
Abstract
Peroxiredoxins constitute a major family of cysteine-based peroxide-scavenging enzymes. They carry an intriguing redox switch by undergoing substrate-mediated inactivation via overoxidation of their catalytic cysteine to the sulfinic acid form that is reverted by reduction catalyzed by the sulfinic acid reductase sulfiredoxin (Srx). The biological significance of such inactivation is not understood, nor is the function of Srx1. To address this question, we generated a mouse line with a null deletion of the Srx1-encoding Srxn1 gene. We show here that Srxn1 −/− mice are perfectly viable and do not suffer from any apparent defects under laboratory conditions, but have an abnormal response to lipopolysaccharide that manifests by increased mortality during endotoxic shock. Microarray-based mRNA profiles show that although the response of Srxn1 −/− mice to lipopolysaccharide is typical, spanning all spectrum and all pathways of innate immunity, it is delayed by several hours and remains intense when the response of Srxn1 +/+ mice has already dissipated. These data indicate that Srx1 activity protects mice from the lethality of endotoxic shock, adding this enzyme to other host factors, as NRF2 and peroxiredoxin 2, which by regulating cellular reactive oxygen species levels act as important modifiers in the pathogenesis of sepsis. Antioxid. Redox Signal. 14, 2071–2080.
Introduction
Typical 2-Cys Prxs carry an intriguing redox switch, undergoing peroxide-mediated inactivation via overoxidation of CysP to the sulfinic acid (R-SO2H) form (11, 41, 42) that is reverted by a slow ATP-dependent enzymatic process catalyzed by the sulfinic acid reductase sulfiredoxin (Srx) (4, 10). Inactivation only occurs during enzymatic cycling and is proportional to the amount of substrate at both non-saturating and saturating conditions. The biological significance of Prx reversible inactivation is not understood. The observation that inactivation is an attribute of eukaryotic but not prokaryotic enzymes has led to suggest that it is an acquired gain of function selected for regulatory purposes (42). Only typical 2-Cys Prx are overoxidation sensitive because they carry an additional helix absent in overoxidation-insensitive enzymes, which slows down the rate at which CysP–SOH condensates with Cys residue, allowing its further oxidation by H2O2 (42). It has thus been proposed that Prx inactivation could act as a “floodgate” that restricts scavenging to the low levels of endogenously produced H2O2, but allows higher levels of H2O2 to signal (42). In S. pombe, the Prx enzyme Tpx1 is the H2O2 receptor that activates the H2O2-stress response regulator Pap1 by catalyzing its oxidation, and its reversible overoxidation constitutes the mechanism that inhibits Pap1 activation when H2O2 levels are high (6, 37). In mammals, however, despite many observations of the involvement of Prxs in H2O2 signaling, the existence of a Prx H2O2 floodgate has never been validated, and no data of a biological utility of Prx inactivation has yet been provided.
Srx appears to be the only known protein capable of catalyzing sulfinic acid reduction; in mammals, Sestrins were also reported to have such activity (9), but a re-examination of their enzymatic properties did not confirm this finding (38). Srx sulfinic reductase activity is dedicated to overoxidized 2-Cys Prxs, and is not known to use any other substrate (39). In yeast, plants, and mammals, a unique gene encodes Srx.
In an effort to understand why are 2-Cys Prx enzymes reversibly inactivated by overoxidation by their substrate, and to gain knowledge of the in vivo functions of mammalian Srx, we generated a mouse line with a null deletion of the Srx-encoding Srxn1 gene. We report here the description of this knockout mouse line. We show that, surprisingly, loss of Srx does not produce any overt phenotype under laboratory growth conditions. Reactive oxygen species (ROS) are thought to have crucial effects during sepsis by both regulating immune cell activation and contributing to tissue injury (1, 22, 26, 36). Further, mice lacking Prx2 (44), Prx3 (25), or the oxidative stress response-regulator NRF2 (34) share the phenotype of a decreased tolerance to sepsis. As Prxs sulfinylation and its reactivation by Srx1 might become important for either mitigating peroxide stress and/or regulating peroxide signaling, we explored the tolerance of srxn1 −/− mice to lipopolysaccharide (LPS)-induced endotoxic shock. We show that loss of Srx causes an abnormal response to LPS, amplifying the innate immune response and decreasing the tolerance to endotoxic shock. Srx1 thus adds to Prx2, Prx3, and NRF2 as host factors that by regulating cellular ROS levels act as important modifiers in the pathogenesis of sepsis.
Materials and Methods
Generation of Srx−/− mice
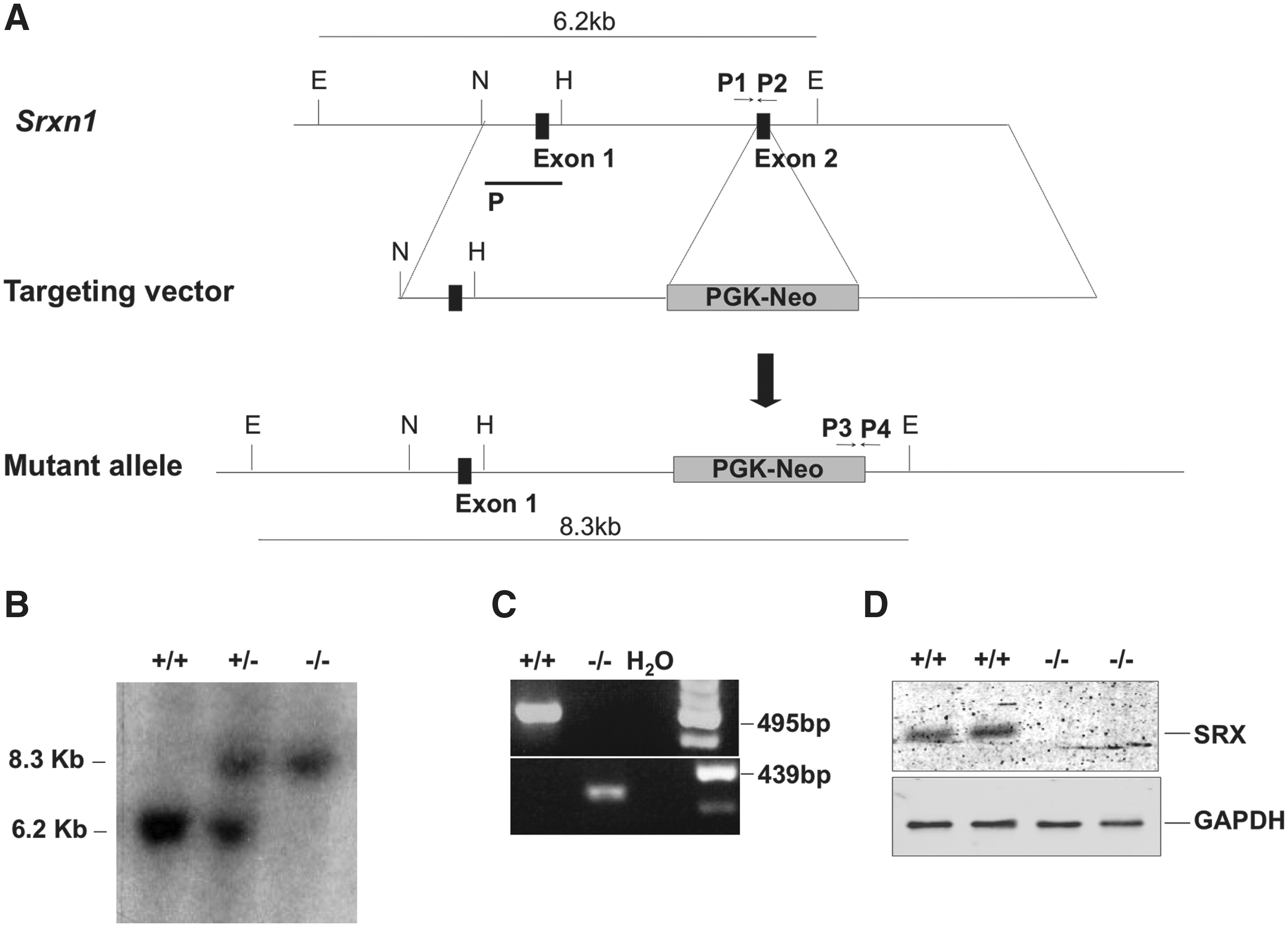
A 3.7 kb fragment 5′ to exon 2 of the Sxrn1 gene locus was polymerase chain reaction (PCR)-amplified from mouse genomic DNA and cloned between the Nhe1 and EcoR1 sites of PSJ4. Downstream to this fragment, a PCR-amplified 3.4 kb genomic fragment 3′ to exon 2 was inserted between the Not1 and Xho1 sites. A 2.1 kb fragment containing a Neomycin-resistance cassette (Neo) was then subcloned at the Not1 site of PSJ4 to generate the targeting vector (Fig. 1A). The vector was introduced into AT-1 mouse embryonic stem (ES) cells (a derivative of 129Sv) (a gift from M. Vernet) (8) by electroporation, and G-418-resistant recombinants containing the mutated allele in one of the Srxn1 loci were identified by Southern blot and PCR (Fig. 1A). ES recombinant clones were injected into blastocysts embryos of C57BL/6 mice to produce chimeric mice, which were subsequently bred with C57BL/6 mice to produce F1 Srxn1+/− heterozygotes. These F1 mice were either mated with each other to produce F2 Srxn1 −/− homozygote mice or were further backcrossed with C57BL/6. One Srxn1 −/− mouse line was established after both four and seven backcrosses. DNA genotyping was performed on mouse-tail biopsies by Southern blot using a 949 kb NheI to HindIII probe spanning Srxn1 exon 1 (Fig. 1A, B), and by reverse transcription-PCR using primers P1, P2, P3, and P4 (Fig. 1A, C). All mice were fed with standard mouse chow and houses in a pathogen-free facility. Animal procedures complied with the guidelines of the French ethics committee for animal research.
Cell cultures
Mouse embryo fibroblast cultures were established by harvesting embryos at embryonic day 13. Head and liver were removed. The remaining embryonic tissue was torn with scissors in phosphate-buffered saline (PBS) containing trypsin (GIBCO–Invitrogen), and incubated 5 min at 37°C. The cells in the supernatant were plated in Dulbecco's modified Eagle's medium + 10% fetal calf serum (FCS), 1% glutamine, 1% penicillin and streptomycin (GIBCO–Invitrogen), and cultured at 37°C in 5% CO2. Cultures were passed 1:3 after reaching confluence and frozen in liquid nitrogen in DMSO + 10% FCS. Cultures were discarded after reaching passage 7. Mouse bone morrow-derived macrophages (BMDMs) were established as described (13), and grown in RPMI + 10% FCS, 2 mM glutamine, and 10% L-929-derived conditioned medium.
Monitoring ROS levels by fluorescence
Mouse BMDMs that were left untreated or were incubated during 3 h with N-acetylcysteine (10 mM) or during 30 min with H2O2 (100 μM) were washed with Ca2+- and Mg2+-free PBS, detached using trypsin-EDTA, washed again, and suspended in PBS containing 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA; 10 mM; Molecular Probes, Invitrogen), and incubated at 37°C for 30 min. ROS levels were indirectly measured by CM-H2DCFDA fluorescence emission on a Beckman Coulter Cytomics FC500 flow cytometer.
Western blot analyses
Cells were washed on ice with PBS, and lysed in RIPA buffer (Tris-HCl pH 8 [50 Mm], NaCl [300 mM], triton X-100 [1%], and EDTA 1 mM). Centrifuged, cleared lysates were diluted with 1/3 final volume of 3× loading buffer (Tris-HCl pH 6.8 [0.2 M], glycerol [45%], SDS [6%], β-mercaptoethanol [6% v/v], and bromophenol blue [0.03%]). After heat-denaturation, proteins were separated by SDS-PAGE, transferred onto a nitrocellulose membrane, and immuno-stained in blocking buffer ODYSSEY (Li-Cor Biosciences) with polyclonal rabbit antibodies specific for Srx (1/2000) (10), the SO2H/SO3H forms of mammalian 2-Cys Prxs (40) (gifts from Dr. S. G. Rhee), or GAPDH (1/10000; AMBION). Membranes were stripped with the NewBlot Nitro Stripping buffer (Li-Cor Biosciences) when needed. Detection was performed after chromophore-coupled secondary-antibody staining, using the LI-COR Odyssey infrared imager.
Treatments
Endotoxic shock was induced by intraperitoneal (IP) injection of LPS (Escherichia coli, serotype 0111:B4, lot number 028K4090; Sigma) at a dosage of 10–100 μg/g of body weight in wild-type and srxn1 −/− mice that were matched for age (10–16 weeks) and gender. PBS (20 μl/g) was injected in control animals. Survival was monitored by inspection every 4 h for 5 days. Data were expressed as means ± standard error of the mean. Blood was withdrawn from the orbital sinus of anesthetized animals and collected in tubes containing EDTA (2 mg/ml blood) and serum collected after centrifugation. Alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine, creatine kinase, glucose, albumin, bilirubin, lactate dehydrogenase, lactate, and alkaline phosphatase were measured on an Olympus AU400 (CEFI, Institut Claude Bernard). Blood analyses (white blood cells and red blood cell counts, hematocrit, hemoglobin, erythrocyte indexes—Mean Corpuscular Hemoglobin, Mean Corpuscular Hemoglobin Concentration, Mean Corpuscular Volume—, platelets) were performed at the Mouse Clinical Institute.
Histological analyses
Skin, mammary glands, heart, aorta, trachea, lungs, pleura, bone marrow, spleen, thymus, mesenteric lymph nodes, esophagus, stomach, small and large intestines, salivary glands, liver, pancreas, kidney, urinary bladder, ovaries, testis, muscle, brain, and eyes were collected from euthanized mice and processed for routine macroscopic and microscopic histological analysis to detect and analyze systematically organ defects and tissue alterations in mice. Organs were fixed in formalin or Bouin's fluid (testis) and decalcified (bones, whole head). A sample of each organ was embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. Analyses were performed at the Mouse Clinical Institute (ICS).
RNA sample preparation for microarray analyses
Groups of three male wild-type or srxn1 −/− mice matched for age (12 weeks old) were used. Mice were subjected to a nonlethal IP injection of LPS at dosage of 12 μg/g of body weight or of PBS (20 μl/g) (control animals). Lung and spleen were isolated 6 and 20 h after LPS challenge, and 6 h after PBS injection. Total RNA was extracted using the RNeasy Mini kit (Qiagen), according to the manufacturer's recommendations. RNA sample quality was evaluated by capillary electrophoresis using the Agilent RNA 6000 Nano Chip kit, and its concentration measured by its absorbance at 260 nm on a NanoDrop 1000 spectrophotometer. To generate cRNAs, 2 μg of RNA was used to generate first-strand cDNAs using a T7-oligo(DT) primer and Superscript II Reverse Transcriptase (Affymetrix). Second-strand synthesis used DNA ligase and DNA polymerase I, Rnase H, and then T4 DNA polymerase. Double-stranded cDNA was cleaned up using the GeneChip Sample Cleanup Module (Affymetrix). In vitro transcription was carried out using T7 RNA polymerase and a biotinylated nucleotide analogue/ribonucleotide mix for cRNA amplification and biotin labeling (GeneChip IVT Labeling Kit). Biotinylated cRNAs were cleaned up using the GeneChip Sample Cleanup Module, quantified, and fragmented into a mean size of 35–200 nucleotides by incubation at 94°C for 35 min in fragmentation buffer (Affymetrix).
mRNA profiling
mRNA profiling was performed on 12 groups of 3 replicates each (36 samples): PBS control, 6 h after injection, in wild-type and srxn1
−/− mice, spleens and lungs (12); LPS, 6 h after injection, in wild-type and srxn1
−/−, spleen and lungs (12); LPS, 20 h, in wild-type and srxn1
−/−, spleen and lungs (12). It used GeneChip mouse genome 430 2.0 arrays (Affymetrix) that cover ∼34,000 well-characterized mouse genes. Fragmented cRNA were hybridized onto arrays for 16 h at 45°C together with internal hybridization controls (bioB, bioC, bioD, cre, and oligonucleotide B2). Washing and staining procedures were performed in the Affymetrix Fluidics Station 450. Probe arrays were exposed to 10 washes in nonstringent wash buffer A (6× SSPE, 0.01% Tween20) at 30°C, followed by 6 washes in stringent buffer B (100 mM MES, 0.1 M [Na+], and 0.01% Tween20) at 50°C. Biotinylated cRNA were stained with a streptavidin–phycoerythrin conjugate (SAPE, 10 μg/ml) and washed again 10 times with buffer A. An amplification step was performed using goat IgG (100 μg/ml), and then biotinylated with antistreptavidin antibody (3 μg/ml), each followed by an additional SAPE staining (5 min at 35°C). Arrays were then washed 15 times in buffer A at 35°C before scanning using the Affymetrix GeneChip Scanner 3000. Probe-level expression data (CEL files) were finally produced using the GeneChip® Operating Software (GCOS). Quality-control and statistical analyses (background adjustment, normalization, and probe-level summarization of data) used the GC-Robust Multi-Array average algorithm (GC-RMA) and the statistical packages tool Bioconductor (
Results
Generation of Srxn1−/− mice
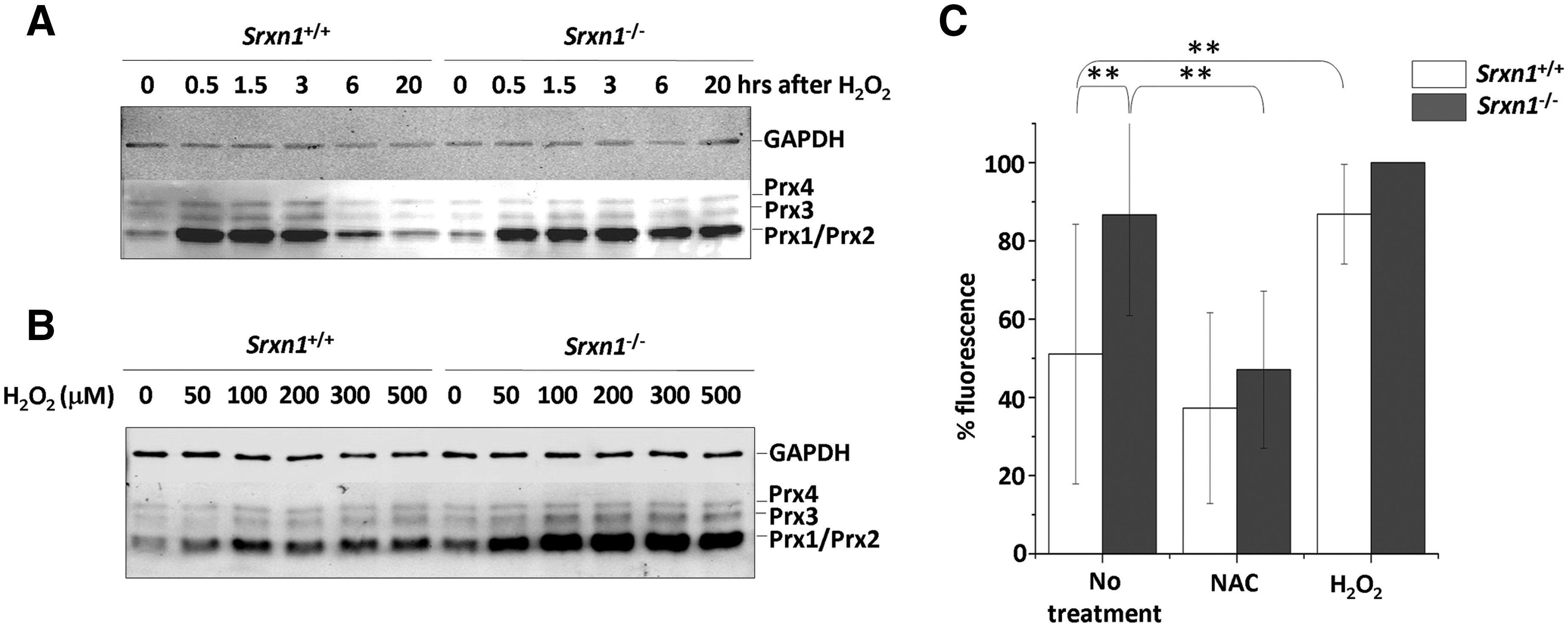
In an effort to understand the physiological function of Srx, we generated a null allele of Srxn1 in AT-1 ES cells by replacing exon 2 that contains the Srx catalytic domain, with a neomycin-resistance (Neo) gene cassette (Fig. 1A). Recombinant ES cells were injected into blastocysts embryos of C57BL/6 mice to produce chimeric mice, from which were derived Srxn1 −/− mice. Srx1 inactivation was established by genotypic analysis (Fig. 1A–C) and confirmed by Western blot analysis, which indicated the absence of Srx (Fig. 1C). Srx inactivation was also confirmed by the accumulation of overoxidized Prxs in H2O2-treated Srxn1 −/− but not Srxn1 +/+ mouse embryonic fibroblasts (MEFs), the biochemical signature of defective sulfinic acid reductase activity (Fig. 2A, B). A Western blot with an antibody specific for the sulfinylated form of the four 2-Cys Prxs (I–IV) revealed four bands in untreated MEF lysates of both genotypes, which based on size correspond to Prx1/Prx2 for the two lower ones, to Prx3 for the upper third one, and to Prx4 for the uppermost band. Exposure to H2O2 (100 μM) led to an increase in the Prx1/Prx2 SO2H signal in both Srxn1 +/+ and Srxn1 −/− MEFs. This signal then declined back to basal levels 6 h after H2O2 exposure in Srxn1 +/+ but not in Srxn1 −/− MEFs, where it remained elevated up to 24 h (Fig. 2A). Compared to wild-type MEFs, Prx1/Prx2 sulfinylation was also much more intense in Srxn1 −/− MEFs incubated with increasing doses of H2O2 (50–500 μM). These data thus not only confirm that Srx1 is inactive in Srxn1 −/− MEFs, but also indicate that there is no other enzyme that can substitute for Srx1 in reducing sulfinylated Prxs.
Srxn1−/− mice do not have any spontaneous phenotype
Homozygous Srxn1
−/− mice were obtained at the expected wild-type:heterozygote:null frequency. They were fertile and healthy, did not display any spontaneous apparent defects, and had a normal life expectancy. The blood cell count (leukocytes, lymphocytes, platelets, and hematocrit) of 35-week-old Srxn1
−/− mice (males and females, n = 10) was not different from that of age-matched Srxn1
+/+ mice (not shown). A necropsy performed at 34–35 weeks of age (n = 3) did not reveal any macroscopic lesions, and a systematic microscopic analysis of these mice (see Materials and Methods) did not reveal either any abnormality that could distinguish Srxn1
−/− and Srxn1
+/+ mice. The pattern of Srxn1 tissue expression, established by real-time quantitative PCR, did not reveal either any hint to a physiological role of Srx1 except for a moderately higher expression in the brain (Supplementary Fig. S1; Supplementary Data are available online at
Mice lacking Srx are highly susceptible to LPS-induced endotoxic shock
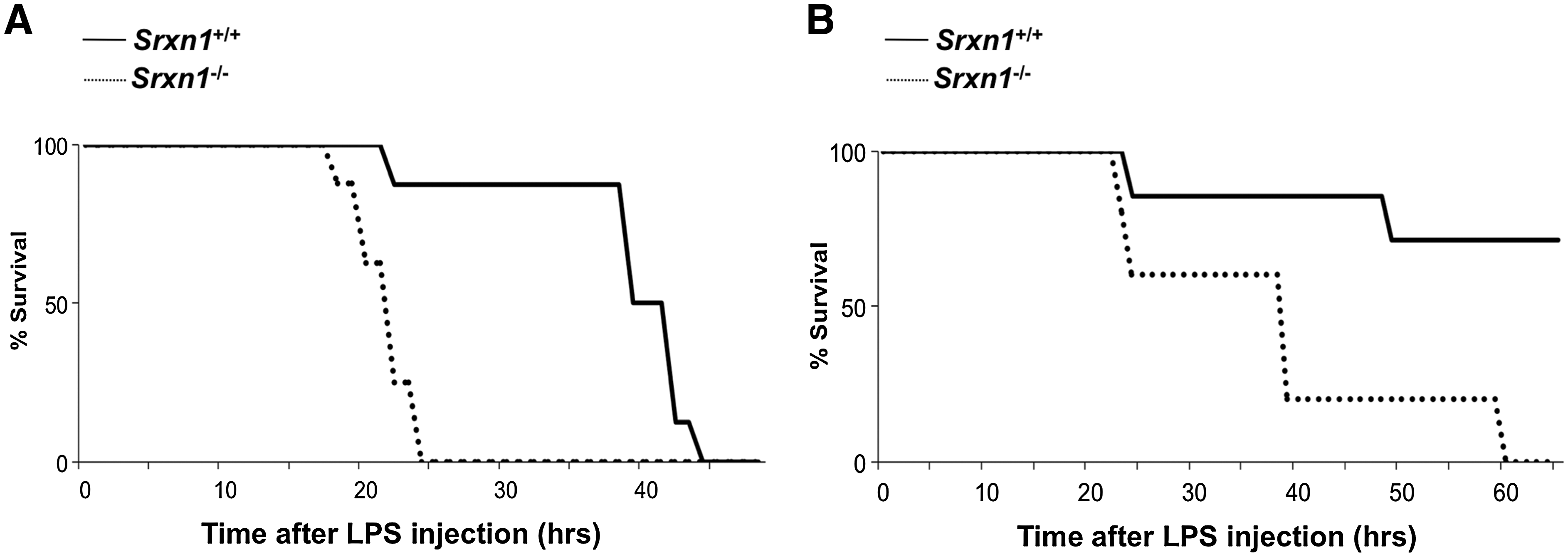
The absence of any apparent phenotype in Srxn1 −/− mice led us to explore experimental conditions that would unravel a phenotype. Prxs sulfinylation is known to occur as a result of Prx enzymatic cycling and of increased peroxide levels and inactivates Prx peroxidase function (18). As Srx1 enzymatic function is to reactivate Prxs by reduction of its sulfinated form, the enzyme could become important for mitigating peroxide stress and/or regulating peroxide signaling. Sepsis provided a good model, as involving the release of ROS by activated phagocytic cells (27), which have microbicidal functions (5), contribute to pathogenesis by causing cellular and tissue damage (1, 36), and amplifies signaling events (22, 29). We used bacterial endotoxin (LPS) administered to mice by IP injection, which replicates the clinical spectrum of septic choc (3), and monitored survival during 5 days. In initial dose–response studies, we established that the dose of 25-to 50 μg/g body weight resulted in the death of 50%–100% of wild-type animals. LPS (25 μg/g) was administered to 3-month-old Srxn1 +/+ (n = 8) and Srxn1 −/− (n = 8) male mice (Fig. 3A). Such dose of LPS was lethal for both groups of animals. However, in the Srxn1 −/− group 100% of the animals had died within the first 24 h after LPS injection, whereas 87.5% of wild-type animals remained alive up to 40 h (p < 0.001). To check whether this phenotype crossed genders, we monitored the effect of LPS (25 μg/g) in 3-month-old Srxn1 +/+ (n = 7) and Srxn1 −/− (n = 5) female mice (Fig. 3B). As previously observed, C57BL/6 female mice were significantly more resistant to the lethal effect of LPS than males, and this difference was also observed for Srxn1 −/− mice. Still, Srxn1 −/− mice were significantly more sensitive to the lethal effect of LPS, as they all died within 3 days, whereas only two Srxn1 +/+ mice died within this period, the remaining five mice being still alive and healthy after 5 days (p < 0.001).
Serum levels of the liver damage markers ALT and AST were measured in 5-month-old Srxn1 +/+ (n = 6) and Srxn1 −/− (n = 6) males that had received an IP injection of a sublethal dose of LPS (10 μg/g) (Table 1). ALT levels remained low in animals of both genotypes at 6 h but then increased in Srxn1 −/− mice, but not in Srxn1 +/+. AST levels were similarly moderately augmented at 6 h in mice of both genotypes, but then significantly increased at 24 h in Srxn1 −/− mice but not Srxn1 +/+ (p < 0.05). No significant difference was observed in the serum levels of creatine kinase, lactate dehydrogenase, lactate, alkaline phosphatase, and creatinine (not shown).
Serums were collected 6 and 24 h after LPS injection (10 μg/g body weight), and alanine aminotransferase and aspartate aminotransferase measured (U/L). Data (mean ± SE) are from 5-month-old Srxn1 +/+ and Srxn1 −/− male mice (n = 6) (p < 0.05).
LPS, lipopolysaccharide.
These results show that Srx1 clearly protects mice from sterile endotoxic shock.
The genomic response to LPS is delayed in Srxn1−/− mice
We searched for transcriptional changes that could explain the differential tolerance of Srxn1 +/+ and Srxn1 −/− mice to LPS-induced endotoxemia by comparing the spleen and lung mRNA profiles of 12-week-old Srxn1 +/+ (n = 3) and Srxn1 −/− (n = 3) males, 6 and 20 h after a nonlethal IP LPS injection (12 μg/g), with those of the same organs from matched animals, 6 h after an IP injection of PBS (see above). Principal Component Analysis (PCA) showed tight triplicate samples clustering, indicating good reproducibility, and clear divergence between conditions (PBS:6H vs. LPS:6H vs. LPS:20H), which reflected the extent of the LPS genomic response (Supplementary Fig. S2).
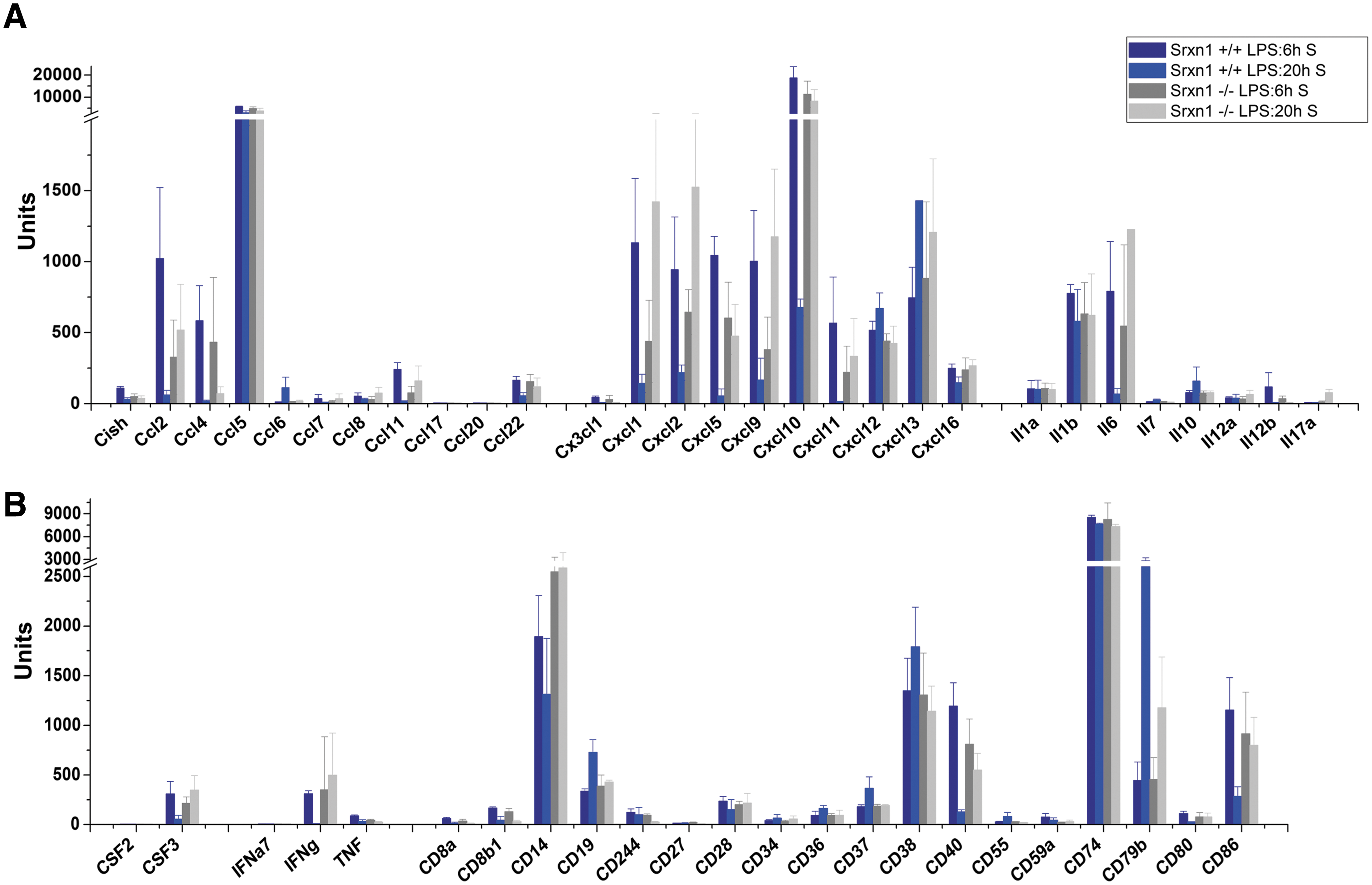
At the 6 h time-point (LPS:6H), wild-type and Srxn1 −/− mice had both typical responses to LPS that spanned all innate immunity—cytokines, chemokines and their receptors, chemokine signaling, Toll-like, NOD-like, and RIG-I-like receptor and cytosolic DNA-sensing pathways, JAK-STAT signaling, and complement and coagulation cascades—and several adaptive immunity pathways—antigen processing/presentation, NK, and T-cell and B-cell signaling pathways (Supplementary Table S2). However, despite the similarity of the responses of Srxn1 −/− and Srxn1 +/+ mice, important differences were seen, not so in the number of genes in each of these functional categories (Supplementary Tables S2 and S3), but in the amplitude of gene expression (Fig. 4, Supplementary Fig. S3, and Supplementary Table S4). Messenger RNA level fold changes were significantly higher for many innate immunity-related transcripts in Srxn1 +/+ mice, even upon taking into account the constitutive upregulation of several of these genes in the spleen of Srxn1 −/− animals. Especially striking was the higher expression in Srxn1 +/+ mice of genes encoding cytokines, chemokines, and costimulatory cell surface molecules (Fig. 4, Supplementary Fig. S3, and Supplementary Table S4), which are crucial effectors of the systemic inflammatory response triggered by LPS. At the 20 h time-point, however, the genomic response was now much more intense in the Srxn1 −/− mice, with regard to both the number of regulated genes (Fig. 5 and Supplementary Tables S5 and S6) and their expression change (Fig. 4, Supplementary Fig. S3, and Supplementary Table S4). The number of regulated genes indeed substantially decreased in Srxn1 +/+ animals, especially in spleen, whereas it increased in Srxn1 −/− mice (Supplementary Table S5). Figure 5 depicts the number of regulated genes in the gene clusters the most representative of innate immunity, indicating that at the 6 h time-point they are comparable between genotypes in spleen, whereas at the 20 h time-point they remain high or further increase in both organs of Srxn1 −/− mice, while they significantly decrease in Srxn1 +/+ mice, especially in spleen (Fig. 5A). Similarly, in lungs the amplitude of gene expression remained high in Srxn1 −/− mice for many innate immunity genes, whereas it decreased in Srxn1 +/+ mice (Fig. 5B). Especially demonstrative of such trend was exhibited by the fold changes of genes encoding cytokines, chemokines, and costimulatory molecules, which were high at 6 h in Srxn1 +/+ and low in Srxn1 −/−, as mentioned above, but then decreased in the former at 20 h, and increased in the latter, in both spleen (Fig. 4 and Supplementary Table S4) and lungs (Supplementary Fig. S3 and Supplementary Table S4).

Discussion
We report here the generation of a mouse line with a null mutation in the gene encoding Srx1, and its description. Srxn1 −/− mice were fully viable and did not suffer from any apparent clinical and histological defects under laboratory conditions. Srxn1 −/−-derived MEFs were totally defective in reducing the sulfinylated form of Prx1 and Prx2 generated by H2O2, which confirmed the Srx1 defect of Srxn1 −/− mice, and also indicated that no other mouse enzyme can substitute for Srx1 in its Prx-specific sulfinic reductase function. Despite lack of apparent defects, microarray-based mRNA profile analysis showed that Srxn1 −/− mice upregulated several genes linked to adaptive and innate immunity under laboratory conditions, which possibly indicates a silent immune defect. Further, compared to their wild-type counterpart, they had an abnormal response to LPS that manifested by a significantly increased mortality during endotoxic shock, and a delayed and prolonged genomic response to LPS. Microarray-based mRNA profiles showed that the response of Srxn1 −/− mice to LPS was in fact typical, in terms of the nature of the genes regulated, which spanned the all spectrum of pathways triggered during activation of innate immunity as well as several pathways of adaptive immunity. However, compared to Srxn1 +/+ mice, this response was delayed by several hours, and remained intense or even increased at late time after LPS injection (20 h), when the response of Srxn1 +/+ mice had already largely dissipated. Such prolonged response was particularly marked with regard to the proinflammatory cytokines IL1β and IL6, which are essential effectors of the inflammatory response (12, 14), and for several chemokines, including Ccl2, Ccl11, Ccl20, Ccl22, Cx3cl1, Cxcl1, Cxcl2, Cxcl5, Cxcl9, Cxcl10, and Cxcl11, that have essential functions in leukocyte chemotaxis at sites of inflammation and injury (24). Such prolonged response was observed for many other genes of immunity as γ-IFN that stimulates inducible nitric oxide synthase-dependent nitric oxide production by macrophages and cell surface MHC molecules expression, the macrophage cell surface costimulatory molecules CD14 that amplifies LPS responsiveness of Toll-like receptor 4 (TLR4), CD40 that activates macrophage upon binding CD154 on activated T cells, and CD86 that activates T cells by binding CD28 receptor, and also for genes encoding innate immunity kinases and transcription factors (Supplementary Fig. S4). Such a delayed and prolonged genomic response to LPS is consistent with the poor clinical tolerance of Srxn1 −/− animals to endotoxic shock, which clearly indicates that Srx activity protects mice from the lethality of endotoxic shock.
What is the basis for the role of Srx during endotoxemia, and how it relates to its enzymatic function? The function of Srx is to reactivate 2-Cys Prxs when in their sulfinylated inactive form. Prxs themselves constitute an important protection against cellular damage caused by peroxide and peroxinitrite, and have also demonstrated functions in H2O2 signaling (7, 18, 35). LPS-induced systemic inflammation is initiated by LPS binding to TLR4 on the surface of monocytic cells, and TLR4-proximal and distal signaling critically involve ROS produced by membrane NADPH oxidase (26). Prx2 was recently shown to exert a negative control on LPS-TLR4 signaling by holding check on the levels of ROS produced by NADPH oxidases (44), which corroborate the fact that mice lacking Prx2, as those lacking Srx1 (this study), are hypersensitive to the lethality of LPS-induced endotoxemia (44). Srx may therefore influence sepsis by controlling the strength of TLR4 signaling through its effect on Prx2. Disruption of NRF2, a transcription factor that controls the cellular redox poise by regulating antioxidants, including Prxs, activities of the thioredoxin and glutathione pathways, and phase II enzymes (15, 20), also amplifies LPS-TLR4 signaling and causes a drastic increase in lethality during LPS-induced endotoxic shock (34), an effect that is dependent on the NADPH oxidase catalytic subunit gp91phox (23). Interestingly, NRF2 regulates not only the expression of Prxs (13) but also that of Srx1 (2, 19, 30, 33). Further, we have recently shown that in LPS/γ-IFN-stimulated mouse macrophages, inducible nitric oxide synthase-derived NO activates NRF2-dependent Srx expression (Abbas et al., manuscript submitted). Nrf2 −/−-derived macrophages were in fact unable to reduce overoxidized Prx1/Prx2, indicating that in these cells Srx expression is strictly dependent upon NRF2. We also showed in this study that NO treatment increases peroxide scavenging and decrease peroxide levels in wild-type-derived but not Srxn1 −/−-derived macrophages, thus linking LPS-induced macrophage NO production and the induction of an important NRF2-Srx antioxidant pathway. This study (Abbas et al., manuscript submitted), together with the data of Thimmulappa (34) and Kong (23) and the data presented here, thus suggests that the abnormal LPS response of mice lacking Srx or NRF2 might be caused by the defective NO-dependent induction of the NRF2-Srx antioxidant axis that exacerbates TLR4 signaling and the ensuing systemic inflammatory response by loss of control on the levels of NADPH oxidase-produced ROS. Prx2 might also be part of this antioxidant axis, but it is not induced by NO (13). The NRF2-Srx antioxidant axis could, however, also be important for protecting macrophages, and possibly other cells, from NADPH oxidase-produced ROS damage. However, the abnormal regulation of many genes of adaptive and innate immunity in untreated Srxn1 −/− mice, and the peculiar delayed and prolonged LPS response of these mice both suggest a defect in a regulatory rather than in an oxidative damage protective mechanism. Whereas loss of Prx, or NRF2, or as shown here Srx decreases the tolerance to LPS-induced endotoxemia, the loss of catalase increases this tolerance (45), which has led these authors to suggest that H2O2 could have anti-inflammatory effects by inhibiting the proteasome, and hence NF-κB activation. Such opposite roles of antioxidants during sepsis might, nevertheless, be reconciled by considering that Prxs (and Srx) are only active under very low levels of H2O2, whereas catalase becomes an efficient antioxidant at much higher H2O2 levels (18). Accordingly during sepsis, the deficiency in Prx or Srx should amplify H2O2 signaling, which presumably proceeds at low levels H2O2, whereas the deficiency in catalase should favor a buildup of H2O2 levels, high enough to prevent NF-κB activation.
In conclusion, the present study further indicates that Srx adds to Prx2, Prx3, and NRF2 as host factors that by regulating cellular ROS levels act as important modifiers in the pathogenesis of sepsis.
Footnotes
Acknowledgments
We greatly acknowledge Patrick Hery for his expert help with mice experiments; Drs. Benoît Biteau, Flora Tomasello, and Agniezska Sekowska for their help in the initial stage of the Srxn1 −/− mouse line generation; and Dr. Sue Goe Rhee for his kind gift of reagents. This work was supported by grants from Association pour la Recherche Centre le Cancer (ARC) and Agence Nationale de la Recherche (ANR) SOLEMA to M.B.T., and ANR NOPEROX to J.-C.D. and M.B.T. A.-G.P. was supported by an ANR fellowship NOPEROX. M.B.T. is the recipient of a fund program, “Equipe Labellisée Ligue 2009,” from La Ligue Contre le Cancer.
Author Disclosure Statement
There are no competing financial interests with regard to the data included in this article for any of its authors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.