
Abstract
Significant amounts of oxygen free radicals (oxidants) are generated during cerebral ischemia/reperfusion, and oxidative stress plays an important role in brain damage after stroke. In addition to oxidizing macromolecules, leading to cell injury, oxidants are also involved in cell death/survival signal pathways and cause mitochondrial dysfunction. Experimental data from laboratory animals that either overexpress (transgenic) or are deficient in (knock-out) antioxidant proteins, mainly superoxide dismutase, have provided strong evidence of the role of oxidative stress in ischemic brain damage. In addition to mitochondria, recent reports demonstrate that NADPH oxidase (NOX), an important pro-oxidant enzyme, is also involved in the generation of oxidants in the brain after stroke. Inhibition of NOX is neuroprotective against cerebral ischemia. We propose that superoxide dismutase and NOX activity in the brain is a major determinant for ischemic damage/repair and that these major anti- and pro-oxidant enzymes are potential endogenous molecular targets for stroke therapy. Antioxid. Redox Signal. 14, 1505–1517.
Introduction
Generation of and Cell Injury by Oxidants After Cerebral Ischemia
Generation of oxygen free radicals
Free radicals are molecular species that contain one or more unpaired valence electrons not contributing to intramolecular bonding. Free radicals are highly active with other molecules, such as DNA and lipids, pairing with their single electrons and causing oxidation of those molecules (15). Several oxygen free radicals (oxidants) and their derivatives are generated after stroke, including superoxide anions (O2 ·−), hydrogen peroxide (H2O2), and hydroxyl radicals (·OH). O2 ·− are formed when oxygen acquires an additional electron, leaving the molecule with only one unpaired electron. O2 ·− are formed within the mitochondria, and ∼2% to 5% of electron flow is used to produce them (Fig. 1) (9). Pro-oxidant enzymes such as xanthine oxidase and NADPH oxidase (NOX) also catalyze the generation of O2 ·−. O2 ·− can react with nitric oxide (NO) to produce peroxynitrite (ONOO−) (NO + O2 ·− → ONOO−), which is a strong oxidative radical that causes protein nitration and dysfunction (5). NO generated from neuronal nitric oxide synthase nitrosylates protein, which leads to cell dysfunction (26). SOD detoxifies O2 ·− to H2O2 (O2 ·− + 2 H+ → H2O2), which is further converted to H2O by catalase or glutathione peroxidase (2H2O2 →2H2O + O2) (Fig. 1) (9). Highly reactive ·OH is produced by H2O2 through the Fenton reaction (H2O2 + Fe2+ → OH− + Fe3+ + ·OH) and the Haber-Weiss reaction (O2·− + H2O2→ ·OH + HO− + O2) or by ONOO−(Fig. 1) (5, 10). A constitutively low concentration of oxidants is needed and they act as signaling molecules for various functions such as regulation of vascular tone, monitoring of oxygen tension, and erythropoietin production (9, 15). However, excessive oxidants may irreversibly oxidize macromolecules such as DNA, lipid, and protein, and cause severe cell injury. Endogenous antioxidant enzymes such as SOD, glutathione peroxidase, and catalase play an important role in the maintenance of low-concentration oxidants and redox homeostasis in the tissue. Besides antioxidant enzymes, other antioxidants, including glutathione, ascorbic acid, and vitamin E, are also involved in the detoxification of oxidants (9, 10). Figure 1 summarizes the generation of oxidants in vivo that leads to brain cell damage under ischemia and reperfusion conditions.
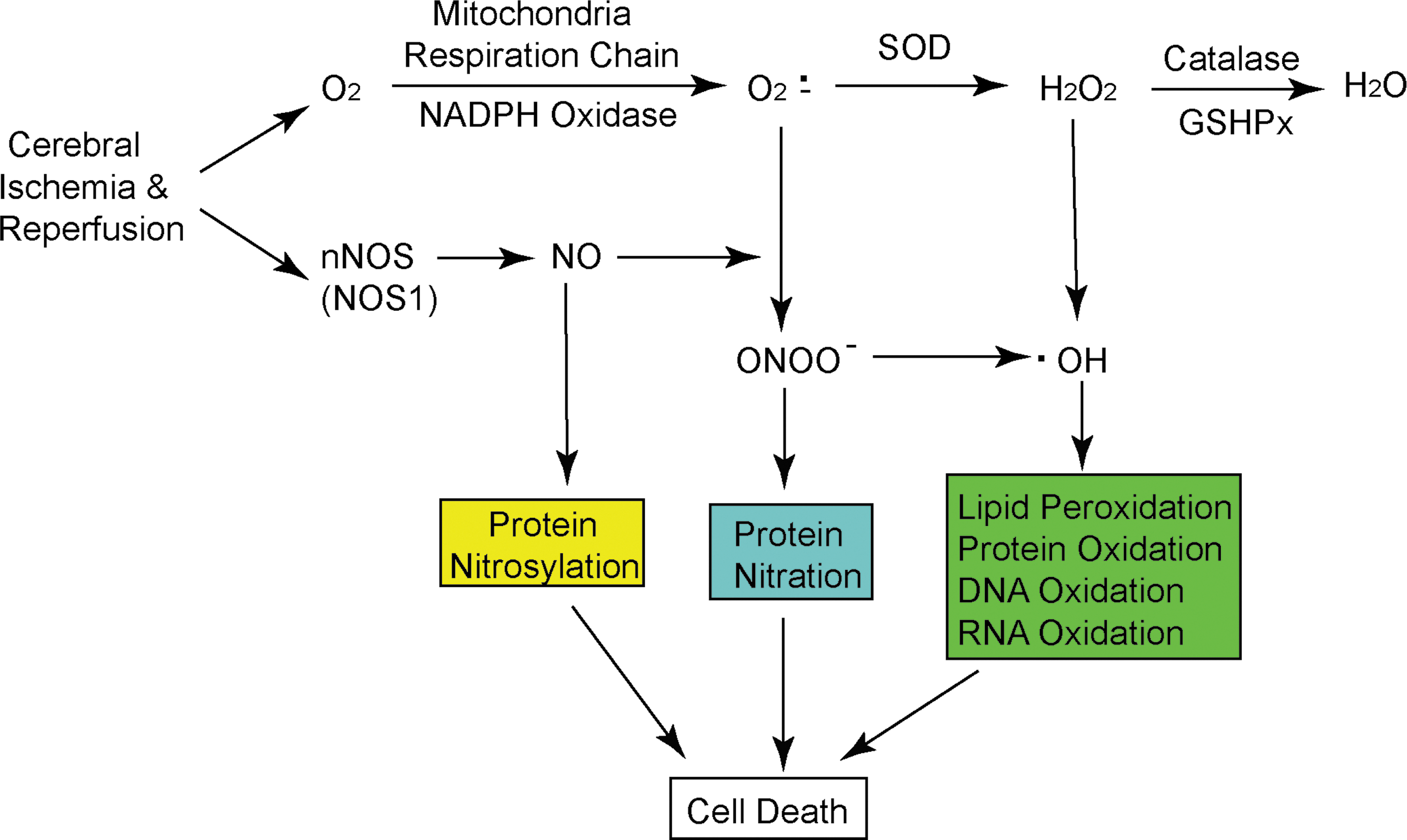
Oxidative damage and detection in vivo
During brain ischemia/reperfusion, multiple detrimental processes take place, including overproduction of oxidants, inactivation of detoxification systems, and consumption of antioxidants. These changes cause the disruption of the normal antioxidative defense ability of brain tissue (9). Oxidants have a very short half-life, which makes them very difficult to measure directly in the laboratory. However O2 ·− production can be assessed by detection of oxidized hydroethidine (HEt) in vivo. HEt, systemically administered, can pass the blood–brain barrier (BBB) and is selectively taken up by cells in the brain. Intracellular HEt is oxidized by O2 ·− and converted to ethidine (Et), which provides a red fluorescence signal (49). Under physiological conditions, Et signals are detected as particles in the cytosol, representing a small amount of O2 ·− production in mitochondria. At 4 h of reperfusion in the focal ischemic area, a diffuse pattern of Et fluorescent signals in neurons suggested the spread of and a massive amount of O2 ·− generation in the cytosol (49). Moreover, global ischemia also caused increased Et signals in the rat hippocampal CA1 region, which exhibited delayed cell death during reperfusion (36). Therefore, oxidative stress plays an important role in delayed neurodegeneration after global ischemia.
In addition to the HEt method, oxidative stress can also be assessed by measuring the oxidized products of macromolecules, including nucleic acid, lipids, and protein, and 8-hydroxy-2-deoxyguanosine (8-OHdG), an oxidized form of deoxyguanosine that is a widely used biomarker of in vivo oxidative DNA damage. After transient focal cerebral ischemia (tFCI), the level of 8-OHdG started to increase as early as 3–6 h during reperfusion. A robust amount of 8-OHdG was detected at 24 h of reperfusion after tFCI (13). Lipid peroxidation is well established after stroke, and it causes alternations in cell membrane fluidity, an increased permeability of membranes, and a decrease in membrane ATPase activity, thus leading to cell injury. Lipid peroxides derived from polyunsaturated fatty acids are unstable and decompose to form a complex series of compounds such as malondialdehyde (MDA). Cerebral ischemia can cause a significant amount of MDA formation in the ischemic hemisphere (13). 4-Hydroxy-trans-2-nonenal (HNE), a derivative of the ω-6 family of polyunsaturated fatty acids caused by a free radical attack, also significantly increases after brain focal or global ischemia (13, 73).
Neuroprotective Effect of the Antioxidant Enzyme SOD
Three major SODs have been identified and each enzyme has a specific cellular distribution and metal cofactors. Copper/zinc SOD (SOD1) is a dimeric cytosolic enzyme that is coded by the human SOD1 gene and that needs copper and zinc cofactors. Manganese SOD (SOD2), which is expressed in mitochondria, requires manganese for enzyme activity. Extracellular SOD is mainly localized in extracellular space, cerebral fluid, and cerebral vessels and its cofactors are copper and zinc (Table 1) (9). Numerous research reports, especially studies of SOD overexpression or in knock-out animals, have proven that SODs are actively involved in neuroprotection after brain ischemia. Recent studies using transgenic (Tg) and mutant SOD approaches are summarized in Table 2 (9, 53).
SOD, superoxide dismutase.
All data from References 9 and 53, unless otherwise noted.
NF-κB, nuclear factor kappa B; pFCI, permanent focal cerebral ischemia; pPRAS, phosphorylated proline-rich Akt substrate; tFCI, transient FCI; tGCI, transient global cerebral ischemia.
Copper/zinc SOD
As a result of the extremely short half-life (6 min) of SOD1 in circulating blood and its failure to pass the BBB and be taken up intracellularly, its direct application is limited in cerebral ischemia (9). However, using modified enzymes such as liposome-entrapped SOD1 or lecithinized SOD1, which has an increased half-life, the BBB becomes permeable, thus alleviating oxidative stress and reducing brain infarct volume by 50% in rodents subjected to tFCI (9).
SOD1-overexpressing Tg mice have been developed as the TGHS/SF-218 strain, which carries the SOD1 gene with a CD1 background (18). Heterozygous Tg mice (SOD1+/− Tg) have a threefold increase in SOD1 activity, and a fivefold increase in SOD1 activity has been reported in homozygous mice (18). The protective effect of SOD1 against ischemia/reperfusion cerebral damage was confirmed in mice subjected to tFCI. Infarction volume in SOD1+/− Tg mice was 26% less than in wild-type mice. The decrease in infarction in the SOD1+/− Tg mice paralleled alleviation of neurological deficits (75). In a permanent middle cerebral artery occlusion model, which did not involve reperfusion, genetic manipulation of SOD1 did not provide protection against brain ischemic damage, suggesting that SOD1 mainly protects against reperfusion damage when oxidants are robustly generated (11). Multiple mechanisms have been identified that lead SOD1 to block mitochondrial-mediated apoptosis (16, 22, 60, 68) and to regulate cell death and cell survival signal transduction pathways (55, 62, 65).
In addition to focal ischemia, SOD1 also protects rodent brains against transient global cerebral ischemia (tGCI) induced by bilateral common carotid artery occlusion and blood withdrawal (12, 51, 68). Global ischemic insult caused significant delayed neuronal death in the hippocampal CA1 region in rodents, as assessed by cresyl violet staining and DNA fragmentation analysis (67, 68). Administration of manganese tetrakis (4-benzoic acid) porphyrin, a SOD mimetic, attenuated tGCI-induced CA1 pyramidal neuronal damage and neurological deficits (40). SOD1+/− overexpression also reduced hippocampal neuronal death by 50% in a global ischemia model (12, 48, 68). No difference in plasticity of the posterior communicating artery was identified between wild-type and SOD1+/− Tg mice. Thus, the protective role of SOD1 overexpression is unlikely due to the modification of blood vessels and increasing blood supply (48).
The role of SOD1 in cerebral ischemia was further investigated by the study of SOD1-deficient mice (36, 39). SOD1−/+ mice, with a 50% reduction in SOD1 activity, demonstrated larger infarct volume and brain swelling, accompanied by increased apoptotic neuronal cell death compared with wild-type controls after tFCI (39). Further, the mortality of homozygous mutant SOD1−/− mice was 100% at 24 h of reperfusion after tFCI, and was significantly higher than in wild-type mice (10%) (39).
Manganese SOD
The protective role of SOD2 against ischemic insult was first identified in SOD2-overexpressing PC6 cells and SOD2+/− Tg mice. Overexpression of SOD2 in PC6 cells prevented apoptosis induced by Fe2+, amyloid β-peptide, or nitric oxide-generating agents. In addition, SOD2 ameliorated membrane lipid peroxidation and protein nitration, and reduced brain infarction volume by ∼25% after tFCI (37). On the contrary, SOD2−/+ mice with less SOD activity exhibited more O2 ·− production, exacerbated cerebral infarction, and worsening neurological deficits after tFCI (45, 46).
Cerebral ischemia/reperfusion also downregulated SOD2 expression (by 60% at 3 h of reperfusion) (32). Signal transducer and activator of transcription 3 (STAT3) is a novel transcription factor of the SOD2 gene promoter. After tFCI, STAT3 levels began to decrease as early as at 1 h of reperfusion, and its recruitment into the SOD2 promoter was completely blocked. STAT3 deactivation caused subsequent downregulation of SOD2 transcription and lower SOD2 protein levels in brain tissue. Therefore, in addition to increased generation of oxidants, cerebral ischemia also impairs endogenous SOD2 generation and further aggravates oxidative brain damage (32).
Redox signaling and SODs
Many signal pathway molecules are known to be regulated by the redox state in cells. Oxidative stress induces the activation/inhibition of those pathways (15). As well as directly alleviating an oxidative attack in macromolecules, SOD also regulates redox-sensitive signal transduction pathways upstream after ischemia (Fig. 2) (9).

The transcription factor nuclear factor kappa B (NF-κB) is composed of a heterodimer with one 50-kDa (p50) and one 65-kDa (p65) polypeptide. NF-κB dimers are retained in the cytosol by interacting with an inhibitor subunit IκB. IκB kinase contains a cysteine residue in its regulatory domain, which is sensitive to modification by oxidants (35). Oxidants activate IκB kinase and subsequently cause the phosphorylation of IκB. Phosphorylated IκB is ubiquitinated and degraded by proteasomes, resulting in dis-inhibition of NF-κB, which then translocates into the nucleus, binds the promoter, and plays a pivotal role in controlling multiple gene expression. NF-κB gene products, such as tumor necrosis factor-α, interleukin (IL)-1β, IL-6, inducible nitric oxide synthase, and matrix metalloproteinase 9 (MMP9), mediate the postischemic inflammatory response, BBB disruption, and secondary neuronal injury (59). Meanwhile, NF-κB also regulates expression of antiapoptotic proteins, including Bcl-2, playing a role in neuronal survival (59). Due to the different gene products of NF-κB, its role in activation and signaling in ischemic brain damage is controversial (59). It is possible that the variable nature of ischemic injuries (permanent vs. transient, duration, and severity of ischemia) and activation of different cell types (neurons vs. microglia) cause different gene product transcription and different roles for NF-κB in cerebral ischemia.
Our studies revealed that phosphorylation and degradation of IκB began to increase at 1 h of reperfusion after 1 h of tFCI, followed by activation of NF-κB as shown by electrophoretic mobility shift assay. However, in SOD1+/− Tg mice, which demonstrated less pronounced oxidative stress, activation of the NF-κB pathway was alleviated compared with wild-type mice (65, 66). Indeed, compared with wild-type mice, SOD1+/− Tg mice demonstrated significantly less upregulation of cytokines and chemokines such as IL-1β, tumor necrosis factor-α, IL-6, monocyte chemoattractant protein 1, and macrophage inflammatory protein 1α (54) (Fig. 2). SOD1 also promoted NF-κB-mediated neuroprotective gene transcription after tFCI, including antiapoptotic factors Bcl-2 and X chromosome-linked inhibitor-of-apoptosis protein (66).
p38, a mitogen-activated protein kinase (MAPK) family member, can be strongly activated by reactive oxygen species (15). Significant activation of the p38 pathway was revealed in postischemic brain tissue, and p38 played important roles in transducing stress-related signals by phosphorylating intracellular enzymes, transcription factors, and cytosolic proteins involved in apoptosis and inflammatory cytokine production (29). Sustained activation of p38 MAPK is associated with neuronal death, and the selective p38 MAPK inhibitor SB239036 can promote survival of a variety of neurons (4, 58). A p38 MAPK pathway substrate, cytosolic phospholipase A2, caused cell membrane phospholipid hydrolysis and mediated arachidonic acid metabolism, leading to brain damage (2). Postischemic activation of p38 activity and cytosolic phospholipase A2 was significantly alleviated in SOD1+/− Tg rats compared with wild-type rats at both 1 and 3 days after reperfusion (55). This was accompanied by 70% less infarction volume and better neurological behavior in the SOD1+/− Tg rats (55). Therefore, SOD1 alleviates p38-mediated cell death pathway activity and confers neuroprotective effects after brain ischemia (Fig. 2).
Cell survival pathways also play an important role in determining the fate of ischemic tissue. In contrast to p38, Akt, a pathway downstream of phosphoinositide 3-kinase, is a key factor in the regulation of cell survival pathways after cerebral ischemia (20). Akt phosphorylated Bad and obviated its inhibitory effect on the antiapoptotic protein Bcl-XL, ultimately inhibiting the release of cytochrome c (8, 14). In addition, Akt phosphorylated pro-caspase-9 and inactivated caspases (8). Activated proline-rich Akt substrate (PRAS), which mediates the neuroprotective effect of nerve growth factor, is also a downstream substrate of the Akt pathway (63). After tFCI, Akt pathways were inhibited, and its downstream products such as phosphorylated PRAS (pPRAS) were all reduced. Inhibition of the Akt pathway shifted the cell fate toward death signal pathways (62). In addition, fluorescence double staining revealed that the reduction in pPRAS was most remarkable in regions with strong oxidized HEt signals, which further indicates that postischemic oxidants inhibit the Akt-pPRAS pathway (62). SOD1 overexpression not only reduced postischemic oxidative stress, but also significantly promoted activation of the Akt pathway and expression of pPRAS (57, 62). Consistent with these results in focal ischemia, SOD1 overexpression also enhanced persistent Akt pathway activation and provided protection against delayed hippocampal CA1 injury in a global ischemia model (17). Taken together, SOD1 can strengthen cell survival by enhancing the Akt pathways after brain ischemia (Fig. 2).
BBB disruption and SOD
The BBB, which is composed of cerebral vascular endothelial cells and astrocytes, strictly controls exchanges between blood and brain compartments. BBB disruption after stroke can worsen ischemic injury and mortality by increasing edema and causing hemorrhage. After tFCI, endothelial cells are also subjected to increased oxidative stress, as revealed by increased O2 ·− signals in vascular endothelial cells in vivo (45). Oxygen-glucose deprivation (OGD) caused DNA damage in a primary endothelial cell culture, showing that cerebral vascular endothelial cells are vulnerable to ischemic insult (50). OGD-induced cell death was significantly alleviated in SOD1+/− Tg endothelial cells, which indicates that oxidative stress plays a role in endothelial cell ischemic death (50).
As well as leading to injury of endothelial cells, ischemia/reperfusion can also induce degradation of the extracellular matrix components in basal lamina, further impairing BBB permeability (46). MMPs, expressed in microvascular endothelial cells, are a family of proteolytic enzymes that cause extracellular matrix degradation. Free radicals generated during cerebral ischemia/reperfusion cause proMMP activation and these activated MMPs cause BBB injury through degradation of the neurovascular matrix, thereby playing a deleterious role in stroke (30, 43). The MMP-9 inhibitor SB-3CT rescued laminin from proteolysis and neurons from apoptosis in tFCI and in MMP-9 knock-out mice and demonstrated less infarction and BBB disruption after focal ischemia (3, 25). After tFCI, activated MMP-9 was observed at 4 h of reperfusion, paralleling the increase in BBB permeability as detected by Evans blue extravasation (45, 46). In addition, double staining revealed the spatial colocalization of MMP-9 and O2 ·− at the level of the capillary walls and astrocytic processes, which implies that oxidative stress promotes MMP-9 activation after ischemia in vivo (23).
Inhibition of MMP activity by overexpression of SOD1 has been revealed in rodent cerebral ischemia models. In postischemic SOD1+/− Tg mice, not only were less oxidants detected by HEt, but significantly less MMP-9 activation was also observed compared with wild-type mice (34). Moreover, SOD1+/− Tg mice exhibited less Evans blue leakage, which indicates better preserved BBB integrity than in wild-type mice (34). On the contrary, more pronounced MMP-9 gene expression and BBB leakage was reported in SOD1−/− mice (23). NF-κB, a main transcription factor and binding in the promoter region of MMP-9, was significantly activated under oxidative stress conditions (see discussion above), and it may mediate MMP-9 activation after cerebral ischemia.
We have reported that mitochondrial SOD2 is also involved in postischemic MMP activation and BBB disruption, using SOD2−/+ heterozygous mice that had 50% SOD activity. At 72 h of reperfusion after 30 min of ischemia, MMP-9-positive vessel counts for SOD2−/+ mice were significantly higher than in wild-type mice (46). Western blot results revealed significantly more MMP-9 protein expression in both cultured endothelial cells and in isolated microvessels from SOD2−/+ mice after ischemia and reperfusion (45). Tight junction proteins, which are anchored into endothelial cells and stitch them together, are crucial for maintaining the close contact of endothelial cells and BBB integrity. After ischemia, tight junction proteins of endothelial cells, such as zonula occludens-1, occludin, and claudin5, exhibited progressive degradation during the evolution of infarction. This was accompanied by increasing BBB disruption and brain hemorrhage. Degradation of tight junction proteins was more severe in SOD2−/+ mice, which indicates that tight junction proteins are molecular targets of oxidants (45). Further, SOD2−/+ mice demonstrated delayed BBB breakdown and high brain hemorrhage rates. These adverse consequences were absent in wild-type littermates or SOD2+/− Tg mice (45). In summary, SOD maintains BBB integrity and reduces brain damage by protecting endothelial cells and inhibiting MMP activity in experimental stroke.
The Pro-Oxidant Enzyme NOX
General characteristics of NOX
NOX is a multisubunit complex composed of membrane-associated subunits, including gp91phox and p22phox (phox: phagocyte oxidase), cytosolic subunits, which include p47phox, p67phox, and p40phox, as well as one of the small Rho GTP-binding proteins Rac1 or 2. The catalytic subunit of the enzyme gp91phox, NOX2, is present with several homologs, including NOX1, NOX3, NOX4, NOX5, and DUOX1 and 2 (6). In this review we focus on NOX2, which is the prototype NOX and is the most extensively studied. As NOX is being activated, p47phox is phosphorylated on serine 8–9 by either the proline-direct kinase or protein kinase C (PKC) (31). p47phox phosphorylation subsequently causes the cytosolic subunits p47phox, p67phox, and p40phox to translocate into membranes and fuse with the catalytic subunit gp91phox. This is followed by interaction between Rac and gp91phox (6). The activated enzyme complex transports electrons from NADPH in the cytosol to oxygen in the extracellular space, thus producing O2 ·− (6) (Fig. 3). gp91phox knock-out (gp91phox−/−) and gp47phox knock-out (gp47phox−/−) mice have been produced and are widely used in NOX studies (6). Apocynin, a NOX inhibitor with a relatively low affinity (IC ∼ 10 μM), is also a commonly used compound in NOX studies (6).

Distribution and regulation of NOX
The gp91phox subunit was initially discovered in neutrophils. It generates O2 ·− for killing bacteria and therefore plays an important role in host defense (6). gp91phox and its homologs including NOX1 and NOX4 were discovered in nonphagocytic cell types and tissue, including the brain. Infanger and colleagues (28) compared expression of NOX1, gp91phox, and NOX4 in different regions of mouse brains using RT-PCR. gp91phox and NOX4 were the dominant homologs expressed in fore-, mid-, and hindbrain of the mice, whereas NOX1 was detectable, but at low levels. In vivo immunohistological studies have demonstrated expression of the gp91phox subunit in various brain regions, including the cortex, hippocampus, medulla, hypothalamus, pons, and cerebellum (6, 28). Ubiquitous expression of NOX in brain tissue indicates its role in brain physiological function, and indeed NOX has been implicated in regulating synaptic plasticity and memory formation. It is also becoming clear that overactivation of NOX plays a role in many neurodegenerative diseases such as stroke and Alzheimer's disease (6).
Expression and activity of NOX in the central nervous system are regulated by ischemia/reperfusion. After 90 min of tFCI, expression of the gp91phox subunit began to increase as early as 2 h, and reached its peak of 3.5-fold of the baseline at 22.5 h of reperfusion (27). Moreover, at 2 h of reperfusion after tFCI, a significant amount of the p47phox subunit translocated to the membrane, further indicating the activation of NOX (27). Consistent with the changes in the subunits, both NOX activity and O2 ·− levels exhibited a remarkable increase after ischemia during reperfusion (27).
Casein kinase 2 (CK2) plays a role in regulating NOX activity after cerebral ischemic injury. Under normal conditions, Rac1 is captured by the subunit CK2α, which prevents the translocation of Rac1 and activation of NOX. The protein levels of the catalytic subunit CK2α and the total activity of CK2 significantly decreased after tFCI. In addition, binding between CK2α and Rac1 was immediately diminished after tFCI. Therefore, translocation of Rac1 to membrane remarkably increased, which eventually led to NOX activation after focal ischemia (Fig. 3) (38).
The role of NOX in cerebral ischemic damage in vivo
In OGD-treated hippocampal and cortical neurons, three distinct mechanisms contributed to generating oxygen radicals and causing oxidative injury, with each mechanism operating at a different stage of ischemia and reperfusion (1). At the early phase during hypoxia, oxidants were produced by mitochondria and xanthine oxidase. A the late phase, generation of oxidants appeared during reoxygenation and they were absent in gp91phox−/− neurons. Therefore, NOX is the main contributor to late-phase oxidant generation (1). Further, inhibition of NOX by diphenyleneiodonium or genetic knock-out of the gp91phox subunit reduced OGD-mediated cell death by 30% to 40% (1). Consistent with the findings in an OGD model, 2 h of glucose deprivation and 1 h of glucose reperfusion caused significant O2 ·− generation in mouse cortical neurons. This phenomenon was abolished by the NOX inhibitor apocynin or genetic knock-out of the p47phox subunit (69). Moreover, after 2 h of glucose deprivation and 22 h of glucose reperfusion, wild-type neurons exhibited 92% cell death, whereas only 8% to 10% cell death was observed in p47phox−/−-treated and apocynin-treated neurons (69). These data further support the evidence that NOX plays a primary role in ischemic neuronal injury.
During brain ischemia, a significant amount of glutamate is released from cells. Levels of extracellular glutamate rose from 50–90 μM to 1–5 mM in the striatal core, by microdialysis (42). A high level of extracellular glutamate caused excitotoxicity, which further exacerbated ischemic brain injury (42). Massive oxidant generation after excitotoxicity is well established and is an important mechanism of excitotoxic cell damage (42). In vitro studies demonstrated that NOX plays a central role in oxidant generation under excitotoxic conditions. Ca2+ influx through N-methyl-
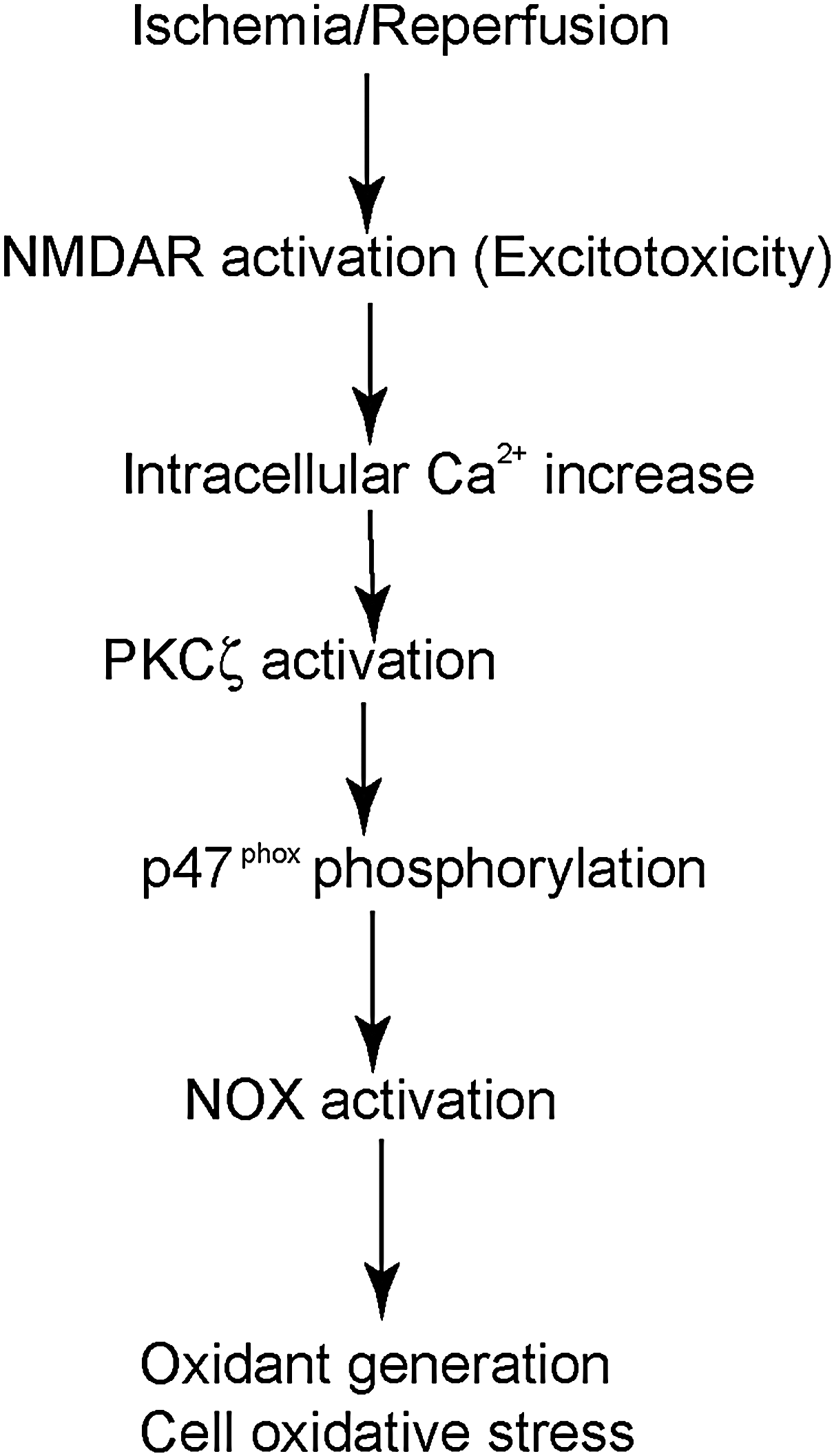
The role of NOX in cerebral ischemic damage in vivo
The role of NOX in focal ischemic brain damage was studied by comparing brain damage between wild-type and gp91phox−/− mice after tFCI (13, 33, 72). Infarct volume at 22 h of reperfusion was significantly less in the gp91phox−/− mice (29.1 mm3) than in their wild-type littermates (54.0 mm3) after 2 h of tFCI. Further, elimination of functional NOX from circulation by bone marrow transplant in the gp91phox−/− mice did not reduce the infarct size induced by tFCI. This suggests that inactivation of circulating NOX in neutrophils does not confer protection against transient ischemia and reperfusion (72). Recently, we investigated the role of NOX in cerebral ischemia using both pharmacological and genetic manipulation approaches (13). NOX inhibition by apocynin reduced postischemic O2 ·− generation in neurons and microglia (13). In addition, the amount of lipid or DNA peroxidation products, including MDA, HNE, and 8-OHdG, was significantly reduced in the gp91phox−/− and apocynin-treated mice. Thus, inhibition of NOX reduces free radical generation and alleviates oxidative damage to brain macromolecules, including DNA and lipids, in vivo (13). Moreover, NOX promotes oxidant formation in cerebral vascular endothelial cells and contributes to the disruption of the BBB, a major component of the neurovascular unit, after ischemia. Genetic ablation of gp91phox or treatment with apocynin alleviated the postischemic BBB disruption and brain hemorrhage at 24 h of reperfusion (33). We further compared brain damage at 72 h of reperfusion after focal ischemia among wild-type, gp91phox−/−-treated, and apocynin-treated mice (13). At 72 h of reperfusion, the brains showed a larger infarction volume than at 24 h (106 vs. 53 mm3), which suggests a delayed cell death process. The gp91phox−/−-treated or apocynin-treated mice still showed 46% to 50% less brain infarction than the wild-type mice at 72 h of reperfusion. This further confirms that NOX inhibition indeed protects the brain, not just delays the progress of brain damage after ischemia (13). Further, apocynin treatment also significantly reduced neurological deficit scores in mice after focal ischemia. Thus, the mice with NOX inhibition not only showed less brain damage, but also maintained better behavioral functions (13, 71).
In addition to focal ischemia, NOX inhibition also protects the brain against forebrain and global ischemic injury. After mice were subjected to transient forebrain ischemia, the brains showed a threefold increase in Et signals in CA1 neurons relative to sham controls. This increase was almost abolished by p47phox−/− or apocynin administration. The number of degenerating neurons identified in the CA1 hippocampus at 3 days of reperfusion was also nearly abolished in NOX-inhibited mice (70). Further, the role of NOX in global ischemia was investigated. Global ischemic insult caused ∼90% hippocampal pyramidal neuronal degeneration in gerbils. Strong HNE immunoactivity was also revealed, indicating lipid peroxidation due to excessive production of oxidants (73). Pretreatment with apocynin significantly attenuated HNE expression and increased CA1 neuronal survival from ∼10% to ∼60% (73). tGCI insult caused injury to selective mouse striatal medium spiny neurons. NOX inhibition significantly attenuated the death of these neurons (77). Therefore, NOX contributes to brain damage in global ischemia as well as in focal ischemia.
Crosstalk between NOX and SOD in cerebral ischemia
Intracellular redox homeostasis is normally maintained by concerted actions of antioxidant systems, including SOD and pro-oxidant enzymes such as NOX. After ischemia, gp91phox protein expression and NOX activity rise significantly, which shifts the balance to increasing oxidative stress in brain tissue (13, 27). The increase in gp91phox in SOD1 Tg mice was much less than that in wild-type mice after tFCI. Further, in SOD1−/− mice, there was significantly more gp91phox upregulation than in wild-type mice after cerebral ischemia (13). This suggests that NOX expression is regulated by the redox state of the brain tissue and endogenous SOD levels (13). In contrast, administration of the NOX inhibitor apocynin reduced infarction by >50% in SOD2−/− mice subjected to tFCI. This further indicates that the elaborate crosstalk between NOX and SOD and the inhibition of NOX can be a new therapeutic target for an antioxidant-enzyme deficit (13).
Oxidative Stress Involving Postischemic Mitochondrial Dysfunction
Cytochrome c–mediated apoptosis after ischemia
Mitochondria play a central role in apoptosis by releasing cytochrome c from the intermembrane space to the cytoplasm. In the cytosol, cytochrome c binds apoptotic protease activating factor 1 and, in the presence of dATP, forms a multimeric complex. Procaspase-9 is then recruited to the complex and activated by autocleavage. Activated caspase-9 is the initiator of and is upstream of the executor caspase, caspase-3, which attacks the cell structure, leading to apoptosis (Fig. 5) (74, 76). Activated caspase-9 directly cleaves procaspase-3, and active caspase-3 triggers apoptosis (74). The inhibitor-of-apoptosis family proteins negatively regulate caspase activation by binding caspases. Second mitochondria-derived activator of caspases, which is also released from mitochondria during cell-death-related stimuli, neutralizes the effect of inhibitor-of-apoptosis family proteins and promotes caspase activation (Fig. 5) (74).
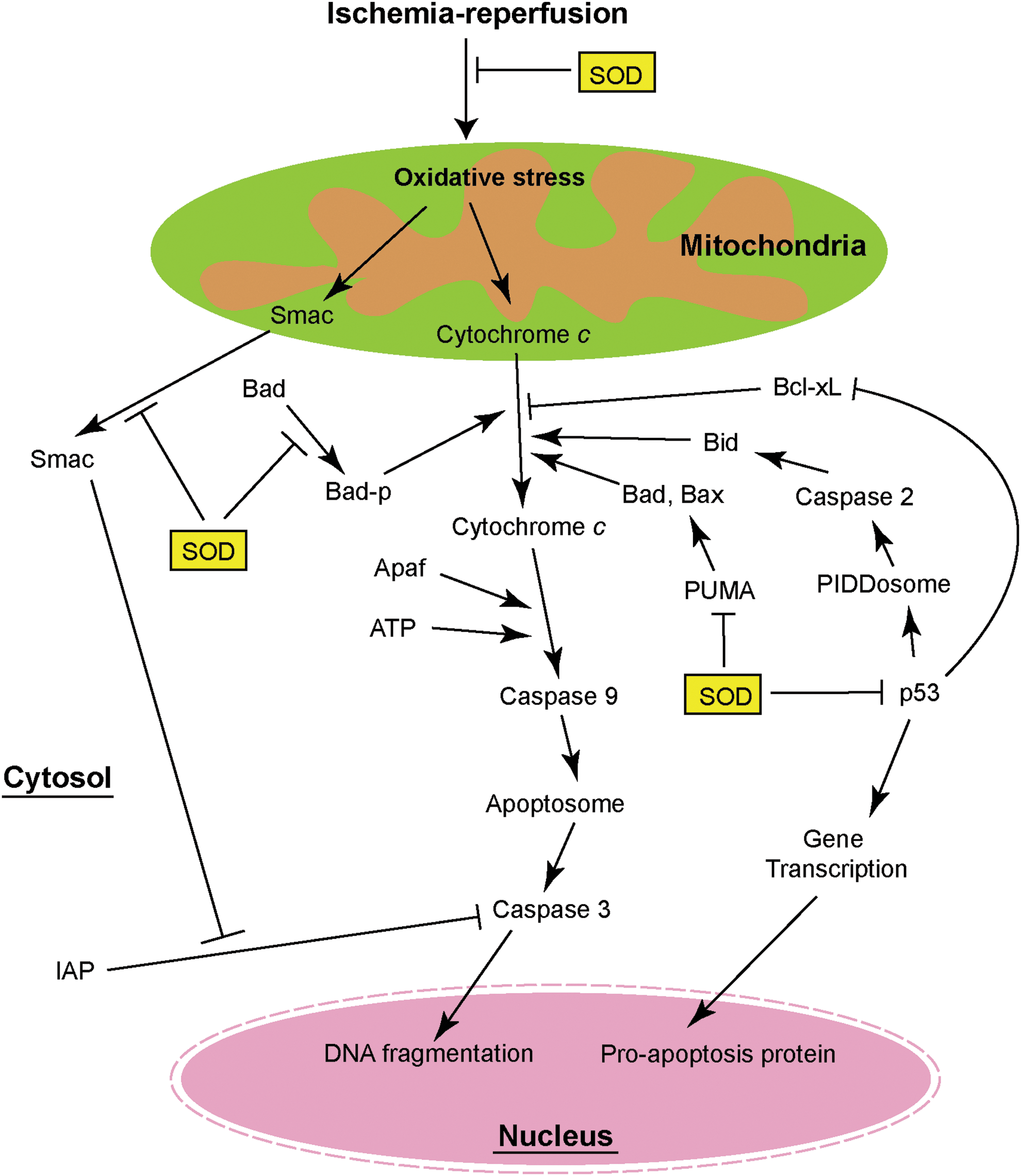
A large amount of literature has demonstrated that oxidative stress causes the release of cytochrome c and subsequent apoptosis after brain ischemia (10, 12, 16, 22, 39). After tFCI, the cytosolic cytochrome c level began to increase 2 h after reperfusion, and correspondingly, mitochondrial cytochrome c was significantly reduced. Cytosolic cytochrome c release was less significant in SOD1 Tg mice compared with wild-type mice. These data suggest that SOD1 blocks release of cytochrome c from mitochondria and could thereby reduce apoptosis after tFCI (22).
A major pro-apoptotic protein, Bad, is important for cytochrome c release. Bad exists as an inactivate complex with the molecular chaperone 14-3-3. Apoptotic stimuli caused the dephosphorylation of Bad and its translocation to the mitochondrial membrane, where it neutralized the antiapoptotic protein Bcl-XL and promoted cytochrome c release (79). SOD1 overexpression may inhibit cytochrome c release through prevention of Bad dephosphorylation and translocation to mitochondria. Further, SOD1 increased the binding between Bad and 14-3-3. Therefore, SOD1 inhibited cytochrome c release from mitochondria and promoted cell survival by shifting the fate of Bad (60) (Fig. 5). Consistent with studies of focal ischemia, SOD1 overexpression also alleviated cytochrome c and second mitochondria-derived activator of caspases release from mitochondria, and reduced cleaved caspase-9 and caspase-3 compared with wild-type mice after global ischemia. Further, 85% of hippocampal CA1 neurons in wild-type rats showed apoptotic DNA damage 3 days after ischemia, which was significantly higher than in their SOD1 Tg littermates (45%) (68).
The role of SOD2 in apoptosis was studied using SOD2−/+ mice. Cytosolic accumulation of cytochrome c and DNA laddering were more severe in SOD2−/+ mice compared with wild-type animals after permanent FCI (21). In addition, more cleaved caspase-9 and cytochrome c proteins were detected in the cytosol of SOD2−/+ mice by Western blotting. Further, the SOD2−/+ mice showed significantly more TUNEL-positive cells than wild-type animals (56). Therefore, SOD2 blocked cytosolic release of cytochrome c, and then cytochrome c mediated the downstream apoptosis pathway, and finally alleviated apoptosis after cerebral ischemia.
The p53 pathway mediated apoptosis after ischemia
The tumor suppressor gene p53 induces apoptosis through transcription-dependent pathways, in which p53 binds to specific DNA sequences and transcripts expression of a number of pro-apoptotic protein genes, including Bcl-2-associated X protein, BH3-interacting domain death agonist (Bid), and p53-upregulated modulator of apoptosis (PUMA) (24) (Fig. 5). Further, p53 is also involved in mitochondrial-mediated apoptosis through transcription-independent pathways. Apoptotic stimuli can cause p53 to translocate into the mitochondrial membrane and form an inhibitory complex with protective Bcl-XL in the mitochondrial fraction. Therefore, the p53 protein can facilitate the release of cytochrome c, leading to apoptosis by inactivation of Bcl-XL (16) (Fig. 5). p53 translocation is facilitated after cerebral ischemia, and inhibition of p53 is neuroprotective (16, 41). As well as alleviating apoptosis, recent studies also demonstrated that inhibition of p53 enhances the survival and proliferation of endogenous neural progenitor cells and functional recovery after stroke in rats (44).
Under physiological conditions, degradation of p53 hindered its role in apoptotic regulation and prevention of cell death. In addition, the transcription regulatory activity of p53 was abolished by phosphorylated murine double minute-2 gene in the nucleus (47). During cerebral ischemia, increased amounts of p53 were attributed to greater accumulation of ubiquitylated p53 and less p53 degradation (61). SOD1 overexpression not only facilitated p53 degradation through reduction of ubiquitylated p53, but also promoted murine double minute-2 gene phosphorylation and nuclear translocation. Therefore, SOD1 strongly inhibits p53-mediated apoptosis by furthering p53 degradation and decreasing p53 activity (61) (Fig. 5).
The p53 gene transcription product, PUMA, is a critical mediator of apoptosis induced by a wide variety of stimuli. It directly binds and antagonizes antiapoptotic Bcl-2 family members such as Bcl-2-associated X protein and Bak. Therefore, it induces mitochondrial dysfunction and caspase activation (78). After global ischemia, p53-mediated PUMA transcription and PUMA translocation to the mitochondrial membrane were significantly increased. Both the p53 inhibitor pifithrin-α and SOD1 overexpression abolished PUMA upregulation (51). The p53 pathway is also involved in neuronal apoptosis through another important downstream protein, the PIDDosome, which includes a protein complex of p53-induced protein with a death domain (PIDD) and procaspase-2. Global ischemia caused p53-mediated/caspase-2 activation through the PIDDosome and caspase-2 promoted neuronal apoptosis by activating Bid in mouse brains (52). It would be interesting to further detect the role of oxidative stress in the p53/PIDD/caspase-2/Bid pathway using SOD mutant mice.
In summary, oxidative stress can induce neuronal apoptosis through specific mitochondrial or p53-mediated pathways (Fig. 5). In addition to the specific pathways discussed above, alleviation of unspecific mitochondrial membrane damage by SOD overexpression cannot be excluded and it may also contribute to reduced cytochrome c release. Alternatively, increased SOD in the cytosol or mitochondria may directly reduce mitochondrial oxidative stress, thereby preventing the release of cytochrome c.
Clinical Trials of Antioxidants and the Challenges of Experimental Animal Studies
Despite our understanding of the pathophysiological role of oxidative stress in ischemic brain damage, and the promising results conferred by antioxidants in experimental stroke studies, a recent randomized, double-blind trial (stroke acute ischemic NXY treatment II) that employed NXY-059, a scavenger of free radicals, failed to demonstrate favorable outcomes when compared with matching placebo groups (64). There may be multiple reasons for the discrepancies between clinical and experimental studies, including but not limited to, the differences in subjects (humans vs. rodents), therapeutic window (within 6 h postischemia vs. pretreatment or post-treatment), age (68.8 years vs. juvenile animals), and comorbidity (77.2% hypertension, 46.1% cardio-embolic stroke, 33% ischemic heart disease, and 24.9% diabetes vs. healthy animals). Future investigation of the therapeutic window and use of aging animals with comorbidities (hypertension, diabetes, etc.) may help the successful translation to clinical usage.
Conclusions
Oxidant generation is a hallmark of the pathological process after cerebral ischemia and reperfusion. Genetic manipulation of pro-oxidant or antioxidant enzymes has provided a powerful tool to study the roles of free radicals in ischemic brain injury. SOD overexpression protects against ischemic brain damage by reducing oxidative injury and modifying redox signals. SOD also alleviates mitochondrial dysfunction and subsequent apoptosis after cerebral ischemia. In contrast, NOX, a pro-oxidant enzyme, exacerbates cerebral oxidative stress and contributes to ischemic brain damage. Comprehensive knowledge regarding the crosstalk between SOD and NOX in postischemic oxidative stress (Yin and Yang) may provide insights into the development of potential therapeutic strategies for stroke.
Footnotes
Acknowledgments
The authors thank Liza Reola and Bernard Calagui for technical assistance and Cheryl Christensen for editorial assistance. This work was supported by Grants PO1 NS014543, RO1 NS025372, RO1 NS036147, and RO1 NS038653 from the National Institutes of Health, and by the James R. Doty Endowment. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.