
Abstract
Autophagy is a basic cell biological process ongoing under physiologic circumstances in almost all cell types of the human organism and upregulated by various stress conditions including those leading to inflammation. Since autophagy affects the effector cells of innate and adaptive immunity mediating the inflammatory response, its activity in these cells influences the antimicrobial response, the development of an effective cognate immune defense, and the course of the normal sterile inflammatory reactions. The level of autophagic activity may determine whether tissue cells die by apoptosis, necrosis, or through autophagy, and, as a consequence, whether the clearance of these dying cells is a silent process or results in an inflammatory response. Loss or decreased autophagy may lead to necrotic death that can initiate an inflammatory reaction in phagocytes through their surface and cytosolic receptors. Engulfment of certain cells dying through autophagy can activate the inflammasome. The intertwining regulatory connections between inflammation and immunity extend to pathologic conditions including chronic inflammatory diseases, autoimmunity and cancer. Antioxid. Redox Signal. 14, 2233–2243.
Introduction
There is much evidence of a complex interaction between the autophagy pathway and diverse aspects of innate and adaptive immunity, including pathogen resistance, antigen presentation, tolerance, and lymphocyte development, as well as the negative regulation of cytokine signaling (reviewed recently by (101)). The present review focuses on the importance of autophagy in the primary inflammatory response, particularly how the level of autophagic activity in the immune competent cells influences the inflammatory reaction and why clearance and pro-inflammatory character of dying cells are determined by the interplay between autophagic and cell death pathways.
The Autophagy Pathway
Autophagy begins when a flat membrane cisterne called the phagophore or isolation membrane, which is thought to originate from the ER (1, 15), the trans Golgi, late endosomes or infrequently from the nuclear membrane, wraps around a portion of the cytosol forming a vacuole called the autophagosome (Fig. 1). Autophagosomes undergo maturation involving fusion with endosomes and eventually with lysosomal vesicles, where they deliver their cargo. The cargo is then digested by lysosomal hydrolases and the catabolites are transported to the cytoplasm for reutilization by permeases and translocases (26, 66). In the nucleation step, a complex containing Beclin-1, the PI3K, Vps34, Atg14, and Atg15 marks the earliest form of the phagophore. Atg5 is conjugated with Atg12 by Atg7 and Atg10. The Atg5–12 complex forms a large multimeric complex at the phagophore with Atg16L1. This localizes mainly to the convex surface of the isolation membrane and it may determine the curvature of the forming autophagosome through a protein lattice (97), however its exact function is not understood. It also marks the site for microtubule-associated protein1 light chain 3 (LC3) lipidation. LC3 (also known as Atg8) is proteolytically activated by Atg4, losing five C-terminal amino acids. The ubiquitin -activating and –carrier proteins, Atg7 and Atg3, couple the exposed terminal glycine of LC3 to phosphatidyl-ethanolamine (PE), which is inserted in the forming autophagosomal membrane or phagophore. The membrane is then elongated and sealed. In the maturation step, autophagic vacuoles fuse with early and late endosomes as well as lysosomes. The process probably begins with homotypic fusion of endosomes and this large multivesicular structure fuses with the autophagosomes, giving rise to what is called the amphisome. The Beclin-1–Atg14 complex and another Beclin-1 complex with UV radiation resistance-associated gene protein (UVRAG) drive this step by activating a complex termed HOPS (homotypic vacuole fusion and protein sorting), which acts as a Rab7 guanine nucleotide exchange factor (54). Finally amphisomes fuse with dense lysosomes acquiring lysosomal membrane proteins and degradation enzymes and their pH becomes slightly acidic (∼5.7). Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins such as vesicle-associated membrane protein 8 (VAMP8) and Vti have been suggested to mediate the actual membrane fusion events.
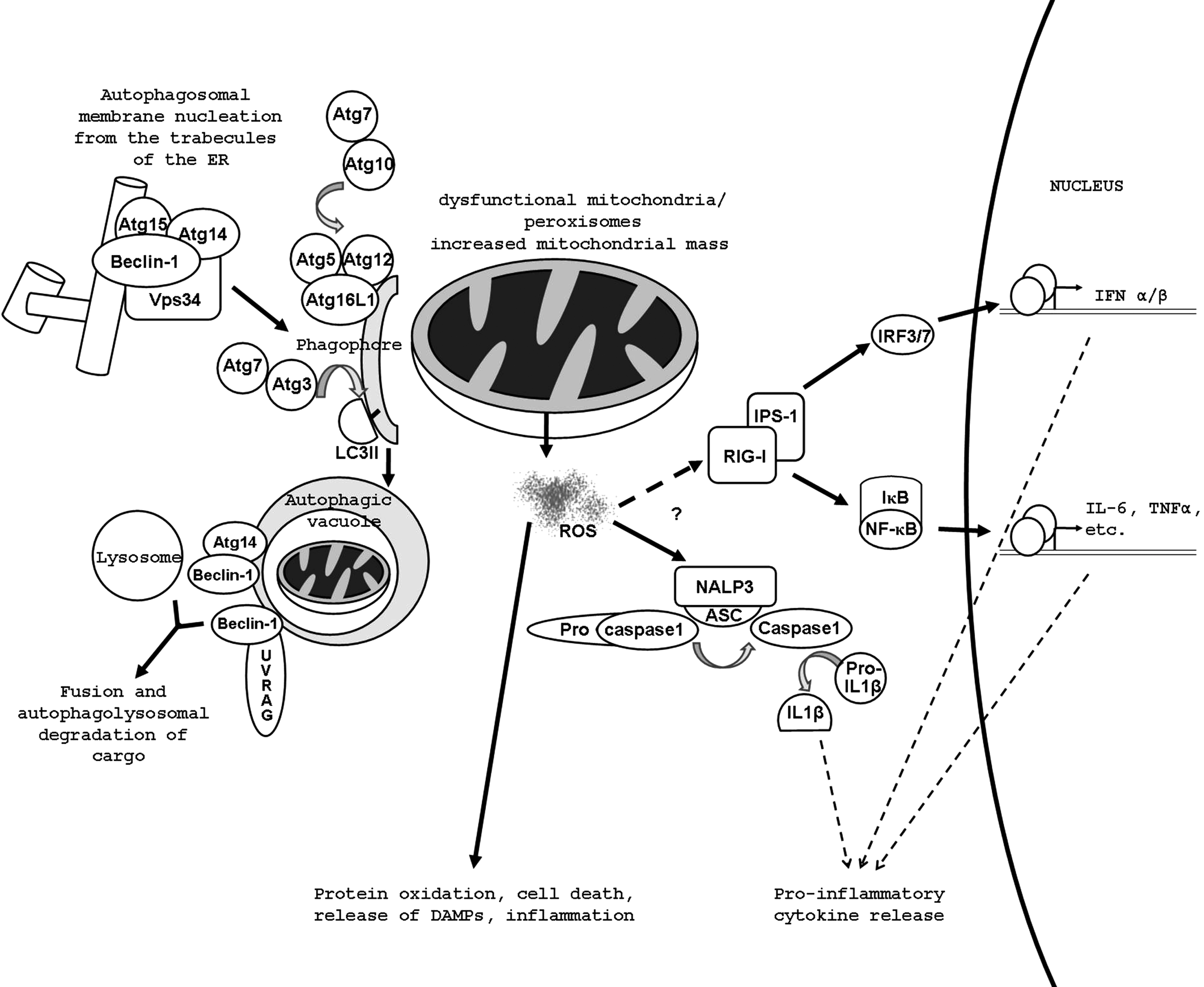
Influence of Autophagy on the Inflammatory Response in the Immune Effector Cells
The antimicrobial response
The most direct way in which autophagy influences inflammation is the autophagic capture of intracellular microbes and their elimination via autolysosomes in the process sometimes referred to as xenophagy (12). There are specific adapter molecules targeting bacteria for capture by the autophagolysosomes (106, 107) and viruses, as well as bacteria-developed factors to counter autophagy (reviewed by (12)). It appears that autophagy may have evolved as one of the first autonomous defense mechanisms in eukaryotes working against invading microbes as phagocytosis of different extracellular pathogens resembles very much the machinery for autophagic clearance of intracellular organelles via the autophagolysosomal system (50). It also has been shown that autophagy can serve also as a back-up mechanism for phagocytosis of Francisella tularensis, Listeria monocytogenes, Mycobacterium tuberculosis, Salmonella enterica, Shigella flexneri, Streptococcus pyogenes, or Toxoplasma gondii (14, 52, 79). TLRs regulate autophagy induction (reviewed by (101)) which is needed for efficient autophagic control of intracellular pathogens. On the other hand, microbial components initiate the signal cascade for cytokine induction through the various TLRs.
One of the functions of autophagy is the delivery of foreign antigen material to innate and adaptive immunity (46, 51, 83). Autophagy may exert cytoprotective and pathogen trafficking functions in infected cells by activating Atg5 which is required for delivery of viral nucleic acids to the endosomal TLR7 which further activates type I interferon (IFN) signaling (52). Autophagy can be utilized in adaptive immunity by the major histocompatibility complex (MHC) class II antigen presentation system for endogenous antigens (e.g., Epstein Barr viral antigens) (51). The latter holds great potential for future vaccine development by which vaccine efficacy would be increased through improvement of the T helper cell response (52). After viral infection, autophagy is activated by the double-stranded viral RNA-activated protein kinase R (PKR) which phosphorylates and therefore activates the eukaryotic translation initiation factor-2α (eIF-2α), this in turn inhibits viral replication (90, 94). Alternatively, the receptor-interacting protein (RIP) pathway can induce autophagy in response to viral infection (7).
Autophagy impinges on inflammation through regulation of intracellular ROS production
A common theme in the background of the phenotypes resulting from the genetic ablation of Atg5, Atg7, Atg12, and Atg16L1 appears to be the accumulation of dysfunctional mitochondria in the absence of autophagy, which results in elevated release of reactive oxygen species (ROS) (Fig. 1). Autophagy has in many instances been reported to regulate ROS production negatively by decreasing the mitochondrial mass and eliminating leaky mitochondria and peroxisomes (3, 81). These radicals are known to be activators both of RIG (retinoic acid inducible gene)-I-like receptors (RLRs) (93) and NLRP3 (61) driving the production of more IFN and interleukins. The RLRs, cytoplasmic RNA-sensors, RIG-I, and melanoma-differentiation associated gene (MDA5), signal through interferon β promoter stimulator 1 (IPS-1), which activates two major signaling pathways. It stimulates the IκB kinase (IKK) complex, thereby eventually activating NF-κB and through the IKK-related tank-binding kinase (TBK) 1 and IKKɛ it activates interferon regulatory transcription factor (IRF) 3/7 and IFN type I production. IPS1 is an outer membrane mitochondrial protein and its amount increases with increased number of mitochondria. This alone is not enough to account for amplified signal transmittance on these pathways. It has been hypothesized that an inhibitor of RLR signaling might become inactivated upon oxidation or, on the contrary, oxidation enables a positive-regulator of RLRs. It is also conceivable that ROS-level sensing proteins are involved in RLR signaling in a presently unknown way (93).
Inflammasome activation is under autophagy control
TLR4 agonists, particularly lipopolysaccharide (LPS), cannot induce inflammasome activation and IL-1β secretion in wild-type macrophages. When genes of the autophagy regulators Atg16L1 or Atg7 are deleted or a chemical inhibitor of autophagy is applied, LPS-dependent inflammasome activation occurs suggesting that autophagy controls inflammasome activation and can limit production of the inflammatory cytokines IL-1β and IL-18 (82). In accordance with this finding, autophagy also inhibits pyroptosis, a specialized form of cell death occurring after Shigella infection and mediated by the NLR family member Ipaf through caspase-1 activation (91). Autophagy induction also associates with transient resistance to pyroptosis, and infection of macrophages with Shigella leads to induction of autophagy, which dramatically increases during absence of caspase-1 or Ipaf, showing an additional level of complexity in the links between inflammasome and autophagy. Though several molecular explanations have been provided, the mechanism of autophagy-dependent inflammasome inhibition is not clear at present. Inflammasomes may be directly degraded through autophagy (31). A more likely explanation is negative regulation of ROS generation by autophagy (81, 82). It should be also noted that ROS stimulate autophagy (8,35) probably effectuating a negative feedback loop.
The transglutaminase connection
In the case of other proteins linked to autophagy and inflammation, the connection between the role(s) of transglutaminase 2 (TG2) is even less elucidated. This multifunctional enzyme has recently been implicated in autophagosome maturation (9). In particular, TG2 -/- mouse embryonic fibroblasts accumulated GFP-LC3 labeled vacuoles representing autophagosomes, likely due to failure to efficiently merge with lysosomes. This was partially dependent on the cross-linking function of TG2. This enzyme had formerly been implicated in liposome fusion (19), though the phenomenon at hand might involve a more intricate interaction between TG2 and autophagosomal proteins. An earlier study found that in pancreatic cancer cells, constitutive activation of protein kinase C (PKC) δ suppressed autophagy through induction of TG2 (72). Using specific inhibitors, these authors suggested that TG2 activates PI3K and mammalian target of rapamycin (mTOR), while inhibiting Beclin-1. The exact mechanism though has not been explained and these two antagonistic observations await reconciliation.
TG2 -/- mice are partially protected from LPS-induced sepsis. The serum level of IL-6 and granulocyte colony-stimulating factor (G-CSF) in these animals fell to many-fold lower than that of their wild-type litter mates by 24 hours following a singe injection of LPS, although their basal or maximal levels and induction kinetics did not differ significantly (17). Contrastingly, others have found that TG2-/- macrophages respond to LPS stimulation with significantly higher IL-6 and TNF production (Zsuzsanna Szondy, University of Debrecen, Debrecen, Hungary; personal communication, unpublished data). Activation of NF-kB plays a key role in the inflammatory process by inducing the transcription of pro-inflammatory mediators. TG2 had been shown to directly activate NF-kB by polymerizing IκBα (48) and also TG2 induced the expression of NF-kB (58). It is therefore possible that the endotoxin resistant phenotype of TG2 -/- animals is related to a role of the protein other than its involvement in autophagy.
TG2-deficient macrophages show defects in phagocytosis of apoptotic cells, which is either due to their defective transforming growth factor beta (TGFβ) secretion (92) or to formation of partially functional phagocyte portals (96). TG2, as an adaptor is supposed to bridge milk-fat globule EGF-factor (MFGE8) to integrin β3 on the surface of macrophages creating a docking site for the apoptotic corpse. It has been attributed to this defect that TG2 -/- animals develop autoimmune glomerulonephritis (92) later in life. As we have seen, improper autophagic capacity can also cause a disturbance in self tolerance. It needs further investigation whether this link is applicable to TG2.
Autophagic death of immune competent cells
Although autophagy is basically a pro-survival mechanism, one of the cell death modalities is initiated by an increased rate of autophagy that may lead to irreversible molecular and structural events triggering caspase-independent autophagic death (27, 45). Autophagic death of immune competent cells, particularly of macrophages, may compromise defense against infection, but could also be beneficial in controlling the level and duration of inflammation. It has been reported that there are conditions when autophagy contributes to caspase-independent macrophage cell death involving TIR-domain-containing adapter-inducing interferon-β (TRIF), RIP1, ROS production, and activation of poly(ADP-ribose)polymerase activation (105). In the adaptive phase of the immune response, activation-induced death of CD4+ T cells is partially mediated by autophagy: IFN-γ may induce autophagic death of these cells (18) and knockdown of beclin-1 or atg5 results in decreased death rate of CD4+ T cells upon growth factor withdrawal (53) or presence of immunodeficiency virus envelope protein (16).
Importance of Autophagy in Dying Cells in Determining Anti- or Pro-Inflammatory Response in Phagocytes
Autophagy during apoptotic death
Autophagy is active during programmed cell death when developmental processes require massive cell elimination and frequently activated in cells dying by apoptosis. It is involved in generation of energy-dependent engulfment signals in apoptotic cells, particularly the secretion of the find-me signal lysophosphatidylcholine and exposure of phosphatidylserine on the surface of dying cells (76). Such a role of autophagy in apoptotic cell clearance may have general significance and in case of deficient or inhibited autophagy, the clearance of apoptotic cells becomes inefficient, leading to secondary necrosis of the dying cells. Necrotic cells, unlike the anti-inflammatory apoptotic ones, are potent inducers of the inflammatory response in nearby immune competent cells.
Autophagy and necrosis
When cells die massively in vivo by necrosis, they stimulate a robust inflammatory response (44, 57). Damaged cells release DAMPS, such as high-mobility group box 1 protein (HMGB1) and heat shock proteins that alert the innate immune system via stimulating TLRs. It has been also reported that necrotic cells produced by pressure disruption, hypoxic injury, or complement-mediated damage can trigger a sterile inflammatory response through activating the NLRP3 inflammasome (36). Cells with active autophagy when killed by epidermal growth factor targeted diphtheria toxin, selective release of the immune stimulator HMGB1 occurs without sign of classical necrosis (95). Autophagy promotes necrosis in apoptosis-deficient cells in response to endoplasmic reticulum (ER) stress (99), a condition often associated with human diseases (60). This probably occurs because in cells where excessive ER stress fails to induce apoptosis, the stressing situation keeps accumulating to the point where autophagy is massively induced, leading to cell damage and necrosis. Under defective apoptosis conditions, a cell death mechanism that required autophagy was mediated through RIP-1 kinase activity inducing programmed necrosis called necroptosis (32). This can be utilized in the treatment of childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance by induction of autophagy-dependent necroptosis with the help of a chemical agent (4) release active beclin-1 and inhibiting the anti-autophagic activity of mTOR.
Clearance of cells dying through autophagy may lead to inflammasome activation
Recently, we discovered that cells undergoing autophagic cell death can induce a pro-inflammatory response in human macrophages (73, 74): it was shown by gene array and cytokine secretion analysis that a pro-inflammatory response involving the secretion of IL-6, TNFα, and IL-8 and the anti-inflammatory cytokine IL-10 is generated in human macrophages engulfing MCF-7 cells undergoing autophagic cell death. This finding was surprising, since autophagic dying cells are morphologically and molecularly very similar to apoptotic ones, but the clearance of apoptotic cells is a silent process (56)—apoptotic cells can inhibit pro-inflammatory responses induced by LPS in phagocytes. We could clarify that while cells dying through autophagy can block LPS-induced release of pro-inflammatory cytokines, they can induce IL-1β production and release, and this initiates further pro-inflammatory cytokine response in a paracrine fashion (Fig. 2). It appears that during phagocytosis of autophagic dying cells, ATP acting though its receptor initiates K+ efflux, inflammasome activation, and IL-1β secretion, which in turn initiates further upregulation and release of pro-inflammatory cytokines, such as TNFα and IL-6. One of the several mutually not exclusive mechanisms of inflammasome activation involves extracellular ATP working through the p2X7 receptor, an ATP-gated ion channel, and rapid K+ efflux—the inflammasome sensing the low intracellular potassium level (84). ATP results in opening of pannexin-1-mediated pore through which intracellular compartments-to-cytosol translocation can occur. Our results show that inflammasome activation by autophagic dying cells could be suppressed by high concentration of K+ (data not shown), which is characteristic for NLRP3 (21, 33). Following the engulfment of autophagic dying cells by macrophages, ATP appears in the extracellular space, resulting in the initiation of the above described sequence of events. Autophagic dying cells also may produce activator molecules such as cathepsin B (33), a known inflammasome activator, while being degraded in the phagolysosomal compartment. In addition, reactive oxygen species (ROS) generated in response to endogenous danger signals from engulfed autophagic dying cells may result in activation of the NLRP3/inflammasome complex (85, 108).
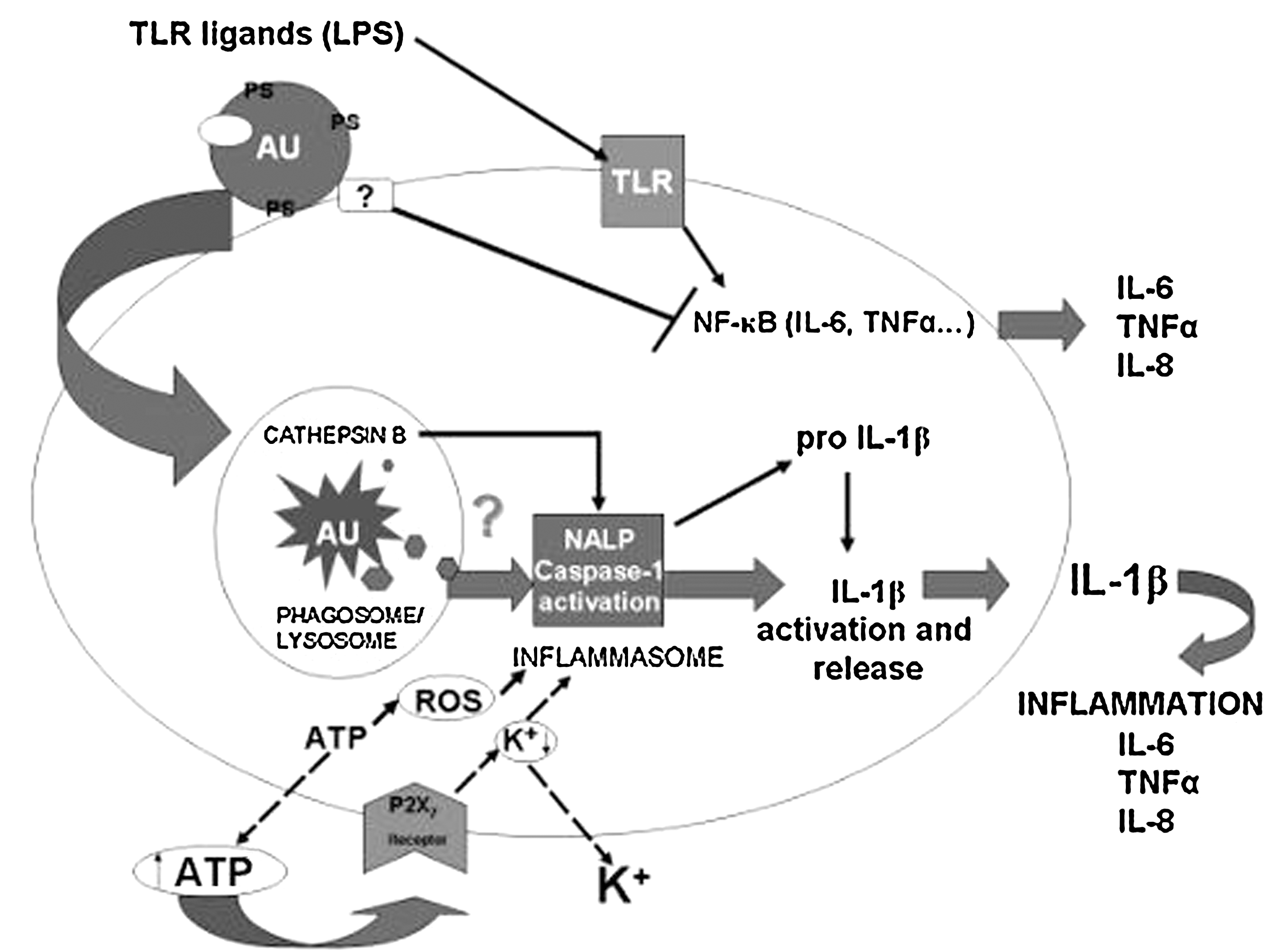
Autophagy in the dying cells promotes cross-presentation
MHC class II presentation of peptides derived from cytosolic proteins results from autophagy (100). Macroautophagy can deliver antigens into autophagosomes for processing by acidic proteases and chaperone-mediated autophagy can translocate endogenous antigens processed by cytoplasmic proteases into the endosomal network to intersect MHC class II (89). In a study by Uhl et al. (98) using virally infected wild-type and apoptosis-deficient Bax/Bak(-/-) fibroblasts as a source of cell-associated antigen, immunization with cells undergoing autophagy before cell death was found to be superior in facilitating the cross-priming of antigen-specific CD8+ T cells and silencing Atg5 expression inhibited priming. They interpreted this autophagic death to be a novel form of 'immunogenic death' with the enhanced priming efficiency being a result of persistent MHC I cross-presentation and the induction of type I interferons. In another study by Lee et al. (47), mice with conditional deletion in Atg5 in dendritic cell (DC) showed impaired CD4+ T cell priming after herpes simplex virus infection and succumbed to rapid disease. The most pronounced defect of Atg5(-/-) DCs was in the processing and presentation of phagocytosed antigens inducing TLR stimuli for MHC class II, the cross-presentation of peptides on MHC I remaining intact in the absence of Atg5. These results suggest that autophagy can influence cross-priming, and targeting the autophagy cascade may provide a better therapeutic/vaccination strategy for achieving potent cross-priming of viral and tumor-specific CD8+ T cells. New vaccination strategies have been based upon this phenomenon, aimed at improving the targeting of antigens to dendritic cells, promoting cross-priming, improving peptide binding to class I molecules, and targeting antigens to both the class I and the class II pathways.
Significance of Autophagy in Human Inflammatory Diseases
Crohn's disease
Crohn's disease (CD) is one of the two major forms of inflammatory bowel disease, which can affect the small bowel and/or the colon. It is characterized by severe chronic inflammation extending across all layers of the intestinal wall. Among etiological factors, genetic predisposition and an infectious cause, particularly an aberrant response to the commensal gut flora, have long been suspected.
Genome-wide association studies have linked two autophagy related genes to CD, Atg16L1 and immunity-related GTPase M (IRGM) (29, 64, 80). The polymorphism associated with CD in Atg16L1 is a single amino acid mutation (T300A) in one of the WD40 repeats, which is unlikely to delete Atg16L1 function. WD40 repeats are propeller like protein–protein interaction platforms and the five of them in Atg16L1 are currently not known what interact with. Atg16L1 T300A has been shown to be less stable in the wake of infection with invasive enteric bacteria. Accordingly, chimeric mice with Atg16L1-deficient hematopoietic cells do not develop spontaneous colitis and survive like their wild-type counterparts in most respects under normal conditions. However, in response to dextran sulfate sodium (DSS)-induced experimental colitis, these chimeric animals die rapidly due to a more severe inflammation and ulceration of the distal colon (82). The levels of IL-1β and IL-18 were significantly elevated in the sera of these animals and their mortality could be reduced by the injection of IL-1β and IL-18 neutralizing antibodies. The importance of Atg16L1 in altered cytokine production in CD is supported by another study, which found enhanced expression of acute phase reactants and adipokynes in Paneth cells of Atg16L1 hypomorphic mice. Paneth cells are found in the crypts of the ileum and sometimes the colon and they perform secretory and phagocytic functions. They are thought to regulate the bacterial flora locally and to systemically influence pro- and anti-inflammatory processes. The polymorphism associated with the immunity related GTPase (also called p47 GTPases), IRGM occurs in the upstream regulatory region of the gene and causes reduced expression of the protein in CD (64). The precise role of IRGM in autophagy is not yet understood, nevertheless it has been shown to play a role in autophagic control of intracellular pathogens, M. tuberculosis, in particular (88). The much more extensive IRG family in mouse has, however, multiple roles, for example in phagosome acidification and in the maturation and the parasitophorous vacuole membrane (34). How analogous functions may contribute to the pathogenesis of CD remains to be deciphered.
Lysosomal storage diseases
Lysosomal storage diseases (LSDs) are a group of over 40 genetic conditions, most of which are caused by deficiency of lysosomal hydrolases. Lysosomal storage diseases are now frequently interpreted as autophagic disorders (77, 87) and in recent years the pathogenesis of many neurodegenerative storage disorders have been rethought with a special focus on autophagy (5, 22, 23, 41). The LSDs in which autophagy has been studied include multiple sulfatase deficiency, mucopolysaccharidosis Type IIIA, GM1-gangliosidosis, Pompe disease, Niemann–Pick disease Type C, neuronal ceroid lipofuscinosis, mucolipidosis Type IV, and Danon disease (77). Impaired fusion between autophagosomes and lysosomes results in the accumulation of polyubiquitinated proteins and lipids and also defective mitochondrial turnover and generation of high levels of ROS, which in turn elicits an inflammatory response and can lead to systemic inflammation (86).
Neurodegeneration and neuroinflammation
Inactivating mutations of parkin-1 are linked to early-onset cases of Parkinson's disease (PD). The specific pathological features of PD are the loss of neurons from the pars compacta of the substantia nigra and the consequent disturbance of dopaminergic signaling in the striatum. The wasting neurons show signs of mitochondrial dysfunction and oxidative stress, contain inclusions of ubiquitinated proteins and organelles (Lewy bodies), and the affected brain territories are surrounded by an infiltrate of neurotoxic microglia and activated astrocytes, indicating a chronic neuroinflammatory process (25). The ubiquitin ligase, Parkin, is a mitochondrial protein that recently has been shown to be directly responsible for mitophagy (a subtype of autophagy in which turnover of damaged mitochondria occurs via the auophagolysosomal pathway) (49). Lee et al. found that cells expressing mutant Parkin failed to recycle their mitochondria when they were damaged. Parkin, an ubiquitin E3-ligase, is selectively associated with depolarized or impaired mitochondria through PTEN-induced putative kinase (PINK) 1 (10, 63) and promotes their autophagy (70, 71). Now we learn that Parkin-mediated ubiquitination of these organelles is required to recruit the autophagy proteins histone deacetylase (HDAC) 6 and p62/SQSTM1 (24). HDAC6 and p62/SQSTM1-dependent transport to perinuclear aggresomes concentrates toxic proteins and dysfunctional organelles for subsequent removal by autophagy and Parkin fails to clear mitochondria in either HDAC6 or p62 deficient cells. Accordingly, Parkin might protect neurons by promoting the clearance of dysfunctional mitochondria, which could produce ROS, oxidative damage, cell death, and inflammation.
Targeted ablation of other autophagy genes in mice also leads to cell death in the central nervous system and neurodegeneration (30, 42) with inflammation. Post mitotic cells such as neurons or muscle cells may be particularly sensitive to accumulation of undegraded material as these cells cannot dilute the accumulated material by cell division.
Paget's disease of bone
Paget's disease of bone (PDB) is a mostly focal, less frequently systemic, chronic condition characterized by excessive resorption of bone by atypical hyperactive osteoclasts. At later stages the resorbed bone is replaced with coarse-fibered, dense trabecular, haphazardily organized bone accompanied by a fibrous, hypervascularized marrow-space, a picture resembling abnormal tissue remodeling after inflammation. Between 10%–50% of patients with familial PDB and between 5–30% of patients with “sporadic” PDB carry mutations in p62/SQSTM1 (78). All PDB-causing mutations impair ubiquitin binding (6). The lack of p62/SQSTM1 severely reduced NF-kB activation in response to the inflammatory cytokines TNFα and IL-1 (69), and the nerve growth factor (NGF), which signal through atypical protein kinase C (aPKC) isoforms, such as ζPKC and ζ/ιPKC. p62/SQSTM1 interacts with RIP and TNF-receptor associated factor (TRAF) 6, which are important adaptor molecules in the signaling pathways activated by TNF-receptor (TNFR), IL-1R, and p75 NGF-receptor (NGFR). In addition, aPKCs interact with p62/Sequestosome 1 (SQSTM1) directly through its (atypical PKC interaction domain) AID region. Inhibition of the p62/SQSTM1-aPKC interaction blocked NF-kB activation in cell cultures, inhibited NGF-induced neuronal survival and differentiation (103), and displayed potent anti-inflammatory and anti-tumor activities in different animal models (69). Although the disruption of the ubiquitin binding activity of p62/SQSTM1 is undoubtedly necessary for PDB, it is still unclear whether the removal of components of the NF-kB signaling pathway through autophagy in osteoclasts is the responsible molecular event.
Arthritis
FLIP (also known as caspase 8 or FADD-like anti-apoptotic molecule) is overexpressed in macrophage-like cells in the synovial tissue of inflamed joints in juvenile idiopathic arthritis (JIA) and its expression positively correlates with the severity of inflammation (104). It has been suggested that upregulation of pro-inflammatory FLIP extends the lifespan of inflammatory cells and thereby procrastinates the resolution of the process. FLIP is reported to promote autophagy and to ameliorate the energy homeostasis of cells (49). It will require further investigation to resolve if and to what extent FLIP's pro-autophagy effect contributes to survival of the pro-inflammatory cell population in the inflamed joints.
Loss of autophagy may promote inflammation and tumorigenesis as a result of a sustained high dead cell burden
Autophagy in tumors is thought to serve the survival of tumor cells (11) by improving stress tolerance, and maintaining viability in an environment where nutrients are sparse due to rapid growth and an insufficiently developed vasculature, and where without autophagy cells would succumb to apoptotic or necrotic demise (37 –39, 102). Autophagic tumor cells can reduce in size and become dormant, although retaining a proliferative capacity for better times. By rescuing tumor cells, autophagy prevents inflammation resulting from necrotic cells (28, 59). This can have ambivalent consequences, considering that many current cancer therapeutics boost autophagy. On one hand, it is harmful as the inflammatory reaction could contribute to the elimination of cancer cells. On the other hand, sustained inflammation inside the tumor would also top up proliferative signals, such as IL-1, IL-6, TNF, IL-11, and IL-23. Tumor-associated inflammatory macrophages also promote neovascularization through the secretion of IL-8, CXCL1, CXCL8, and VEGF. Inactivation of a number of pro-autophagy genes (beclin-1, bif-1, atg4) renders mice tumor prone (55, 75). Apoptosis-defective tumors in which autophagy was simultaneously inactivated by Akt activation or allelic disruption of beclin-1 display chronic necrosis, inflammation, dramatic macrophage infiltration, NF-κB activation, and cytokine production as opposed to tumors in which autophagy is functional (11). Chronic cell death causes inflammation in autophagy-defective tissues, for example in the liver of Atg7 null mice (43).
Conclusion
As it has been discussed above in detail, autophagy can influence inflammatory reactions by several ways in a cell-intrinsic manner, affecting either positively or negatively pro-inflammatory signaling within immune effector cells when they encounter PAMPS or DAMPS. In the context of the inflamed normal tissue, where nutritional supplies are running short, the inflammatory cells themselves might just as well fall back on supporting their functioning by means of autophagy. Its effect on cellular energy homeostasis, tolerance against cellular stress, and prevention or provocation of pro-inflammatory cell death can potentially determine the degree and duration of inflammation. Under certain conditions autophagic dying cells and autophagy-driven necrotic ones can provoke NLRP3 inflammasome activation in macrophages with concurrent caspase-1 activation and secretion of IL-1β, suggesting that some forms of autophagic death are pro-inflammatory and immunogenic in sharp contrast to immune-suppressing apoptotic cell death. This may have particular significance in tumor tissues where loss of apoptosis genes with a shift toward autophagic death frequently occurs, provoking inflammatory reaction which, in turn may contribute to either tumor cell proliferation or development of anti-tumor immune response. During the last couple of years, it has been revealed that polymorphism of autophagy genes and those which regulate autophagy is linked to susceptibility toward various inflammatory disorders including Crohn's disease, Paget's disease, and arthritis. With current technologies, animal models of such diseases can be developed to learn the molecular details of the specific pathogenetic processes and to understand how autophagy contributes to them. These efforts will lead to novel autophagy-targeted therapeutic interventions in cancer and chronic inflammatory disorders.
Footnotes
Acknowledgments
This work has been funded by grants from the Hungarian Scientific Research Fund [OTKA NI 67877, K 61868], an ETT grant from the Hungarian Ministry of Health and EU [MRTNCT-2006-036032, MRTN-CT 2006-035624, LSHB-CT-2007-037730].