, a recycling that has been shown essential for effective autophagy (24). Atg8 mediates membrane tethering and hemifusion (74). To date, at least four mammalian Atg8 homologs have been identified: microtubule-associated protein 1 light chain 3 (LC3/MAP1-LC3/LC3B), GABAA receptor-associated protein (GABARAP), Golgi-associated ATPase enhancer of 16 kDa (GATE-16), and mAtg8L. LC3 is the best characterized of these proteins and is regarded as a functional homolog of yeast Atg8 in autophagy (31). LC3 is synthesized as proLC3, which is cleaved by Atg4B to form LC3-I, with the carboxyl terminal Gly exposed (31). LC3-I is activated by Atg7, transferred to Atg3, and finally conjugated to PE (106, 107). The LC3-PE conjugate has been designated LC3-II (31). The carboxyl terminal Gly of LC3 is also essential for the formation of a thio-ester bond with the active site Cys residues of Atg7 and Atg3, as well as being essential for the formation of an amide bond with PE (94, 108). As LC3-I is localized in the cytosol and LC3-II is localized to autophagosomes (31), LC3-II (LC3-PE conjugate) is a promising autophagosomal marker in mammals. LC3-II on the cytoplasmic surface of autophagosomes is delipidated by Atg4B to recycle LC3-I for further autophagosome formation.
The other Atg8 homologs are also modified by Atg4B (Atg4A for GATE-16), Atg7, and Atg3 (32, 94, 101, 104). The carboxyl termini of the proforms of GABARAP, GATE-16, and mAtg8L are cleaved by Atg4B (or Atg4A for proGATE-16) to expose Gly, and the resulting GABARAP-I, GATE-16-I, and mAtg8L-I are modified to GABARAP-II, GATE-16-II, and mAtg8L-II, respectively (101, 104, 105). Membrane-localized GABARAP-II on the cytoplasmic surface is also delipidated by Atg4B. Little endogenous GATE-16-II (i.e., the lipidated form) has been observed in rat and mouse tissues and cell lines (110). The physiological function of GATE-16-II is not yet known, whereas GATE-16 is an essential component of intra-Golgi transport and postmitotic Golgi reassembly independent of its lipidation (86). Although mApg8L-II was not observed during starvation-induced autophagy, it was identified during the differentiation of C2C12 cells to myocytes (105).
Atg12 conjugation is closely interrelated to Atg8/LC3 lipidation. Atg5 deficiency results in a defect of Atg8/LC3 lipidation (67, 71). The yeast Atg12-Atg5 conjugate functions in vitro as an E3-like enzyme for Atg8 lipidation (18). Mammalian Atg16L determines the site of LC3-lipidation, that is, autophagosome formation (15). Therefore, the Atg16/Atg12-Atg5 complex may function as an E3-ligase complex to facilitate LC3-lipidation complex and may also act to determine the site of autophagy. Lack of Atg3 in mammals leads to a decrease in the Atg12-Atg5 conjugate as well as impairing LC3 lipidation (95). Further, loss of Atg3 has been associated with a defective process of autophagosome formation, including defects in the elongation and complete closure of the isolation membranes, resulting in malformed autophagosomes. In cells lacking Atg3, Atg16L and Atg5 are localized to elongated isolation membranes/incomplete autophagosomes, suggesting that elongation of the isolation membrane can occur in the absence of LC3 lipidation (95). Thus, in addition to its E3-like function, the Atg16/Atg12-Atg5 complex may function in the elongation of isolation membranes.
Initial Step of Autophagy: Omegasomes and the Initiation of Isolation Membranes
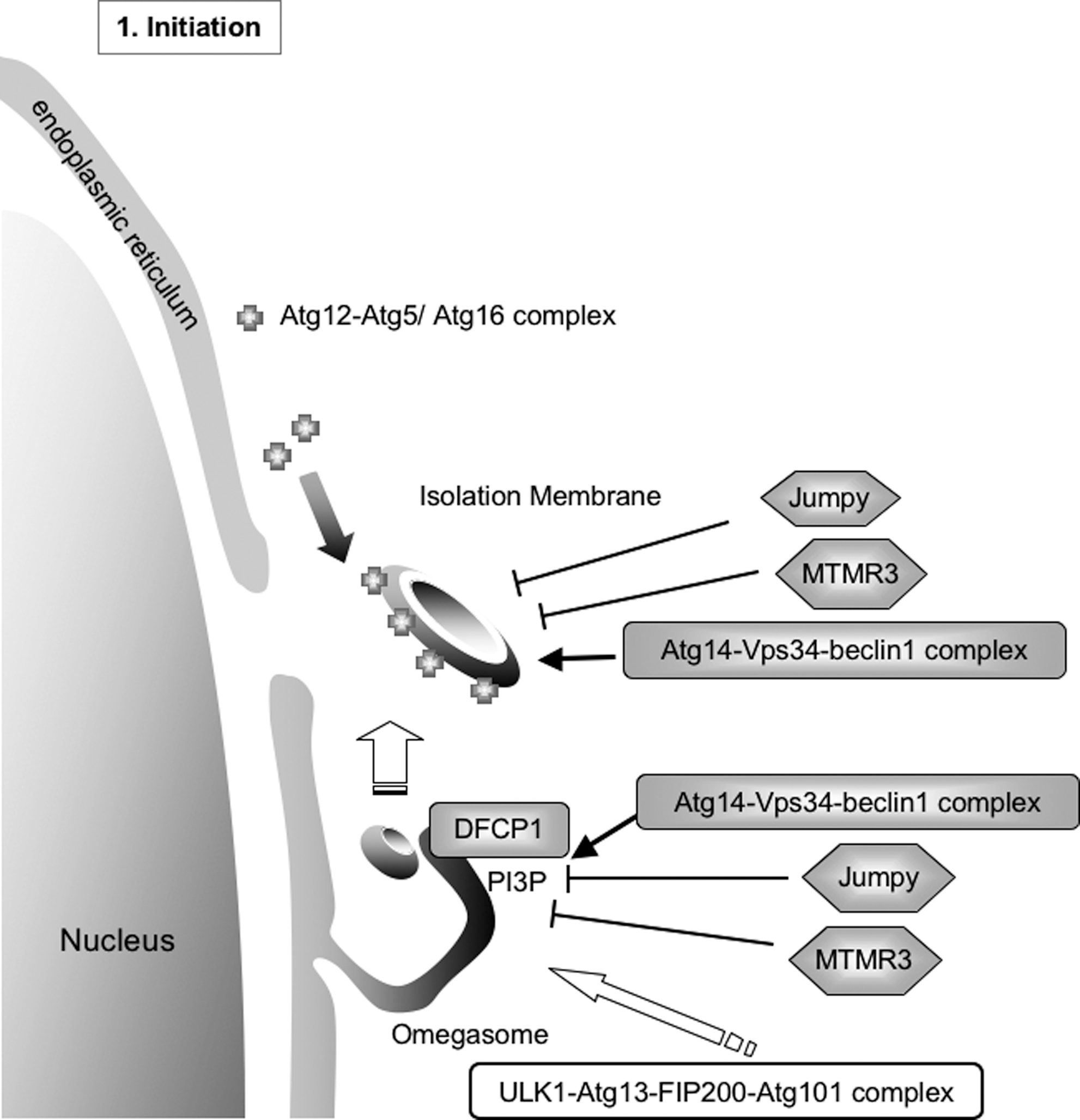
Autophagy accompanies dynamic membrane formation and membrane fusion (Fig. 3). During the first step of autophagy, the ULK1-Atg13-FIP200-Atg101 kinase complex activates autophagic signaling via the mTor-signaling pathway. The omegasome, which is shaped like the Greek letter omega, is associated with the ER (Fig. 5) (4). A PI(3)P-binding protein, double FYVE domain-containing protein 1 (DFCP1), is localized to PI(3)P on the omegasome under starvation conditions, but localizes to the ER and Golgi under nutrient-rich conditions. The Atg14-Vps34-beclin1 PI3-kinase complex positively regulates the DFCP1-positive omegasome formation. Thereafter, the isolation membrane is formed inside the ring of the omegasome, and the Atg12-Atg5/Atg16 complex is localized to the isolation membrane (47, 62, 67). The protein mAtg9, the ULK1-Atg13-FIP200-Atg101 kinase complex, and the Atg14-Vps34-beclin1 PI3-kinase complex also localize to the isolation membrane. DFCP1 itself, however, is likely not required for autophagosome formation. Two PI(3)P phosphatases (Jumpy/MTMR14 and MTMR3) negatively regulate the formation of omegasomes and isolation membranes by decreasing the level of PI(3)P (98, 114).
Elongation of the Isolation Membrane and Formation of Autophagosomes
Following the formation of the isolation membrane, it elongates to engulf cytoplasmic components (Fig. 6). The Atg12-Atg5/Atg16 complex is localized to the cup-shaped structure. In the later stages of isolation membrane elongation, the Atg12-Atg5/Atg16 complex progressively dissociates from the isolation membrane, whereas LC3-II gradually localizes to both sides of the isolation membrane (see Fig. 6, Cross-section 1) (67). During this process, LC3-I, a cytosolic form of LC3, is progressively lipidated by Atg7, Atg3, and the Atg12-Atg5/Atg16 complex to form LC3-II. Finally, the isolation membrane closes to form the autophagosome (Fig. 6, Maturation). Although LC3-II is localized to autophagosomes, most of the Atg12-Atg5/Atg16 complex dissociates from the autophagosome (67). The autophagosome is a double membrane structure, with LC3-II present on both the cytosolic and the intra-autophagosomal surfaces (Fig. 6, Cross-section 2). Therefore, LC3-II increases during autophagosome formation. Rab32 and Rab33 also contribute to the elongation of the isolation membrane (21, 26). Following autolysosome formation, Atg4B delipidates LC3-II on the cytoplasmic surface to recycle LC3-I (Fig. 7).
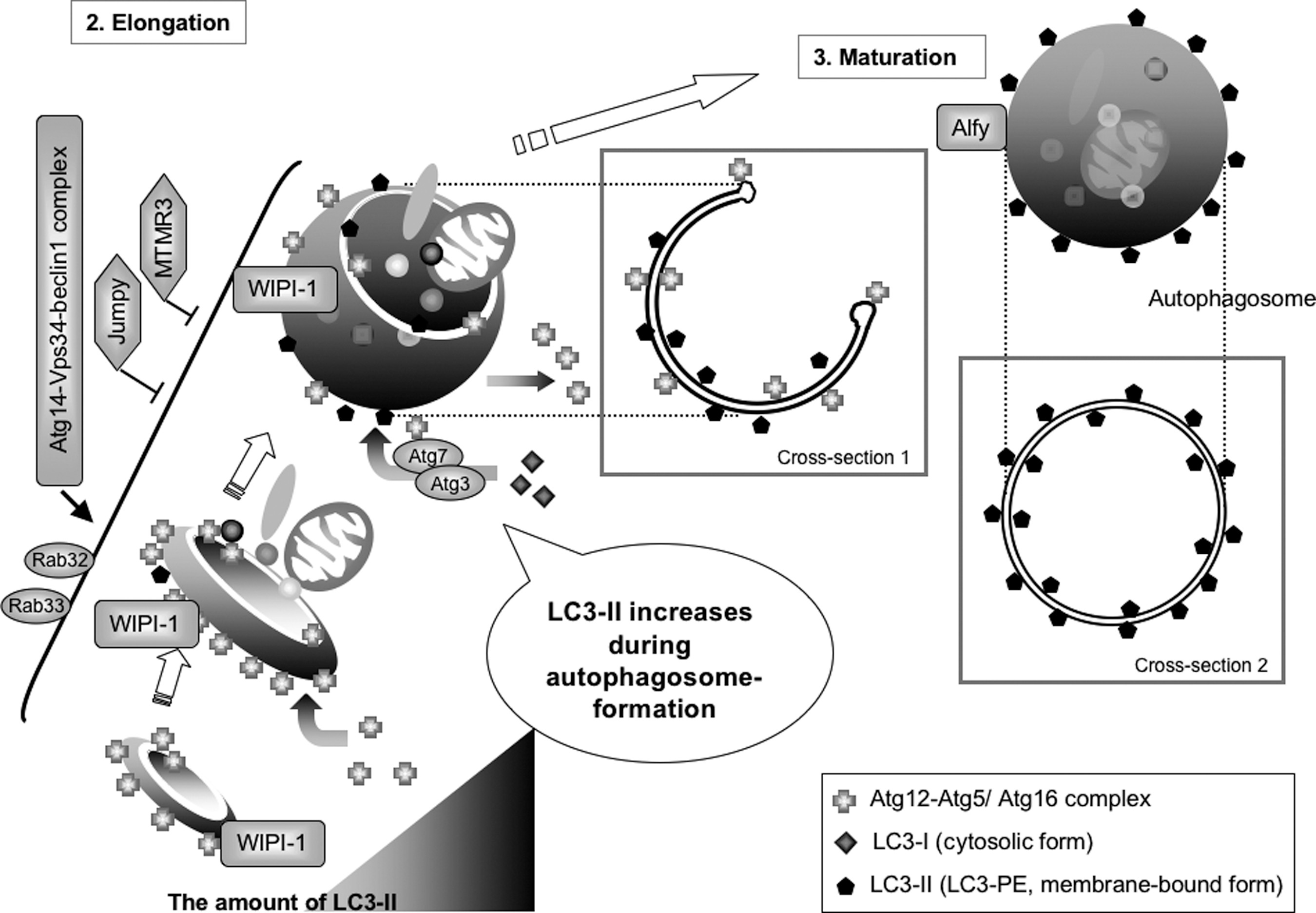

Autophagosome–Lysosome Fusion and Degradation
Soon after autophagosome formation, its outer membrane fuses with lysosomes to form autolysosomes, a process requiring Rab7 (17, 28). A FYVE and coiled-to-coil domain-containing protein FYCO1 functions as a Rab7 effector, binding to LC3 and PI(3)P, and mediating microtubule plus end-directed vesicle transport (82). The fusion of autophagosomes and lysosomes is positively regulated by the UVRAG-Vps34-beclin1 PI3-kinase complex and negatively regulated by the Rubicon-UVRAG-Vps34-beclin1 PI3-kinase complex (25, 50, 55, 99, 121). At present, it is unclear how and what controls the composition of these Vps34-beclin1 complexes. Alfy, a PI(3)P-binding FYVE domain-containing protein, has been found to localize with autophagosomes and protein granules (91). Functional multivesicular bodies are required for autophagic Alfy-mediated clearance of protein aggregates (13).
Following autolysosome formation, the lysosomal hydrolases, including cathepsins and lipases, degrade the intra-autophagosomal contents, whereas cathepsins degrade LC3-II on the intra-autophagosomal surface (109, 111). In yeasts, Atg15, a vacuolar lipase, and Atg22, a vacuolar membrane protein, are indispensable for the specific degradation of autophagic bodies (9, 112, 117). At present, mammalian homologs of yeast Atg15 and Atg22 have not been identified.
Selective Autophagy
In addition to classical or nonselective autophagy, recent findings have shown that processes of selective or chaperone-mediated autophagy are physiologically important (Table 1). These processes are mediated by p62/SQSTM1, a ubiquitin- and LC3-binding protein (5, 45, 83) that also binds to LC3A (one of the other LC3 isoforms), GABARAP, GATE-16 (GABARAPL2), and mAtg8L (GEC1/GABARAPL1) (83). Following their binding to p62, these ubiquitylated protein aggregates are sequestered into autophagosomes. Therefore, when autophagy is impaired, p62- and ubiquitin-positive aggregates accumulate in cells.
Mitophagy is a process of mitochondria-selective autophagy that functions in the clearance of damaged mitochondria (42). This process is mediated by the Parkin and PTEN-induced putative kinase 1 (PINK1) proteins, which are encoded by Parkinson disease-linked genes (8, 54). Parkin is a ubiquitin E3 ligase, and PINK1 contributes to the translocation of Parkin into damaged mitochondria, inducing turnover of damaged mitochondria via autophagy. Parkin mediates the ubiquitylation of voltage-dependent anion channel 1 and mitofusin (16), and p62 contributes to Parkin/PINK1-mediated mitophagy. Uth1 and Atg32 have been identified as mitophagy-specific proteins in yeasts (34, 41), but their mammalian homologs have not been identified yet.
Pexophagy is a process of peroxisome-specific autophagy, which has been investigated in the methylotrophic yeast Pichia pastoris (87). In addition to most of the “core” Atg proteins, which are required for pexophagy, this process requires the proteins Atg24, Atg25, and Atg26, which are specific to and indispensable for pexophagy (2, 72, 116). In mammals, Atg7 deficiency leads to a defect in pexophagy (27), and Pex14 contributes to pexophagy (20).
The cytoplasm-to-vacuole targeting (Cvt) pathway is a unique protein transport system in yeasts (53). Aminopeptidase I and α-mannosidase are synthesized in the cytosol and selectively packaged into Cvt vesicles during their formation. The Cvt vesicles fuse with the vacuoles, in which the transported proteins are processed to their active forms. Although the Cvt pathway is selective and nondegradative, it includes several Cvt pathway-specific Atg genes, including Atg11, Atg19, Atg20, and Atg21 (48, 56, 88, 96, 119). At present, there have been no reports of a Cvt pathway in mammals.
ER-specific autophagy is designated for ERphagy or reticulophagy and contributes to the protein quality control process in the ER (40, 46). In addition to the ubiquitin–proteasome-mediated ERAD process (ERAD I), there is a stress-induced ER response of autophagy, ERAD II. At present, the molecules contributing to ER-specific recognition of autophagy have not been identified.
Estimation of Autophagic Activity
Because of the pathophysiological importance of autophagy, it is necessary to estimate autophagic activity accurately and quantitatively. However, as autophagy is a dynamic and complicated process, it must be analyzed correctly. Two classical but definitive methods, conventional morphometry of autophagosomes and autolysosomes by electron microscopy and measurement of autophagic flux by assessing long-lived protein degradation, are still useful. Among these conventional methods are the LC3-II turnover assay using lysosomal inhibitors to measure autophagic flux, measurement of the number of LC3-positive puncta (or green fluorescent protein [GFP]-LC3 puncta), and the tandem fluorescent protein-LC3 (tfLC3/monomeric red fluorescent protein [mRFP]-GFP-LC3) color change assay. Critical issues and guidelines for monitoring autophagy have been described (43, 69).
The LC3-II turnover assay is a measure of autophagic flux, in which LC3-II is assayed by immunoblotting with anti-LC3 antibody in the presence and absence of lysosomal inhibitors. During autophagosome formation, cytosolic LC3-I is conjugated to PE, with the resulting LC3-II localized to autophagosomes (31). These autophagosomes fuse with lysosomes to form autolysosomes, in which sequestered intra-autophagosomal components (including intra-autophagosomal LC3-II) are degraded. Therefore, LC3-II itself is used as a marker of autophagosome formation and its degradation to monitor late autophagic flux (102). A mixture of E64d (a membrane-permeable inhibitor of cathepsins B, H, and L) and pepstatin A (a membrane-permeable inhibitor of cathepsins D and E) is used to inhibit lysosomal function (102). Treatment of cells with this inhibitor cocktail results in significant accumulation of autolysosomes (and LC3-II dots) because there is little degradation of their contents. Thus, the accumulation of LC3-II reflects the activity of the delivery process of LC3-II into lysosomes, that is, autophagic flux. Fluorescent microscopic assays of LC3-positive puncta in the presence and absence of the lysosomal inhibitor cocktail may also be useful. Methods for immunoblotting and immunostaining of endogenous LC3 have been described (109). Bafilomycin A1 (an inhibitor of V-ATPase) is also used to inhibit autophagy and to estimate the autophagic flux of LC3-II. As V-ATPase contributes to the acidification of other organelles, including the Golgi and endosomes, bafilomycin A1 may show multiple off-target effects (44, 93).
Cells with defective autophagy show increased concentrations of ubiquitin and p62 (45). A dominant negative mutant of Rab7, Rab7T22N, inhibits autophagosome–lysosome fusion (17, 28). Thus, overexpression of this Rab7 mutant under starvation conditions results in the augmented accumulation of autophagosomes. Increases in p62- and ubiquitin-positive inclusions in tissues may indicate insufficient autophagy.
Although LC3 fused to GFP (GFP-LC3) is useful for in vivo imaging of autophagosome formation (61, 68), caution must be exercised because of the limitations of GFP-LC3 (6, 35). GFP-LC3 tends to form puncta in cells independent of autophagy, and GFP fluorescence in lysosomes may occur even after degradation of the LC3 moiety. Therefore, this method tends to overestimate the number of autophagosomes. These problems may be avoided by using a mutant, GFP-LC3ΔG, as a negative control. GFP-LC3 transgenic mice, however, can be used to study autophagy in many tissues outside the brain, where few GFP-LC3 puncta are observed (58, 68). As the brain is one of the organs most protected against starvation, most of the endogenous LC3 in the brain is in the cytosolic form, LC3-I.
The mRFP-GFP-LC3 color change assay is based on the difference in pH stability between GFP and mRFP. Autophagosomes have a pH similar to that of the cytosol, whereas autolysosomes have acidic pH. At acidic pH, the fluorescence of mRFP is stable, whereas that of GFP decreases. Therefore, the merged color of mRFP-GFP-LC3 in autophagosomes is yellow, whereas that in autolysosomes is red (39). This assay is suitable for real-time (and short-term) monitoring of autophagy, but care should be taken for use in long-term monitoring of autophagy. The fluorescence derived from GFP in the lysosomes has been observed even after the degradation of LC3 (35).
The enzyme-based pho8Δ60 assay system has been used for the quantitative estimation of autophagy in yeasts (78). At present, however, an enzyme-based quantitative assay for autophagy in mammals has not been established yet (69).
Concluding Remarks
Research on autophagy is expanding because of findings of its increased pathophysiological importance in mammals. We have reviewed the “core” Atg complexes essential for autophagosome formation, their functional roles during autophagy, selective autophagy, and assays of autophagy. Most autophagy-related proteins are conserved from yeasts to mammals; these proteins have been characterized and divided into five subgroups. Mammalian cells have mammalian-specific Atg proteins and more complicated mechanisms, probably because mammalian cells utilize autophagic machinery for tissue- and cell-specific functions in addition to self-defense mechanisms against intracellular and extracellular stresses. In addition to the dynamic and complicated processes of autophagy in mammalians, mammalian-specific functions during autophagy or autophagic mechanisms may complicate the analysis of autophagy in mammals. Although LC3-II is a promising autophagosomal marker, it is necessary to carefully interpret the results of LC3-II immunoblotting and fluorescence analyses (69). Future pathophysiological and clinical applications based on autophagy will require the use of autophagy-specific inhibitors and/or activators. Atg4B, a cysteine protease, and autophagy-specific class III PI3-kinase complex are candidates for drug screening.
Footnotes
Acknowledgments
This study was supported in part by grants-in-aid from the Ministry of Health, Labor, and Welfare of Japan and by Grants-in-Aid for Scientific Research on Priority Areas “Proteolysis in the Regulation of Biological Processes” from the Ministry of Education, Science, Sports, and Culture of Japan.