
Abstract
The endothelium is immunoregulatory in that inhibiting the function of vascular adhesion molecules blocks leukocyte recruitment and thus tissue inflammation. The function of endothelial cells during leukocyte recruitment is regulated by reactive oxygen species (ROS) and antioxidants. In inflammatory sites and lymph nodes, the endothelium is stimulated to express adhesion molecules that mediate leukocyte binding. Upon leukocyte binding, these adhesion molecules activate endothelial cell signal transduction that then alters endothelial cell shape for the opening of passageways through which leukocytes can migrate. If the stimulation of this opening is blocked, inflammation is blocked. In this review, we focus on the endothelial cell adhesion molecule, vascular cell adhesion molecule-1 (VCAM-1). Expression of VCAM-1 is induced on endothelial cells during inflammatory diseases by several mediators, including ROS. Then, VCAM-1 on the endothelium functions as both a scaffold for leukocyte migration and a trigger of endothelial signaling through NADPH oxidase-generated ROS. These ROS induce signals for the opening of intercellular passageways through which leukocytes migrate. In several inflammatory diseases, inflammation is blocked by inhibition of leukocyte binding to VCAM-1 or by inhibition of VCAM-1 signal transduction. VCAM-1 signal transduction and VCAM-1-dependent inflammation are blocked by antioxidants. Thus, VCAM-1 signaling is a target for intervention by pharmacological agents and by antioxidants during inflammatory diseases. This review discusses ROS and antioxidant functions during activation of VCAM-1 expression and VCAM-1 signaling in inflammatory diseases. Antioxid. Redox Signal. 15, 1607–1638.
II. VCAM-1 Regulation of Leukocyte Recruitment and Inflammation in Several Diseases
V. VCAM-1 Signals Through ROS During Leukocyte Transmigration
VII. Antioxidant Regulation of VCAM-1 Signals in In Vivo Models
VII. Clinical Implications for Vitamin E Regulation of Inflammation Involving VCAM-1
I. Introduction to Leukocyte Recruitment
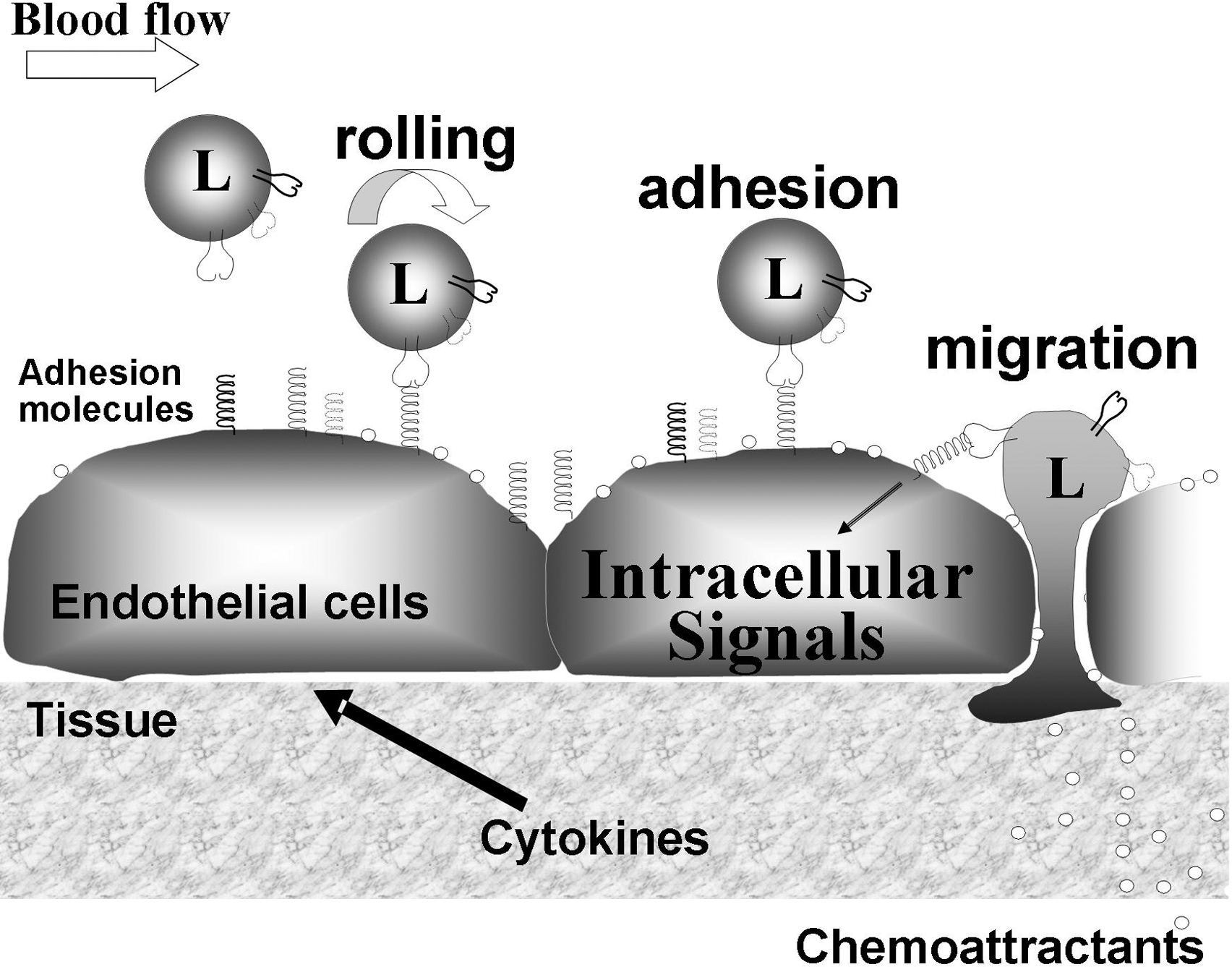
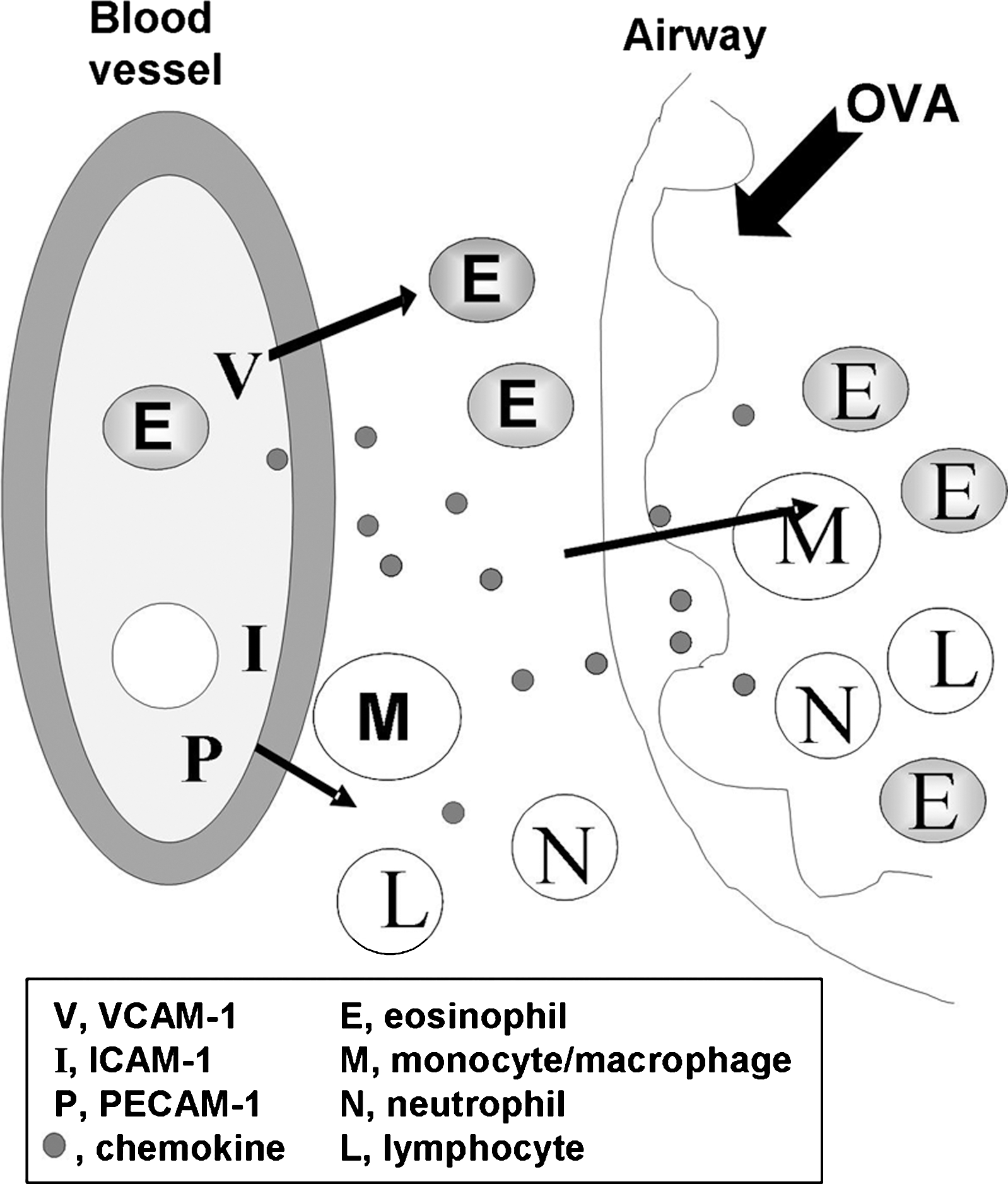
Leukocyte binding to the adhesion molecules activates signals within the endothelial cells that allow opening of narrow vascular passageways as small intercellular gaps through which leukocytes migrate (Fig. 1) (64, 184, 201). Leukocyte movement through these passageways is stimulated by chemokines that are produced by the endothelium and the tissue (Fig. 1). The majority of leukocyte migrate through intercellular gaps, but under conditions of high levels of inflammation, a small percentage of leukocytes can also migrate through individual endothelial cells by transcellular migration (50, 180, 195). When there is inhibition of the endothelial cell adhesion molecule signals, leukocytes bind to the endothelium but do not complete transendothelial migration (2). The cells that bind to the endothelium but do not complete transendothelial migration are often released from the endothelium and continue in the blood flow as demonstrated by intravital microscopy. Thus, the endothelial cell adhesion molecules and their intracellular signals are a source for intervention in leukocyte recruitment.
The vascular recruitment of leukocytes is a three-step process involving rolling of leukocytes on the endothelium followed by arrest of the leukocyte on the endothelium through high affinity adhesion, and then transmigration of the leukocyte through the endothelium (Fig. 1). The rolling of leukocytes on the luminal side of the endothelium is mediated by the low affinity receptors, selectins and addressins (188, 298). In lieu of the selectin interactions with addressin, rolling can also be mediated by leukocyte α4β1-integrin in its low affinity state interacting with vascular cell adhesion molecule-1 (VCAM-1/CD106) on the endothelium (15). Binding of selectins on leukocytes stimulates “outside-in” signals in leukocytes, increasing the affinity of the integrin family of receptors that then bind to the endothelial cell adhesion molecules intercellular adhesion molecule-1 (ICAM-1/CD54) or VCAM-1 (13, 45, 262). The high affinity integrin binding by blood leukocytes mediates arrest of the leukocytes on the endothelium. Then, the arrested leukocytes migrate into the tissue.
The affinity of leukocyte integrins for vascular adhesion molecules is also rapidly increased by “inside-out” signals from chemokine receptors on leukocytes (14, 48, 52, 131, 160, 161, 307). Chemokines have specificity for leukocyte cell types that express the chemokine receptors. This chemokine-specific activation results in increased integrin affinity on those leukocyte subsets that are responding to chemokines in the microenvironment. Thus, T cells, B cells, mast cells, eosinophils, monocytes, and stem cells migrate on VCAM-1, but their activation for binding to VCAM-1 depends on cell-type-specific chemokines in the microenvironment (4, 5, 8, 10, 23, 36, 57, 107, 108, 123, 138, 218, 243, 250, 268). The chemokine-activated leukocytes are selected for migration by their integrin-mediated high affinity adhesion. The adherent leukocytes then migrate on chemokine gradients into the tissue (195). It has also been reported that α4β1-integrin binding affinity on CD34+ bone marrow-derived cells or eosinophils is enhanced by ligand binding to the coexpressed adhesion receptor platelet-endothelial cell adhesion molecule-1 (PECAM-1), implicating signals transmitted from PECAM-1 as determinants of α4β1-integrin affinity (56, 162). The binding of leukocytes to the endothelium and the specificity of these interactions have been discussed in previous reviews (52, 83, 131, 160, 161, 170, 181, 201, 269, 270). This review will focus on VCAM-1 expression and function during VCAM-1 regulation of leukocyte transendothelial migration as it is regulated by ROS and antioxidants. Also discussed are the important regulatory roles for VCAM-1 signals and antioxidants during VCAM-1-dependent inflammation in vivo.
II. VCAM-1 Regulation of Leukocyte Recruitment and Inflammation in Several Diseases
A. VCAM-1 expression and shedding
VCAM-1 functions in combination with other adhesion molecules to regulate immune surveillance and inflammation. VCAM-1 expression is induced by cytokines produced in the tissue, high levels of ROS, oxidized low density lipoprotein (oxLDL), 25-hydroxycholesterol, turbulent shear stress, high glucose, and microbial stimulation of endothelial cell TLRs (49, 121, 124, 167, 177, 179, 182, 208, 210, 212, 229, 230, 242, 310, 322). This activation of VCAM-1 gene expression is regulated by the transcription factors nuclear factor kappa B (NFκB), SP-1, Ap-1, and interferon regulatory factor-1 (68, 163, 167, 182, 230, 287). For example, VCAM-1 expression is induced by the cytokines tumor necrosis factor (TNF)α and interleukin (IL)-1β, the adipokine Visfatin, the proatherogenic amino acid homocysteine, and proatherogenic hyperglycemia (46, 142, 144, 182, 196, 230). The mechanism of action of these stimulants is through induction of ROS generation for the stimulation of NFκB (46, 142, 144, 182, 196). However, the concentrations of endothelial cell ROS generated in response to these stimulants are not known. It is reported that high concentrations of ROS (400 μM hydrogen peroxide) can activate NFκB and consequently VCAM-1 expression in aortic endothelial cells (166). TNFα-induced VCAM-1 expression is blocked by scavenging superoxide by overexpression of superoxide dismutase but not blocked by scavenging hydrogen peroxide by overexpression of catalase in endothelial cells (54). Consistent with this finding, the TNFα-induced expression of VCAM-1 by NFκB binding to the VCAM-1 promoter is blocked by nitric oxide, which is known to react with superoxide (142). Conversely, the nitric oxide synthase inhibitor N-monomethyl-
VCAM-1 can also be released from the endothelial surface through cleavage by a disintegrin and metalloprotease 17 (ADAM17) (97) and, although less characterized, may be released by ADAM8 (185, 186) or ADAM9 (103, 222). Therefore, VCAM-1 is present in the plasma in a soluble form (sVCAM-1) and is used as predictive biomarker of disease (17, 127, 154, 297, 319). Levels of sVCAM-1 in plasma increase with activation of the endothelium in multiple diseases (44, 55, 89, 98, 133, 154, 206, 219, 227). This sVCAM-1 is thought to either limit leukocyte integrin binding to endothelial VCAM-1 by binding to leukocytes or stimulate leukocyte chemotaxis (147, 282, 288).
B. VCAM-1 function in the bone marrow and lymph nodes
VCAM-1 is expressed in lymph nodes and the bone marrow for the regulation of leukocyte homing (Table 1). The function of VCAM-1 in the bone marrow has been demonstrated in a mouse model with a conditional deletion of murine VCAM-1. In these mice, deletion of VCAM-1 results in reduced B cell homing to the bone marrow (243). In the bone marrow, it has also been reported that VCAM-1 regulates proplatelet formation in the osteoblastic niche (221). VCAM-1 expression has also been reported to be induced on mesenchymal stem cells by cytokine stimulation (313). This mesenchymal stem cell expression of VCAM-1 is reported to participate in immunosuppression of T cell responses (243). Further, VCAM-1 regulates hematopoietic stem cell recruitment to injured liver and melanoma metastasis to the liver (138, 255, 299). In lymph nodes and tonsils, VCAM-1 is expressed by postcapillary high endothelial venule cells and follicular dendritic cells (151, 187, 317). VCAM-1 on the lymph node follicular dendritic cells mediates B cell binding (21, 151). Thus, VCAM-1 has a role in the bone marrow, lymph nodes, and liver.
VCAM-1, vascular cell adhesion molecule-1.
C. VCAM-1 regulation of inflammatory diseases: treatment of clinical disease with natalizumab
VCAM-1 has a regulatory role in peripheral tissue inflammation in several diseases (Table 1). In these diseases, there are different leukocyte cell types that bind VCAM-1 via the leukocyte ligand α4β1-integrin (Table 2). This is, at least in part, a result of leukocyte-specific chemokine activation of α4β1-integrin into the integrin's high affinity conformation (52, 131, 160, 161). The cell types with high affinity integrin migrate on VCAM-1. In allergic disease, blocking VCAM-1 by intravenous injection of anti-VCAM-1 blocking antibodies inhibits eosinophil recruitment in asthma models in several species (57, 107, 250). Further, in allergic disease, blocking VCAM-1 or using VCAM-1 knockout mice inhibits mast cell precursor binding to endothelium and inhibits recruitment of mast cell precursors to antigen-stimulated lungs and intestine (4, 5, 8, 36, 108). In a mouse model of atopic dermatitis, VCAM-1 blockade reduces severity of inflammatory disease and delays the onset of disease (53). In inflammatory bowel disease, antibody inhibition of VCAM-1 blocks T cell infiltration into the intestine (268). In an experimental model of multiple sclerosis, blocking VCAM-1 inhibits T cell infiltration into the brain (23). Consistent with this, multiple sclerosis patients have elevated VCAM-1 but not mucosal addressin cell adhesion molecule-1 expression in brain tissue (10). In clinical trials, blocking the VCAM-1 ligand, α4-integrin, with antibodies (natalizumab) reduces disease severity in multiple sclerosis and Crohn's disease (61, 211). Unfortunately, treatment of multiple sclerosis with natalizumab is complicated by the rare occurrence of progressive multifocal leukoencephalopathy (61, 211). Thus, due to the side effects of natalizumab, there is a need for alternative targets to limit VCAM-1-dependent inflammation. These alternative targets are VCAM-1 itself or VCAM-1 signaling intermediates that are discussed in this review.
D. VCAM-1 regulation of inflammation during infection
VCAM-1 also has a role in regulation of inflammation during infection (Table 1). During infections, microbial TLR ligands and the cytokines of the immune response likely stimulate VCAM-1 expression. VCAM-1 expression is induced by stimulation of TLRs on endothelium, dendritic cells, and fibroblasts (82, 121, 295, 313). During lymphocytic choriomeningitis virus infections, VCAM-1 expression by the endothelium mediates CD8+ T cell infiltration into the brain (218). Moreover, deletion of VCAM-1 blocks disease severity and blocks monocyte/dendritic cell migration into the brain during lymphocytic choriomeningitis virus infections (218). In experimental visceral leishmaniasis, VCAM-1 interaction with α4β1 integrin regulates the production of dendritic cells since antibody inhibition of VCAM-1 or α4β1-integrin blocks the dendritic cell response in the spleens in these mice (271). Thus, VCAM-1 has regulatory functions in infection-induced inflammation.
E. VCAM-1 function in cardiovascular diseases
It has been reported that VCAM-1 has an important role in cardiovascular diseases and in the embryonic development of the cardiovascular system (Table 1). VCAM-1 is required for development of the heart since the VCAM-1 knockout mouse is an embryonic lethal due to malformation of the heart (105). In atherosclerosis, VCAM-1 is the first adhesion molecule expressed before atherosclerotic plaque development (125). In the carotid artery, neointimal formation is reduced by VCAM-1 siRNA or by antibody blockade of α4β1-integrin in rodents (27, 236). In advanced stages of atherosclerosis, VCAM-1 can be expressed by smooth muscle cells (38, 110). VCAM-1 is also linked to calcification of aortic stenosis in patients with coronary artery disease (173). In the atherosclerotic carotid arteries, VCAM-1 mediates monocyte adhesion as demonstrated with anti-VCAM-1 blocking antibodies (123). In cardiac allografts, lower levels of VCAM-1 expression are indicative of a reduction in rejection (41, 320). In ischemia-reperfusion of the liver, blocking VCAM-1 inhibits leukocyte recruitment and injury (138). Also of interest, VCAM-1 expression is induced in the aorta in HIV transgenic rats (106). Thus, VCAM-1 has an important regulatory role in cardiovascular diseases.
Thus, in VCAM-1-dependent inflammatory diseases, the specificity of cell types recruited in inflammatory diseases is dictated by the combination of adhesion molecules and by the specific chemokines for activation of integrins on leukocyte cell types. VCAM-1 binding and intracellular signaling are potential targets for intervention in several diseases. Therefore, it is important to understand the mechanisms for VCAM-1 functions so that approaches can be developed to modulate VCAM-1-dependent inflammation during disease. In addition, selective inhibition of VCAM-1 function would block excess inflammation in VCAM-1-mediated inflammatory disease while maintaining the beneficial antimicrobial immune responses that utilize the vascular adhesion molecules ICAM-1 or PECAM-1.
III. VCAM-1 Structure/Function
A. VCAM-1 structure
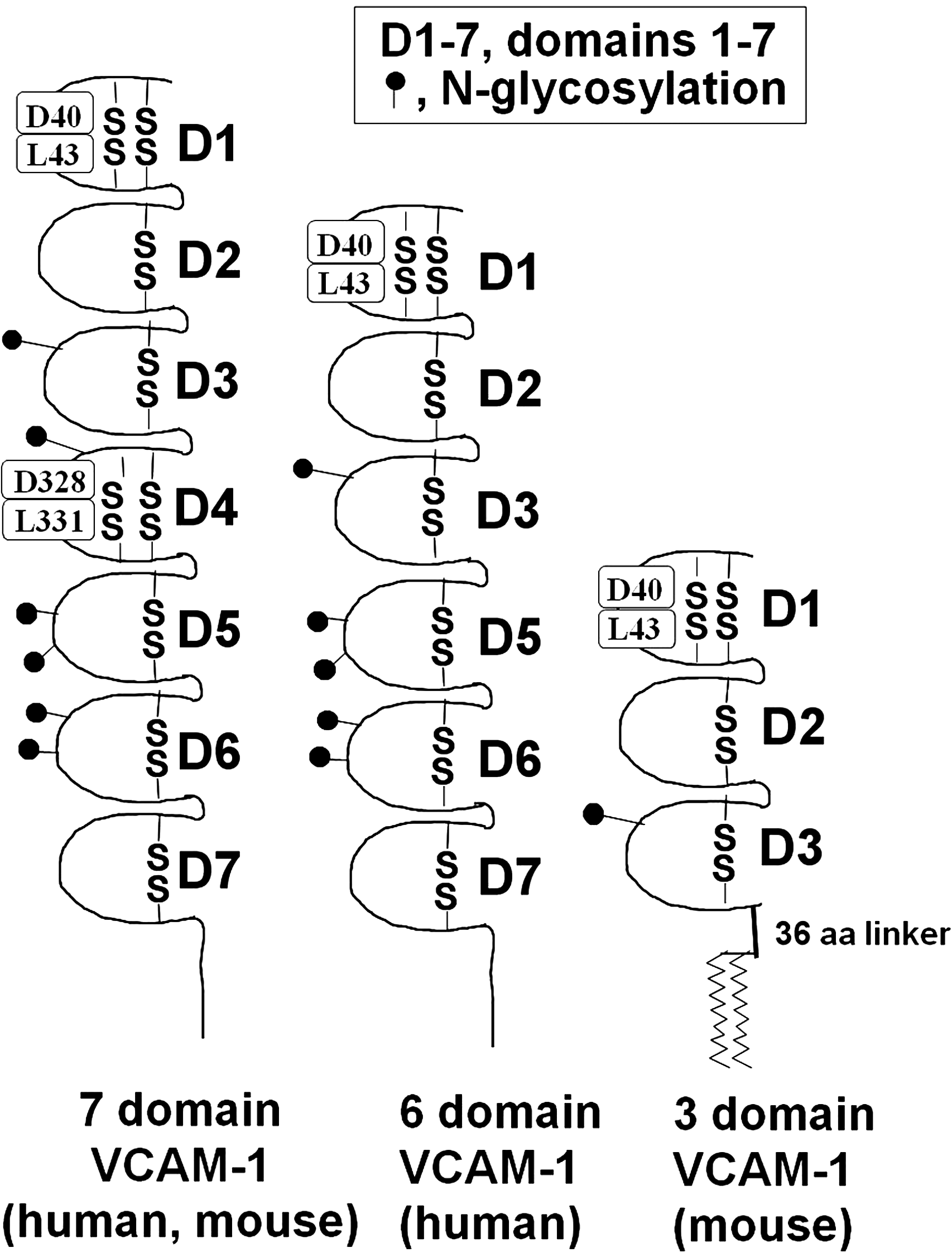
VCAM-1 is a member of the immunoglobulin (Ig) superfamily of proteins. VCAM-1 is comprised of several extracellular Ig-like domains that contain disulfide-linked loops, a single type I transmembrane domain, and a 19 amino acid carboxyl-terminus cytoplasmic domain (157, 215) (Fig. 2). Interestingly, the amino acid sequence of this cytoplasmic domain is 100% identical among several species, including rat, mouse, human, and rabbit (22, 100, 118, 224, 232, 233). The extracellular region of the full-length form of VCAM-1 contains seven Ig-like domains (Fig. 2). There is homology within these Ig-like domains, such that domains 1 and 4 have sequence homology, domains 2 and 5 have sequence homology, and domains 3 and 6 have sequence homology (69, 119, 233). In addition, there are splice variants of the full-length form of VCAM-1. There are two human forms and two mouse forms of VCAM-1 (Fig. 2). Human VCAM-1's two splice variants result in a receptor with either a seven Ig-like domain protein or a six-domain VCAM-1 that lacks domain 4 (Fig. 2) (69, 70). Mouse VCAM-1 also has a full-length seven-domain form as well as a truncated form with only the first three domains (Fig. 2). This mouse three-domain form of VCAM-1 is linked to glycophosphatidylinositol (GPI) for insertion in the plasma membrane (146, 157, 200, 280). Moreover, this three-domain variant has a 36 amino acid GPI-linker that is a unique sequence not found in the six- or seven-domain VCAM-1 molecules or in other members of the Ig superfamily (Fig. 2) (200). It has been speculated that the GPI link might enable the three-domain VCAM-1 to move through the plasma membrane faster than the other VCAM-1 variants, thereby quickening the endothelial response to tethering a rolling leukocyte (146).
B. Ligands and VCAM-1 binding regions
All of the variants of VCAM-1 have been shown to bind to α4β1 integrin (VLA-4) (15, 85, 209, 215, 309) (Fig. 3). α4β1-integrin also binds to fibronectin, heparin, and junction adhesion molecule-B in endothelial junctions (176, 235, 258). In addition to α4β1-integrin, VCAM-1 can bind to other integrins such as α4β7 integrin and αdβ2 integrin (51, 102, 247). These integrins are expressed by eosinophils, basophils, lymphocytes, mast cells, and monocytes (5, 33, 36, 174, 197, 307). The binding domains of VCAM-1 have been identified using a combination of domain truncations, substitutions with ICAM-1 sequences, and amino acid mutations. It has been shown that α4β1 integrin binds to Ig-like domains 1 and 4 (216, 223, 244, 301). Antibody inhibition of either domain 1 or 4 partially blocks Ramos cell α4β1 integrin binding to VCAM-1 (216) (Fig. 3). However, inhibition of both domains 1 and 4 completely blocks binding (216). In both domains 1 and 4, a mutation of either an aspartate (domain 1 amino acid 40 or domain 4 amino acid 328) or a leucine (domain 1 amino acid 43 or domain 4 amino acid 331) to an alanine results in a significant reduction of Ramos cell binding to these VCAM-1 mutants expressed in COS cells (Fig. 3) (244, 301). Mutating several other amino acids in domain 1, including R36, Q38, I39, P42, L70, or T72, reduces binding of α4β1 integrin and α4β7 integrin (58). However, mutations in domain 1 at N44 or E66 specifically reduce binding of α4β7 integrin but not α4β1 integrin (58). Of these mutations, D40 and L70 mutations are reported to inhibit binding while not perturbing the gross structure of VCAM-1 (58, 244). In addition, domain 2 is necessary for the binding function of domain1 (244). Domain 1 of VCAM-1 also binds α4β7 integrin as demonstrated using anti-VCAM-1 domain 1 blocking antibodies or domain 1 blocking peptides (216, 247, 321).
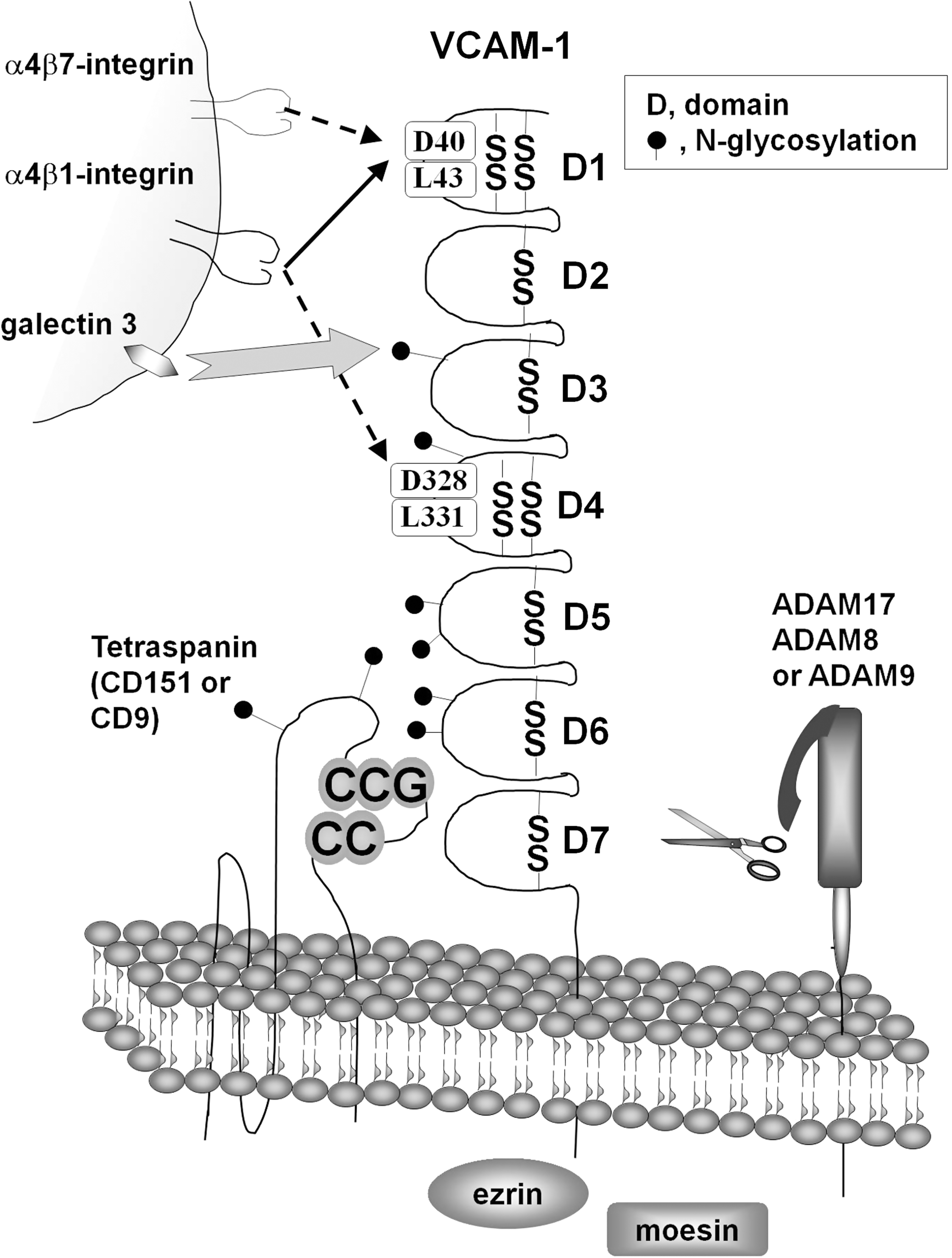
The binding to VCAM-1 is regulated by the activation state of the integrins (51, 143). Integrins at low affinity roll on VCAM-1, whereas the high affinity conformation of the integrins mediates firm adhesion to the endothelium that can withstand the force of the blood flow (15, 99, 307). In addition, integrin binding to domains 1 versus domain 4 of VCAM-1 is modulated by the degree of activation of the α4β1 integrin. The α4β1 integrin binding to domain 4 has a higher requirement for activation than for binding to domain 1 (Fig. 3) (143). Moreover, α4β1 integrin versus α4β7 integrin differ in their activation requirements for binding to domains 1 and 4 of VCAM-1 (143). For half maximal binding to domain 4 of VCAM-1, α4β1 integrin requires significantly higher activating concentrations of divalent cations than α4β7 integrin (Fig. 3) (143). The binding activity of α4β1 integrin to domain 1 of VCAM-1 is also regulated by CD24 expression (143). Moreover, α4β7 integrin binding to VCAM-1 requires a higher activation state than for its binding to the mucosal addressin cell adhesion molecule-1, an endothelial cell adhesion molecule (Fig. 3) (31). Thus, the α4-integrins bind to two domains of VCAM-1 and this binding to VCAM-1 domains is regulated by the activation state of the integrins.
In addition to integrins, VCAM-1 can bind galectin-3 (Fig. 3). It has been reported that recombinant galectin 3 columns bind several proteins from BALB/3T3 cells, and the major band, at 100 kD, was identified as VCAM-1 by mass spectrometry (278). Moreover, VCAM-1 is immunoprecipitated from proteins bound to a recombinant galectin-3 column (278). This galectin-3 binding to VCAM-1 is lost by treatment of VCAM-1 with N-glycanase, indicating that VCAM-1's N-glycans bind to galectin-3 (278). There are six N-glycosylation sites on VCAM-1 and these are located in domains 3–6 (Fig. 3) (198). Galectin-3 has been implicated in eosinophil binding to VCAM-1 (239). Eosinophil binding to endothelial cells expressing VCAM-1 is blocked to an equal extent with anti-α4β1-integrin or antigalectin-3 antibodies (239). Treatment with these two antibodies together does not exhibit further inhibition of adhesion. Moreover, the interactions with galectin-3 are complicated since it has been demonstrated by ELISA that α4β1-integrin binds directly to galectin-3, that galectin-3 can bind to galectin-3, and that endothelial cells express galectin-3 in addition to VCAM-1 (239). In this study of eosinophil binding to endothelial cells, it was not demonstrated whether galectin-3 on eosinophils directly binds VCAM-1 (239). Thus, during eosinophil interactions with endothelium, there are several galectin-3 ligands expressing N-glycans, including VCAM-1.
C. VCAM-1 is a part of the tetraspanin-enriched microdomains
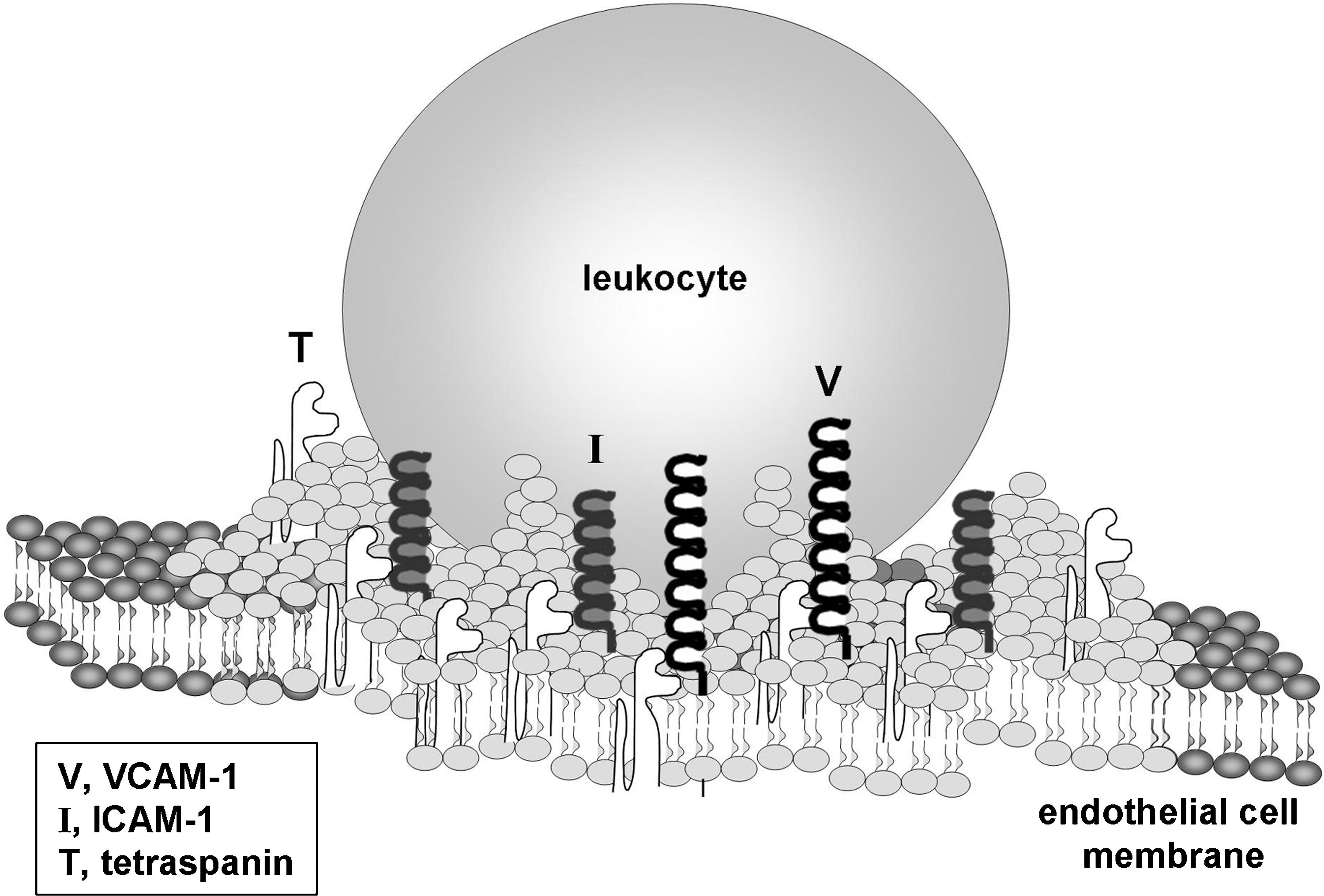
Several studies have examined the role of VCAM-1 in cell adhesion and migration. In activated endothelial cells, VCAM-1 is found in a lipid-raft-like platform containing ICAM-1 and the tetraspanins CD9, CD81, and CD151, known as the tetraspanin-enriched microdomain (Fig. 4) (24, 26, 113). Fluorescent microscopy shows that when T cells adhere to activated human umbilical vein endothelial cell (HUVEC) monolayers, VCAM-1, ICAM-1, CD9, CD81, and CD151 all colocalize to rings surrounding the lymphocyte (24). The specific interactions between ICAM-1, VCAM-1, and the tetraspanins within the tetraspanin-enriched microdomain were demonstrated using coimmunoprecipitation and fluorescence resonance energy transfer (FRET) analysis studies. Coimmunoprecipitation studies demonstrate that VCAM-1 associates with CD151 and CD9 and that ICAM-1 associates with CD9 (25). FRET-fluorescence lifetime imaging microscopic analysis in resting HUVECs reveal that VCAM-1 does not homodimerize nor does it form a heterodimer with ICAM-1. However, there is a low incidence of ICAM-1 homodimerization (26). The FRET-fluorescence lifetime imaging microscopic analysis also confirmed that VCAM-1 interacts with CD151 and that ICAM-1 interacts with CD9 [19, 22]. When CD151 and CD9 expression is reduced by siRNAs, the surface expression of VCAM-1 and ICAM-1 is similarly reduced, thereby suggesting a role for tetraspanins in structurally supporting the surface expression of VCAM-1 and ICAM-1. Under static binding conditions, the siRNA reduction of CD9 or CD151 in HUVECs does not alter the level of lymphocyte adhesion as compared to scrambled siRNA. In addition, the siRNA does not alter paracellular permeability (24). In contrast, under physiological vascular shear stress of 5–15 dyn/cm2, siRNA reduction of CD9 or CD151 significantly decreases lymphocyte adhesion and lymphocyte transmigration across HUVECs (24). Therefore, tetraspanins CD9 and CD151 are important for VCAM-1 expression and function.
D. VCAM-1 in apical cup-like structures
When a leukocyte binds to the endothelium, the plasma membrane of the endothelial cell forms an apical cup-like structure (also referred to as the endothelial adhesive platform) to surround the rolling leukocyte (Fig. 4) (24, 26, 47). Fluorescent microscopy shows that these cup-like structures contain VCAM-1 and ICAM-1 but not ICAM-2, vascular endothelial-cadherin, or PECAM-1 (47). It is likely that the cup-like structure is important in mediating firm adhesion between the leukocyte and endothelial cell and enabling transmigration. Confocal microscopy indicates that this apical cup-like structure is surrounded by polymerized actin that is associated with vinculin and VASP but is not connected to basal stress fibers or tubulin (25). VCAM-1's cytoplasmic domain is not required for the formation of the apical cups. In resting HUVECs transfected with a cytoplasmic tail-truncated VCAM-1, T lymphocyte binding to the endothelial cells still leads to the formation of an apical cup containing both the transfected VCAM-1 and endogenously expressed ICAM-1 (26). This finding suggests that proteins within the apical cup-like structure are recruited through extracellular interactions and not through their cytoplasmic domains (26).
E. Cytoplasmic domain of VCAM-1
The amino acid sequence of the cytoplasmic domain of VCAM-1 is 100% identical among many mammalian species, including the human, mouse, rat, rabbit, Sumatran orangutan, chimpanzee, common shrew, and microbat (22, 100, 118, 224, 232, 233) (NCBI NP001126200.1, NCBI XP001135527.1, Ensemble ENSSARP00000011070, Ensemble ENSMLUP00000011488). The protein sequence for the cytoplasmic domain of VCAM-1 in guinea pig and dolphin differs from the above species by only one conserved amino acid substitution (Ensemble ENSTTRP0000001370, Ensemble ENSCPOP00000006062). This high degree of identity suggests that the cytoplasmic domain is important for VCAM-1 expression or function. VCAM-1 has been shown to coimmunoprecipitate with ezrin and moesin, two structural proteins in the cytosol that are known to bind to actin (Fig. 3) (25). This was supported by confocal microscopy showing the colocalization of VCAM-1 with ezrin and moesin (25). The structure and function of the cytoplasmic domain of VCAM-1 during VCAM-1 signaling are currently under investigation.
IV. Overview of a Model for VCAM-1 Signaling
A. Model of VCAM-1 signaling through ROS
During inflammation, VCAM-1 expression is induced on endothelial cells by cytokines or turbulent shear stress. The cytokines and turbulent shear stress signal through high levels of short-lived ROS to induce NFκB-dependent activation of VCAM-1 expression in endothelial cells (46, 54, 142, 144, 166, 182, 196, 276). This VCAM-1 protein synthesis requires several hours. Then, ligand binding to VCAM-1 induces rapid transient signaling through low levels of ROS that induce signals for the support of leukocyte transendothelial migration. The activated endothelial cells in lymph nodes and inflammatory sites express VCAM-1 on their luminal surface and their lateral surface but not on their basal surface (Fig. 5B). Therefore, at these endothelial surfaces, VCAM-1 activates intracellular signals through ROS (Fig. 5A). Localized VCAM-1 signals that induce changes in endothelial cell shape during leukocyte transendothelial migration are important since endothelial cell shape changes are confined to the site of leukocyte binding to the endothelium during leukocyte rolling and transendothelial migration. The signals in this pathway are transient and occur within minutes, consistent with the transient, rapid nature of leukocyte transendothelial migration.

An overview of VCAM-1 signals is introduced here before specifically discussing each of these signals. It has been reported that activation of VCAM-1 stimulates calcium channels, intracellular calcium release, the G protein Gαi2, and the low-molecular-weight G protein Rac1 (65, 184, 225). The calcium flux and Rac1 activate the NADPH oxidase NOX2 (65, 184). VCAM-1 does not activate other enzymes that generate ROS (184). The activated NOX2 generates superoxide that then dismutates to hydrogen peroxide (H2O2), generating 1 μM H2O2 during VCAM-1 signaling (65, 286). This 1 μM H2O2 is relatively low as compared to the 50–200 μM H2O2 produced by macrophages or neutrophils (66, 74). It is also much lower than the exogenous 100–1000 μM H2O2 added to endothelial cells in studies on oxidative damage of endothelium or the exogenous 400 μM H2O2 added to endothelial cells in studies for ROS induction of VCAM-1 expression (20, 116, 120, 165, 289). These differences in H2O2 levels are important in understanding functions of oxidation, as we and others reported that 1 μM H2O2 and >50 μM H2O2 have opposing effects on signal transduction (1, 73, 90, 238).
During VCAM-1 signaling, the 1 μM H2O2 oxidizes the pro-domain of matrix metalloproteinases (MMPs), causing autocatalytic cleavage of the pro-domain and activation of the MMPs. The 1 μM H2O2 also diffuses through cell membranes at 100 μm/s (183). In contrast, superoxide remains primarily extracellular as it has a relatively low diffusion rate across membranes. These intracellular ROS activate endothelial cell p38MAPK for the regulation of endothelial cell gap formation (294). The 1 μM H2O2 also directly oxidizes and transiently activates intracellular protein kinase Cα (PKCα) in endothelial cells (1). This activated PKCα induces phosphorylation and activation of protein tyrosine phosphatase 1B (PTP1B) (72). Interestingly, the PTP1B that has an oxidizable cysteine in its catalytic domain is not oxidized during VCAM-1 signaling in endothelial cells (72), indicating specificity of targets for oxidation by the low concentrations of ROS generated during VCAM-1 signaling. The signals downstream of the PTP1B that regulate endothelial cell junctions are currently under further investigation. Most importantly for this signaling pathway, the signals in Figure 4A have been demonstrated to function in regulation of VCAM-1-dependent leukocyte transendothelial migration in vitro and in vivo (1, 2, 30, 65, 72, 73, 140, 184, 225). Thus, VCAM-1 is not simply a scaffold for leukocyte adhesion, since it also activates “outside-in” signal transduction in endothelial cells. Several of these VCAM-1 signals are also activated by other adhesion molecules. For example, VCAM-1 and ICAM-1 both activate calcium fluxes, PKC, p38MAPK, and cytoskeletal changes in endothelial cells (88, 305, 306).
B. Cell models for VCAM-1 signals
To examine VCAM-1 signaling, both cytokine-activated primary cultures of endothelial cells and endothelial cell lines are necessary, since there are distinct advantages to each of these approaches. Primary cultures of cytokine-activated endothelial cells have the advantage of being primary cells that can be specifically stimulated by crosslinking VCAM-1 with anti-VCAM-1 antibody-coated beads (Fig. 6). However, activated primary cultures of endothelial cells express multiple adhesion receptors for leukocytes and thus are difficult to use to examine functions specific to VCAM-1 during leukocyte transendothelial migration (Fig. 6). The advantage of the murine endothelial cell line lymph node-derived high endothelial venule-like (mHEV) cells is that the mHEV cells express VCAM-1 but not multiple other receptors for leukocyte migration (Fig. 6, Table 3) (285). Thus, leukocytes migrate specifically on VCAM-1 on the mHEV cells, without complications due to leukocyte binding to many other vascular adhesion molecules (285). The leukocyte adhesion to the mHEV cells is blocked by function blocking antibodies to VCAM-1 and its ligand α4-integrin but not other adhesion molecules (Fig. 6, Table 3) (184, 285). Further, VCAM-1 is constitutively expressed by the mHEV cells; therefore, analysis of VCAM-1 signaling is not complicated by signals from cytokine induction of VCAM-1 expression (64, 285). Thus, the mHEV cells provide a model to test the functional outcome of VCAM-1 signals on VCAM-1-dependent leukocyte migration. In addition, the migration of leukocytes across the mHEV cells is induced by the chemokine monocyte chemoattractant protein-1, which is constitutively expressed by the mHEV cells (Fig. 6) (237). When examining the migration of spleen cells, the cells that migrate across the mHEV cells are >90% lymphocytes (286). Eosinophils also migrate on VCAM-1 in this mHEV model (unpublished data). Moreover, antibody crosslinking of VCAM-1 on mHEV cells or activated primary cultures of endothelial cells generate the same time course and magnitude of signals. Thus, the mHEV cell lines and primary cultures of activated endothelial cells provide models with unique assets to examine VCAM-1 signals during VCAM-1-dependent leukocyte migration (Fig. 6).

ICAM-1, intercellular adhesion molecule-1.
For activation of VCAM-1 by antibody crosslinking, anti-VCAM-1 antibodies are either used to coat 10 μm beads that are the size of leukocytes (Fig. 6) or are used in antibody complexes composed of anti-VCAM-1 and a secondary antibody (1, 65, 72, 73). VCAM-1 signals are not activated by primary anti-VCAM-1 antibodies alone (184), indicating that crosslinking is necessary. Leukocyte binding to the mHEV cell lines or antibody crosslinking of VCAM-1 on mHEV cell lines activates signals with the same magnitude and time course (65, 184). Further, antibody crosslinking of VCAM-1 on cytokine-activated primary cultures of human endothelial cells (HUVECs or human microvascular endothelial cells from lung) stimulates VCAM-1 signaling with the same magnitude and time course as the mHEV cell lines, indicating that the VCAM-1 signals are consistent for these endothelial cells. In addition to activation by VCAM-1 directly, the VCAM-1 signaling intermediate 1 μM H2O2 (Fig. 6) is sufficient to activate the downstream signals with the same time course as crosslinking VCAM-1 (65, 184). Therefore, for the study of VCAM-1 function, a combination of cell approaches is used to identify VCAM-1 “outside-in” signals and, importantly, used to define whether the VCAM-1 signals have a functional role in VCAM-1-dependent leukocyte migration. Approaches for examining the in vivo function VCAM-1 signals are included later in this review in section VII discussing the in vivo role of VCAM-1 signals.
V. VCAM-1 Signals Through ROS During Leukocyte Transmigration
A. VCAM-1 activates calcium fluxes, Rac1, and Gαi
Ligand binding to VCAM-1 activates rapid signals in endothelial cells (Fig. 5). Lymphocyte binding to VCAM-1 or anti-VCAM-1-coated 10 μm beads stimulates a calcium flux in 30 s in endothelial cells. This calcium flux is mediated by verapamil-sensitive calcium channels and the release of intracellular calcium, which are required for the production of 1 μM H2O2 in mHEV cells (65). A calcium flux is also induced by VCAM-1-dependent monocyte adhesion or antibody cross-linking of VCAM-1 on lipopolysaccharide-activated HUVECs (175). In addition, anti-VCAM-1-coated 10 μm bead stimulation of endothelial cells activates Rac1 that is required for the production of the 1 μM H2O2 by the endothelial cells (Fig. 5) (65). Transfection with dominant negative Rac1 prevents the anti-VCAM-1-stimulated generation of H2O2 (65). Dominant negative Rac-1 also blocks VCAM-1-dependent migration of lymphocytes across mHEV cell monolayers (65) and blocks migration of U-937 cells across cytokine-activated HUVECs (294). Thus, since Rac1 is involved in the assembly of the active NOX2 complex and, as discussed below, VCAM-1 stimulation of H2O2 generation occurs through NOX2, endothelial Rac1 likely promotes assembly of NOX2 complex formation during VCAM-1 signaling (Fig. 5) (34, 77, 168). In addition to VCAM-1 signaling through the low-molecular-weight G protein Rac1, VCAM-1 also signals through Gαi. In in vitro assays for VCAM-1-dependent transmigration, leukocyte transmigration across monolayers of endothelial cells was blocked by the Gαi inhibitor pertusis toxin without altering the VCAM-1-dependent binding to the apical surface of the endothelial cells (225). Thus, VCAM-1 activates calcium fluxes and the G proteins Rac1 and Gαi. The mechanisms for VCAM-1 activation of these G proteins are under investigation.
B. VCAM-1 activation of endothelial cell NOX2 during leukocyte transmigration
1. VCAM-1 activates NOX2
The VCAM-1 stimulation of calcium fluxes and Rac-1 activates the membrane complex NADPH oxidase in endothelial cells for the production of ROS (Fig. 5) (65, 294). NADPH oxidase catalyzes the production of superoxide from oxygen using the cofactor NADPH. Then, superoxide dismutates to H2O2. NADPH oxidases consist of two transmembrane subunits and three cytoplasmic subunits that are recruited to the membrane to form the active NADPH oxidase complex (7, 192). There are several forms of NADPH oxidase that differ in their catalytic subunit and their cell-specific expression (65, 74, 132, 169, 192, 290). Endothelial cells express the NADPH oxidase subunits gp91 phox, p22 phox, p47 phox, and p67 phox (132, 192). Lymphocyte binding to VCAM-1 or anti-VCAM-1-coated beads activate the NOX2 form of NADPH oxidase, which utilizes the gp91phox catalytic subunit (2, 63, 65, 73, 140, 184). The VCAM-1 activation of NOX2 has been demonstrated in several studies using antisense for the catalytic subunit of NOX2 (gp91phox), pharmacological inhibitors, extracellular scavengers of superoxide and H2O2, and chimeric gp91phox deficient (CYBB) mice (2, 63, 65, 73, 140, 184). Antisense for gp91 phox blocks expression of gp91 phox in endothelial cell lines and blocks VCAM-1-induced H2O2 production in endothelial cells (65). The pharmacological inhibitor of NADPH oxidase apocynin blocks VCAM-1-stimulated H2O2 generation in endothelial cell lines and primary cultures of endothelial cells (184). Exogenous addition of the scavenger of superoxide, superoxide dismutase, or the scavenger of hydrogen, catalase, scavenge these species of extracellular ROS and block VCAM-1-dependent leukocyte transendothelial migration in vitro (184). Importantly, anti-VCAM-1-coated beads stimulate ROS generation in primary cultures of endothelial cells and in mHEV cells with the same time course and magnitude, indicating that these signals are consistent for endothelial cells (184, 294). In contrast to VCAM-1 activation of NADPH oxidase, it has been reported that antibody crosslinking of the endothelial cell adhesion molecules ICAM-1 and PECAM-1 does not activate endothelial cell NADPH oxidase (184, 291). In support of NOX2 activation by VCAM-1 but not ICAM-1 or PECAM-1, mice deficient in nonhematopoietic NOX2 exhibit a reduction in VCAM-1-dependent recruitment of leukocytes but no effect on ICAM-1-dependent or PECAM-1-dependent recruitment of leukocytes (2).
2. VCAM-1 signals through NOX2 mediate VCAM-1-dependent leukocyte transmigration
VCAM-1-dependent lymphocyte transmigration requires NOX2-generated ROS. It is reported that VCAM-1-dependent lymphocyte transendothelial migration in vitro is blocked by pharmacological inhibition of NADPH oxidase with apocynin, blocked by inhibition of endothelial cell flavoproteins with diphenyliodonium, and blocked by extracellular scavenging of ROS with superoxide dismutase or catalase (2, 184). These inhibitors block lymphocyte transmigration without affecting VCAM-1-dependent adhesion of leukocytes to the endothelial cells (2, 184). In contrast, VCAM-1 does not activate other ROS generating enzymes for VCAM-1-dependent lymphocyte migration because VCAM-1-dependent migration is not affected by pharmacological inhibition of xanthine oxidase, nitric oxide synthase, or cytochrome P450 (184). In addition, inhibition of endothelial cell PI3 kinase and tyrosine kinases does not block VCAM-1-dependent leukocyte transendothelial migration (184). In contrast to the function of endothelial cell NADPH oxidase, lymphocyte flavoproteins, including NADPH oxidase, are not required for VCAM-1-dependent lymphocyte transendothelial migration (184). Thus, NADPH oxidase in endothelial cells but not lymphocytes is required for VCAM-1-dependent lymphocyte migration. Moreover, pharmacologic and antisense inhibition of NADPH oxidase or scavenging of ROS in endothelial cells blocks VCAM-1-dependent ROS generation and VCAM-1-dependent leukocyte migration.
3. VCAM-1-induced NOX2 generates low concentrations of ROS with specific signals that are distinct from signals by high levels of ROS
The level of VCAM-1-stimulated ROS production is much lower than the level of ROS that cause oxidative damage in tissues. To measure these low levels of VCAM-1-stimulated ROS, the ROS-sensitive probe dihydrorhodamine 123 has been used (65, 184). This probe becomes fluorescent when oxidized by H2O2, but not by superoxide, in the presence of cellular peroxidases (114). In addition, although xanthine oxidase-generated ROS can oxidize dihydrorhodamine 123, VCAM-1 does not activate xanthine oxidase signaling (184). Importantly, only 1 μM H2O2 is produced by the endothelial cells when lymphocytes bind to VCAM-1 or when VCAM-1 is crosslinked by anti-VCAM-1-coated beads (63, 65, 73, 184, 286). This is in contrast to the 50–200 μM H2O2 released by neutrophils and macrophages for the destruction of pathogens (66, 74) or released in disease states for oxidative damage such as atherosclerosis, pulmonary fibrosis, ischemia-reperfusion syndrome, and neurodegenerative diseases (241, 281). The oxidative damage to endothelial cell functions and junctions by large amounts of H2O2 (200–1000 μM) (28, 93, 116, 120, 141, 165, 193) are not consistent with the signals that occur during VCAM-1 signaling and leukocyte transendothelial migration. During VCAM-1 signaling, 1 μM H2O2 directly activates MMPs (73, 238), directly activates PKCα (1), and indirectly stimulates an increase in PTP1B activity (1). In contrast, high levels of H2O2 (>50 μM) directly inhibit MMPs (73, 238), inhibit PKCα (1), and inhibit tyrosine phosphatases (90). The function of the low levels of ROS for the generation of rapid, transient, and reversible signals is important because once a leukocyte reaches an endothelial cell junction, the process of transmigration occurs within a couple of minutes. The mechanisms for VCAM-1/ROS-induced activation of MMPs, PKCα, and changes in endothelial cell shape are discussed below.
C. VCAM-1 signals through NOX2 modify endothelial cell actin polymerization and intercellular gap formation
VCAM-1 signals mediate changes in endothelial cell actin structure. At the site of VCAM-1 binding, the endothelial cell actin coalesces at the endothelial cell surface, forming a cup-like structure in endothelial cell lines and cytokine-activated primary endothelial cells (25, 26, 47, 184). This VCAM-1-stimulated change in actin structure is mediated by endothelial cell NADPH oxidase (184, 294). VCAM-1-stimulated Rac1 and ROS also induce intercellular gap formation and loss of β-catenin at the gaps in IL-1β-activated HUVECs (294). The VCAM-1-activated intercellular gaps require VCAM-1 activation of Rac1 and ROS since the VCAM-1-induced gap formation is blocked by a dominant negative Rac1 or the ROS scavengers N-acetyl-
D. VCAM-1-induced ROS activate MMPs
1. Rapid activation of endothelial cell-associated MMPs
ROS production by VCAM-1-stimulated-endothelial cells activates MMPs (Fig. 5) (73). MMPs are held at the cell surface by membrane type-MMPs (MT-MMPs) and adhesion molecules. MMP-2 binds to transmembrane MT1-MMP (MMP14) (329). MMP-9 and MMP-7 bind to cell surface CD44 (3, 205, 324), and pro-MMP-9 can bind to ICAM-1 (92). It is also reported that MMP-1, MMP-2, and MMP-9 bind to α2β1 integrin on keratinocytes, αvβ3 integrin on endothelial cells, and α2 integrin on epithelial cells, respectively (260). The MMPs bound to the endothelial cell surface can have local functions, whereas MMPs released by endothelial cells are washed away by the flow of blood. Endothelial cell-associated MMP2 and MMP9 are activated by lymphocyte binding to VCAM-1 or antibody crosslinking of VCAM-1 as determined by gelatin zymography (73). This occurs without altering levels of cell-associated MMPs or tissue inhibitors of MMPs (TIMPs) as determined by western blot (73). The time course and magnitude of this MMP activation is the same for lymphocyte binding to VCAM-1 and for antibody crosslinking of VCAM-1 on either endothelial cell lines or IL-4-activated primary cultures of endothelial cells (73). Moreover, this activation of endothelial cell-associated MMPs occurs within minutes, which is consistent with the 2 min leukocyte transendothelial migration process once a leukocyte reaches a site for migration. The anti-VCAM-1-coated bead activation of the endothelial cell MMPs is not altered by laminar flow, at the rate found in postcapillary venules (2 dynes/cm2) (73). Thus, this force of “tugging” on VCAM-1 does not influence the signaling for activation of MMPs, whereas signaling by other adhesion molecules is influenced by the force on the receptor (12). The VCAM-1-stimulated activation of endothelial cell-associated MMP2 and MMP9 is mediated through ROS because the MMP activation is blocked by pretreatment of endothelial cells with antisense against the NADPH catalytic subunit gp91 phox, pharmacologic inhibitors of NADPH oxidase or scavenging of H2O2 with exogenous catalase (73). Interestingly, the same magnitude and rapid time course for activation of endothelial-associated MMPs also occurs when VCAM-1 is bypassed by addition of the VCAM-1 signaling intermediate, exogenous 1 μM H2O2 (73).
In contrast to the VCAM-1-mediated activation of MMPs by 1 μM H2O2, high concentrations of exogenous H2O2 (>50 μM H2O2) induce oxidative damage and inhibit basal endothelial cell-associated MMP activity (73). This is consistent with a report by Rajagopalan et al. (238), indicating that purified MMPs are inhibited by H2O2 at concentrations >50 μM, whereas 1 μM H2O2 activates purified MMPs (238). Thus, low concentrations of H2O2 activate MMPs, whereas high levels of H2O2 inhibit MMP enzymatic activities and induce oxidative damage (20, 165, 289). The data in these reports emphasize the opposing regulatory functions of low versus high levels of ROS.
The mechanism for ROS activation of MMPs is conserved among the MMPs. Briefly, MMPs are synthesized in a nonactive form, containing a conserved propeptide cysteine that is bound to the conserved zinc atom in the active site of the MMPs. ROS oxidize the cysteine in the propeptide domain that opens the propeptide arm and exposes the MMP active site (204). This opening of the propeptide arm stimulates autocatalytic removal of the arm, forming an active MMP (293). Thus, H2O2 does not have specificity for MMP isozymes given the conserved cysteine-zinc bond in pro-MMPs (204) and the rate of diffusion of H2O2 at 100 μm/s (183). Therefore, H2O2 activates those MMP isozymes expressed at the sites of VCAM-1-stimulated ROS generation.
The MMPs that are activated during VCAM-1 signaling degrade extracellular matrix and may cleave endothelial cell junction molecules. It has been reported that MMPs can cleave the endothelial cell junction molecule vascular endothelial-cadherin (117). This degradation by MMPs likely participates in opening endothelial cell–cell adhesions for the formation of passageways through which leukocytes can migrate. Consistent with this, it is reported that the VCAM-1-stimulated ROS-activation of endothelial cell-associated MMPs regulates VCAM-1-dependent leukocyte transmigration. In these studies, endothelial cells on transwells were pretreated with the MMP inhibitors GM6001 or BB3103 and washed before the migration assay (73). MMP inhibitor pretreatment of endothelial cell lines blocked VCAM-1-dependent lymphocyte migration in a dose-dependent manner without affecting cell viability (73). The last wash, from cells that had been pretreated with inhibitor, did not affect migration of untreated cells, indicating that the inhibitor-treated cells were sufficiently washed and that the effect of the inhibitor was on the endothelial cells (73). In addition, the MMP inhibitor blocked anti-VCAM-1 stimulated endothelial cell-associated MMP activity (73). Romanic et al. (245) also demonstrated that TIMP-2-mediated inhibition of MMP activity blocks T cell transmigration (245), although they did not determine whether inhibition with TIMP-2 was mediated by blocking lymphocyte or endothelial cell MMPs. Thus, endothelial cell-associated MMP activity is necessary for VCAM-1-dependent lymphocyte transendothelial migration. Further, the requirement for VCAM-1-stimulated endothelial cell ROS generation and endothelial cell-associated MMP activity during lymphocyte migration indicate that the endothelial cell has an active role in VCAM-1-dependent lymphocyte migration (Fig. 5).
2. Delayed activation of lymphocyte-associated MMPs
Since H2O2 diffuses rapidly (183), endothelial cell H2O2 that is generated during VCAM-1 signaling has the potential to also very rapidly activate MMPs on the surface of leukocytes when leukocytes are bound to the endothelium. However, several reports indicate that upon binding to VCAM-1, lymphocyte MMPs are activated but only after prolonged periods of 2–12 h (73, 87, 245, 318). This delay in lymphocyte MMP activation is a consequence of the high levels of TIMPs expressed by the leukocytes. Deem et al. reported that endothelial cell-derived ROS generated during lymphocyte binding to VCAM-1 activates lymphocyte MMPs at 2 h (73). In these studies, lymphocytes were incubated with monolayers of endothelial cells, nonbound lymphocytes were removed by washing, and bound lymphocytes were released from the monolayers by reversing lymphocyte binding with soluble anti-VCAM-1. Lymphocyte MMP9 was activated at 2–5 h (73). This activation of lymphocyte MMPs is mediated by endothelial cell-derived ROS, as the activation is blocked by pharmacological inhibition of endothelial cell ROS generation with diphenyliodonium or apocynin but not by pharmacologic inhibition of ROS-generating enzymes in lymphocytes (73). Moreover, exogenous addition of 1 μM H2O2 to purified lymphocytes induces activation of lymphocyte-associated MMPs that is also delayed for 2 h, indicating that 1 μM H2O2 activates the lymphocyte MMPs with the same time course and magnitude as ligand binding to VCAM-1 (73). Interestingly, after a 5 h treatment of purified lymphocytes with 1 μM H2O2, the expression of lymphocyte MMP9 is not altered, but the expression of tissue inhibitor of MMP (TIMP)-1 and TIMP2 by lymphocytes is reduced by 60%–80% as determined by western blot (73). Thus, H2O2 activates lymphocyte MMPs by oxidation and loss of TIMPs, the endogenous inhibitors of MMPs. The mechanism for this VCAM-1/ROS-induced loss of TIMPs is, at least, through proteosome degradation of the TIMPs because the 1 μM H2O2-induced loss of TIMP expression is blocked by the proteosome inhibitor MG132 (Fig. 7). Thus, the ROS-induced reduction in TIMPs on lymphocytes results in a threefold increase in the MMP9/TIMP ratio, reflecting a net increased MMP activity at 2–5 h. Thus, H2O2 activation of the lymphocyte-associated MMPs is mediated by the downregulation of the expression of the relatively high levels of TIMPs on lymphocytes without altering expression of lymphocyte MMPs (73). Thus, the mechanism for the activation of lymphocyte MMPs is through ROS inactivation of the TIMPs. This is in contrast to a direct effect of H2O2 on endothelial cell-associated MMPs for the rapid activation of endothelial cell MMPs within minutes. The activation of lymphocyte-associated MMPs is too late for lymphocyte MMP function during transmigration because transmigration occurs within a few minutes. Moreover, pretreatment of lymphocytes with the MMP inhibitors GM6001 or BB3103, followed by a wash, does not alter VCAM-1-dependent transmigration (73).
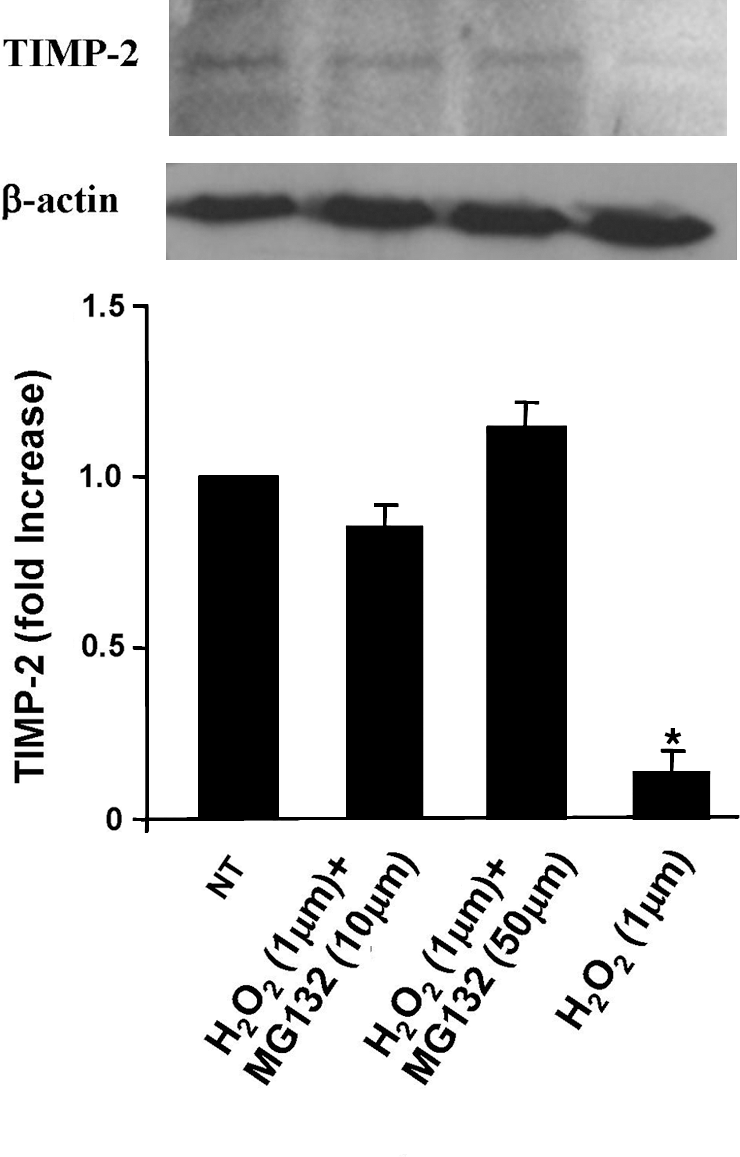
3. The antioxidant bilirubin inhibits VCAM-1-induced MMP activation
The in vitro studies on VCAM-1 activation of ROS demonstrate that endothelial cell production of ROS rapidly activates endothelial cell-associated MMPs that are required for VCAM-1-dependent lymphocyte migration. The antioxidant bilirubin blocks this VCAM-1-dependent lymphocyte migration in vitro and blocks VCAM-1 activation of MMPs (140). Bilirubin is generated from heme by hemoxygenase-1 (140, 249). After generation of bilirubin, it can undergo redox cycling such that oxidation of bilirubin converts it to biliverdin (249, 259, 272, 273). Bilirubin and biliverdin are membrane permeable (217, 273). Biliverdin is recycled back to bilirubin by biliverdin reductase and the cofactor NADPH (140). Hemoxygenase-1 and biliverdin reductase are expressed by endothelial cells and endothelial cell lines (140). Further, bilirubin is taken up by endothelial cells, and thus bilirubin in endothelial cells can function as an antioxidant (140). Bilirubin acts as an antioxidant in that it reduces oxidized phospholipids with the approximate rate of antioxidant vitamins (274). Concentrations of bilirubin in the upper physiological range block anti-VCAM-1 activation of endothelial cell-associated MMP2 and MMP9 without affecting cell viability (140). Further, bilirubin blocks VCAM-1-dependent migration of lymphocytes across endothelial cells in vitro without affecting cell viability (140). Consistent with an antioxidant function for bilirubin, VCAM-1-dependent lymphocyte migration is not blocked by the stable bilirubin conjugate ditaurobilirubin that cannot scavenge ROS (140). The bilirubin inhibition of lymphocyte migration results from an inhibition of migration rather than inhibition of lymphocytes available for migration as the number of lymphocytes bound to the endothelial cell monolayer is unaffected by bilirubin (140). Therefore, the antioxidant bilirubin blocks VCAM-1-dependent lymphocyte migration across endothelial cells, at least by, blocking the ROS-mediated activation of MMPs.
To summarize VCAM-1 activation of MMPs, lymphocyte binding to VCAM-1 activates endothelial cell generation of ROS, which induces a delayed activation of lymphocyte MMPs. This delay in ROS-induced MMP activity in lymphocytes is a consequence of the time required for a reduction in TIMP expression. The 2–5 h delay in activation of the lymphocyte-associated MMPs is too late for transendothelial migration but likely regulates migration of lymphocytes through extravascular tissues. In contrast, pretreatment of endothelial cells with these MMP inhibitors blocks leukocyte transendothelial migration. Thus, the delayed activation of lymphocyte MMPs is consistent with the requirement for endothelial cell MMPs, but not lymphocyte MMPs, during VCAM-1-dependent lymphocyte migration.
E. VCAM-1 signals oxidize and activate PKCα
VCAM-1-induced H2O2, which can diffuse through membranes at 100 μm/s (183), stimulates intracellular signals through PKCα (Fig. 5) (1). Ligand binding to VCAM-1 activates autophosphorylation of PKCα Thr638 in endothelial cell lines or primary cultures of endothelial cells (1). This activation of PKCα is blocked by inhibition of NADPH oxidase or scavenging of ROS (1). Moreover, this VCAM-1 activation of PKCα is induced by direct oxidation of PKCα cysteines (1). Bypassing VCAM-1 by exogenous addition of the VCAM-1 signaling intermediate, 1 μM H2O2, also stimulates activation of PKCα with the same time course as that for ligand binding to VCAM-1, indicating that 1 μM H2O2 is sufficient for activation of endothelial cell PKCα (1). Importantly, VCAM-1 activation of PKCα is required for VCAM-1-dependent leukocyte transmigration because VCAM-1-dependent leukocyte transendothelial migration is blocked by dominant negative PKCα or selective pharmacological inhibitors of PKCα without affecting leukocyte binding to the endothelial cells (1). Thus, VCAM-1 stimulates oxidative activation of endothelial cell PKCα, which is required for VCAM-1-dependent leukocyte transendothelial migration.
F. VCAM-induced PKC activates PTP1B
VCAM-1 signaling via ROS and PKCα activates downstream signals in endothelial cells through PTP1B (Fig. 5) (72). Ligand binding to VCAM-1 increases PTP1B serine phosphorylation and the phosphatase activity of PTP1B in endothelial cells (72). This activation of PTP1B is blocked by inhibition of endothelial cell NADPH oxidase or inhibition with the PTP1B inhibitor, CinnGEL-2ME (72). CinnGEL-2ME specifically inhibits PTP1B because it has a side chain that binds to a site on PTP1B specific for PTP1B and it also binds to and inhibits the active site of PTP1B (199, 326). Bypassing VCAM-1 by exogenous addition of 1 μM H2O2 to the endothelial cells increases endothelial cell PTP1B activity that is similar in magnitude and time course as that observed with ligand binding to VCAM-1. Further, VCAM-1 activation of PTP1B is downstream of PKCα during VCAM-1 signaling (72) since anti-VCAM-1-stimulated serine phosphorylation of PTP1B, the active form of PTP1B (40), is blocked when endothelial cells are transfected with a plasmid containing dominant negative PKCα or treated with the PKCα inhibitor Gö-6976 (72).
Interestingly, during VCAM-1 signaling, PTP1B is activated and not inhibited by oxidation (72). Although it has been reported that PTP activity can be inhibited by high levels of oxidants (50–200 μM H2O2) (94, 267, 281) through oxidation of the conserved cysteine in the PTP1B catalytic site (39, 90, 91, 94, 111, 234, 267, 281), VCAM-1 stimulates the production of only 1 μM H2O2 (65, 73), which activates PTP1B rather than inhibits it (72). Moreover, analysis of PTP1B for oxidation of cysteines revealed that PTP1B in endothelial cells is not oxidized after VCAM-1 signaling or after exogenous addition of 1 μM H2O2 (72). However, purified PTP1B is susceptible to oxidation upon addition of 1 μM H2O2 (72). Thus, within VCAM-1-stimulated endothelial cells, there is compartmentalization of targets for oxidation by low levels of NOX2-generated ROS because PKCα is oxidized, but PTP1B is not oxidized (72). This occurs even though H2O2 diffuses through membranes at 100 μm/s (183). Therefore, it is possible that the low concentrations of H2O2 are readily consumed as they oxidize targets, diffusion lowers the H2O2 below a threshold for oxidation of PTP1B, or local antioxidant mechanisms protect PTP1B. Thus, compartmentalization of ROS may limit the proteins that are modified by ROS. Forman et al. (94) proposes that low levels of ROS function as signaling molecules because (i) they have a restricted location of action, (ii) their signals are transient, and (iii) their oxidation reactions are reversible. ROS modify thiolate anions (-S−) to form sulfenate (-SO−) as well as react with disulfide linkages (281). These can be reduced back to their native state by intracellular thiols in the cell such as thioredoxin, peroxiredoxins, and glutathione (94). Thus, PTP1B may be protected from oxidation by antioxidants or its compartmentalization to the endoplasmic reticulum (ER).
PTP1B is located on the ER membrane with its catalytic domain external to the ER (Fig. 5). An important question regarding compartmentalization is how PTP1B, which is localized to the ER, mediates VCAM-1 signaling. Studies have demonstrated that the ER membranes containing PTP1B reach the plasma membrane and that the PTP1B in this ER membrane can dephosphorylate receptors in the plasma membrane without receptor internalization (16, 263). Thus, during VCAM-1 signaling, endothelial cell PKCα phosphorylates and activates PTP1B, which then modulates localized signals in the endothelial cells. These signals need to have localized functions because the endothelial cell changes are limited to the site of leukocyte binding without retraction of the rest of the endothelial cell.
Most importantly, PTP1B participates in the active function of the endothelial cell during VCAM-1-dependent leukocyte transmigration because inhibition of PTP1B blocks VCAM-1-dependent lymphocyte transmigration without altering adhesion (72). Interestingly, when both the extracellular signals through MMPs and the intracellular signals through PTP1B are blocked, there is a greater inhibition of VCAM-1-dependent leukocyte transendothelial migration.
PTP1B is interesting as it has been a target for drug development. PTP1B is a potential target because PTP1B-deficient mice (without foreign antigen challenge) are physiologically normal and have normal body weight, making PTP1B a potentially promising drug target (29, 71, 84). PTP1B is most studied in diabetes because inhibitors of PTP1B block PTP1B dephosphorylation of the insulin receptor and block the development of diabetes in animal models (29, 79, 84, 104, 246, 253, 328). Moreover, PTP1B-deficient mice do not develop tumors, and do not develop diabetes in response to high fat diet, although there is a small effect on the immune system (29, 79, 84, 104, 112, 246, 253, 328). This has led to PTP1B inhibitors that have been in phase II clinical trials for diabetes (42, 79). Thus, whether clinical PTP1B inhibitors alter leukocyte recruitment during inflammation has clinical implications.
VI. VCAM-1 Signals in In Vivo Models
A. Gαi2 regulation of VCAM-1-dependent leukocyte recruitment in vivo
Gαi2 functions in VCAM-1-dependent leukocyte recruitment in vitro and in vivo (Fig. 5). In in vivo studies using Gαi2−/− mice, it was demonstrated that a signaling event in a nonlymphohematopoietic compartment of the lung is required for the recruitment of leukocytes during inflammation (225). This was examined in a model of allergic inflammation in which airway challenge with chicken egg ovalbumin (OVA) induces VCAM-1-dependent recruitment of eosinophils. In OVA-challenged Gαi2−/− mice, VCAM-1-dependent eosinophil recruitment is inhibited (225). In addition, the inhibition of leukocyte recruitment was specific to the G protein Gαi2−/− since mice deficient in Gαi3 do not have altered leukocyte recruitment in response to OVA (225). Gαi2 function in eosinophils was not required for eosinophil chemotaxis since Gαi2−/− eosinophils responded to chemotactic factors (225). Consistent with a function for nonhematopoietic Gαi2, Gαi2−/− eosinophils adoptively transferred into wild-type mice are able to be recruited during allergic responses. In contrast, there is a reduced recruitment of wild-type eosinophils in Gαi2−/− mice, indicating that signaling in a resident cell of the lung is required for the accumulation of eosinophils (225). Moreover, in the OVA-challenged Gαi2−/− mice, there are elevated blood leukocyte numbers and an accumulation of leukocytes on the luminal surface of the blood vessels (225). Thus, the blood leukocytes are available for migration and capable of chemotaxis, but their transmigration is blocked. Interestingly, this occurs without altering the lung levels of the Th2 cytokines IL-4 and IL-5, and without inducing the Th1 cytokine interferon γ, indicating that the Gαi2−/− deficiency did not alter regulatory inflammatory cytokines during allergic responses (225). In vitro, inhibition of endothelial cell Gαi blocks VCAM-1-dependent leukocyte transendothelial migration (225). These reports are consistent with specific Gαi2-mediated signaling in endothelial cells for the extravasation of leukocytes and for tissue-specific leukocyte accumulation. The mechanism for Gαi2 function in the VCAM-1 signaling pathway is under investigation. Nevertheless, since Gαi2-deficient mice are viable and Gαi2 regulates leukocyte recruitment, it may be a potential target for clinical intervention in inflammatory diseases.
B. NOX2 regulation of VCAM-1-dependent leukocyte recruitment in vivo
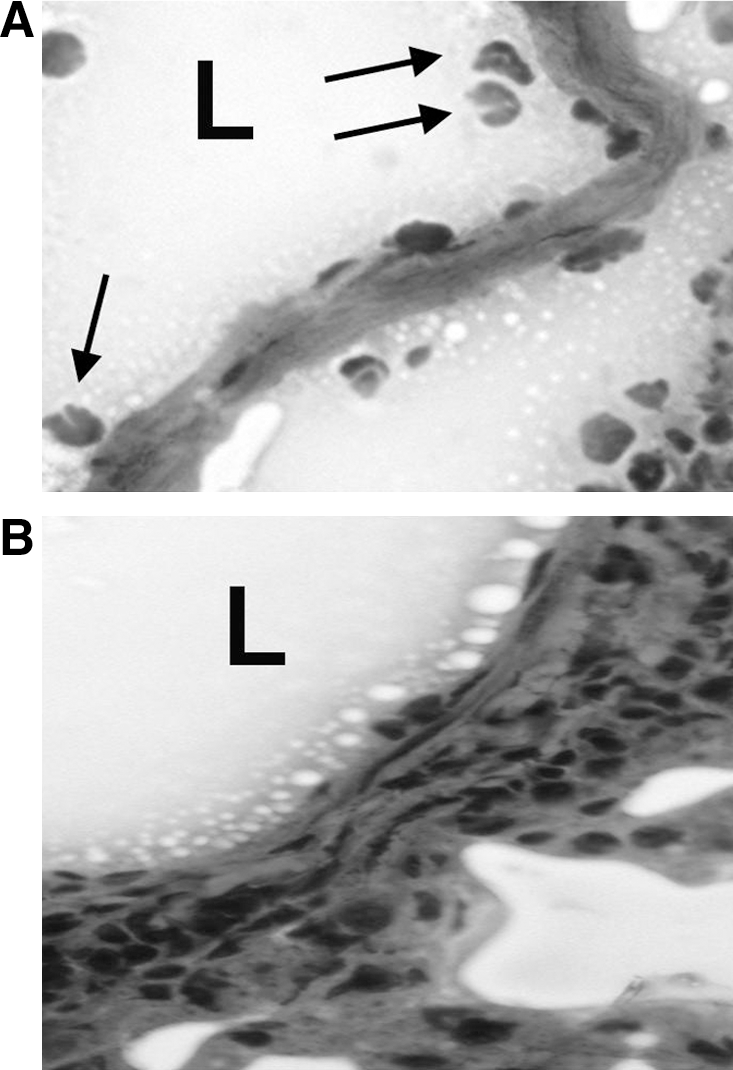
VCAM-1 signals through NOX2 in vivo. The gene that encodes the catalytic subunit of NOX2, gp91phox, is CYBB. CYBB-deficient mice have been used to examine VCAM-1-dependent eosinophil recruitment in response to OVA, a model of allergic inflammation. To examine the function of NOX2 in nonhematopoietic cells and avoid effects of NOX2 deficiency in leukocytes, green fluorescent protein C57BL/6J mouse bone marrow cells, which express wild-type gp91 phox, were transplanted into irradiated CYBB mice and into control irradiated C57BL/6 wild-type mice (2). To induce VCAM-1-dependent eosinophil infiltration into the lung (2), the chimeric CYBB mice and chimeric wild-type mice were sensitized intraperitoneally with the antigen OVA in the adjuvant alum and then the lungs were challenged by intranasal administration of OVA (Fig. 8) (2). It is well established that OVA-stimulated eosinophilia in the lung as well as OVA-stimulated eosinophilia in the skin requires adhesion to VCAM-1 as antibodies to VCAM-1 block the eosinophilia (Fig. 8) (57, 107, 250). Interestingly, eosinophil infiltration into the bronchoalveolar lavage is inhibited by 68% in the OVA-challenged chimeric CYBB mice, but the infiltration of other leukocytes, which migrate on other adhesion molecules, is not altered (2). This is consistent with reports that 70% of OVA-induced eosinophil infiltration is VCAM-1-dependent (57, 107, 250). Most interestingly, there is an accumulation of eosinophils on the luminal surface of the endothelial cells in lung tissue of OVA-challenged chimeric gp91 phox-deficient mice, suggesting that the eosinophils bound to endothelium but that they could not undergo VCAM-1-dependent transmigration (Fig. 9) (2).
In the CYBB chimeric mice, there are sufficient mediators present for induction of eosinophilia since there was no difference between the OVA-challenged chimeric wild-type and OVA-challenged chimeric deficient mice for cytokines, chemokines, VCAM-1 expression, or blood eosinophil numbers (2). Further, in these mice, there is no effect on the initial sensitization from the intraperitoneal administration of OVA since there is no effect on OVA-specific IgE in the OVA-challenged mice (2). The chimeric CYBB mice also exhibit a 70% reduction in airway hyperresponsiveness (AHR) (2). Moreover, intratracheal administration of purified eosinophils into the chimeric CYBB mice recovers the AHR (2), suggesting (i) that bypassing the endothelium overcomes the reduced AHR in the chimeric CYBB mice and (ii) that gp91 phox expression by other cells of the lung such as fibroblasts are not critical for the reduced AHR in the nonhematopoietic gp91phox deficient mice. These studies provide support for the in vivo relevance of VCAM-1 signals through ROS for eosinophil recruitment in experimental allergic asthma.
VII. Antioxidant Regulation of VCAM-1 Signals in In Vivo Models
A. The antioxidant bilirubin inhibits VCAM-1-dependent inflammation
Consistent with the inhibitory function of bilirubin on VCAM-1-dependent leukocyte transmigration in vitro (140), it has been reported that bilirubin blocks VCAM-1-dependent leukocyte migration in vivo. This was examined in a model of VCAM-1-dependent leukocyte infiltration into the lung in response to the antigen OVA. In studies examining bilirubin regulation of recruitment of eosinophils during allergic inflammation, mice were sensitized by intraperitoneal injection of OVA in alum and then challenged by intranasal inhalation of OVA (140). At the time of intranasal OVA challenge, mice also received either intraperitoneal injections of bilirubin at upper physiological concentrations or vehicle control (140). The treatment with bilirubin inhibits eosinophil infiltration into the bronchoalveolar lavage by >90% and inhibits lymphocyte infiltration by 60% (140). The migration of eosinophils into the tissue is also reduced by 90% as determined by immunohistochemistry for the eosinophil granule component, major basic protein (140). The reduction in eosinophil and lymphocyte infiltration is consistent with the VCAM-1 dependence of eosinophil migration and the partial VCAM-1 dependence of lymphocyte migration in this lung response to OVA (57, 107, 250). As anticipated, there is no effect of bilirubin administration on the OVA-induced infiltration of monocytes or neutrophils (140), which is independent of binding to VCAM-1 in this model of allergic inflammation. Although there is reduced eosinophilia with the administration of bilirubin, there are sufficient numbers of eosinophils available for migration as there is not a reduction in blood eosinophils in the bilirubin-treated group compared to the nontreated group (140). In fact, there is a threefold increase in blood eosinophil numbers with bilirubin administration, which is consistent with inhibition of blood eosinophils transendothelial migration (140). VCAM-1 is available for eosinophil binding to the endothelium since bilirubin treatment does not alter the induction of endothelial VCAM-1 expression in OVA-treated mice (140). It is also reported that other antioxidants such as vitamin E also do not alter VCAM-1 expression but do block VCAM-1-dependent leukocyte recruitment (30). Therefore, although ROS can induce VCAM-1 expression, the lack of antioxidant effect on VCAM-1 expression is consistent with compensatory mechanisms for induction of VCAM-1 expression by the many pro-inflammatory mediators that induce VCAM-1 expression.
The infiltration of eosinophils in response to OVA is regulated by cytokines and chemokines. However, bilirubin treatment does not alter the OVA-induced increase in Th2 cytokines (IL-4, IL-5, IL-6, or IL-10) in lung lavage fluid or in OVA-restimulated draining lymph node cells (140). Since IL-5 was not altered, this suggests that IL-5 was sufficient for bone marrow recruitment of eosinophils. In addition, bilirubin does not increase expression of Th1 cytokines (IL-2, IL-12, interferon γ, or TNFα), which are not expected to be upregulated by OVA stimulation (140). Bilirubin also does not alter the OVA-induced increase in the chemokines monocyte chemoattractant protein-1 or eotaxin (140). Thus, in this report, VCAM-1-dependent eosinophil and lymphocyte infiltration into the lung is reduced by the antioxidant bilirubin without altering the expression of the VCAM-1, cytokines, or chemokines that regulate eosinophil infiltration in response to OVA. These data are consistent with bilirubin scavenging of endothelial-cell derived ROS generated during VCAM-1 signaling. Moreover, it is reported that in vitro, bilirubin blocks VCAM-1 activation of MMPs in endothelial cells and blocks VCAM-1-dependent leukocyte transendothelial migration (140). Therefore, bilirubin, which blocks VCAM-1 signals through ROS in vitro, also inhibits VCAM-1-dependent eosinophilia in allergic responses in mice.
Bilirubin may also regulate VCAM-1-dependent inflammation in cardiovascular disease. It has been reported, in a patient population in China, that low bilirubin associates with increased cardiovascular disease risk factors, including older age, higher body mass and systolic blood pressure, increased glycated hemoglobin, fasting and 2 h insulin, triglyceride, very-low-density lipoprotein, apolipoprotein B concentrations, and lower high-density lipoprotein concentrations (149). They suggest in their report that abnormal intermediate bilirubin metabolism and antioxidant deficiency may be linking factors in cardiovascular disease (149). In another report on a prospective clinical study in Korea, low serum bilirubin is an independent predictor of stoke incidence, suggesting that bilirubin may have a protective function against stroke risk (145). Thus, bilirubin has a regulatory function in limiting leukocyte recruitment during inflammatory diseases, since reports indicate that bilirubin protects against cardiovascular disease and asthmatic inflammation, that inflammation in these diseases is dependent on VCAM-1, and that bilirubin blocks VCAM-1-dependent inflammation by blocking VCAM-1 signaling.
B. Vitamin E regulation of VCAM-1-dependent inflammation
1. Introduction to vitamin E isoforms
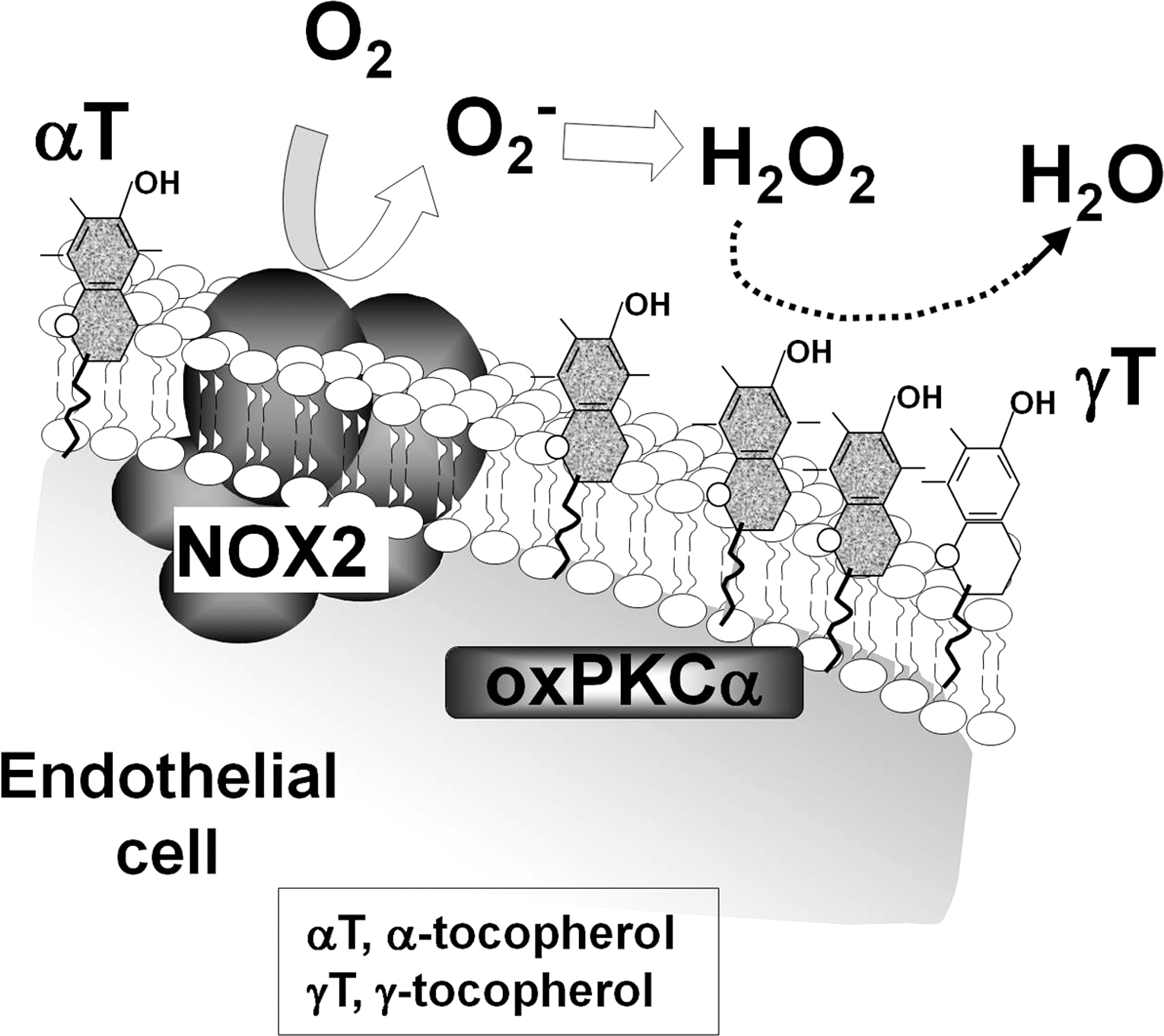
Vitamin E is commonly used as an antioxidant to try to limit oxidative damage and inflammatory disease. Vitamin E has been used in diseases that involve VCAM-1-mediated leukocyte recruitment such as asthma, arthritis, and atherosclerosis. However, there are contradictory outcomes for vitamin E in clinical studies of asthma and atherosclerosis. In addition, there are contradictory outcomes for vitamin E supplementation in animal models of inflammation. These clinical and experimental studies have focused on analysis of one form of vitamin E, α-tocopherol, even though multiple forms of vitamin E are present in the studies. Our recent report on novel properties of tocopherol isoforms suggests that the reported contradictory outcomes of the previous studies are consistent with new expectations for the combination of isoforms of vitamin E that were present in these reported studies.
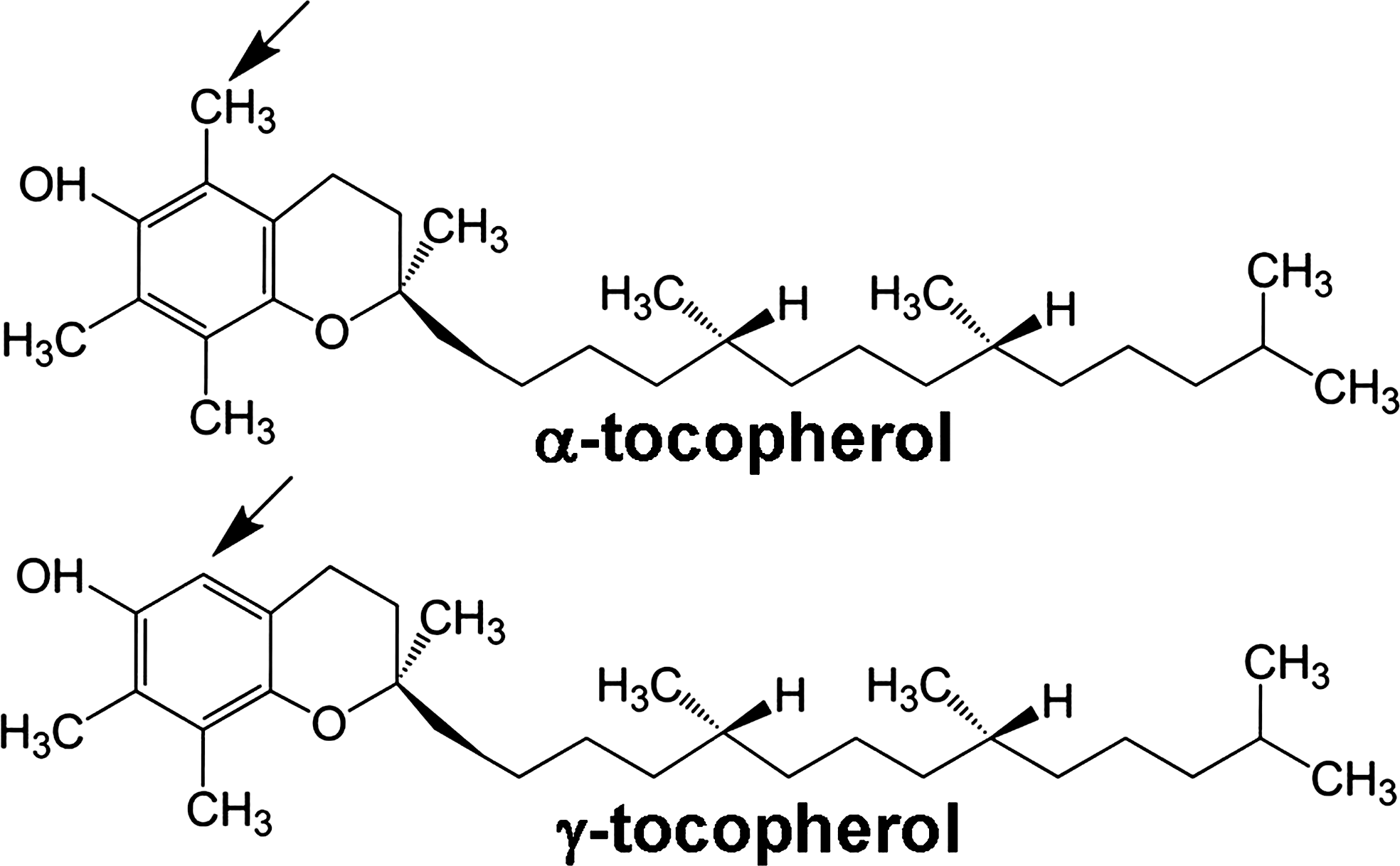
Vitamin E is an antioxidant lipid vitamin that consists of multiple natural and synthetic forms. The natural forms of vitamin E include α-tocopherol, β-tocopherol, γ-tocopherol, and δ-tocopherol as well as the tocotrienol forms of each of these. The α-tocopherol and γ-tocopherol isoforms (Fig. 10) are the most abundant in diets, supplements, and tissues. However, the α-tocopherol isoform in tissues is about 10-fold higher than γ-tocopherol since there is preferential transfer of the α-tocopherol isoform of vitamin E to lipid particles by liver α-tocopherol transfer protein (312). At equal molar concentrations, the α-tocopherol and γ-tocopherol isoforms have relatively similar capacity to scavenge ROS during lipid oxidation (18, 323). Thus, in vivo, there is likely more ROS scavenging by α-tocopherol than γ-tocopherol because it is at a 10-fold higher concentration in the tissues. However, γ-tocopherol, in contrast to α-tocopherol, also reacts with reactive nitrogen species such as peroxynitrite forming 5-nitro-γ-tocopherol (60, 311). When tocopherols are oxidized, they are recycled by reduction by vitamin C (43, 109, 122). Importantly, besides the antioxidant capacity of the tocopherols, it has been reported that tocopherols also have nonantioxidant functions (19, 30, 327).
2. Vitamin E isoforms regulate VCAM-1-dependent leukocyte transmigration through antioxidant and nonantioxidant mechanisms
In vitro, α-tocopherol blocks, whereas γ-tocopherol elevates, VCAM-1-dependent leukocyte transmigration at physiological concentrations (30, 322). Moreover, treatment with γ-tocopherol ablates the inhibition by α-tocopherol such that the leukocyte transmigration is the same as the vehicle-treated control (30). Interestingly, this occurs at physiological concentrations. Thus, γ-tocopherol ablates the effects of α-tocopherol even though it is at a concentration that is 1/10 that of α-tocopherol (30). These regulatory functions of the tocopherols on leukocyte transmigration are through a direct effect of the tocopherols on endothelial cells because pretreatment of the endothelial cells with α-tocopherol or γ-tocopherol overnight inhibits or elevates, respectively, leukocyte transmigration (30). In contrast, pretreatment of the leukocytes with physiological concentrations tocopherols has no effect on VCAM-1-dependent leukocyte transmigration (30). The γ-tocopherol elevation of transmigration is VCAM-1 dependent since anti-VCAM-1 blocking antibodies inhibit the leukocyte transmigration (30). The tocopherols do not modulate leukocyte-endothelial cell binding, because there is no effect of the tocopherols on VCAM-1-dependent adhesion of the leukocytes to the endothelium when either the endothelial cells or the leukocytes are pretreated with tocopherols (30). The tocopherols modulate endothelial function during VCAM-1-dependent transmigration by altering VCAM-1-induced oxidative activation of endothelial cell PKCα (Fig. 11) (30). Specifically, the VCAM-1-induced activation of PKCα is inhibited by α-tocopherol and the effect of α-tocopherol is ablated by γ-tocopherol. Therefore, the tocopherols have opposing regulatory functions on VCAM-1 signaling during leukocyte transmigration in vitro.
3. Vitamin E isoform-specific regulation of leukocyte recruitment in vivo
In vivo, α-tocopherol and γ-tocopherol also have opposing regulatory functions on leukocyte accumulation during VCAM-1-dependent allergic lung inflammation (30). The studies in this report focused on supplementation with tocopherols after OVA antigen sensitization to determine whether tocopherols modulate the OVA antigen challenge phase. This is important because patients are already sensitized. Supplementation with the tocopherols after OVA sensitization, such that the tissue tocopherols are raised 5–7-fold higher than mice consuming control rodent chow, does not affect body weight or lung weight (30, 191). Consistent with the in vitro studies with tocopherol regulation of leukocyte migration, d-γ-tocopherol elevates leukocyte accumulation in the bronchoalveolar lavage and lung tissue in response to OVA challenge. In contrast, d-α-tocopherol inhibits this inflammation. However, d-γ-tocopherol, at as little as 10% the concentration of d-α-tocopherol, ablates the anti-inflammatory benefit of the d-α-tocopherol isoform in vivo in response to OVA. Further, the levels of tocopherols in this study do not alter the blood eosinophil numbers, indicating that eosinophils were available for recruitment. It is also reported by Okamoto et al. (213) that mice fed with α-tocopherol starting 2 weeks before sensitization with OVA had reduced number of eosinophils in the bronchoalveolar lavage, even though the form or purity of α-tocopherol was not indicated. Therefore, α-tocopherol and γ-tocopherol have opposing functions in vivo.
The opposing functions of purified d-α-tocopherol or d-γ-tocopherol in vivo are not through modulation of expression of several cytokines, chemokines, prostaglandin E2, or adhesion molecules that regulate leukocyte recruitment since these are not altered with tocopherol supplementation (30). The tocopherol modulation of leukocyte infiltration in allergic inflammation, without alteration of adhesion molecules, cytokines or chemokines, is similar to several previous reports of in vivo inhibition of intracellular signals in endothelial cells without alteration of expression of these immune modulators of leukocyte recruitment (2, 140, 225). Therefore, the tocopherol regulatory function in allergic responses is, at least in part, by regulation of endothelial cell VCAM-1 activation of PKCα and leukocyte transendothelial migration. Moreover, natural d-α-tocopherol and natural d-γ-tocopherol differ in structure by only one methyl group (Fig. 10) but at physiological concentrations have opposing regulatory functions in endothelial cells that modulate inflammation. The opposing functions of tocopherol isoforms have important implications for the interpretation of clinical reports and animal studies of vitamin E regulation of inflammation.
C. Reinterpretation of reports on vitamin E regulation of inflammation in experimental models
Our data on tocopherol isoform regulation of inflammation alter interpretations of animal studies with tocopherol modulation of VCAM-1-dependent inflammation. Many reports with animal studies indicate that vitamin E was administered to animals, but the form, source, and purity of tocopherols are often not reported. In addition, the tissue levels of tocopherol isoforms after administration are sometimes not determined. Further, since tocopherols are lipids, there needs to be consideration for tocopherol isoforms that are present in the oils in animal and human diets or in the oil vehicles used for delivery of the tocopherols. We and others have determined the levels of α-tocopherol and γ-tocopherol in dietary oils (Fig. 12) (30, 130, 304). In rodent studies, rodent chow contains α-tocopherol but low to no γ-tocopherol. However, in some reports for allergic inflammation, α-tocopherol is administered in oil vehicles that contain other tocopherol isoforms. In a report by Suchankova et al. (275), purified α-tocopherol was administered in soy oil by gavage and they found no major effect of α-tocopherol on immune parameters or lung airway responsiveness in mice challenged with OVA. However, the soy oil vehicle used in this study contains an abundance of γ-tocopherol (Fig. 12) and they did not measure tissue tocopherol levels or vehicle tocopherol levels. Our interpretation of this study is that γ-tocopherol in the soy oil antagonized the function of the α-tocopherol that was administered. In another report, γ-tocopherol in tocopherol-stripped corn oil was administered daily by gavage to rats 2 weeks after one OVA sensitization and then the rats received two OVA challenges (302). In this report, there was a reduced number of eosinophils and lymphocytes in the bronchoalveolar lavage of the γ-tocopherol-treated mice after OVA challenge (302). However, the purity of the γ-tocopherol in the corn oil vehicle was not reported. Further, the leukocyte infiltration in the OVA response in these rats was predominantly neutrophils rather than the expected predominant eosinophil infiltration (302).
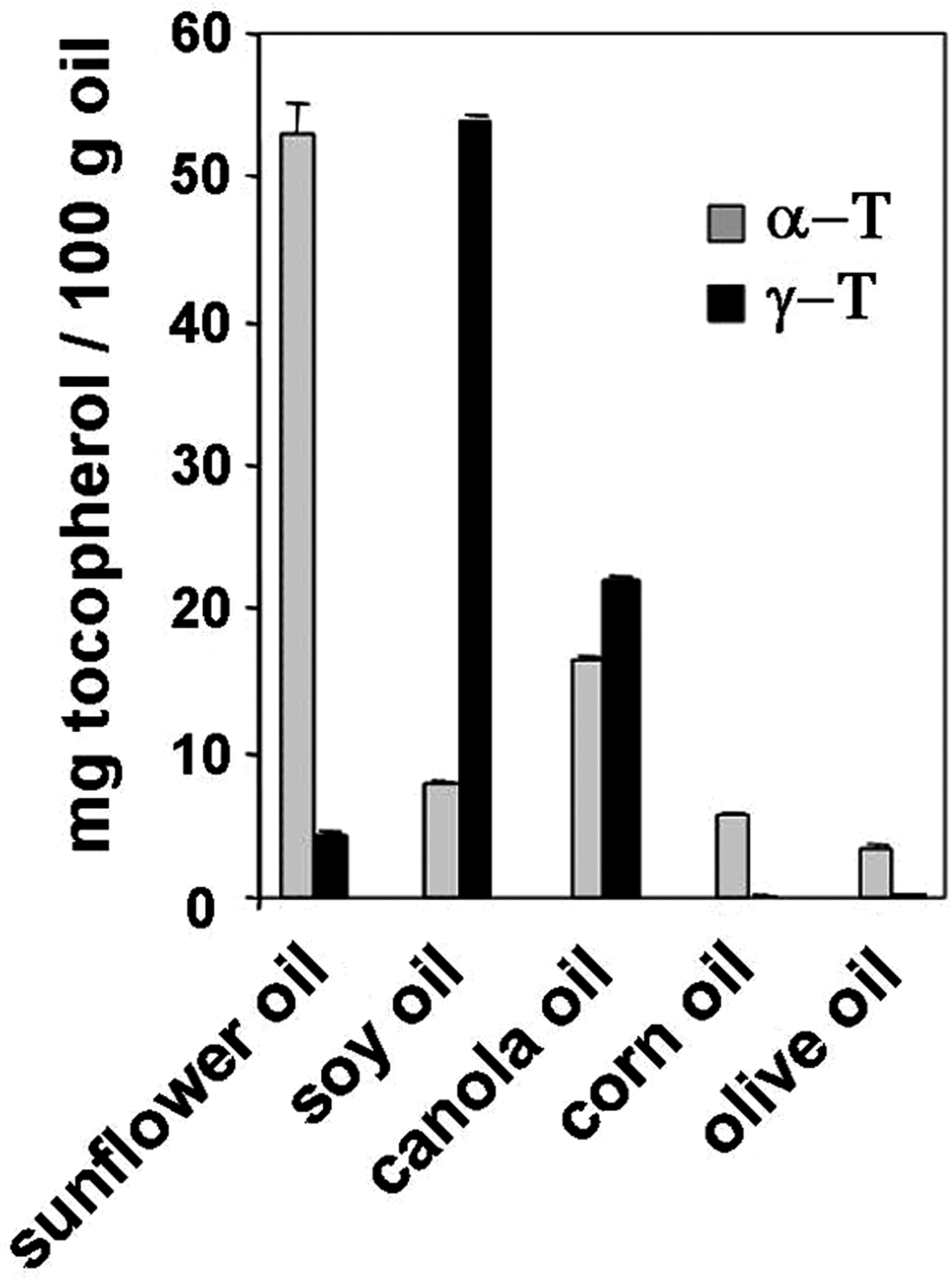
Tocopherols have also been used to scavenge ROS after ozone challenge to the lung. In a study examining γ-tocopherol modulation of ozone exposure after OVA challenge, control rats, which did not receive ozone but received γ-tocopherol for 4 days beginning after the last OVA challenge, had reduced lung eosinophils at day 4 after OVA challenge (303). However, since it takes a few days to raise tissue tocopherol levels, which, in this protocol, is after the peak of eosinophil infiltration, the effect on eosinophils at 4 days after the last OVA challenge was during the resolution phase of eosinophil inflammation. It has also been reported that mice deficient in liver α-tocopherol transfer protein exhibit severe deficiency in tissue α- and γ-tocopherol as well as reduced IgE and reduced IL-5 after OVA challenge to the lung (172). In these mice, it is not known whether severe tocopherol deficiency during mouse development alters leukocyte hematopoiesis or leukocyte responsiveness. The differences among the reports of tocopherol regulation of responses to OVA for VCAM-1-dependent eosinophil recruitment likely reflect differences in the forms of tocopherols, tocopherol concentrations, and time of administration of tocopherols in these studies.
Reports also conflict as to whether the antioxidant tocopherols modulate mediators of leukocyte recruitment during inflammation, including prostaglandins, cytokines, chemokines, and adhesion molecules (81, 86, 101, 128, 129, 135, 150, 164, 203, 213, 228, 251, 252, 296, 310, 314 –316, 325). With regard to adhesion molecules, in vitro, α-tocopherol is reported to block IL-1β-induced ICAM-1 expression on human aortic endothelial cells but not on human umbilical vein endothelial cells, and then, in another report, α-tocopherol does not inhibit TNF-α-stimulated ICAM-1 expression on human umbilical vein endothelial cells (314, 325). oxLDL or 25-hydroxycholesterol-induced VCAM-1 expression on human aortic endothelial cell is blocked by 200 μM α-tocopherol or 10 μM tocotrienols in vitro (208, 322). In vivo, we reported that purified natural d-α-tocopherol and d-γ-tocopherol at physiological levels do not alter OVA-induced VCAM-1 expression on lung venules (30). We suggest that variations in reports on outcomes of tocopherol treatments in vitro and in vivo result, at least in part, from differences in isoforms and purity of tocopherols, in concentrations of the tocopherols within different cells, and in experimental systems. This is important considering our report indicating that forms of tocopherols have cell-type-specific opposing regulatory functions on leukocyte recruitment since tocopherols directly affected endothelial cells but not leukocytes during leukocyte transmigration.
VIII. Clinical Implications for Vitamin E Regulation of Inflammation Involving VCAM-1
Reports of clinical studies on vitamin E primarily focus on the α-tocopherol isoform without adjustment for the dietary contribution of γ-tocopherol to the outcomes of these studies. For interpretation of the clinical studies, it is especially important to take into consideration the dietary contribution of tocopherols because γ-tocopherol is more abundant in western diets. The average plasma concentration of α-tocopherol is the same among many countries (304). However, the American diet is rich in γ-tocopherol found in soy oil, the major form of vegetable oil in the United States. In contrast, γ-tocopherol is low in other oils (sunflower and olive oil) commonly used in some of the European countries (Fig. 12) (30, 130, 304). Consistent with this, in the United States and the Netherlands, the average plasma γ-tocopherol level is 2–6 times higher than that reported for six European countries, Japan, and China (Table 4) (304). This fold increase in plasma γ-tocopherol is similar to fold increase in plasma γ-tocopherol in the rodent studies in which γ-tocopherol opposed the regulatory functions of α-tocopherol, even at 1/10 the concentration of α-tocopherol (30).
A consistent feature of inflammation in allergic asthma is the recruitment of eosinophils and mast cells. The recruitment of these cells is regulated by VCAM-1 and tocopherols (4, 5, 8, 30, 36, 57, 107, 108, 250). In clinical studies of asthma, it is reported that α-tocopherol supplementation of asthmatic patients is beneficial in Italy and Finland, but disappointingly α-tocopherol is not beneficial for asthmatic patients in studies in the United States or the Netherlands (78, 266, 277, 284, 308). These clinical outcomes are consistent with an interpretation that there is little benefit of α-tocopherol for inflammation in the presence of elevated plasma γ-tocopherol because γ-tocopherol is elevated 2–6-fold in people in the United States and the Netherlands (Table 4). Therefore, differences in outcome of the clinical reports on vitamin E modulation of asthma in European countries and the United States may, in part, reflect the opposing regulatory functions of α- and γ-tocopherol forms of vitamin E consumed in diets and supplements. Although there are many other differences regarding the environment and genetics of the people in these countries and it is acknowledged that other dietary factors, including unsaturated fatty acids may modulate asthma (11, 126, 148, 189, 194, 266), the clinical data are consistent with the animal studies demonstrating opposing functions of the tocopherol isoforms on leukocyte recruitment (30).
It has also been suggested that changes in environmental factors, including vitamin E consumption, may contribute to the increased incidence of asthma. The incidence of asthma in several countries, including the United States and the Netherlands, has dramatically increased in the last 40 years (95, 292, 300). It is thought that there must be environmental factors contributing to this increase since it is too rapid for genetic changes. The prevalence of asthma is higher in the United States than in Western Europe, Mediterranean countries, Japan, and China (6, 35, 158, 178). The World Health Organization has reported that the prevalence of asthma from 1950 to the present has increased in many countries, including countries with high rates of asthma, intermediate rates of asthma, or low rates of asthma (35). The increases in prevalence occur as countries assume Western lifestyles (35). The dietary changes in the United States in the last 40 years with increased consumption of γ-tocopherol in vegetable oil may, in part, be a contributing factor to changes in asthma prevalence. In addition, in a Scottish cohort, it is reported that reduced maternal intake of vitamin E (likely referring to α-tocopherol) is associated with increased asthma and wheezing in children up to 5 years old (75). Then, in this same report, it was discussed that from 1967 to 2004, there was a significant increase in vegetable oil intake by Scottish (75), which we interpret as indicative of an increase in dietary γ-tocopherol since vegetable oil is rich in γ-tocopherol (Fig. 12). Therefore, since α-tocopherol levels are low in asthmatics (136, 139, 257, 264) and since α-tocopherol can reduce inflammation, an increase in α-tocopherol in the presence of low γ-tocopherol may be necessary to promote optimal health in asthmatics in combination with other regimens to treat inflammation.
The tocopherol isoform levels may also affect the inflammation in other diseases that involve VCAM-1 such as osteoarthritis and atherosclerosis (125, 171, 173, 207, 256). Although there are conflicting reports for tocopherol regulation in these diseases, we suggest that this may result from opposing functions of tocopherol isoforms present in the subjects in these studies. It has been reported that plasma γ-tocopherol is positively associated with osteoarthritis, whereas plasma α-tocopherol is negatively associated with osteoarthritis (134). In contrast, in another report on knee osteoarthritis, vitamin E supplementation (α-tocopherol) did not relieve symptoms but they did not measure α-tocopherol or γ-tocopherol levels (37). With regard to coronary heart disease and stroke, the benefits of tocopherols are also inconsistent among the studies (76, 265); further, measurements of levels of both α-tocopherol and γ-tocopherol are commonly not reported (76, 80, 190, 202, 203, 265). Studies of tocopherols and heart disease are complex since different dietary oils contain not only different forms of tocopherols but also different lipids that affect heart disease. It has been reported that plasma γ-tocopherol levels are not associated with heart disease or in other reports are associated with an increase in relative risk for myocardial infarction [reviewed in (76)]. In contrast, α-tocopherol intake is either not associated with heart disease or, in other reports, is associated with reduced death from heart disease (80, 190, 202, 265). Therefore, although the clinical reports on vitamin E association with heart disease are inconsistent, for those reports with an effect on heart disease, γ-tocopherol is associated with an increase, whereas α-tocopherol is associated with a decrease in parameters of heart disease. Similar to the antioxidant protective function forα-tocopherol on cardiovascular function, the safflower seed-derived antioxidants N-(p-coumaroyl)serotonin and N-feruloylserotinin reduce parameters of cardiovascular disease, including high-glucose-induced VCAM-1 expression and monocyte adhesion, arterial stiffness in hypercholesterolemic rabbits, atherosclerotic lesions in apolipoprotein-deficient mice, and cardiovascular risk factors (sVCAM-1 and oxLDL) in patients (137, 153, 154, 229). In summary, the opposing functions of α-tocopherol and γ-tocopherol in animal models (30) are consistent with the different outcomes for the clinical studies of tocopherols in heart disease. Future clinical studies of vitamin E regulation of inflammatory diseases should include a systematic design to examine opposing functions of the isoforms of vitamin E on inflammation, leukocyte recruitment, and disease parameters.
IX. Concluding Remarks
During several inflammatory diseases, VCAM-1 expression is induced on peripheral tissue endothelium by several mediators, including cytokines or turbulent shear stress, that signal through ROS. Once VCAM-1 is expressed, it is a scaffold on which leukocytes migrate but VCAM-1 also activates signals in endothelial cells that are required for VCAM-1-dependent leukocyte migration (1, 2, 62, 63, 72). Thus, the endothelium plays an active role in the regulation of VCAM-1-dependent leukocyte migration. Crosslinking of VCAM-1 activates calcium fluxes and Rac-1, which then activates endothelial cell NOX2 (a form of NADPH oxidase). Nox2 catalyzes the production of superoxide from oxygen using the cofactor NADPH. The superoxide dismutates to H2O2. Moreover, VCAM-1 induces the production of only 1 μM H2O2 (65, 73, 184), which is a 100–1000-fold lower concentration of ROS than the concentration of ROS produced by leukocytes or that induce oxidative vascular damage. The VCAM-1-induced H2O2 diffuses rapidly activating endothelial cell-associated MMPs and a delayed activation of lymphocyte-associated MMPs. The endothelial-associated MMPs but not leukocyte-associated MMPs are required for VCAM-1-dependent transendothelial migration. These endothelial cell-associated MMPs degrade matrix and endothelial cell surface receptors in cell junctions (9, 260). In addition to the MMP extracellular targets of oxidation, H2O2 diffuses through membranes at 100 μm/s (183) to oxidize and transiently activate endothelial cell PKCα (1). PKCα then phosphorylates and activates endothelial cell PTP1B (1, 72). As PTP1B is not oxidized, it indicates specificity for ROS targets during VCAM-1 signaling. The rapid activation of VCAM-1 signals is consistent with the local rapid process of leukocyte transendothelial migration. Further, these signals through ROS, MMPs, PKCα, and PTP1B are required for VCAM-1-dependent leukocyte transendothelial migration in vitro (1, 57, 72, 73). Both the MMPs and the PKCα/PTP1B pathways contribute to the VCAM-1-dependent leukocyte transendothelial migration, because inhibition of both pathways exhibits a greater inhibitory effect on migration than inhibition of either pathway alone (72). Since VCAM-1 is located on both the apical and lateral surface of endothelial cells, VCAM-1 may continue to provide localized endothelial cell signals as leukocytes migrate through the lateral junctions. The VCAM-1 signaling through NOX2 in endothelial cells also has a role in leukocyte recruitment in vivo since the nonhematopoietic knockout of NOX2 blocks VCAM-1-dependent eosinophil recruitment in allergic lung inflammation with accumulation of eosinophil binding to the luminal surface of venules (2, 30, 140). VCAM-1-dependent leukocyte recruitment is also regulated in vivo by the antioxidants bilirubin and vitamin E isoforms. Moreover, isoforms of vitamin E have novel opposing regulatory functions during VCAM-1-mediated leukocyte recruitment in vivo and in vitro. These opposing regulatory functions of isoforms of vitamin E are consistent with the seemingly disparate outcomes of vitamin E in clinical studies and animal studies of inflammation. The differential tocopherol isoform regulation of inflammation provides a basis toward designing drugs and diets that more effectively modulate these pathways and improve health. Moreover, VCAM-1 signals provide targets for designing approaches to modulate VCAM-1-dependent processes during inflammatory disease.
Footnotes
Acknowledgments
This study was supported by Grants HL069428 and AT004837 from the National Institutes of Health and by the American Heart Association Grant 0855583G.