
Abstract
Introduction
Influenza virus replication has been studied in depth, and several antiviral compounds have been developed, but their long-term efficacy is often limited by toxicity and the almost inevitable selection of drug-resistant viral mutants (4, 42). The drugs used today all target viral structures, such as the M2 ion channel or neuraminidase (NA), but preclinical research is looking at several means for blocking viral replication by interfering with pathogen-exploited host-cell machinery. One of the more promising and innovative strategies involves inhibiting intracellular pathways that are specifically activated by influenza virus to ensure its replication (15, 29, 34). This approach would offer important advantages, including broad-spectrum efficacy that would be independent of virus type, strain, or antigenic properties and reduced probability to select drug-resistant viral strains. Many of these intracellular pathways are highly sensitive to changes in the intracellular redox state (31).
Viral infection is often associated with oxidative stress (reviewed in 31), and decreased intracellular levels of the antioxidant glutathione (GSH) have been described in influenza virus infection (6, 30). The administration of exogenous GSH inhibits the replication of DNA and RNA viruses by exerting direct effects on the expression of envelope glycoproteins (18, 33, 35). The molecular mechanism underlying this effect has not been fully elucidated, but the maturation of these proteins depends on the formation of disulfide bonds (5), which is strongly affected by reducing agents such as GSH (46).
Cellular glycoproteins (or secretory proteins) undergo folding in the endoplasmic reticulum (ER), a process that involves sequential steps, some of which are unique to the ER (i.e., N-linked glycosylation and disulfide bond formation), and the assistance of diverse folding factors. The oxidoreductases of the protein disulfide isomerase (PDI) family, for example, play major roles in disulfide bond formation, and their activity is influenced by the oxidative environment of the ER (8). The secretory pathway is commandeered by the influenza virus to produce its own glycoproteins, and the decreased intracellular GSH levels found during infection (6, 30) would be expected to favor HA folding and maturation.
A series of GSH derivatives with aliphatic chains of different length coupled to peptides bound to the α-NH2 group of glutamine have been recently synthesized. One in particular, GSH-C4, a hydrophobic molecule that enters cells more easily than GSH, has proved to be more effective than GSH in inhibiting the replication of Sendai and Herpes simplex 1 viruses (17, 18, 35). On the basis of these observations, we hypothesized that GSH-C4 might exert anti-influenza effects by blocking redox-sensitive pathways involved in HA maturation. The present study shows that GSH-C4 strongly diminishes influenza virus replication, an effect associated with retention of immature monomeric HA in the ER and reduced plasma-membrane targeting of the mature glycoprotein. The antiviral activity was mediated by increased intracellular GSH levels, which affected the redox state of PDI in infected cells. In in vivo studies, GSH-C4 also improved the survival of mice lethally infected with influenza A virus. This effect was associated with decreased pulmonary viral titers and increases in the alveolar area compared with those observed in placebo-treated mice.
Results
GSH-C4 inhibits influenza A virus replication in Madin Darby canine kidney cells
To identify the effects of GSH-C4 on influenza virus propagation in cultured cells, we infected confluent monolayers of Madin Darby canine kidney (MDCK) cells with PR8 at a low multiplicity of infection (MOI: 0.01) and incubated them with various concentrations of GSH-C4 for 48 h. Viral production in cell supernatants was measured in hemagglutinating and plaque-forming units (HAU and PFU, respectively) at 24 and 48 h postinfection (p.i.). As shown in Figure 1a and b, viral replication assayed at 24 h p.i. was dose-dependently inhibited by GSH-C4 concentrations of 1–15 mM. Significant reductions were achieved with concentrations of 5 mM or more, and almost complete inhibition was observed with 10 and 15 mM (HAU reductions of−90% to −98% vs. untreated controls [p<0.05]; 3 log inhibition in PFU assays vs. untreated cells [p<0.0001]). These results were confirmed by real-time (RT)–polymerase chain reaction (PCR) assays of viral RNA in supernatants from infected cells (6.6×102 copies/mL for those treated with 10 mM GSH-C4 vs. 2.6×108 in untreated controls). Similar degrees of inhibition were observed at 48 h p.i. (end of experiment), indicating that the treatment effect was not merely the result of a delayed virus burst (data not shown). Similar results were achieved with 10 mM GSH-C4 in MDCK cells infected with influenza A/NWS/H1N1 virus or avian influenza A/Parrot/ULSTER/73 (H7N1) virus (Fig. 1c) or in a subsequent experiment performed with a higher MOI of PR8.
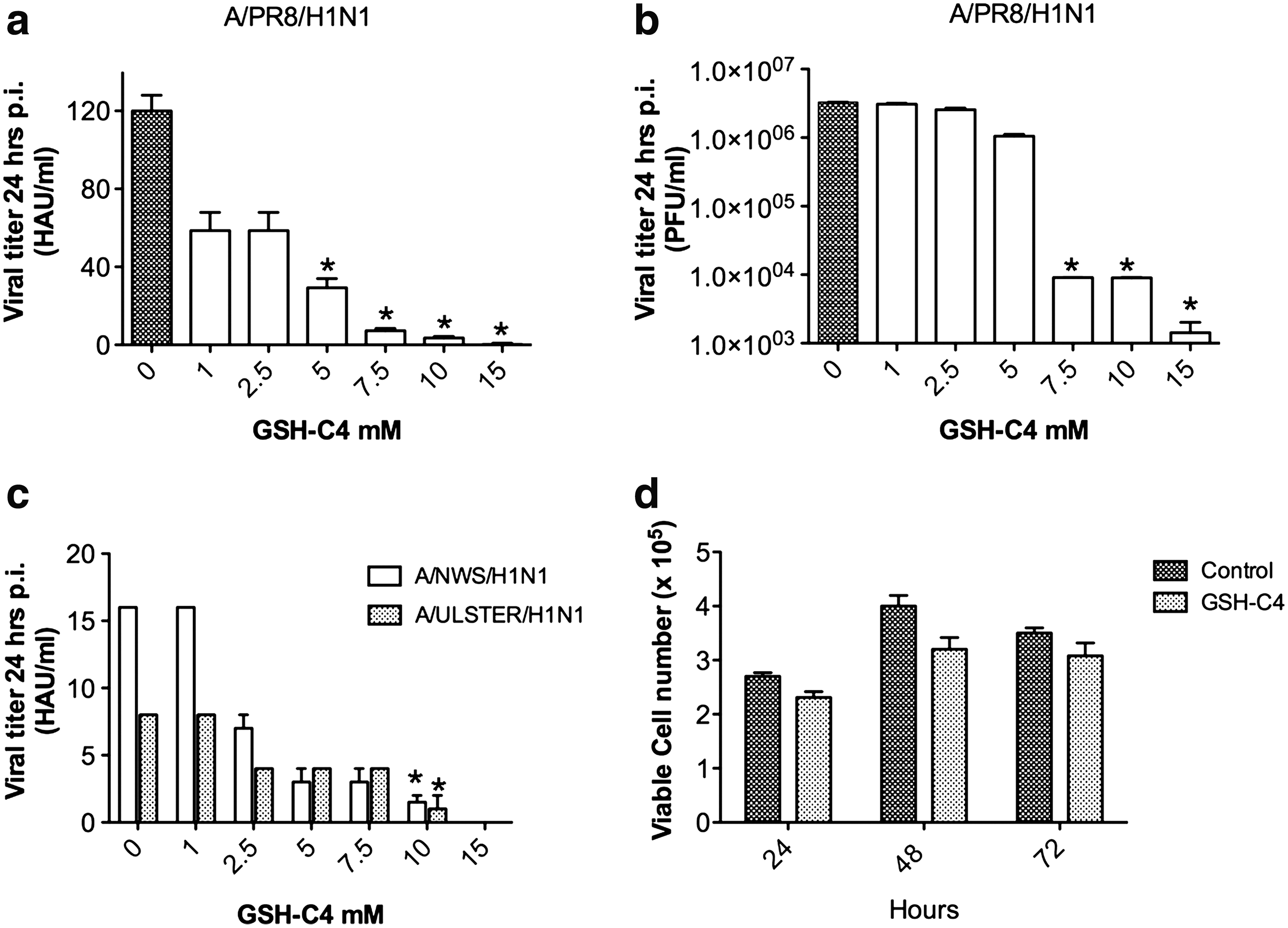
As previously reported (32), exposure of uninfected MDCK cells to antiviral concentrations (10 mM) of GSH-C4 had no significant toxic effects on the uninfected cells. To better investigate the eventual effect of the compound on proliferation and viability of exponentially growing cells, we exposed MDCK cells (plated at the concentration of 2×105 cells/mL) to 10 mM GSH-C4. Counts of viable cells performed at 24–48–72 h after drug addition revealed mild, nonsignificant differences in cell proliferation and mortality rate between GSH-C4–treated cells and untreated control (Fig. 1d). Exposure to 15 mM GSH-C4 caused minor morphological changes typically found in more differentiated MDCK cells (38) (data not shown), so all subsequent experiments were performed with 10 mM GSH-C4.
GSH-C4 impairs PR8 HA maturation and surface expression in NCI cells
Our previous findings suggested that intracellular GSH levels affect the expression of viral envelope glycoproteins (18, 30, 33, 35), so we tested the hypothesis that GSH-C4's antiviral activity was related to the effect on PR8 HA expression. NCI cells were infected with PR8 at an MOI of 1.0 to allow single-cycle replication and exposed to different concentrations of GSH-C4 for 8 h p.i. Cell lysates were then analyzed with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions and gels were immunostained with anti-HA antibody (Ab). GSH-C4 treatment produced dose-dependent decreases in HA expression (Fig. 2a) without significantly affecting nucleoprotein (NP) or matrix protein (M) expression (Fig. 2b).

The reducing conditions used for SDS-PAGE in these studies yield a pool of HA monomers representing different stages in the glycoprotein maturation process (44). The doublet bands in Figure 2a thus correspond to two distinct monomeric species that have been described by Segal et al. (44). The upper component of the doublet represents the mature, properly folded 78-kDa HA0 protein mainly derived from the reduction of dimers and trimers that have been terminally glycosylated in the Golgi complex. The lower component is a lighter-weight monomer that retains the 14-oligosaccharide core unit that is attached to nascent polypeptides in the ER. GSH-C4 treatment had no significant effects on levels of mRNA for HA (or NP) (Fig. 2c), but it dose-dependently diminished the expression of both HA monomers with particularly marked effects on the mature protein.
These findings suggested that the anti-influenza activity of GSH-C4 was related specifically to posttranscriptional effects on viral HA, which cause it to be retained in the ER without completing the maturation process. After core glycosylation, nascent polypeptides normally undergo (i) trimming of terminal glucose and mannose residues by ER glucosidases and mannosidases; (ii) folding, which involves disulfide bond formation catalyzed by ER oxidoreductases; (iii) oligomerization; (iv) transport to the Golgi apparatus for terminal glycosylation; and (v) transfer to their final destination in the cell (11, 13). The next set of experiments was conducted to determine whether GSH-C4 was interfering with one or more of these steps.
First, we compared GSH-C4's effects with those produced by inhibition of the trimming enzymes, schematically shown in Figure 3a (39, 41). PR8-infected NCI cells were treated with 1-deoxinojirimicin (dNM) and castanospermine (CST), both of which inhibit glucosidase activity, and with the mannosidase inhibitor, swainsonine (SW). As observed in previous studies (41), none of these agents produced significant cytotoxicity (data not shown). Each caused a characteristic reduction in the molecular weight of the HA monomer (Fig. 3b), but all of these effects differed from the changes produced by GSH-C4, and none reduced viral yields to the extent that the GSH analog did (Fig. 3b, bottom panel). We also treated infected cells with monensin (MON) and brefeldin A (BFA), which alter cis- and trans-Golgi transport of HA, respectively, after it has undergone folding (12, 40). As shown in Figure 3c, neither of these treatments affected the expression of HA, which was in minor size with respect to that observed in GSH-C4–treated cells. This could be related to the known actions of these drugs mentioned earlier. Indeed, in the presence of BFA or MON, HA undergoes disulfide bond formation in the ER. Thereafter, several oligosaccharide residues are removed, first by glucosidases II and mannosidases in the ER and later by other trimming enzymes in the cis-Golgi. This process leads to a protein whose molecular weight is lower than that of ER core-glycosylated form.

These findings strongly suggest that GSH-C4 does not directly interfere with the glucose trimming or ER-Golgi transport of the HA protein.
We therefore turned our attention to the disulfide bond formation that occurs during protein folding. This complex process, which includes oxidation, reduction, and isomerization reactions catalyzed by PDI-family oxidoreductases, generates a glycoprotein that is fully folded (oxidized) and ready for oligomerization and transfer to the Golgi complex (5). To evaluate GSH-C4's effects on disulfide bonding in HA, we analyzed infected NCI cell extracts with nonreducing SDS-PAGE. The denatured, unreduced HA migrated as three distinct species as already reported by Segal et al. (44), and most probably, they correspond to intermediate species of the HA maturation process (44). Treatment with GSH-C4 profoundly decreased the high-molecular-weight content of HA species while the HA monomer was slightly increased (Fig. 4a). To characterize the nature of the high-molecular-weight HA forms, we performed an additional experiment by treating the samples with a thiol alkylating agent N-ethyl maleimide (NEM). Figure 4b shows that the high-molecular-weight species were present upon NEM treatment, indicating that these forms were not the result of manipulation processing of the sample before western blot analysis but rather intermediate species of HA maturation process (44). Further, GSH-C4 treatment decreased the presence of these species even under NEM treatment (Fig. 4b).

Next, we performed reducing SDS-PAGE on cell extracts treated with endo-β-N acetylglucosaminidase H (Endo H), which selectively removes N-linked carbohydrates that have not been terminally glycosylated. Preliminary time-course experiments showed that by 8 h p.i. most HA is normally Endo H resistant, indicating that it has already undergone terminal glycosylation in the Golgi complex (Fig. 4c), so this time point was used for collection of PR8-infected cells used in subsequent experiments. As expected, in cells that had not been treated with GSH-C4 (Fig. 4d), the ratio of Endo H-resistant HA to Endo H-sensitive HA (HAres/HAsens ratio) was ∼1:1, but this ratio dropped to 0.3:1 in GSH-C4–treated cells, reflecting markedly decreased expression of terminally glycosylated HA (−99% vs. control). In Endo H-treated samples, there were two additional bands. The larger (55 kDa) corresponds in size to the HA1 subunit of mature HA, which is proteolytically cleaved in the trans-Golgi and plasma membrane of the cell (49). The smaller band (40 kDa) might represent an HA0 degradation product, but it could also be a fraction of HA0 with some unglycosylated sites (49) or a deglycosylated form of HA1 (47). Both forms were clearly present in the untreated PR8-infected cells shown in Figure 4d, but their expression was markedly decreased after GSH-C4 treatment (−99% vs. untreated cells).
Collectively, these data suggest that when infected cells are treated with GSH-C4, most of the HA fails to reach the Golgi apparatus and does not undergo final maturation.
Confocal laser-scanning microscopy of infected NCI cells confirmed this interpretation. Specific labeling of trans-Golgi structures with wheat germ agglutinin (WGA) (25) clearly colocalized with HA labeling in untreated cells, but in those treated with GSH-C4, the HA and WGA signals were quite distinct (Fig. 5b).
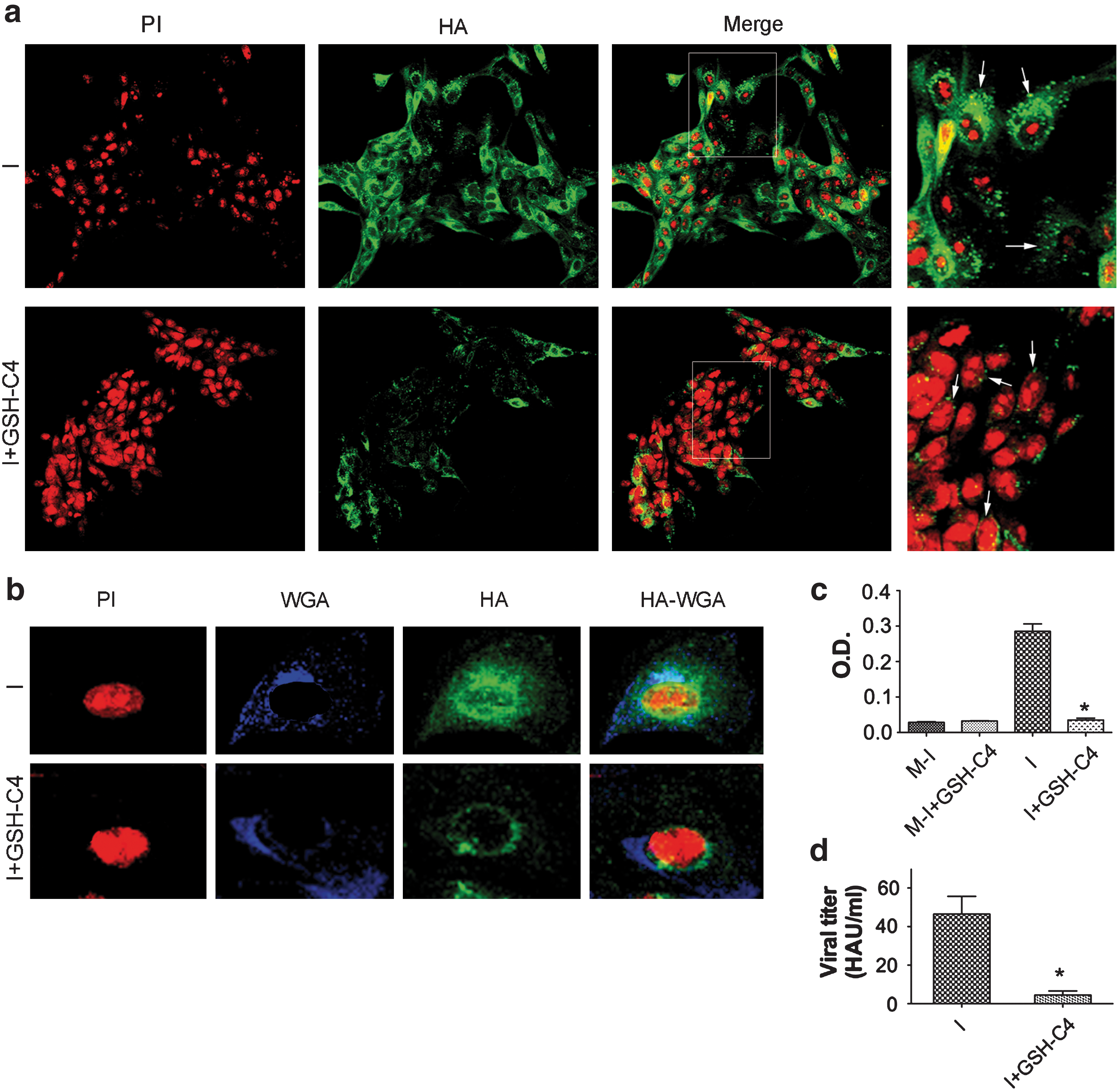
Only properly folded, terminally glycosylated HA trimers are inserted into the plasma membrane of the host cell and utilized for the assembly of the mature virions. Therefore, it was not surprising to find that HA expression in GSH-C4–treated cells was clearly reduced and confined mainly to the perinuclear regions, whereas in untreated cells it was distributed throughout the cytoplasm and cell plasma membranes (Fig. 5a). The scarcity of HA in the cell plasma membranes of treated cells was confirmed by the results of hemadsorption assays (Fig. 5c), and this effect was associated with a 90% reduction in viral replication (Fig. 5d).
Collectively, these findings indicate that GSH-C4 blocks HA maturation at a pre-Golgi level, thereby diminishing its cell plasma-membrane expression. Our next experiments focused on the mechanisms underlying this effect.
GSH-C4 influences the redox state of PDI in PR8-infected cells
The redox state of PDI determines its ability to form (oxidation) and isomerize (reduction) disulfide bonds in cargo proteins (27), and PDI oxidation is one of the rate-limiting steps in oxidative protein folding (8). To investigate the redox state of PDI in our model, we performed gel-shift assays under nonreducing conditions on cell homogenates alkylated with polyethylene glycol-conjugated maleimide (Mal-PEG). Mal-PEG binds to each available sulfhydryl group in a protein, producing shifts in its molecular mass. The number of Mal-PEG shifts observed in PDI thus reflects the degree to which the enzyme is oxidized (2). As shown in Figure 6a (upper panel), PDI was found in both reduced and oxidized forms in untreated mock-infected cells, and this situation was not significantly changed by GSH-C4 treatment. Surprisingly, PR8-infected cells displayed markedly enhanced overall expression of PDI, probably in response to the increased demands related to viral protein production. GSH-C4 treatment clearly decreased the levels of oxidized PDI (Fig. 6a, upper panel) in infected cells but had no significant effect on the proportions of reduced and oxidized enzyme in uninfected cells. A similar pattern emerged when we analyzed the oxidation status of HA: compared with untreated cells, those exposed to GSH-C4 exhibited substantially lower levels of oxidized HA (Fig. 6a, bottom panel). Monomeric HA contains six intrachain disulfide bonds. The absence of multiple bands in Mal-PEG–treated samples suggests that only one free cysteine is accessible for alkylation (43). Both the effects on redox state of HA and PDI effects seemed to be related to intracellular GSH levels. Consistent with our previous findings (30), PR8 infection diminished GSH levels by 55% (vs. uninfected cells), and an even more dramatic drop (−75% vs. uninfected cells) was observed when GSH synthesis in infected cells was blocked by buthionine sulfoximine (BSO) pretreatment. These changes were completely reversed by GSH-C4 treatment (Fig. 6b). Interestingly, after BSO treatment, both HA expression and viral replication were clearly enhanced (Fig. 6c).
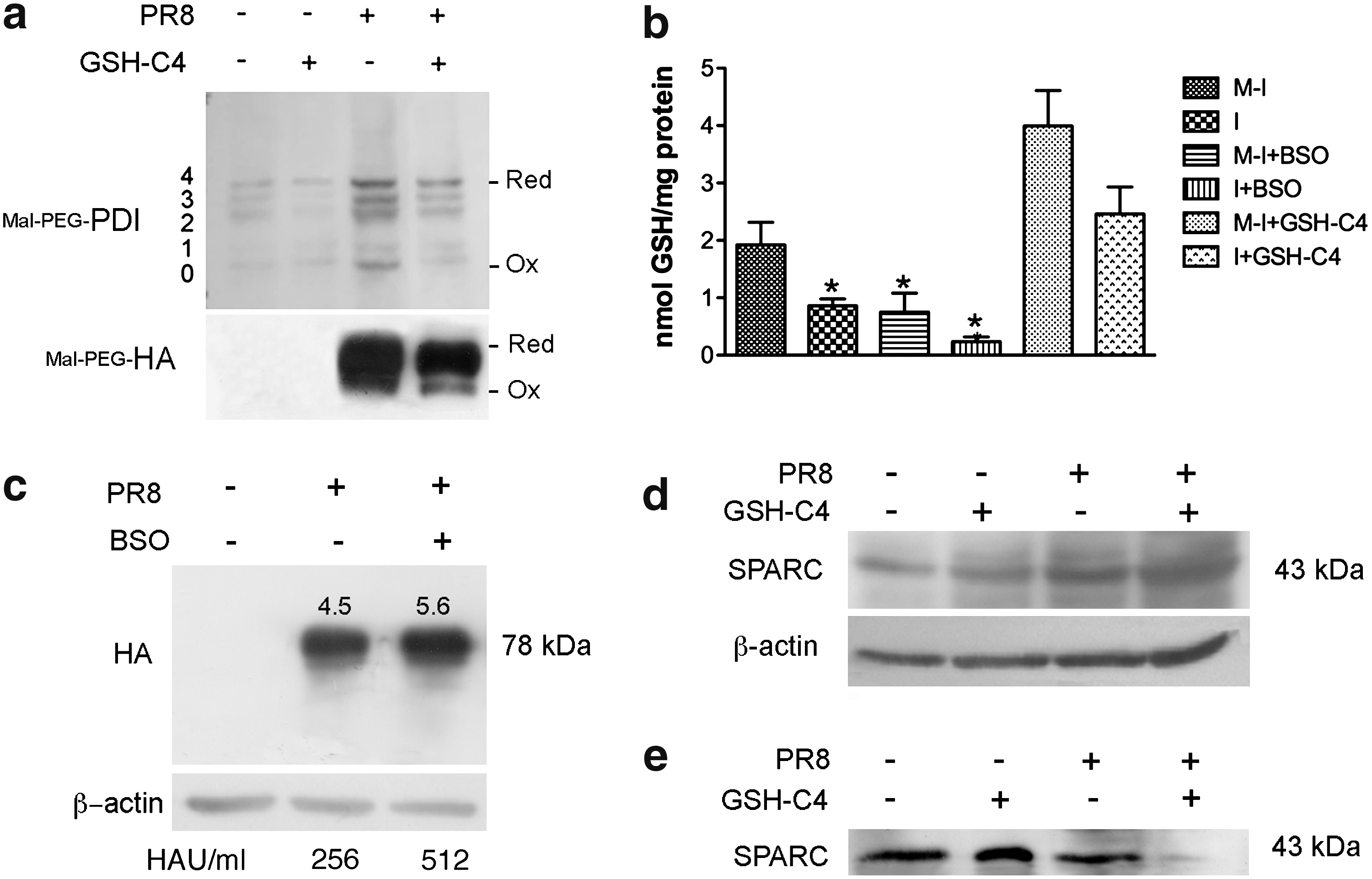
The folding, assembly, and processing of several cellular glycoproteins are also PDI dependent (20), so GSH-C4 could conceivably affect the maturation of host-cell glycoproteins as well. To investigate this possibility, we treated uninfected and infected cells with GSH-C4 and evaluated the expression and secretion of SPARC (secreted protein acidic and rich in cysteine), a widely expressed matricellular glycoprotein whose maturation process closely resembles that of HA (9). As shown in Figure 6d and e, uninfected cells treated for 24 h with GSH-C4 exhibited no significant changes in the expression of SPARC or its secretion into the supernatant. After PR8 infection, SPARC expression increased markedly (paralleling the effects of infection on PDI expression shown in Fig. 6a, upper panel), and its secretion into the supernatants was strongly reduced by GSH-C4 treatment (Fig. 6e).
Collectively, these findings indicate that (i) GSH-C4 reduces PDI oxidation in infected cells by increasing intracellular GSH levels that have been depleted by PR8 infection; (ii) the decreased oxidation of PDI reduces the enzyme's ability to catalyze disulfide bond formation during HA folding; and (iii) in uninfected cells, where the redox state of the cell is “normal” and GSH levels have not been depleted, GSH-C4 has no effect on PDI oxidation or glycoprotein folding.
GSH-C4 increases survival in PR8-infected mice
The in vivo antiviral activity of GSH-C4 was evaluated in a well-established murine model of influenza virus infection (10, 34). Animals were treated for 7 days with daily intraperitoneal injections of 7.4 mg GSH-C4, a dose related (at least theoretically, considering the blood volume in mice) to that which produced maximal antiviral effects in vitro (10 mM). Measurement of plasma GSH levels in uninfected mice revealed significant increases few minutes after GSH-C4 treatment (Fig. 7a) and peak effects (an ∼4-fold increase over control levels) at 30 min after treatment. GSH levels decreased progressively thereafter, but at 60 and 120 min after treatment they were still higher than those found in control mice. The kinetics of the drug were similar during chronic treatment (daily injections for 7 days) (Fig. 7b).

This chronic treatment regimen had no negative effects on the health of uninfected female BALB/c mice. Compared with animals receiving placebo, those treated with GSH-C4 exhibited no significant change in body weight (Fig. 7c) or temperature (Fig. 7d), and animals sacrificed at 5 and 10 days after treatment presented no gross pathological alteration of the liver or spleen (data not shown).
We also compared the in vivo antiviral efficacy of chronic GSH-C4 treatment and placebo in two groups of lethally infected mice (Table 1A). By p.i. day 10, 52% of the GSH-C4–treated animals were still alive (vs. only 13% of those that received placebo; p=0.001). None of the mice that survived to p.i. day 10 showed any sign of disease for the remainder of the observation period (p.i. days 10–40) and were therefore considered cured.
Intranasal inoculation with 1 plaque-forming unit (10 MLD50) of PR8 virus.
p< 0.05 and ** p=0.001 compared with placebo-treated control group—log-rank (Mantel-Cox) test.
GSH, reduced glutathione; p.i., postinfection; NI score, necroinflammatory score.
The effects of GSH-C4 on pulmonary viral titers were examined in other groups of mice. Animals were killed at 6 days after lethal PR8 challenges, and lung homogenates were subjected to TCID50 assay. Compared with those of placebo-treated animals, pulmonary virus titers for GSH-C4–treated animals were reduced by ∼2 logs (Table 1A).
Next we examined lung tissues from uninfected and infected mice treated with GSH-C4 or placebo and sacrificed at 6, 9, 18, and 40 days p.i. No lesions were found in any of the uninfected animals' lungs (Fig. 8, first column), and the mean±SE free alveolar area for these animals was 59.55±1.3 (Table 1B). In infected mice treated with placebo (Fig. 8, second column), the lungs presented thickened alveolar septa, vascular congestion, and areas of inflammation and necrosis characterized by interstitial and alveolar exudates, inflammatory cells, fibrin, and cellular debris. By p.i. day 6, these lesions already involved over 25% of the examined parenchyma, and similar involvement was observed in survivors sacrificed 40 days p.i. (Table 1B).
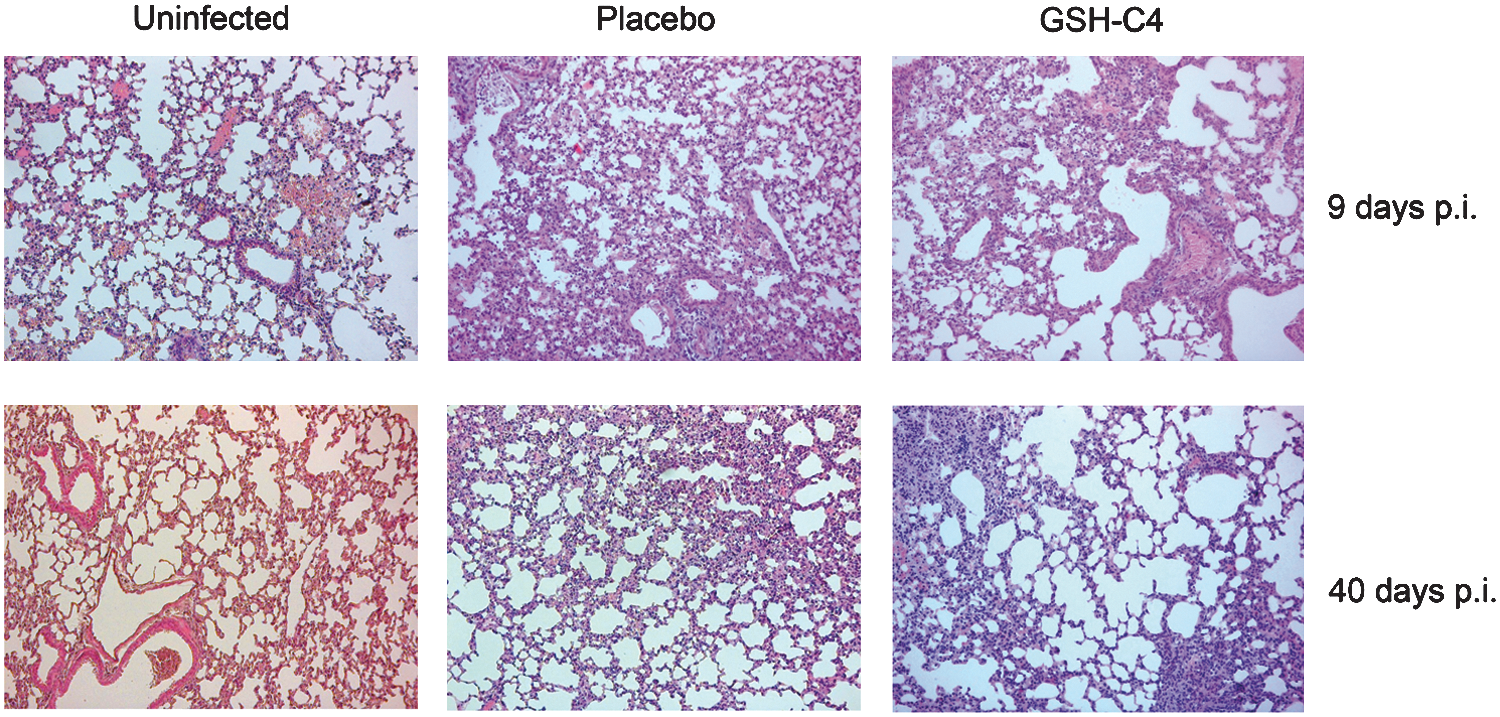
GSH-C4–treated mice sacrificed at 6 or 9 days p.i. displayed inflammatory changes in the lungs similar to those found at these time-points in placebo-treated mice. Later (p.i. days 18 and 40), however, less-extensive lung involvement (<25% of the examined parenchyma) was observed in animals treated with GSH-C4 (Fig. 8, third column).
Placebo-treated mice killed on p.i. days 6 and 9 also had significantly reduced alveolar areas (p<0.05 vs. mock-infected controls) (Table 1B). In contrast, the alveolar areas for GSH-C4–treated infected mice killed on p.i. day 9 were significantly higher (p<0.05 vs. placebo-treated mice) (Table 1B), and this difference persisted for the duration of the experiment, indicating that the virally induced inflammation had a lower impact on lung function.
Discussion
The alarming unpredictability of the influenza A virus has been highlighted by the emergence of highly pathogenic avian strains and, more recently, by the ongoing A H1N1 swine flu pandemic. Vaccination is still a key component of anti-influenza defense strategies, but safe, effective vaccines cannot be produced quickly enough to deal with emergency threats. Our current anti-influenza drug arsenal includes the adamantanes, which target the matrix protein (M2) and inhibit viral uncoating, and NA inhibitors, which impair release of virus from the infected cells. Unfortunately, single-amino-acid substitutions in the M2 and NA proteins have resulted in the development of high-level resistance to both types of drugs (28). Consequently, there is an urgent need for drugs that act on novel molecular targets, providing safe and effective protection against the influenza virus.
In this study, we have shown that GSH-C4, a butanoyl GSH derivative, possesses potent anti-influenza activity that is dependent on neither virus strain nor host cell type: it is based on interference with the maturation of viral HA, which is largely mediated by redox-sensitive, host-cell PDI activity. Mature HA is a homotrimeric glycoprotein with seven N-glycosylation sites and six disulfide bonds. Folding of the HA precursor begins in the ER. Early in the maturation process, a core unit of 14 oligosaccharides is transferred to the nascent polypeptide chain. Immediately thereafter, this core is trimmed by glucosidases I and II, an essential step for the glycoprotein's entry into the calnexin/calreticulin cycle, wherein oxidoreductases catalyze the formation of disulfide bonds (13). The folded oxidized HA undergoes trimerization and is then transferred to the Golgi apparatus for terminal glycosylation. At that point only it is inserted into the cell plasma membrane and utilized to assemble viral particles.
Our experiments indicate that GSH-C4 does not interfere directly with the glycosylation or ER-Golgi transport steps of this process. Its effects on HA expression and electrophoretic mobility are clearly different from those of compounds that block these processes (Fig. 3). GSH-C4–treated cells exhibited accumulations of core-glycosylated HA with marked downregulation of mature 78-kDa protein expression, and these effects were associated with substantially reduced viral replication (Fig. 1). In contrast, the glucosidase inhibitors used in this study keep the HA oligosaccharides in di- or triglucosylated forms, thereby interfering with subsequent steps in the glycosylation process. This causes decreases in the molecular weight of HA, which vary with the type of inhibitor used, but it does not prevent oligomerization of the protein or its transport to the cell surface (41). In accordance with these reports, the inhibitors we used had no significant effect on viral production.
Using different biochemical approaches, we demonstrated that GSH-C4 causes HA to be retained in the ER as a reduced, incompletely folded monomer, which cannot be used for oligomerization. Disulfide bond formation is promoted by ER oxidoreductases (Ero) and PDI-family oxidoreductases, including PDI itself, but also ERp57, ERp61, and ERp72. PDI, the only conserved substrate for Ero in mammalian cells, is generally believed to be the primary acceptor for disulfide bonds from Ero1 (1). The activation state of Ero1α oxidase is controlled by the redox state and availability of its substrate. As for PDI, its catalytic activities are mediated by two active domains, each containing a pair of cysteine residues, and its ability to form (oxidation) and isomerize (reduction) disulfide bonds in cargo proteins is determined by its redox state (27). Indeed, PDI oxidation is a rate-limiting step in oxidative protein folding (24). The ER lumen is ∼20–100 times more oxidized than the cytosol (22), and this feature may serve to keep the ER oxidoreductases in an oxidative state (8). Some earlier studies showed that Ero-mediated oxidation of folding substrates involves protein-based relay that is largely independent of the bulk GSH redox buffer (45), but more recent work indicates that oxidoreductive pathways in the ER are indeed influenced by intracellular GSH levels, either directly or indirectly (24). Moreover, Molteni et al. (2004) have shown that ERO1 and PDI activities are both affected by cytosolic levels of reducing GSH and that lowering these levels in living cells by treatment with BSO and diethylmaleimide also accelerates disulfide bond formation. Appenzeller-Herzog et al. (3) have now shown that ER redox homeostasis largely depends on a dynamic equilibrium between Ero1- and GSH disulfide-mediated oxidation of PDIs. The authors found that PDI is the main substrate of Ero1a, and its oxidation level is dependent on ER GSH levels.
In this study, we demonstrated that intracellular GSH levels decrease during influenza virus infection and that this effect can be reversed by the addition of GSH-C4. In treated cells, PDI was present mainly in the reduced form, suggesting that GSH acts as a direct or indirect reductant of PDI and thereby limits its function in disulfide bond formation. The more oxidative environment established by the influenza virus' depletion of GSH would increase the expression and oxidation of PDI in the ER, accelerating disulfide bonding and enhancing viral glycoprotein maturation. This hypothesis is supported by the fact that BSO treatment significantly decreased intracellular GSH levels and increased HA expression and viral titers in cell supernatants. Collectively, these data indicate that GSH-C4 restores intracellular GSH to preinfection levels, thereby diminishing the oxidizing capacity of PDI and impairing HA maturation.
Interestingly, the addition of GSH-C4 to uninfected cells had no effect on the expression or secretion of SPARC, a host-cell glycoprotein that is also rich in disulfide bonds, and these findings were supported by the absence of significant toxicity in uninfected cells treated with GSH-C4. In contrast, SPARC secretion by infected cells was clearly decreased by GSH-C4. This finding reflects differences between the redox environments of infected and uninfected cells. The pro-oxidant state induced by PR8 infection can be expected to facilitate the work of PDI, an advantage that would disappear with the restoration of physiological reducing conditions in the cytoplasm. When reducing conditions prevail (as they do in uninfected cells), GSH-C4 treatment seems to produce no effect. Indeed, the drug clearly “corrected” the excess of oxidized PDI found in infected cells but had no significant effect on the PDI profiles of uninfected cells. Accelerated protein folding stimulated by the virally induced pro-oxidant conditions might also enhance the synthesis of PDI itself, and this might explain the general upregulation of PDI expression observed in infected cells.
Interestingly, the nonreducing SDS-PAGE analysis of HA showed higher-molecular-weight species that have been already demonstrated to be physiological transient species in the HA maturation (44). An intriguing aspect in this article is that GSH-C4 profoundly inhibits the production of these species. At present, we are not able to dissect the mechanism underlying this phenomenon and whether this result is the consequence of a decrease in PDI oxidation and/or in an increase of the intracellular reduced environment.
In conclusion, our results show that by increasing the reducing environment of the cell, the redox state of PDI and HA shifts versus the reduced form not allowing the oxidation of these proteins, fundamental for the proper activity and maturation, respectively. Further studies are in progress to better characterize the higher-molecular-weight forms of HA and their role in the maturation process.
All these findings are in line with our previous demonstration that exogenous GSH blocks the replication of several viruses (Parainfluenza, HSV-1, HIV) by impairing the surface expression of viral glycoproteins (18, 30, 33, 35). Therefore, the molecular mechanism by which GSH-C4 selectively blocks HA maturation might be exploited to develop an innovative antiviral strategy that targets host-cell enzymes necessary for viral replication. Interestingly, the thiazolides also inhibit HA maturation by interfering with the phase of terminal glycosylation. This also may prove to be a cell-mediated effect (although in this case other mechanisms, such as direct binding between the drug and the viral glycoprotein, have not been excluded) (39). The in vitro concentration of GSH-C4 required to produce antiviral effects is higher than that of most antiviral drugs, including thiazolides. At this purpose, it has to be noted that its antiviral effects are based on its ability to counteract the GSH depletion induced in the host cell by several viruses. Cells generally contain millimolar concentrations of GSH, so the GSH-C4 dose we used (10 mM) is by no means nonphysiological but it completely restores the normal intracellular redox state of infected cells without producing toxic effects in cells or animals.
HA folding is not the only process that would be affected by virally induced redox changes. The pro-oxidant state could also activate redox-sensitive pathways involved in inflammatory cytokine production, such as those involving MAP kinases (14) and transcription factors (NF-κB) (23). High viral replication rates and increased inflammatory responses with cytokine dysregulation are thought to play central roles in the pathogenesis of human disease. The increased mortality in humans infected with highly pathogenic H5N1 viruses and the poor responses of these infections to oseltamivir have been attributed to uncontrolled, virus-induced cytokine storms (48). In our in vivo experiments, lung inflammation was still detectable at 40 days after infection in all animals, but survival, pulmonary virus titers, and alveolar air space were all markedly improved in those treated with GSH-C4.
In conclusion, GSH-C4 emerges from this study as a promising new weapon for combating the growing threat of influenza. The fact that it targets a host-cell pathway involved in viral replication instead of a viral structure offers at least two important potential advantages: (i) broader applicability that is not limited by the strain or antigenic properties of the virus and (ii) lower probability of selection of resistant viral strains. For these reasons, the antiviral potential of GSH-C4 merits further investigation. Research is already underway in our laboratory to define the efficacy of locally administered GSH-C4 in improving lung function in infected animals.
Materials and Methods
Cell lines, toxicity, and viral infections
MDCK and NCI lung carcinoma cells were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS), glutamine 0.3 mg/mL, penicillin 100 U/mL, and streptomycin 100 mg/mL. Cell proliferation and viability were estimated by cell count performed at different times after plating and treatment with Trypan blue (0.02%) exclusion. All reagents were obtained from Invitrogen. Influenza A/PR8/34 virus (PR8) was grown in the allantoic cavities of 10-day-old embryonated chicken eggs. Cells were challenged at 24 h after plating with PR8 at an MOI of 0.01 or, when single-cycle replication was required, 1.0. Mock infection was performed with the same dilution of allantoic fluid from uninfected eggs. Infected and mock-infected cells were incubated for 1 h at 37°C, washed with phosphate-buffered saline (PBS), and incubated with medium containing 2% FBS. Hemagglutination and plaque-forming assays were performed according to standard procedures (19). Hemadsorption was assayed as described elsewhere (10).
Reagents
GSH-C4 was dissolved in RPMI 1640 medium with 2% FBS (final concentration: 1–15 mM). Glycosylation and protein-transport inhibitors were purchased from Sigma-Aldrich. Drugs used to treat cells were diluted in RPMI 1640 with 2% FBS to the following final concentrations: 1 mM for CST and dNM, 10 μM for SW, 1 μM for MON, 1 μg/mL for BFA, and 1 mM for BSO (Sigma-Aldrich).
Western blotting
Cell lysates were resuspended in reducing or nonreducing SDS sample buffer (i.e., with or without dithiothreitol), separated on SDS–polyacrylamide gels, and blotted onto nitrocellulose membranes. Goat polyclonal anti-influenza A virus (Chemicon), mouse monoclonal anti-HA H1N1 (Santa Cruz Biotechnology), anti-SPARC (Santa Cruz Biotechnology), and rabbit polyclonal anti-PDI (Cell Signaling) were used as primary Abs. Secondary Abs were horseradish-peroxidase–conjugated Abs (Jackson). Blots were developed with an enhanced chemiluminescence system (GE-Healthcare).
Mal-PEG treatment
Cell monolayers were washed with PBS-NEM 20 mM at 6 h p.i. and incubated in the same buffer on ice for 20 min. Cells were pelleted and homogenized in 50 μL of the same buffer. After addition of 5 μL of 10% SDS, the samples were denatured for 10 min in a heat block at 97°C. Lysates were reduced with 200 mM Tris(2-carboxyethyl)phosphine and alkylated with Mal-PEG for 1 h. Excess Mal-PEG was removed by methanol/chloroform protein precipitation. A sample of nonreducing loading buffer was added and samples resolved by SDS-PAGE (7.6% acrylamide).
Endo H digestion
Protein lysates were resuspended in 0.1% SDS and 0.1 M β-mercaptoethanol, heated at 95°C for 5 min, cooled, and divided into two equal-sized aliquots. Endo H (4 mU) (Sigma-Aldrich) was added to one aliquot. The sample was digested for 24 h at 37°C, heated for 5 min at 95°C, and analyzed with reducing SDS-PAGE. To suppress any residual protease activity, we added a protease inhibitor cocktail (Sigma Aldrich) and 1 mM phenylmethylsulfonyl fluoride to the lysis buffer used on Endo H–digested samples.
SPARC secretion assay
SPARC secretion was studied in NCI-H292 lung carcinoma cells infected with PR8 (MOI: 0.1) and treated with 10 mM GSH-C4. Twenty-four hours p.i., medium (2 mL) was collected from each culture and loaded into separate Centricon-30K centrifugal concentrators. The sample was spun in a refrigerated centrifuge at 4000 g for 15 min. The retentate was then analyzed by SDS-PAGE and transferred onto nitrocellulose membranes.
Immunofluorescence assays
Cells fixed with 4% paraformaldehyde and permeabilized with 0.01% Triton X-100 were incubated with monoclonal anti-HA and probed with the appropriate Alexa Fluor488–conjugated secondary Ab (Molecular Probes). Nuclei were stained with propidium iodide, and fluorescence was visualized with a confocal laser-scanning microscope (Nikon). Trans-Golgi structures were stained with Alexa Fluor350 WGA.
GSH assay
Intracellular GSH was assayed upon formation of S-carboxymethyl derivatives of free thiols with iodoacetic acid, followed by the conversion of free amino groups to 2,4-dinitrophenyl derivatives by reaction with 1-fluoro-2,4-dinitrobenzene, as previously described (30). GSH and GSSG were used as external standards. Values were expressed as nanomoles of GSH per milligram of protein in the original cell extract. Aliquots of cell lysates were used for total protein determination.
RT-PCR
The RNA of the progeny particles was extracted from the culture medium with a QIAamp Viral RNA Mini kit (Qiagen) for RT-PCR. Complementary DNA (cDNA) was synthesized with the iScriptcDNA Synthesis kit (BioRad), an oligo-dT primer, and random hexamer and then subjected to PCR with the following primer set: HA sense (5′-TGTATAGGCTAC CATGCGAAC-3′) and HA antisense (5′-TTCCGTTGTGGC TGTCTTC-3′); NP sense (5′-CGTCCCAAGGCACCAAAC-3′) and NP antisense (5′-AATCGTCCAATTCCACCAATC-3′).
Mice and treatments
The in vivo antiviral activity of GSH-C4 was evaluated in a well-established murine model of influenza infection (10, 34). Harlan BALB/c mice (6–8-week-old females; average weight: 20 g) were housed and studied under Institutional Animal Care and Use Committee–approved protocols. The MLD50 (50% mouse lethal virus dose) was determined in four groups of mice (each containing five mice) inoculated with serial 10-fold dilutions of virus and followed for 21 days p.i. The in vivo antiviral activity of GSH-C4 was tested in mice that had been intranasally inoculated with 1 PFU (10 MLD50) of PR8 diluted in 50 μL PBS. One hour p.i., infected mice were randomly assigned to treatment groups (10 animals each), which received daily intraperitoneal injections of GSH-C4 (7.4 mg in 100 μL of 0.9% NaCl, ∼370 mg/kg/day) or placebo (100 μL 0.9% NaCl). Treatments were repeated daily for the next 7 days. Body temperature, motor activity, and body weight were monitored daily through p.i. day 15 or 40. Uninfected control groups received identical GSH-C4 and placebo treatments and were monitored daily for signs of toxicity (death, decreased motor activity, and weight loss).
Pulmonary viral titers
Four groups of PR8-infected mice (n=5 per group) treated daily with GSH-C4 or placebo were killed at 6 days p.i. (when pulmonary viral titers were found to peak in preliminary experiments). Each lung was removed and weighed, and homogenates (in RPMI 1640 medium) were subjected to TCID50 assay on MDCK cells. The number of wells showing positive cytopathic effects was scored, and the titer (TCID50) per gram of lung tissue was calculated as previously described (21).
Light microscopy
Lung histology was evaluated in two groups (n=13/group) of mice infected with 1 PFU (10 MLD50) of PR8 and treated with GSH-C4 or placebo, as described earlier. Uninfected control groups received identical placebo treatments. Mice were sacrificed at 6, 9, 18, and 40 days p.i., and survival, body weight, and body temperature were recorded daily for 40 days. Each death or sacrifice was followed by complete necroscopy with macroscopic and microscopic examinations of the lungs. For the histopathological and morphological examination, fragments from each lobe of both lungs were fixed in buffered formalin for 48 h and embedded in paraffin with a melting point of 55°C–57°C. Sections (5-μm-thick) were stained with hematoxylin and eosin and Masson's trichrome.
The samples were evaluated independently and blindly by three investigators (C.L.M., A.F., and G.C.), and necroinflammatory changes were scored as follows (26): 0=no lesions; 1=mild focal inflammation; 2=moderate–severe inflammation or necrosis affecting less than 25% of lung tissue examined; 3=severe inflammation with necrosis or severe inflammation affecting 25%–50% of lung tissue examined; 4=severe inflammation with necrosis affecting more than 50% of the lung tissue examined.
For morphometric studies, images were acquired with a light microscope (Leica DM 4500B) connected to a Videocam (ProgRes C10 plus) and equipped with an image analysis system (Delta Sistemi), as described elsewhere (7). For each lung, we analyzed at least five slides (ten 10×fields per slide). The alveolar area was calculated as a percentage according to the following formula: (lung area surface occupied by alveoli/total lung area surface)×100.
Plasma GSH concentration after GSH-C4 treatment
Harlan BALB/c mice (6–8-week-old females; average weight: 20 g) were treated for 7 days with daily intraperitoneal injections of GSH-C4 or placebo, as described earlier. Animals were divided into six groups (each containing three mice treated with GSH-C4 and three treated with placebo), which were sacrificed at 0, 15, 30, 60, and 120 min after the first treatment and at 30, 60, and 120 min after the final treatment (day 7). Blood from the heart was collected in heparinized tubes and centrifuged at 2000 g for 15 min at 4°C to obtain plasma. Protein was precipitated by adding metaphosphoric acid (final concentration: 5%). After centrifugation at 22,300 g for 30 min, the low-molecular-weight thiols of the supernatant were derivatized and measured by HPLC with a μBondapak NH2 column (Waters).
Statistical analysis
Susceptibility of influenza A virus to GSH-C4 in the treatment and control groups was compared by an unpaired t-test or analysis of variance wherein the distributions were approximately log-normal. The log-rank test was used to compare the survival distributions of the placebo- and GSH-C4-treated groups. A p value of<0.05 was considered significant.
Footnotes
Acknowledgments
This work was partially supported by the Italian Ministry of Instruction, Universities, and Research (Special Project “Fund for Investments on Basic Research”-FIRB RBIP067F9E and Reti FIRB RBPR05NWWC_006), the Italian Ministry of Health-ISS, and Fondazione Roma grants 2009. The authors thank Ineke Braakman and Mieko Otsu for their helpful suggestions; Drs. Ignacio Celestino and Dolores Limongi for their excellent technical assistance; and Marian Everett Kent for editing the manuscript.
Author Disclosure Statement
No competing financial interests exist.