
Abstract
Nitric oxide (NO) mediates cellular signaling pathways that regulate a plethora of physiological processes. One of the signaling mechanisms mediated by NO is through S-nitrosylation of cysteine residues in target proteins, which is now regarded as an important redox-based physiological action. Deregulation of the protein S-nitrosylation upon nitrosative stress, however, has also been linked to various human diseases, such as neurodegenerative disorders. Between these physiological and pathophysiological roles, there are mechanisms whereby a milder level of nitrosative stress provides S-nitrosylation of some proteins that counteracts the pathological processes, serving as a negative feedback mechanism. In addition, NO has recently emerged as a mediator of epigenetic gene expression and chromatin changes. In this review, these molecular mechanisms, especially those in the central nervous system and neurodegenerative disorders, are described. Antioxid. Redox Signal. 14, 1493–1504.
Introduction
NO participates in cellular signaling pathways that regulate broad aspects of brain function physiologically, including neurotransmission per se, modulation of synaptic plasticity, and neurodevelopment (11, 33, 37, 39, 52, 68, 103). Excessive generation of NO and NO-derived reactive nitrogen species (nitrosative stress) (115), however, has also been implicated in the pathophysiology of neurodegenerative disorders (11, 18, 19, 39, 99, 100). These effects were largely achieved by activation of guanylate cyclase to form cyclic guanosine-3′,5′-monophosphate (37, 72). Emerging evidence has suggested that a more prominent action of NO is via posttranslational modifications, that is, reversible modification such as S-nitrosylation of thiol groups in regulatory proteins (50, 77) and irreversible modification such as protein tyrosine nitration (55, 114) (Fig. 1). Tyrosine nitration is a covalent addition of a nitro group (-NO2) to one of the two equivalent ortho-carbons of the aromatic ring in tyrosine residues, which affects protein structure and function (55, 56, 74, 85, 114, 120, 131). Elevated levels of protein tyrosine nitration, which are reported in several neurodegenerative diseases, may be utilized as a pathological marker under nitrosative stress (56, 74, 120, 131).

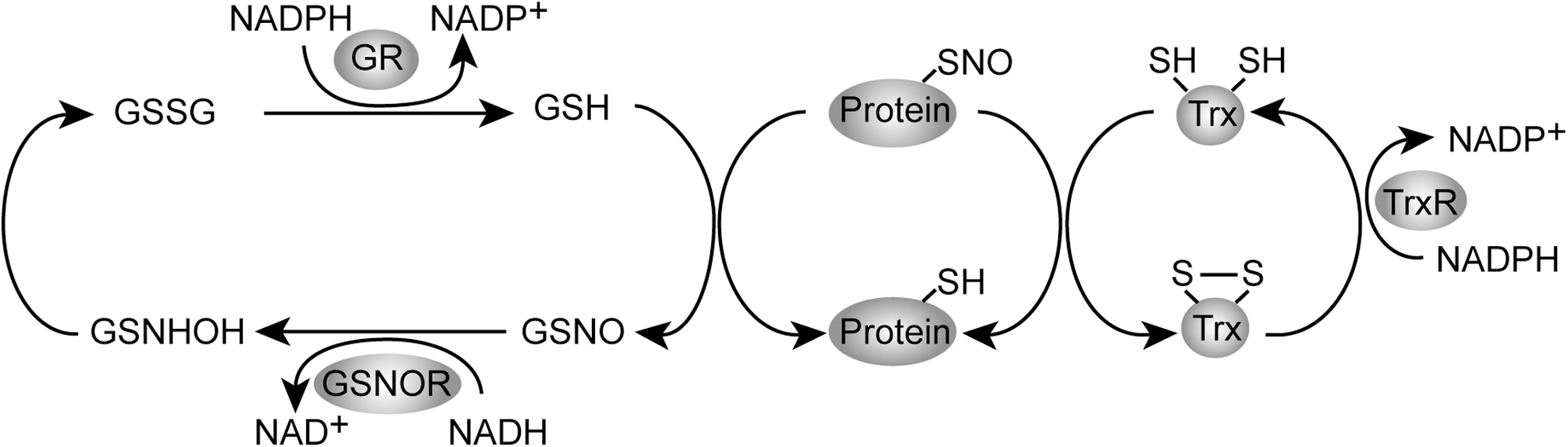
S-nitrosylation is a covalent addition of an NO group to a cysteine thiol/sulfhydryl (RSH or, more properly, thiolate anion, RS-), which results in formation of an S-nitrosothiol derivative (35, 50). S-nitrosylation is likely a prototypic redox-based signaling mechanism (134), because the S-nitrosothiols not only can be reduced to form thiols, but also can be oxidized to form either S-glutathionylation (–SSG), cysteine sulfenic acid, cysteine sulfinic acid, or cysteine sulfonic acid (38, 133, 145, 148). S-nitrosylation of cysteines is readily reversible with high spatial and temporal specificity. In addition, there are two major mechanisms to remove NO group from S-nitrosylated Cys thiol side chains (S-nitrosoglutathione reductase system and thioredoxin system) (6, 34) (Fig. 2). In the former mechanism an NADH-dependent oxidoreductase S-nitrosoglutathione reductase specifically catalyzes the denitrosylation of GSNO, by which protein S-nitrosylation is regulated in the cellular equilibrium between S-nitrosylated proteins and GSNO. The latter mediates direct denitrosylation of multiple S-nitrosylated proteins. The temporal and spatial regulation of S-nitrosylation and denitrosylation confers specificity to the NO-based cellular signaling (6). In this review article, we focus on the role for S-nitrosylation in the physiology and the pathophysiology in the central nervous system (Fig. 3).
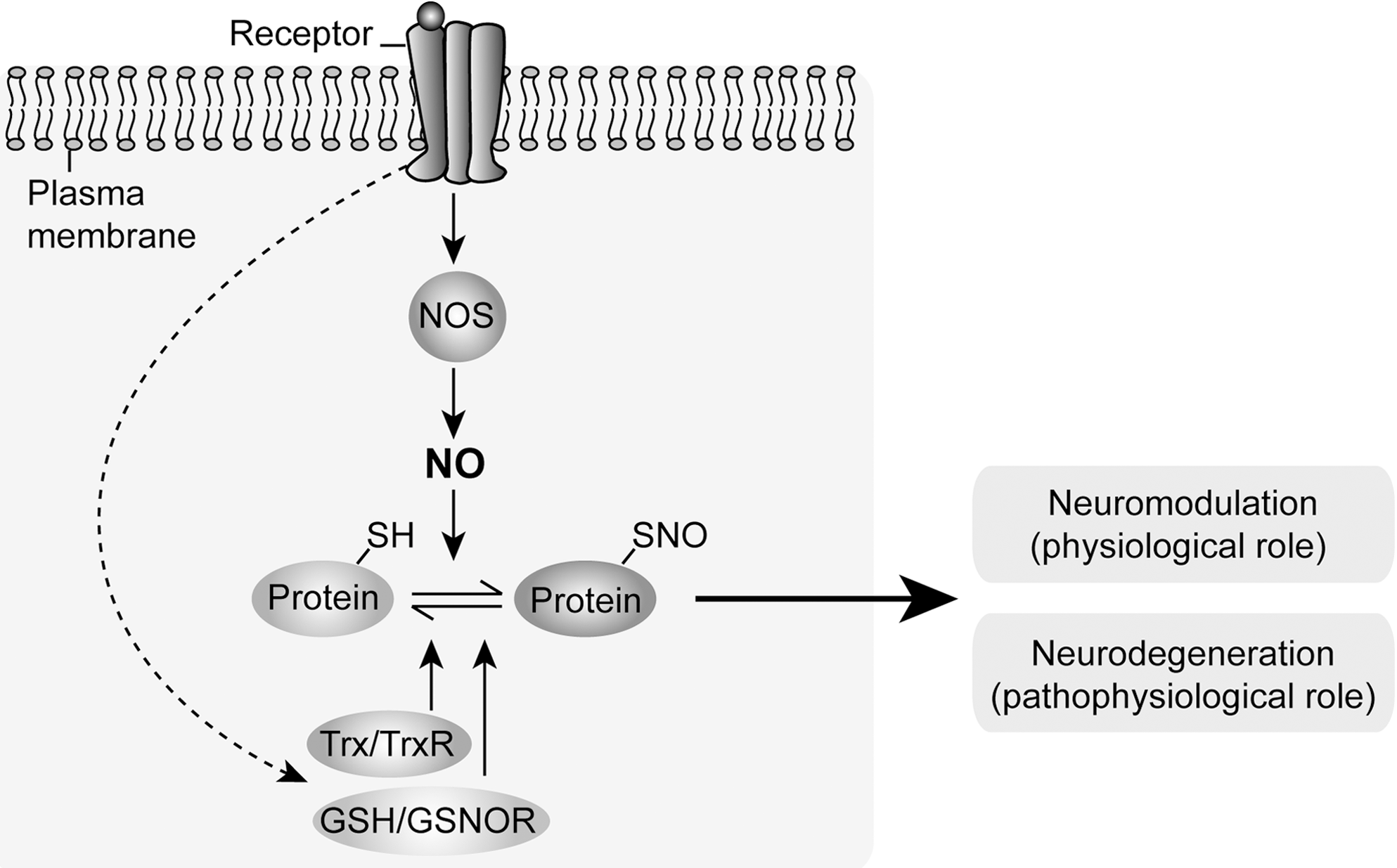
One model for understanding both NO-mediated neuromodulation (physiological role) and neurodegeneration (pathophysiological role) involves a central role for the N-methyl-
A potential new role for protein S-nitrosylation is its influence on the cascades that regulate epigenetic regulation of gene expression (104). Epigenetic mechanisms have both physiological and pathophysiological implications. In the last section of this review, we discuss this new topic.
S-Nitrosylation and Neuromodulation
Through S-nitrosylation NO modifies a variety of proteins that regulate various cellular and physiological pathways (Fig. 4). In this section, we highlight two representative pathways that play key roles in neuromodulation.

S-nitrosylation and glutamate receptors
Glutamate neurotransmission via several types of glutamate receptors, including NMDA-R and alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPA-R), mediates many physiological events, such as neuronal development, neuronal connectivity, and synaptic plasticity.
Regulation of AMPA-R trafficking to the cell surface is a key mechanism that underlies synaptic plasticity in long-term depression and long-term potentiation, as models of learning and memory (65, 128). Once NO is generated physiologically by nNOS after NMDA-R activation, this promotes S-nitrosylation of N-ethylmaleimide sensitive factor, which enhances surface expression of AMPA-R (53). Another mechanism by which NMDA-R activation enhances AMPA-R surface expression is through S-nitrosylation of stargazin (125). Stargazin is a transmembrane AMPA-R regulatory protein, which is also known to modulate AMPA-R surface expression. A recent report indicates that activation of NMDA-R lead to S-nitrosylation of stargazin at cysteine 302, which increases the stargazin-AMPA-R protein interaction and surface expression of AMPA-R (125).
Excessive activation of NMDA-R results in neuronal cell death, and thus tight regulation of NMDA-R activity is crucial for physiological maintenance of the synapse (46). NO also S-nitrosylates NR1 and NR2 subunits of NMDA-R at specific cysteines/thiol groups and decreases calcium entry via this ionotropic receptor (14, 61, 78). This is an important negative feedback mechanism that avoids overactivation of NMDA-R. In addition, NMDA-R receptor enhances S-nitrosylation of serine racemase, resulting in a decrease of its catalytic activity in generating D-serine (96), a coagonist of NMDA-R (94, 109). These mechanisms are likely to be required for proper neuronal development and adult brain function. Further, physiological levels of synaptic NMDA-R activity boost intrinsic antioxidant defenses that are important for neuronal longevity (45, 46, 110).
In analogy to NMDA-R and serine racemase, S-nitrosylation of caspases, main executors of cell death signaling, inhibits their enzymatic proteolytic activities (63, 76, 138, 141, 143). Cortical neurons treated with several NO donors, including S-nitrosothiols, exhibited a significant reduction in staurosporin-induced caspase-3 and caspase-9 activation, probably owing to the NO-mediated S-nitrosylation of the cysteine residue at the catalytic site of these caspases (152).
S-nitrosylation and transcription factors
Several studies have demonstrated that NO affects a wide variety of transcription factors, including estrogen receptor (ER), hypoxia-inducible factor (HIF), nuclear factor-kappa B (NF-κB), early growth factor-1 (egr1), and cAMP response element binding (CREB) [reviewed in (24)].
ERs and HIF are directly S-nitrosylated by NO, which attenuates their ability to activate gene transcription (36, 75). Estrogens are important for synaptic plasticity and memory processes, and their actions are mediated via two distinct receptors (ERα and ERβ), both of which are widely expressed in the central nervous system (88, 132). HIF-1α expression is induced by hypoxic conditions in neurons, astrocytes, and ependymal and endothelial cells, which is crucial for neuronal cell viability and function under physiological and pathophysiological conditions [reviewed in (25)]. NO also S-nitrosylates egr1 and NF-κB (p50) in the zinc–sulfur clusters, which inhibits binding of zinc to the domains and in turn blocks these transcription factors from binding with DNA (24, 82). In addition, IκB kinase β is S-nitrosylated and enzymatically repressed, leading to decreases in IκB phosphorylation and NF-κB nuclear translocation (116).
Several studies have shown that NO modifies the activity of CREB in regulating neuronal survival, differentiation, and plasticity (21, 79, 97, 112, 117). There is no evidence that CREB is directly S-nitrosylated. Nonetheless, protein S-nitrosylation is involved in the effects of NO on CREB (104): S-nitrosylation of histone deacetylase 2 (HDAC2) at cysteines 262 and 274 upon exposure to brain-derived neurotrophic factor (BDNF) detaches this molecule from the chromatin and facilitates acetylation of histones, which in turn promotes CREB-dependent gene transcription.
Of note, S-nitrosylation also occurs in bacteria. In Escherichia coli, S-nitrosylation of the transcription factors OxyR and SoxR enhances their transcriptional activity and modulates bacteria response to oxidative and nitrosative stress (30, 47).
S-Nitrosylation and Neurodegeneration
Although NO has many physiological functions, it also has pathological roles. Once excess NO is generated (nitrosative stress), it reacts with oxygen to form very toxic reactive nitrogen species, such as NO2, N2O3, and ONOO− (11). In addition, recent studies have shown aberrant levels of protein S-nitrosylation in neurodegenerative disorders, including Alzheimer's, sporadic Parkinson's, diffuse Lewy body diseases, and stroke (18, 35, 99). In these disorders, proteins with abnormal S-nitrosylation include GAPDH, parkin, protein disulfide isomerase (PDI), Prx2, X-linked inhibitor of apoptosis (XIAP), dynamin-related protein 1 (Drp1), heat-shock protein 90 (HSP90), and matrix metalloproteinase-9 (MMP-9) (13, 20, 32, 38, 83, 143, 145, 148) (Fig. 5). Among them a novel role for an old enzyme, GAPDH, which has been characterized by many investigators (22, 42, 43, 130), is described in a subsection below.

Mutation in parkin, an E3 ubiquitin ligase, is known to cause an autosomal recessive juvenile parkinsonism (67). Zinc-binding cysteine residues in the RING domain of parkin have been identified as targets of S-nitrosylation (148). S-nitrosylation of parkin increases its E3 ligase activity (148) and promotes its auto-ubiquitination, ultimately inhibiting its enzymatic activity (20, 148). PDI is an endoplasmic reticulum (ER)-associated chaperone protein that prevents neurotoxicity caused by ER stress and protein misfolding (145). PDI is S-nitrosylated at one or both of the cysteine thiols in each of its dithiol (Cys-Gly-His-Cys) active sites, resulting in the inhibition of its disulfide isomerase activity and accumulation of misfolded protein in the ER (145). Cumulative S-nitrosylation of parkin and PDI might underlie the accumulation of misfolded and ubiquitylated proteins that ultimately leads to cell death.
S-nitrosylation of XIAP, a well-known antiapoptotic protein, has been reported to increase in patients with Parkinson's disease (143). S-nitrosylation of cysteine residues within the baculoviral IAP-repeat motifs of XIAP was observed and compromises its antiapoptotic function (143). S-nitrosylated XIAP fails to bind to caspase-3 and thus fails to prevent caspase-3 activation (143). This suggests that S-nitrosylation of XIAP can compromise neuronal survival in Parkinson's disease.
Prx2, a 2-Cys Prx, a member of a family of abundant antioxidants is known to protect against oxidative stress in neurons (113). Prx2 is inactivated by S-nitrosylation of both its catalytic and resolving cysteine residues (C51 and C172, respectively), sensitizing dopaminergic neurons to H2O2-dependent cell death (32). Increased nitrosative stress and Prx2 S-nitrosylation might contribute to the loss of dopaminergic neurons in Parkinson's disease (32).
S-nitrosylation of Drp1 is enhanced in brains from patients with Alzheimer's disease (13). The mitochondrion undergoes consistent fusion and fission to maintain its proper function, and Drp1 is one of the important proteins that regulate mitochondrial fission. S-nitrosylation of Drp1 at C644 promotes its multimerization and thus mitochondrial fission, which causes neuronal damage (13). Further, exposure of nNOS-expressing cells to β-amyloid protein results in Drp1 S-nitrosylation, suggesting a role for S-nitrosylation in neurodegeneration associated with Alzheimer's disease (13).
HSP90, a chaperone protein and coactivator of eNOS, is another S-nitrosylated protein. This posttranslational modification decreases HSP90 ATPase activity and its positive effect on eNOS activity (83). Postmortem brains from patients with Alzheimer's disease exhibit increased levels of HSP90 (31, 62), and S-nitrosylation of HSP90 is likely to contribute to accumulation of tau and β-amyloid aggregates in the disease (98).
MMP-9, a protein involved in remodeling of extracellular matrix, is induced (93, 118) and S-nitrosylated during ischemic brain injury (stroke) (38). S-nitrosylation activates MMP-9, which leads to neuronal apoptosis (38).
S-nitrosylated GAPDH
GAPDH, which was once considered to be a simple housekeeping protein, has been proven to be involved in processes far beyond glycolysis (16, 22, 42, 43, 130) (Fig. 6). Among its multifunctional roles, GAPDH plays a role in stress sensing and it mediates cellular dysfunction (42, 43). GAPDH has received particular attention as one of the major targets of NO in cells after it was shown that NO induced ADP ribosylation (29, 150) and S-nitrosylation of GAPDH (92). S-nitrosylation of GAPDH further facilitates covalent linkage with NAD (89, 91). These posttranslational modifications inhibit its glycolytic activity (29, 90 –92). Thus, loss of GAPDH glycolytic activity could be a potential mechanism of NO-induced cell death (90, 102). In contrast to this loss-of-function theory, however, several groups have reported that gain-of-function of GAPDH may also play a role in cell death. This novel concept has been supported by many reports in which treatment with antisense oligonucleotides or RNAi to GAPDH reduces neuronal and non-neuronal cell death, as long as energy loss by GAPDH is supplemented (41, 57 –60, 122, 124). In the course of addressing the mechanisms for this gain of toxic function of GAPDH, we found that a small pool of GAPDH is translocated to the nucleus during cell dysfunction and death (124). Other groups have also replicated this observation (60, 66, 73, 84, 86, 87, 123, 137, 139). Thus, we asked, what is the molecular mechanism underlying this nuclear translocation?
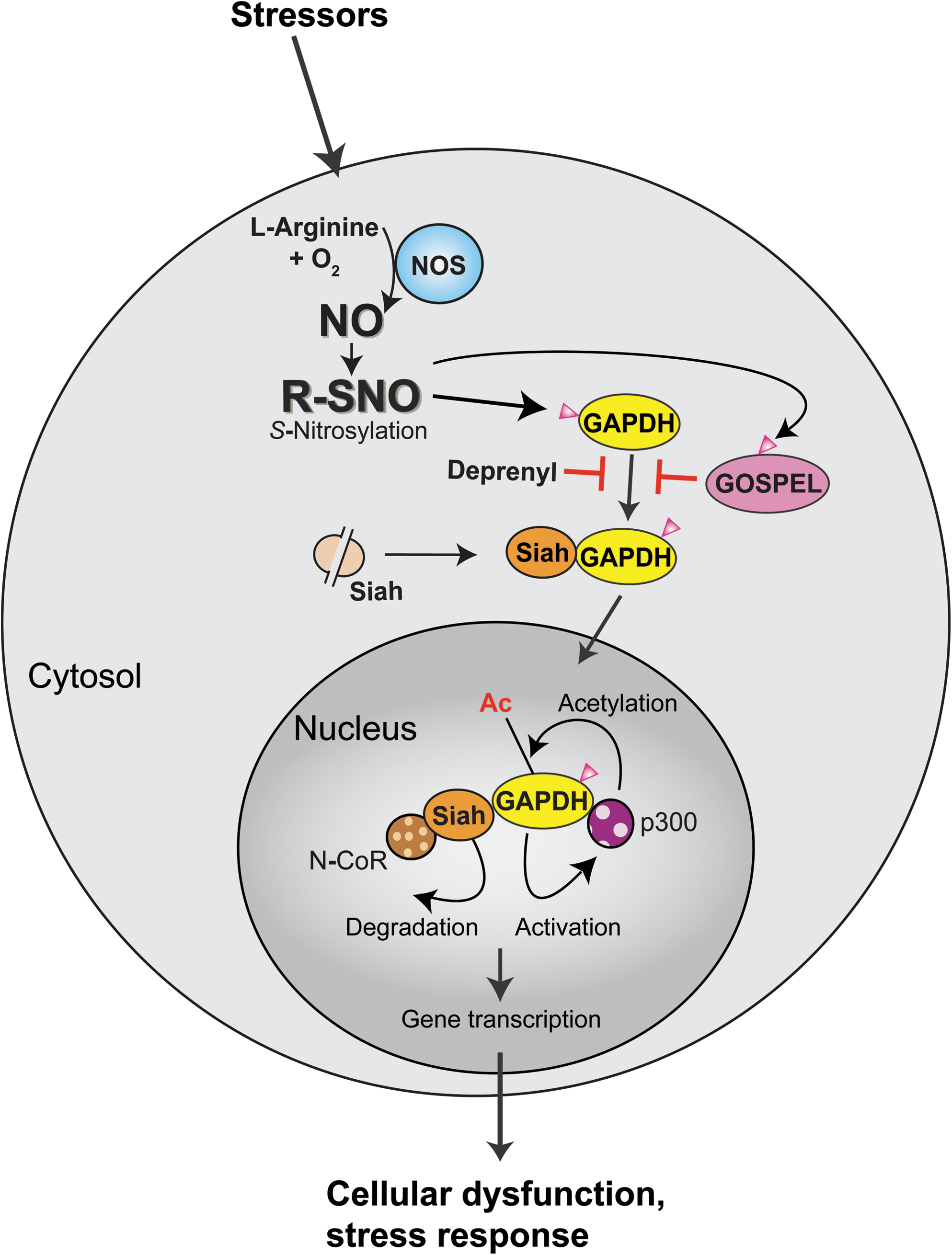
Stimulation of nNOS in neurons or of iNOS in macrophages triggers S-nitrosylation of GAPDH at cysteine 150, which allows GAPDH to bind to the E3 ubiquitin ligase Siah with a strong nuclear localization signal. Consequently, nuclear translocation of GAPDH occurs together with Siah (41). Stabilized Siah in the protein complex with S-nitrosylated GAPDH seems to facilitate ubiquitination and degradation of the nuclear corepressor (N-CoR) (41, 151). S-nitrosylation (followed by sulfonation shown in Fig. 7) on only a few percentages of GAPDH is sufficient to activate this cascade. Further studies have shown that nuclear-translocated GAPDH is further acetylated at lysine160 by the histone acetyltransferase (HAT) p300/CBP via direct protein interaction, which in turn stimulates the catalytic activity of p300/CBP. This nuclear event leads to the acetylation of downstream targets, including the tumor suppressor p53 (127). By both of these mechanisms, the nuclear GAPDH–Siah complex may regulate gene expression, which results in cellular dysfunction and death (Fig. 6).

Accumulating evidence suggests that nuclear GAPDH may be involved in several neurodegenerative disorders (16). Nuclear GAPDH has been found in fibroblasts and in postmortem brains from patients with polyglutamine diseases (such as Huntington's disease or dentatorubral-pallidoluysian atrophy) (86, 129), Parkinson's disease (139), and Alzheimer's disease (87, 144). Some studies suggested that binding of GAPDH occurs with β-amyloid peptides, mutant huntingtin, androgen receptor, and atrophin-1 (4, 10, 27, 70, 146). In an experimental model of brain ischemia, accumulation of nuclear GAPDH is also reported (137). Moreover, promising pharmacological evidence further supports a role for nuclear GAPDH in cell dysfunction and death: deprenyl used for symptomatic amelioration for patients with Parkinson's disease potentially may block GAPDH–Siah binding, in addition to its classic action as a monoamine oxidase B inhibitor (106, 136, 140, 147). Some of structural derivatives of deprenyl, even lacking this inhibitory action, are still neuroprotective (81, 106, 111, 136, 140, 147, 149). Among them, TCH346 shows neuroprotective action largely via blockade of GAPDH–Siah binding and nuclear translocation of the GAPDH–Siah protein complex (44), and rasagiline has shown neuroprotective effects in ethanol-induced cell death mediated by a novel GAPDH-monoamine oxidase B pathway (107, 108)
Deprenyl and TCH346 provide evidence that there are exogenous interventions that interfere with GAPDH–Siah binding, which is crucial in initiating a cell death cascade. We recently reported that there is an endogenous inhibitor against GAPDH–Siah binding and its sequential death cascade (126). An interactor of GAPDH, GAPDH's competitor of Siah protein enhances life (GOSPEL), is also S-nitrosylated at cysteine-47. This S-nitrosylation augments binding of GOSPEL with GAPDH, competing with binding of GAPDH with Siah (Fig. 6). Once nitrosative stress exceeds threshold, GAPDH–Siah binding seems to overwhelms GAPDH–GOSPEL binding; whereas, owing to more rapid kinetics of S-nitrosylated GAPDH binding with GOSPEL compared with that with Siah, GAPDH–GOSPEL binding inhibits GAPDH–Siah interaction at the initial phase of nitrosative stress. This is analogous to S-nitrosylation of NMDA-R as described above (14, 61, 78): activation of NMDA-R at a modest level has a protective mechanism resulting from S-nitrosylation (a type of negative feedback), by inhibiting the overactivation of this receptor that results in massive activation of nNOS, nitrosative stress, and cell death/dysfunction. Likewise, we reported that overexpression of GOSPEL is neuroprotective, whereas mutant GOSPEL lacking the S-nitrosylation site cannot bind to GAPDH and thus fails to block cell death in primary neuron cultures (126). This neuroprotective action of GOSPEL was further validated in a model with NMDA insult in vivo (126).
S-Nitrosylation and Epigenetics in Nervous System
Epigenetic chromatin remodeling, such as modifications of DNA and histones, is a central mechanism for regulation of gene expression (7, 71, 135) (Fig. 8). Among chromatin modifications, histone acetylation plays a pivotal role in the epigenetic regulation of transcription and other functions in cells, including neurons (3, 7, 12, 17, 26, 80, 85, 121, 142). HATs (12, 119) and HDACs (26, 101, 142) catalyze the acetylation and deacetylation, respectively, of histones at lysine residues. The interplay between HATs and HDACs alters the net balance of histone acetylation levels, thereby remodeling chromatin structure. In general, an increase in protein acetylation at histone tails results in a more open and relaxed chromatin conformation, thus facilitating transcription factor interaction with specific gene promoters and activating gene expression. HDACs often function as a component of the transcriptional repressor complex to silence gene expression and induce chromatin compaction through histone protein deacetylation. HATs are grouped into three distinct families of Gcn5-related N-acetyltransferases, MYST (
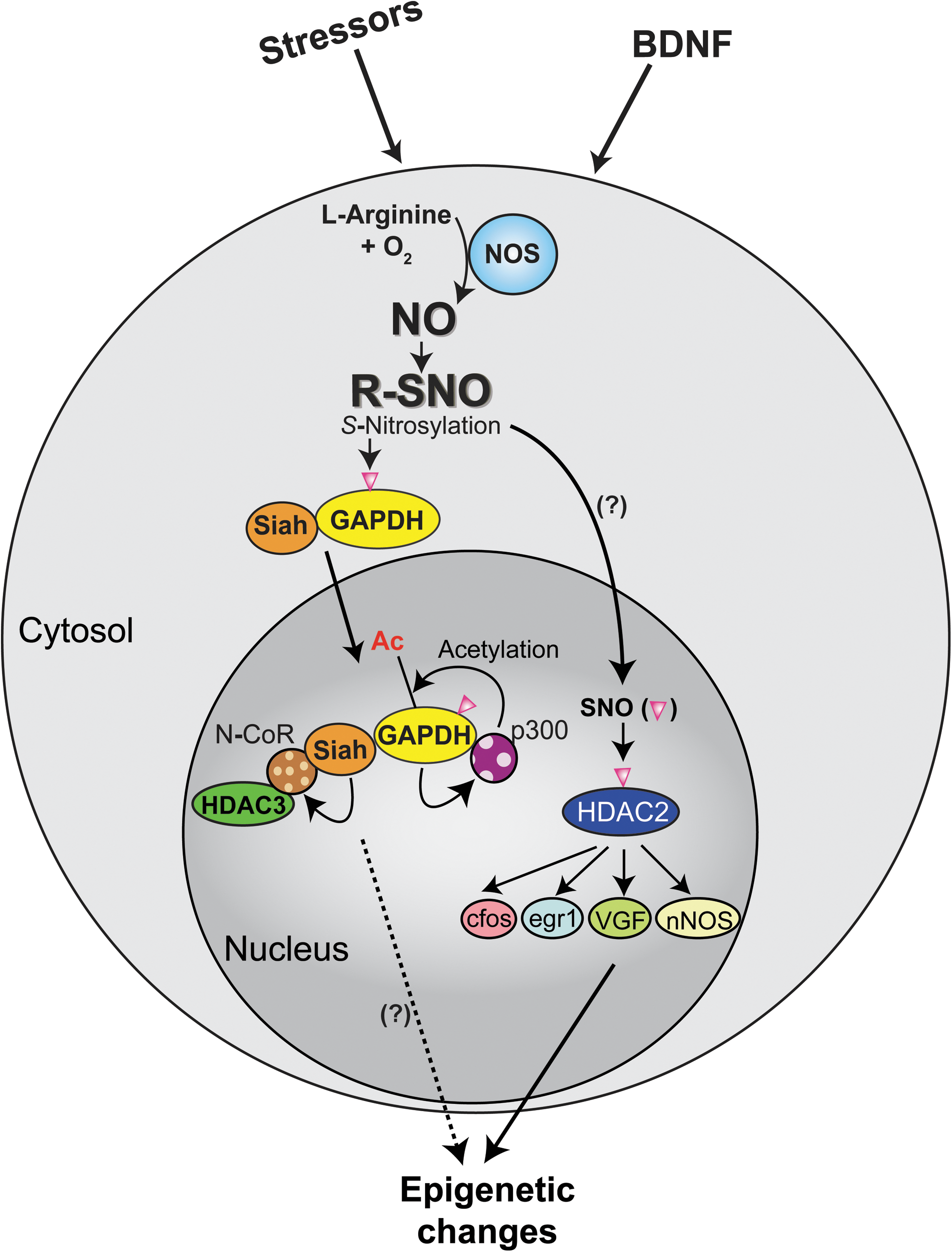
HDAC2 may be a representative target of S-nitrosylation in epigenetic control of neuronal gene expression (Fig. 8). Stimulation of cortical neurons with BDNF increases expression of nNOS, which in turn results in S-nitrosylation of HDAC2, an HDAC that is highly expressed in neurons, at cysteine residues 262 and 274 (104). This posttranslational modification induces dissociation of HDAC2 from CREB-regulated gene promoters, and increases histone acetylation at specific promoter regions and gene transcription, including c-fos, egr1, VGF, and nNos (104, 117). HDAC2 S-nitrosylation seems to be necessary for BDNF-dependent neuronal dendritic growth and branching, suggesting a key role of S-nitrosylation in neuronal development (104). S-nitrosylation of HDAC2 is also reported in muscles, especially those from a mouse model of Duchenne muscular dystrophy (23).
Is there any other important S-nitrosylated protein that may possibly play a key role in epigenetic control of neuronal gene expression? As described above, once S-nitrosylated, GAPDH translocates to the nucleus together with Siah (41). This GAPDH–Siah complex in the nucleus is known to affect p300/CBP and N-CoR. Considering that N-CoR interacts with HDAC3 (a class I HDAC) and SIRT1 (a class III HDAC), nuclear GAPDH–Siah may influence both HATs and HDACs and possibly play a role in epigenetic regulation of neuronal gene expression (Fig. 8). Neural precursor cells from N-CoR gene-disrupted mice display spontaneous differentiation into astroglia-like cells (49). SIRT1 is known to regulate neuronal differentiation (51). p300/CBP has also been shown to play an important role in development and memory consolidation as demonstrated in knockout mouse models (1, 64, 69, 105). Thus, as a working hypothesis we propose that a pool of GAPDH localized in the synaptic compartments can act as a sensor for neuronal activity and NO generation, conveys the signal to the nucleus, and then may modulate epigenetic regulation of gene expression via histone modifications contributing to neurodevelopment and plasticity.
Conclusions
Physiological S-nitrosylation modifies a number of proteins that regulate various cellular functions. On the other hand, excess S-nitrosylation that generates nitrosative stress contributes to activation of several cell death cascades, including the GAPDH cascade. Between these physiological and pathological roles, there are mechanisms whereby a milder level of nitrosative stress provides S-nitrosylation of target proteins that counteracts the death cascades, including S-nitrosylation of NMDA-R, GOSPEL, and caspases. Once the stress levels become catastrophic, NO may kill the unhealthy cells to protect the whole body. It remains to be elucidated how the balance of protein S-nitrosylation and denitrosylation is maintained under nitrosative stress. The overall mechanism of NO and nitrosative stress may consistently contribute to a general homeostasis of the organism. The brain is a key organ that contributes to the maintenance of the general homeostasis for the organism. Thus, the role for NO signaling in the neurons and neuronal networks will continuously be an important topic in biological science.
Footnotes
Acknowledgments
We thank Ms. Y. Lema for preparation of figures and Dr. P. Talalay for critically reading the manuscript. This work was supported by Silvo O. Conte Center MH-084018 (A.S.), MH-069853 (A.S.), and MH-088753 (A.S.), as well as by grants from Stanley (A.S.), CHDI (A.S.), HighQ (A.S.), S-R (A.S.), and NARSAD (N.S. and A.S.).