
Abstract
5′-Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a key sensor of cellular energy status. AMPK signaling regulates energy balance at the cellular, organ, and whole-body level. More recently, it has become apparent that AMPK plays also an important role in long-term decisions that determine cell fate, in particular cell cycle progression and apoptosis activation. Here, we describe the diverse mechanisms of AMPK activation and the role of AMPK in the regulation of cellular energy balance. We summarize recent studies implicating AMPK activation in the regulation of neuronal survival and as a key player during ischemic stroke. We also suggest that AMPK activation may have dual functions in the regulation of neuronal survival: AMPK provides a protective effect during transient energy depletion as exemplified in a model of neuronal Ca2+ overloading, and this effect is partially mediated by the activation of neuronal glucose transporter 3. Prolonged AMPK activation, on the contrary, can lead to neuronal apoptosis via the transcriptional activation of the proapoptotic Bcl-2 family member, bim. Molecular switches that determine the protective versus cell death-inducing effects of AMPK activation are discussed. Antioxid. Redox Signal. 14, 1863–1876.
Compromised Cellular Bioenergetics During Ischemic Conditions
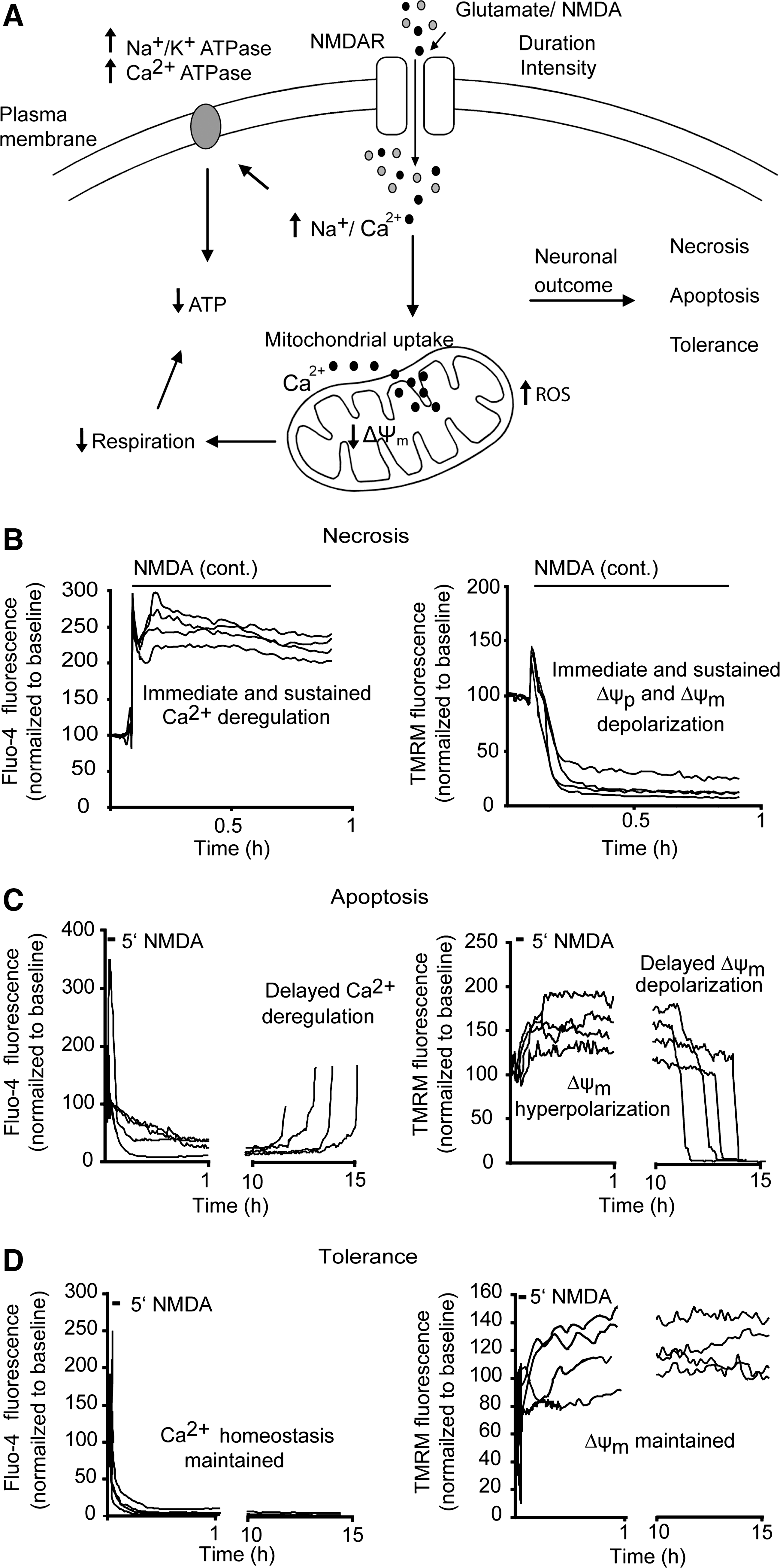
Several in vitro and in vivo models have been employed to understand the molecular events leading to neuronal injury after prolonged glutamate receptor overactivation, with N-methyl-D-aspartate receptors playing the most prominent role in mediating injury (23, 92, 93). The extent and type of neuronal injury induced by glutamate is dependent on the duration and severity of the glutamate exposure (4, 73, 113 –115) (Fig. 1) with clinical studies linking higher plasma glutamate concentrations to larger lesions in stroke patients (19). Prolonged glutamate exposure or high concentrations of synaptic glutamate causes a rapid necrotic injury coupled to irreversible cytosolic Ca2+ overloading (108, 109). Continual uptake of excessive cytosolic Ca2+ by mitochondria is linked to a rapid collapse of mitochondrial bioenergetics (ATP depletion) with immediate Ca2+ deregulation, ionic imbalance, and loss of neuronal integrity (4, 12, 22, 73, 108, 111, 113 –115) (Fig. 1). In contrast, transient glutamate receptor overactivation results in a delayed apoptotic neuronal injury triggered by a secondary failure in mitochondrial bioenergetics, release of proapoptoptic factors from mitochondria, and a secondary failure of neuronal Ca2+ homeostasis termed delayed Ca2+ deregulation several hours postglutamate exposure (4, 13, 26, 65, 73, 83, 84, 113 –115) (Fig. 1). Transient exposure of neurons to glutamate can also lead to induction of neuronal tolerance characterized by full recovery of mitochondrial bioenergetics and cytosolic Ca2+ levels postinitial glutamate insult (113, 116) (Fig. 1). Transient glutamate excitation may more accurately reflect the neuronal damage occurring in the ischemic penumbra, the part of the brain surrounding the ischemic core, and if perfusion is improved, it may have the capacity to recover (53, 90). However, despite the significant evidence linking glutamate release and excitotoxicity to neuronal loss during stroke, several clinical trials utilizing different glutamate receptor antagonists have proved unsuccessful (82).
5′-Adenosine Monophosphate-Activated Protein Kinase: A Primary Stress Sensor in the Control of Cell Metabolism
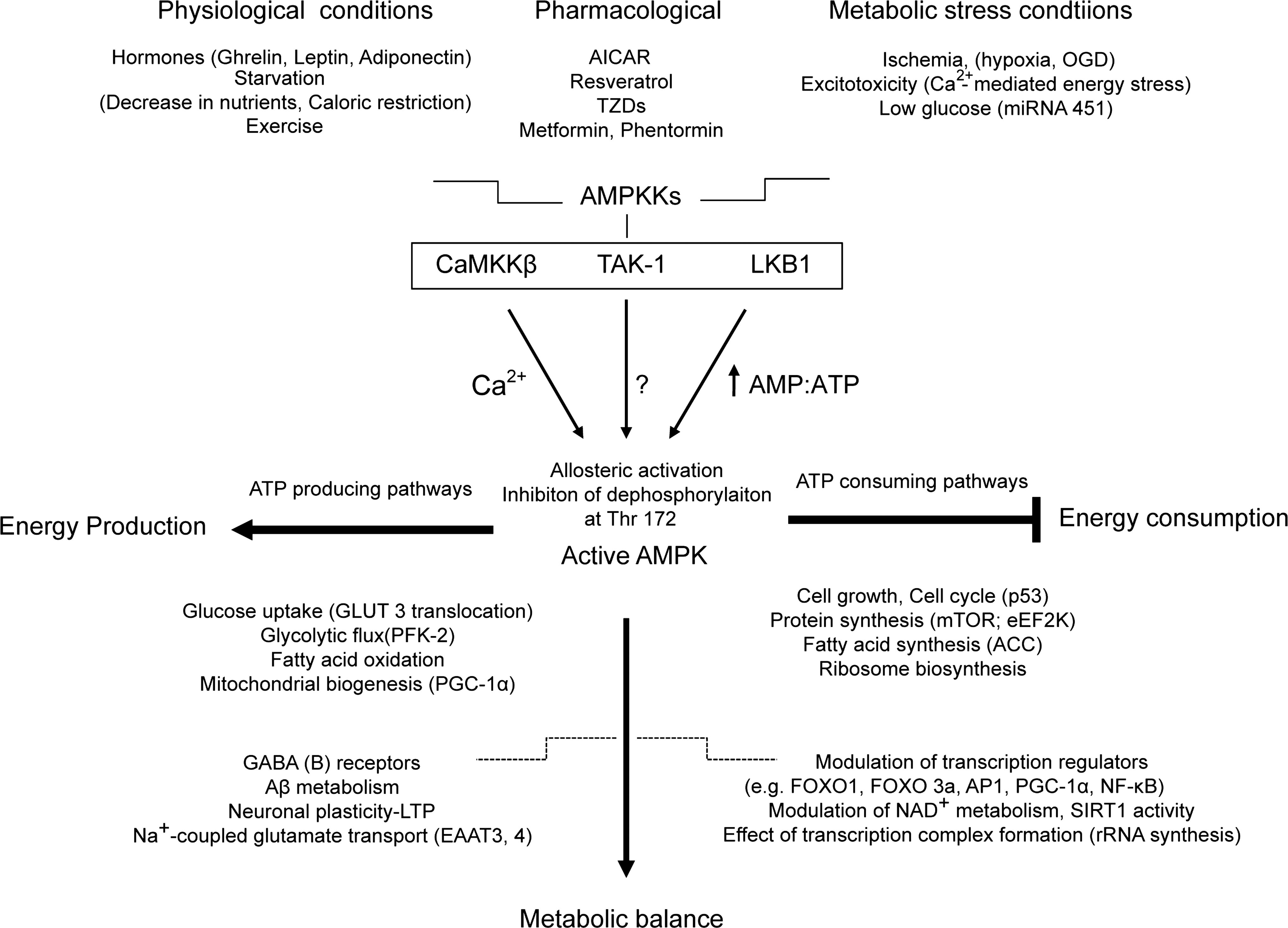
Maintenance of energy homeostasis under physiological or metabolic stress conditions is a fundamental requirement for all living cells. Mitochondrial metabolism in neurons is dynamic and can be altered in response to ischemic events. Increased demands for ATP in energy compromised neurons can lead to acute increase in 5′-adenosine monophosphate (AMP) levels with subsequent activation of cellular energy sensor AMP-activated protein kinase (AMPK) (17, 47) (Fig. 2). AMPK is an evolutionarily conserved kinase that acts as an ancient sensor of cellular energy status. Initially, AMPK signaling was primarily considered as a regulator of energy balance at the cellular level. Recently, it has become apparent that AMPK plays also an important role in the maintenance of the energy balance at the whole-body level as it responds to signals from hormones and cytokines such as leptin, adiponectin, and ghrelin (80). AMPK is activated in conditions of compromised energy availability (decrease in ATP levels coupled with an increase in AMP:ATP ratio) to regulate a wide range of cellular events and to re-establish metabolic balance within the cell (44). Once AMPK signaling is activated, it promotes the rapid phosphorylation of various downstream targets, resulting in inhibition of ATP-consuming anabolic pathways and stimulation of pathways that generate ATP (17, 69, 91) (Fig. 2). Activation of AMPK may have also long-term effects. AMPK may alter gene expression via the modulation of transcription regulators, including forkhead box class O 3a (FOXO3a), peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), and nuclear factor-kappa B, and may have effects on transcription complex formation by adapting rRNA synthesis to changes in cellular energy availability (29, 32, 52, 71, 118) (Fig 2).
The adenylate kinase reaction (2 ADP↔ATP + AMP) is in close equilibrium, making the resultant AMP:ATP ratio a more sensitive indicator of cellular energy conditions than ADP:ATP ratio (46). Thus, during energy demanding conditions when the ATP:ADP ratio in cells is decreased, adenylate kinase potentially amplifies the signal into a higher increase in AMP:ATP ratio (40).
Structure of AMPK and Regulation of AMPK Activation
AMPK exists in cells typically as a heterotrimer with its subunits highly conserved through evolution. This heterotrimeric complex consists of a catalytic α-subunit and two regulatory subunits (β and γ). In mammalian cells each subunit is encoded by a number of distinct genes (α1, α2, β1, β2, γ1, γ2, and γ3) (17, 47) with differential tissue expression. The two α-subunit (α1 and α2) isoforms contain conventional serine/threonine domains, including a Thr-172 residue that is phosphorylated by upstream kinases (50). The β-subunit (β1 and β2) contains C-terminal domains that appear to act as scaffold to form the AMPK complex with α- and γ-subunits (54). The β-subunits also contain a carbohydrate-binding domain (glycogen-binding domain) that allows the AMPK complex to associate with glycogen (54, 76, 77). Recently, the glycogen-binding domain has been proposed to be a regulatory domain that allows AMPK to function as a sensor of cellular reserves of energy in the form of glycogen (76, 77). However, importance of glycogen-binding domain of AMPK for AMPK effects in the nervous system has not been analyzed in detail. The γ-subunits, also referred to as AMP/ATP binding subunits, are responsible for the binding of the regulatory nucleotides. High amounts of ATP antagonize an AMP-mediated activation of AMPK (98).
Activation of AMPK by AMP is complex and proceeds via a two-step mechanism. First, allosteric activation of AMPK occurs by AMP binding to the γ-subunit, which stimulates the kinase activity residing in the α-subunit. Second, binding of AMP to the γ-subunit potentiates the phosphorylation of AMPK at Thr172 residue within the catalytic α-subunit, and this phosphorylation has been shown to be essential for AMPK activity (106). Activation of AMPK by phoshorylation is much more significant than by allosteric activation; however, the combination of both effects may cause >1000-fold increase in AMPK activity (106). Some recent studies emphasized the importance of AMP binding to the γ-subunits (98) in inhibiting the dephosphorylation of Thr172 by protein phosphatase-2Cα, thus maintaining AMPK active (96). This suggests that the effect of AMP on phosphorylation described in previous studies may have been due to a reduction in dephosphorylation rather than increase in phosphorylation (18, 96).
Activation of the AMPK Signaling Cascade and Relevance to the Central Nervous System
AMPK kinases are upstream kinases responsible for phosphorylation and activation of AMPK. Identification of potential upstream AMPK kinases has revealed the existence of at least three potential candidates: LKB1 (serine threonine kinase 11), transforming growth factor-β-activated kinase (TAK-1), and Ca2+/calmodulin-dependent protein kinase kinase (CaMKK) (42, 48, 120, 121). LKB1 is a tumor suppressor kinase that in vitro has been linked to phosphorylation and activation of AMPK (43, 51, 99). In a number of models deletion of LKB1 in specific tissues almost entirely abolished AMPK activity (94, 95), suggesting that the LKB1 complex is the predominant regulator of AMPK activation in several mammalian cell types and tissues (18). It also appears that this complex is constitutively active, contributing to a low level of basal phophorylation on Thr172. Interestingly, increases in the phoshorylation status of LKB1 have been demonstrated in ischemic brain (70). MicroRNA-451 has recently been identified as a new regulator of the LKB1/AMPK pathway in glioma cells under conditions of energetic stress (34). TAK-1, a member of mitogen-activated protein kinase kinase family, has been identified as a functional member of the AMPK kinase family that also phosphorylates Thr172 in vitro (81). The exact role of TAK-1 in the control of AMPK activity, in particular in the central nervous system, warrants further investigation.
Increases in intracellular Ca2+ trigger the activation of Ca2+-sensitive enzymes, including a family of Ca2+/calmodulin-dependent, serine/threonine protein kinases, the CaMKKs (100). CaMKKs may act as alternative upstream AMPK kinases (18, 49, 120). Studies in rat brain tissue reported that AMP promotes the phosphorylation of AMPK by activating CaMKKs (50). Recent studies reported that CaMKKβ, rather than CaMKKα, is the major isoform of the kinase that mediates the effects on AMPK (49, 120). CaMKKβ is activated by increased levels of cytosolic Ca2+; AMPK may be activated physiologically to prepare the cell for an increased metabolic demand (18). This may also represent a beneficial mechanism in cells exposed to pathophysiological Ca2+ overloading. In this context, a recent study demonstrated stimulation of CaMKKβ and AMPK activity after administration of kainic acid into the hippocampus (66).
Physiological and Pharmacological Activation of AMPK
Under physiological conditions, AMPK is an important mediator of metabolic responses to exercise in skeletal muscle (39). The activation of AMPK inhibits ATP-consuming processes and activates carbohydrate and fatty acid metabolism to restore ATP (58). AMPK downregulates biosynthetic pathways such as fatty acid and cholesterol biosynthesis, protein synthesis, and glycogen synthesis, and upregulates pathways such as fatty acid oxidation, glycolysis, glucose uptake, and mitochondrial biogenesis (42). Physiological modulation of AMPK activity by signals from hormones (leptin, adiponectin, and ghrelin) that control food intake and starvation (fasting) has also been described, and emphasize the important role of AMPK in the maintenance of the energy balance at the whole-body level (3, 79) (Fig 2).
Given that AMP is the allosteric activator of AMPK, analogs of AMP have been developed that are effective AMPK activators. The AMP analog 5-aminoimidazole-4-carboxamide riboside (AICAR) is a valuable experimental tool for the modulation of AMPK function both in vitro and in vivo (38). AICAR is taken up into the cell by adenosine transporters (33) and converted by adenosine kinase into the monophosphorylated nucleotide, zeatin ribose-5-monophosphate, which mimics effects of AMP binding on AMPK (27). Although AICAR has been an important experimental tool in the activation of AMPK, it has several limitations due to its lack of specificity and low potency (5). In addition, the zeatin ribose-5-monophosphate product mimics the effects of AMP such as stimulation of other AMP-sensitive enzymes and may alter the function of adenosine receptors and transporters (33). Therefore, it has been proposed that the use and interpretation of data obtained in neurons from experiments with AICAR should be not considered as conclusive and should be re-confirmed by other molecular approaches (33).
Pharmacological Activation of AMPK and Future Prospects
Given the central role of AMPK as a metabolic sensor, it has become a pharmacological target for various diseases involving conditions of metabolic dysregulation. Of note, different currently used pharmacological agents have been reported to activate AMPK. These include drugs used for the treatment of type 2 diabetes, in particular thiazolidinediones and metformin. Thiazolodinediones have multiple pharmacological actions, including the inhibition of complex I activity of the mitochondrial electron transport chain. These drugs hence have the ability to indirectly activate AMPK by increasing the AMP:ATP ratio (39). Metformin is selectively taken up into the liver and is believed to activate AMPK in a nucleotide-independent manner (17). Natural compounds extracted from various plants that have healthy-giving properties such as resveratrol (present in grapes and red wine), berberine (present in oregon grape and barberry), and (−) epigallocatechin-3-gallate (present in green tea) may also stimulate AMPK activity; however, their exact mechanisms of action are not yet fully understood (7, 55, 68). Notably, the results obtained from studies in which pharmacological agents activating AMPK have been used should be interpreted with caution because of various off-target effects mediated in an AMPK-independent manner. Therefore, the development of alternative agents that are effective and act directly on AMPK can help to clarify the therapeutic potential of this metabolic sensor in the treatment of stroke.
AMPK Activation in Cerebral Ischemia and Excitotoxicity
Various metabolic, traumatic, or toxic insults to the nervous system have the capacity to activate AMPK, including cerebral ischemia, hypoglycemia, or exposure to metabolic toxins (17, 40, 42). The α1/2, β1/2, and γ1-subunits of AMPK are highly expressed in neurons (107). Expression of AMPK subunits is less abundant in astrocytes, suggesting that the main role of AMPK in the central nervous system is restoring energy levels within neurons.
AMPK activity has been shown to increase during glucose deprivation, metabolic stress, ischemia, and hypoxia in neurons both in vivo and in vitro (28, 33, 78). AICAR has been shown to be neuroprotective in some models of cerebral ischemia (25). It would appear that AMPK activation for a limited period promotes the recovery of neuronal energy level, for example, by increasing glucose utilization (116). These findings are supported by a study demonstrating that AMPK activation with low concentrations of AICAR (0.1 mM) is neuroprotective against glucose deprivation, metabolic, excitotoxic, and oxidative insults (28). Importantly, this protection after incubation of primary neurons with low concentrations of AICAR correlated with a transient increase in AMPK activity (28). A study by Moss and coworkers suggested that AMPK binds directly to GABAB receptors and phosphorylates S783 in the cytoplasmic tail of the R2 subunit, which in turn may potentially stimulate GABA-dependent inhibition of presynaptic Ca2+ channels by inhibiting presynaptic neurotransmitter release and/or promote activation of K+ channels by blocking postsynaptic depolarization in neurons under metabolic stress (45, 64). Activation of AMPK has also been shown to occur in studies of ischemic preconditioning in both the liver and the heart after energetic stress (85, 86, 105); however, preconditioning studies in neurons have to our knowledge not yet been performed.
Nevertheless, there is a paradox concerning AMPK activation in mediating neuronal tolerance or apoptosis (28, 69, 78, 87, 91, 103). In contrast to the AMPK protective effect, some studies recognized that prolonged activation of AMPK can be detrimental via the activation of cell death machinery (15, 61). Moreover, the pharmacological inhibition or gene deletion of AMPK can be neuroprotective in stroke (70, 78). The discrepancies between in vivo and in vitro studies elucidating the role of AMPK in neuronal survival (28, 78) could be due to differences in glucose concentrations, which has been previously shown to be critical for determining neuronal survival and the levels of AMPK activation (62, 91). Therefore, by using the similar metabolic conditions our laboratory has further explored the protective versus cell death-inducing effects of AMPK activation in neurons exposed to metabolic stress, as exemplified in a model of excitotoxic Ca2+ overloading.
Transient Glutamate Exposure Results in Rapid and Transient AMPK Activation with an Increase in Glucose Transporter 3 Trafficking That Mediates Neuroprotection
Transient exposure of primary neurons to excitatory neurotransmitter glutamate can lead to a delayed apoptotic neuronal injury; however, many neurons exposed to the same excitotoxic insult can tolerate the event with their survival linked to an alteration in mitochondrial bioenergetics (hyperpolarization of ΔΨm) (113, 116) (Fig. 1). In line with previous studies, primary neurons (murine neocortical and cerebellar granule neurons) exposed to a 5 or 10 min stimulation with N-methyl-D-aspartate or glutamate had populations of neurons that underwent apoptotic injury (50%–60% of neurons having pycnotic nuclei), whereas the rest of the neurons in the same population of cells could tolerate the excitotoxic insult (26, 114 –116). Although several signaling pathways have been implicated in the excitotoxic process, it appears now that a loss of mitochondrial function and a rapid decrease in neuronal ATP levels are key elements in dictating neuronal outcome to an excitotoxic event (1, 6, 26, 113, 116). Depletion of cellular ATP levels leads to an acute increase in AMP:ATP ratio, which in turn triggers the activation of the energy sensor AMPK (17, 48). Indeed, we have previously described a rapid increase in AMPK phosphorylation (26, 116) at early time points (30–60 min) postexcitotoxicity (Fig. 3A). Of particular note is that the duration of AMPK activity was transient in nature with a detectable decrease in its activity at 4 h postexcitation (Fig. 3A). We have also previously described an inverse correlation between cellular ATP levels and AMPK activity, with significant depletion of ATP levels evident within 5 min of glutamate stimulation, persisting for up to 30–60 min, and coupled to both an accumulation of AMP and an activation of AMPK (26, 116). Further, this adaptive response of ATP recovery was coupled to increased glucose uptake and NAD(P)H availability in primary neurons postexcitation (113).

Given that increased glycolytic flux can be particularly important as energy compensatory mechanism when mitochondrial function is compromised (72, 113, 116), we further investigated glucose regulation in response to the energy crisis initiated by transient glutamate receptor overactivation. Neuronal glucose uptake is facilitated by a family of glucose transporters (GLUTs), with the GLUT 1 and 3 isoforms considered as major GLUTs within the brain (101). Moreover, the GLUT 3 isoform has a higher affinity for glucose and greater transport capacity in comparison to other GLUTs (101) and its abundance in primary neurons has been previously demonstrated by our group (116). Employing immunofluorescence analysis, a significant increase in GLUT 3 expression at the plasma membrane was detected, with maximal expression levels identified 60 min after the excitation event (116) (Fig. 3B). The potent role of AMPK signaling in the regulation of glucose transport has been highlighted in a number of models of energetic stress (2, 24, 37, 74, 116). Glutamate-induced GLUT 3 activation was inhibited by gene silencing of the AMPK α 1/2 subunit and by the AMPK inhibitor, compound C (116). To see if the activation of AMPK had a direct effect on the trafficking of GLUT 3 to the plasma membrane in primary neurons, we exposed the neurons to AMPK agonist AICAR. In a line with our previous work, an early increase in GLUT 3 cell surface expression could be identified in neurons incubated with AICAR (116) (Fig. 3C). These data indicate that the short-term pharmacological stimulation of AMPK activity is sufficient to increase GLUT 3 trafficking to the plasma membrane in neurons. Previously, we defined a correlation between hyperpolarization of mitochondria and neuronal survival postexcitotoxicity (113). Increased translocation of GLUT 3 to the cell surface results in increased glucose transport, thus enhancing the glycolytic capacity providing mitochondrial substrates [NAD(P)H, FADH2] for ATP production. This may coincide with elevations in ΔΨm mediated by the increased proton efflux. In this context, gene silencing of GLUT 3 reduced the mitochondrial hyperpolarization in response to glutamate, and potently sensitized neurons to excitotoxic injury (116), indicating that GLUT 3 activation by AMPK exerts a neuroprotective response.
Delayed Apoptotic Injury in Response to Glutamate Receptor Overactivation Is Associated with AMPK-Mediated Activation of Proapoptotic BH3-Only Protein Bim
Primary neurons have proven to be a very versatile in vitro model for the characterization of the molecular events that lead to delayed apoptotic neuronal injury in response to an excitotoxic stimulus (4, 13, 65, 73, 113, 115). On further investigation of the molecular events that underlie many of the physiological processes, we recently identified that the Bcl-2 homology domain 3 (BH3)-only protein Bim was a central factor in coupling a sustained energetic stress to the induction of neuronal apoptosis (26). The expression levels of the Bim protein (Fig 4A) are significantly increased at a time point postexcitation (16 h) when most primary neurons underwent an apoptotic cell death, and gene knockout or gene silencing of bim protects against excitotoxic apoptosis, delayed mitochondrial depolarization, and delayed Ca2+ deregulation (26).

Prolonged AMPK Activation Triggers bim-Dependent Apoptosis in Neurons
Indeed, further studies indicated that prolonged activation of the energy sensor AMPK is detrimental and sufficient for upregulation of Bim. To address the paradox and potential dual role that AMPK may play in cell death or cell survival, we chronically stimulated AMPK with the agonist AICAR. A chronic exposure of primary neurons to the AMP mimetic AICAR resulted in a continuous activation of AMPK (increase in the phosphorylation state at Thr 172) demonstrated by Western blot (26, 116) (Fig. 4C) with a delayed apoptotic neuronal evident injury 48 h post-AICAR addition (116) (Fig. 4B). As the pharmacological activation of AMPK with AICAR was reported to be nonspecific (33, 38), we also employed the constitutively active AMPK-α1 (CA-AMPK) plasmid to examine this phenomenon (26, 119). In a similar fashion to the AICAR response (Fig. 4B), the long-term overexpression of the CA-AMPK induced significant cell death (over 50%) within 24–48 h time frame (26) (Fig. 5A).
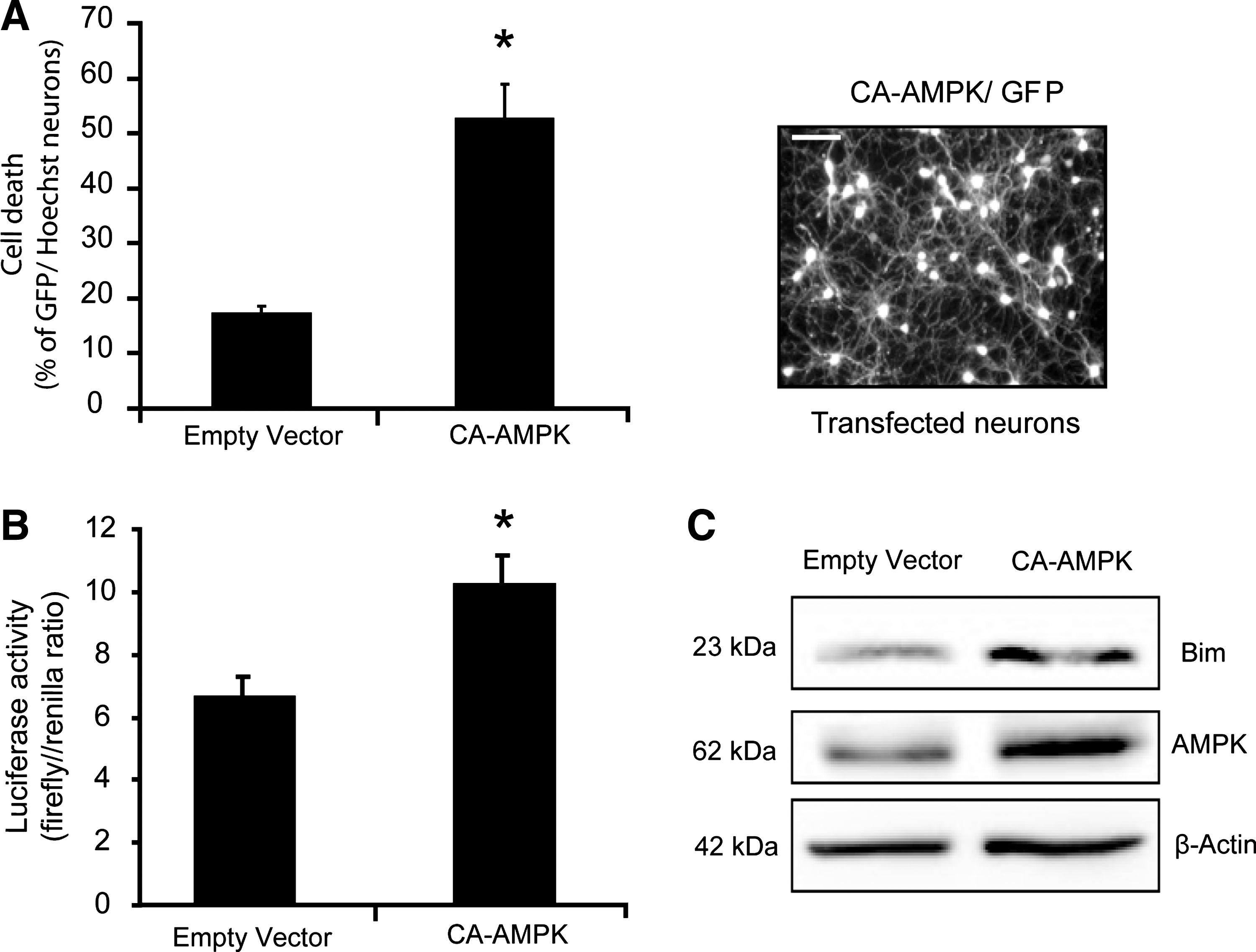
We also examined if Bim played a significant role in mediating the apoptotic neuronal injury associated with chronic AICAR stimulation or CA-AMPK expression (26) (Figs. 4C and 5B, C). We detected a marked increase in Bim expression evident after chronic AICAR exposure and CA-AMPK overexpression (Figs. 4C and 5C). bim gene deletion also rescued neurons against the apoptosis-inducing effects of prolonged CA-AMPK overexpression (26). Reporter studies confirmed increased bim promoter activity after prolonged CA-AMPK overexpression (Fig. 5B).
Molecular Mechanisms That May Determine Cell Survival and Cell Death Responses After AMPK Activation
Previous findings as well as findings from our group suggested that AMPK activation may exert both protective and cell death-inducing effects in neurons (26, 28, 64, 69, 78, 87, 91, 103, 116). Our data indicate that AMPK signaling has a dual role in neurons (Fig. 6) with early activation regulating GLUT 3 trafficking, glucose uptake, and energy recovery, and with the prolonged activation associated with direct induction of Bim and the activation of apoptotic pathways. Thus, the duration of the AMPK activity as a consequence of the energy deprivation induced after an insult may be pivotal in the life-or-death decisions within a cell.
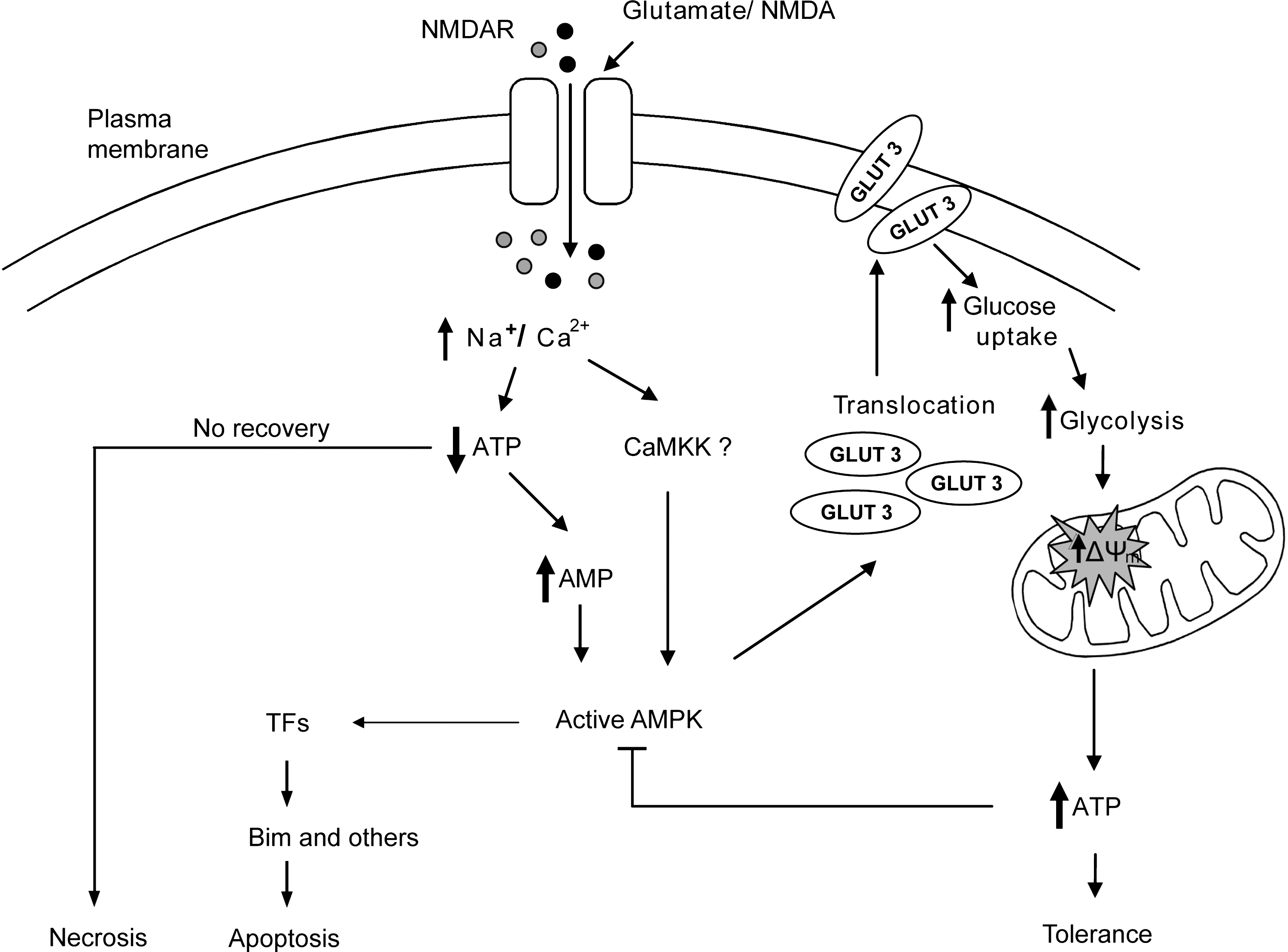
To address the paradox concerning the role of AMPK activation in cell death and cell survival, we employed the pharmacological activator AICAR (Fig. 4) or overexpression of CA-AMPK (Fig. 5) to stimulate AMPK in primary neurons. In line with previous studies in non-neuronal cells (89), we observed that the chronic exposure of cells to AICAR or prolonged overexpression of a CA-AMPK-α1 isoform induced a prolonged increase in the phosphorylation state of AMPK, and was sufficient to lead to a progressive loss of neuronal viability that was associated with increased bim promoter activity. These results raised the question as to the signaling pathways that are modulated after chronic AMPK activation to stimulate bim gene expression. AICAR studies in non-neuronal models demonstrated a correlation between chronic activation of AMPK and the c-Jun N-terminal kinase (JNK) (59, 124), a stress response kinase that also stimulates Bim expression (89). Both excitotoxic injury and AMPK have been shown to increase activator protein 1 (AP1) activity (26, 30), one of the transcription factors that control bim expression in neurons (9, 117). Supporting a downstream role of JNK in this scenario, AMPK can stimulate JNK phosphorylation in HepG2 cells (67). However, AMPK is also able to inhibit JNK activity in other cell lines (97, 123), illustrating that regulation of JNK by AMPK is complex and may depend on cell type and stress stimulus.
Biswas and coworkers showed that bim promoter activation in neurons is highly complex, depending on the simultaneous binding to its promoter of three different transcription factors: AP1, FOXO3a, and c-Myb. c-Myb activation is associated with an inappropriate start of the neuronal cell cycle (35), but there is current no experimental evidence for an interaction between AMPK and c-Myb. However, several lines of evidence indicate that FOXO3a can be regulated by AMPK to induce bim expression. FOXO3a inactivation depends on its phosphorylation by the prosurvival Akt kinase (11, 14). AMPK activation can downmodulate Akt activity by controlling IRS-1 (110), the upstream kinase complex mammalian target of rapamycin C (mTORC) (10), and phosphatase PP2A (60). This suggests that AMPK could activate FOXO3 dephosphorylation, its subsequent nuclear translocation, and the activation of bim gene expression. This hypothesis is in line with findings in models of excitotoxicity and metabolic stress that are associated with AMPK activation and a downmodulation of Akt activity (20, 110). Besides this regulation, several studies have shown that AMPK can control FOXO3 transcriptional activity, by direct phosphorylation (36), or by stimulation of its deacetylation (16). In addition AMPK may activate the transcription factor p53 (57), which can also modulate FOXO3 activity (14).
From our findings it is evident that the duration of AMPK activity may determine whether AMPK exerts protective or cell death-inducing effects on neurons. Molecular switches must allow AMPK to activate different downstream effectors to trigger GLUT 3 translocation as a protective response (among other protective responses), or to induce bim gene expression. Stress conditions, including prolonged energy depletion, have been shown to increase levels of AMPK-α1/α2 and -β1/β2 in the nuclei (63). It is therefore possible that nuclear presence of AMPK may depend on the duration of its activation, and will eventually enable the above-described transcriptional regulation of the bim gene. Nuclear presence of AMPK may control the binding of specific transcription factors to their corresponding DNA promoters. In line with this hypothesis, AMPK can control the DNA-binding of transcription factors by direct phosphorylation, such as in the case of FOXO3a or AREBP (36, 41, 56), or by phosphorylation of their coactivators, such as p300 (41, 122).
We should also consider that AMPK heterotrimers can be constituted from different isoforms of the AMPK subunits (α1, α2, β1, β2, γ1, γ2, and γ3) (104). α1 and α2, which hold the AMPK catalytic domain (41), could have specific affinity for different substrates. There is also evidence for different expression patterns and cellular localization (107), which could allow for stress- and cell type-dependent effects of AMPK activation.
Conclusion
New data have emerged supporting the concept that AMPK activation, in particular the duration of its activation, is a pivotal factor in the decision between cell death and cell survival signaling in neurons subjected to metabolic stress, such as cerebral ischemia. However, additional in vivo studies and translational work is necessary to carefully validate the dual role of AMPK in controlling neuronal survival during and postischemia. Therefore, both the modulation of AMPK activity in neurons and its downstream effects warrant further investigations.
Footnotes
Acknowledgments
This study was supported by grants from Science Foundation Ireland (08/IN1/1949), the Health Research Board (RP/2006/333), and the European Union FP7 (Marie Curie Intra-European Career Development Fellowship PIEF-GA-2009-237765).