
Abstract
In their natural environments, cells are regularly exposed to oxidizing conditions that may lead to protein misfolding. If such misfolded proteins are allowed to linger, they may form insoluble aggregates and pose a serious threat to the cell. Accumulation of misfolded, oxidatively damaged proteins is characteristic of many diseases and during aging. To counter the adverse effects of oxidative stress, cells can initiate an antioxidative response in an attempt to repair the damage, or rapidly channel the damaged proteins for degradation by the ubiquitin-proteasome system (UPS). Recent studies have shown that elements of the oxidative stress response and the UPS are linked on many levels. To manage the extra burden of misfolded proteins, the UPS is induced by oxidative stress, and special proteasome subtypes protect cells against oxidative damage. In addition, the proteasome is directly associated with a thioredoxin and other cofactors that may adjust the particle's response during an oxidative challenge. Here, we give an overview of the UPS and a detailed description of the degradation of oxidized proteins and of the crosstalk between oxidative stress and protein degradation in health and disease. Antioxid. Redox Signal. 15, 2265–2299.
I. Introduction
In eukaryotic cells the majority of intracellular proteins are degraded by the ubiquitin-proteasome system (UPS) (99, 117, 269). This system relies on a cascade of three enzymes, termed E1, E2, and E3, that conjugate the small protein ubiquitin to specific target proteins (185, 269). In some cases the ubiquitin-chain on the target protein may be elongated by an E4 ubiquitin-chain elongation factor (184). Subsequently, the proteins that have been marked with polyubiquitin are targeted to the 26S proteasome, a large and abundant proteolytic particle found in the nucleus and cytosol of all eukaryotic cells (99). At the 26S proteasome the ubiquitin chains are released, whereas the substrate is degraded into shorter peptides that can be displayed on the cell surface for immuno surveillance (181, 334) or further degraded to free amino acids by various amino peptidases.
Bioinformatic analyses have shown that a significant fraction of the eukaryotic genome is dedicated to the UPS (304). In plants such as Arabidopsis thaliana, UPS components occupy more than 5% of the genome (83). This large number of genes of the UPS is mainly attributed to the many different E3 ubiquitin-protein ligases (83, 304) that are the main factors in determining the specificity in conjugation of ubiquitin to target proteins.
In parallel with the UPS, the autophagy-lysosome system also provides the cell with means to destroy intracellular proteins. Autophagy is a self-degradative process that plays a regulatory role and maintains housekeeping functions in removing misfolded or aggregated proteins, clearing damaged organelles, including mitochondria, endoplasmic reticulum (ER), and peroxisomes, as well as eliminating intracellular pathogens (189). Interestingly, recent studies have shown that autophagy also relies on the ubiquitin system and the two systems are connected on many levels (189, 191).
In their normal environment, cells are often exposed to chemical and physical insults that may lead to protein misfolding and ultimately protein aggregate formation and cell death. Among such stress conditions, the presence of oxidizing agents, leading to oxidative stress, plays a prominent role. Since reactive oxygen species (ROS) emerge as a side product during respiration in mitochondria (197), all aerobic organisms will experience mild oxidative stress. However, ROS are also produced during oxidative folding in the ER (91). In addition, higher eukaryotes produce ROS in response to infection and during signaling events (86, 357). Finally, cells may also encounter exogenous ROS derived from pollution, medications, or UV irradiation (357).
The presence of ROS may induce various modifications in proteins, such as carbonylation of side chains (325). In addition, the sulfur-containing amino acids are highly susceptible to oxidation. Methionine residues are oxidized to methionine sulfoxides or further to methionine sulfones. Oxidation of cysteine residues can lead to formation of disulfide bonds or various cysteic acids. Finally, oxidized proteins may become misfolded, leading to an increased hydrophobicity and aggregate formation (72).
In cases where the oxidative stress is not too severe, the cell relies on enzymes such as glutaredoxins, thioredoxins, and methionine sulfoxide reductase to reduce the oxidized proteins (357) (Fig. 1); if the oxidized proteins have also become misfolded, molecular chaperones may subsequently refold and salvage them (Fig. 1). Moreover, induction of enzymes that catalyze ROS detoxification such as superoxide dismutases (SODs), catalases, and peroxidases may counter further damage to the cell. However, when proteins become more severely or even irreversibly damaged by an oxidative insult, these proteins are instead directed for degradation by the proteasome and/or the autophagy system (Fig. 1) (38, 61).

In the present article, we provide a general overview of the UPS followed by a review of how the UPS is regulated by redox signaling and how the UPS functions to maintain a proper intracellular milieu by degrading oxidatively damaged proteins and specific redox-relevant signaling molecules.
II. The Ubiquitin System
The process of ubiquitin conjugation, occurring in both the nucleus and cytosol, is connected with a range of cellular functions such as DNA repair, transcription regulation, signaling, endocytosis, and autophagy, but is best known for its function in intracellular protein degradation via the UPS (185, 269).
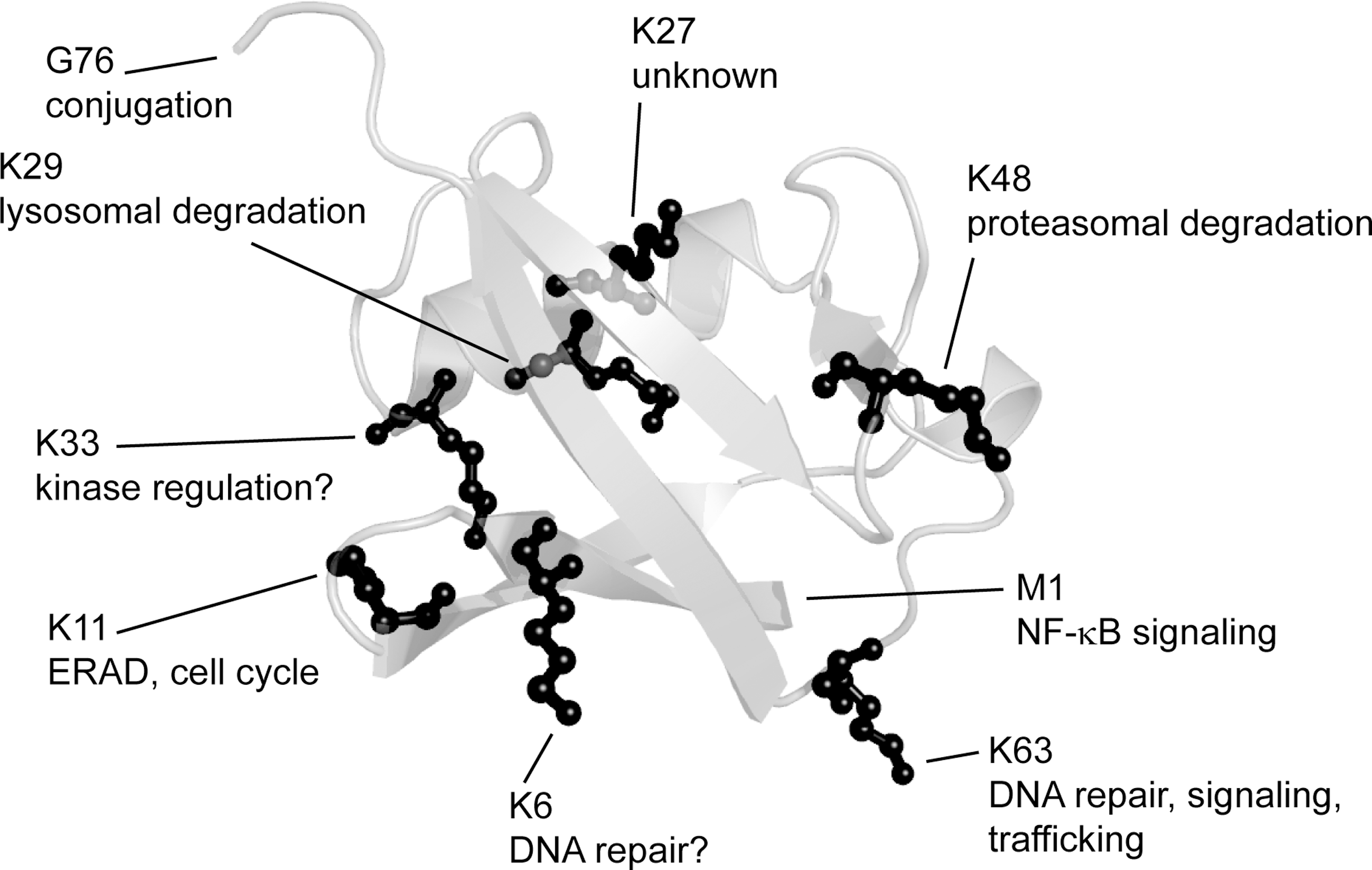
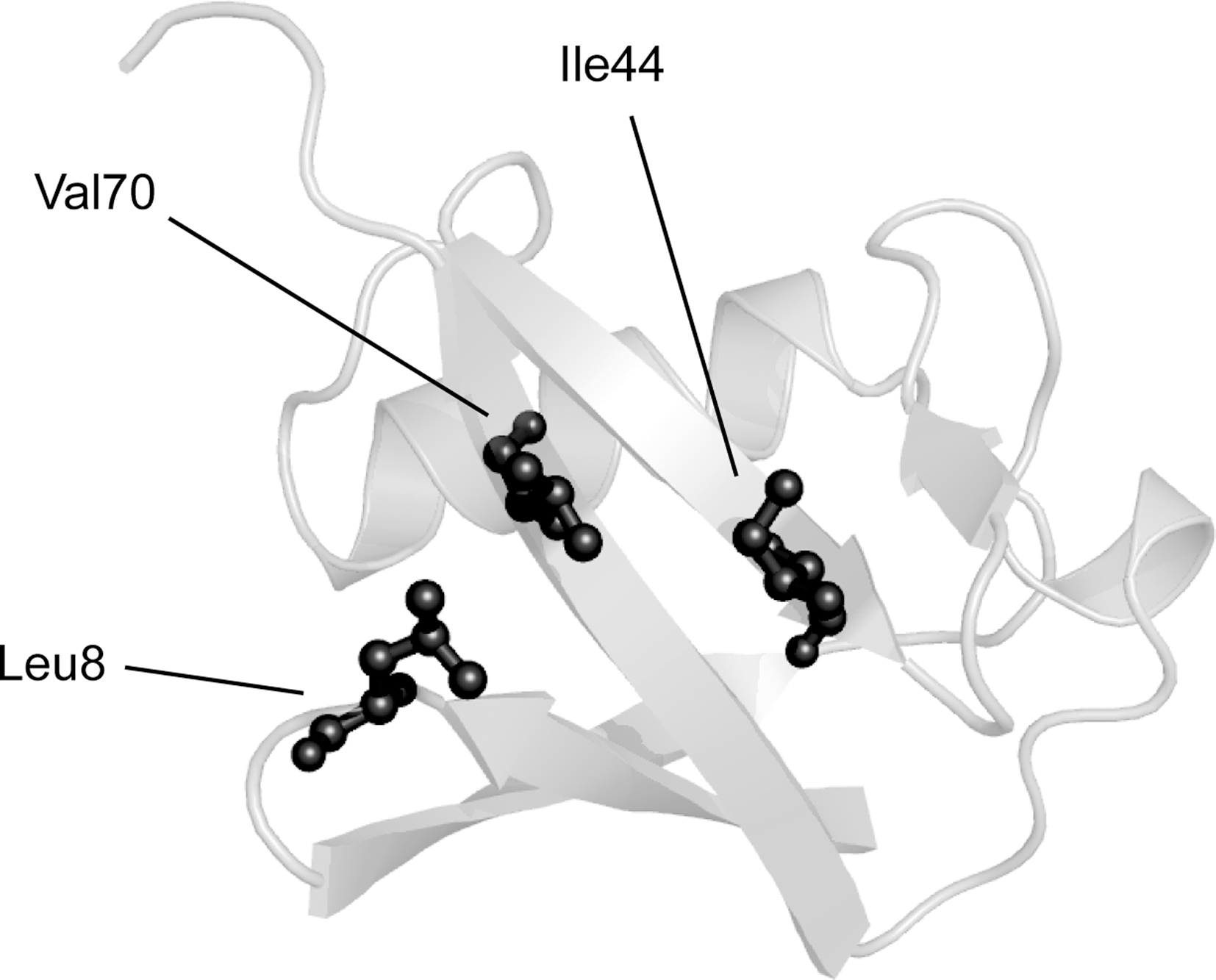
Ubiquitin is a small and essential protein of only 76 residues that is phylogenetically conserved in all eukaryotic cells (269). Structurally, ubiquitin is compact and adopts a β-grasp fold, composed of a five-stranded antiparallel β-sheet traversed by a single helix (364). There are several functionally important sites within ubiquitin. The C-terminal carboxyl group of G76 and the primary amino group in each of the seven lysine residues function in conjugation of ubiquitin to target proteins and to other ubiquitin molecules (Fig. 2) (185, 270). Ubiquitin also features a hydrophobic surface patch consisting of residues L8, I44, and V70 (Fig. 3) (18). This area forms the binding surface for the noncovalent interactions between ubiquitin and ubiquitin-binding proteins (81).
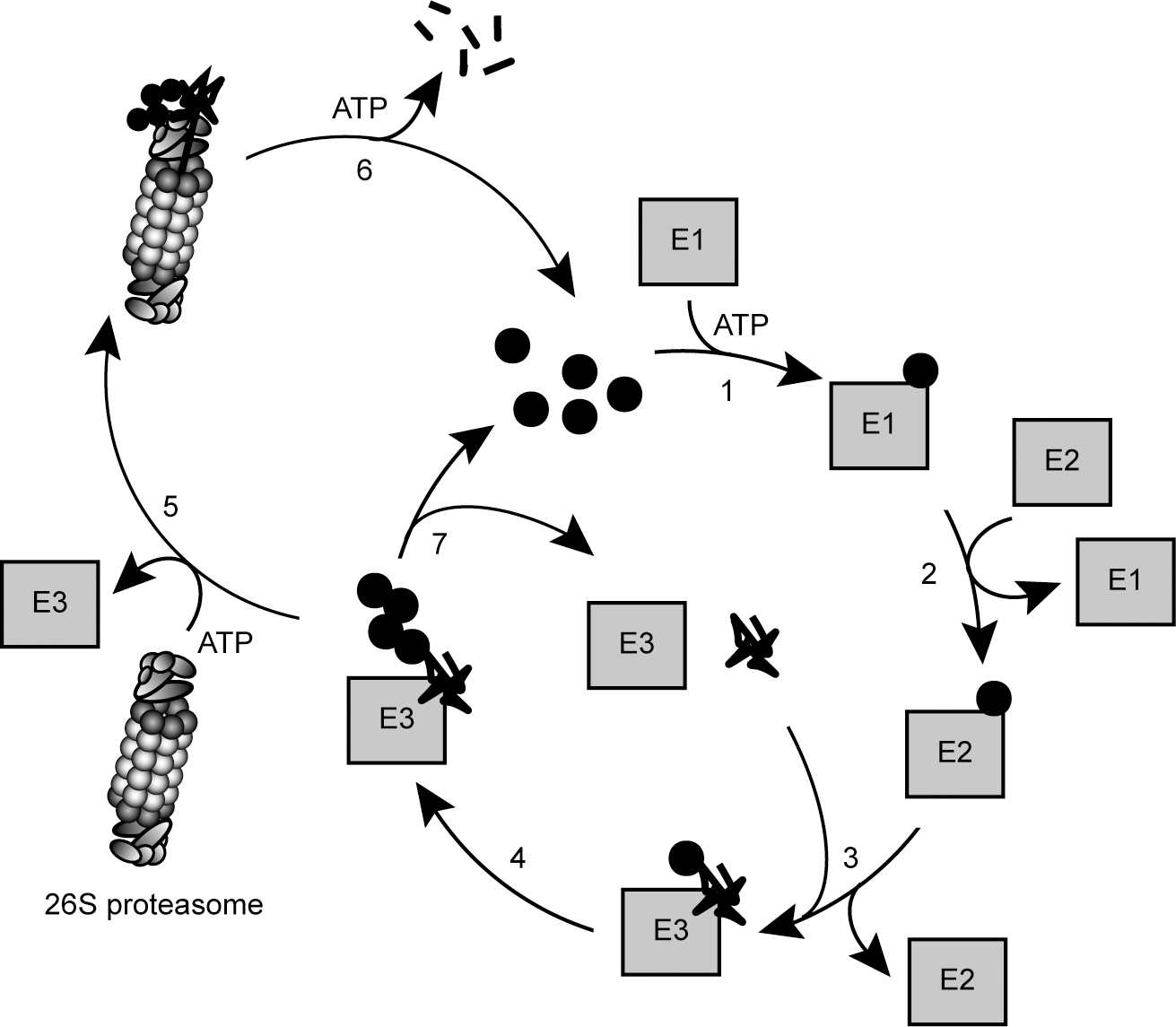
The process of ubiquitin polymerization (Fig. 4), in which a substrate becomes labeled with a chain of ubiquitin moieties, requires the sequential action of three types of enzymes: E1, E2, and E3 (269). First, ubiquitin is bound and activated by the E1 ubiquitin-activating enzyme in an ATP-dependent reaction. As a result, ubiquitin is covalently attached to the E1 via a thioester bond between the E1 active site cysteine residue and the C-terminal carboxyl (G76) of ubiquitin. The ubiquitin-loaded E1 then transfers the activated ubiquitin to the active site cysteine in an E2 ubiquitin-conjugating enzyme. Finally, ubiquitin is transferred from the E2 to the substrate by an E3 ubiquitin-protein ligase (269) (Fig. 4). The conjugation between ubiquitin and the substrate occurs via an isopeptide bond between the ɛ-amino group on a lysine residue in the substrate and the C-terminal carboxyl group in ubiquitin. On the rare occasions where the substrate protein does not contain any accessible lysine residues, ubiquitin is instead conjugated to the α-amino group in the N-terminus of the substrate (55). More recently, other examples of nonlysine ubiquitylation were reported. For instance, the viral E3 ligase mK3 and its E2 partner conjugate ubiquitin also to serine residues, thus forming an ester bond (369).
In successive rounds of ubiquitylation, a lysine residue in the substrate-bound ubiquitin is conjugated to another ubiquitin unit yielding substrates that carry covalently linked ubiquitin chains. In some cases, the ubiquitin-chain elongation factor, E4, binds to the ubiquitin moieties of preformed conjugates and catalyses further ubiquitylation, resulting in extension of the ubiquitin chain (184).
A. E3 ubiquitin-protein ligases
The human genome encodes two ubiquitin-specific E1s and about 50 E2s (304). However, in general the specificity of the ubiquitylation process lies with the substrate binding regions in the E3s. Accordingly, the human genome encodes hundreds of E3s. These E3 enzymes are split between two different families (185), known as homologous to E6-associated protein (E6AP) carboxy terminus (HECT) and really interesting new gene (RING). Database searching indicates that the RING family is by far the largest. In humans about 600 genes are predicted to encode RING domain E3s (77, 208), whereas the HECT family is comprised of about 30 members (288).
Catalytically, members of the HECT family employ a unique reaction mechanism among the E3s (288). While the HECT E3s form a thioester intermediate with ubiquitin, similar to the E1 and E2 enzymes, the RING family E3s facilitates ubiquitin conjugation by acting as bridging factors between the E2s and the substrates, allowing transfer of ubiquitin from the E2 protein to the substrate (288).
At its core, the RING domain consists of a short motif, rich in cysteine and histidine residues (105). The spacing of the residues is conserved and functions to coordinate the bound zinc ions (27). Most RING domain E3s function as single subunit enzymes, but some important members are components of larger protein complexes such as the cullin-RING ligase (CRL) complexes. The canonical example of a CRL E3 is the so-called Skp1/cullin/F-box (SCF) complex (317) that mediates the degradation of a wide variety of proteins, including cell cycle components (317), transcription factors (365), and misfolded glycosylated proteins (394). In the SCF complex, the cullin component acts as a scaffold that binds Skp1 and the RING protein Rbx1, which in turn recruits the E2. Skp1 functions as a bridge to a variable F-box protein. The F-box protein is responsible for substrate binding and thus determines the specificity of the ubiquitin conjugation reaction (153). The human genome encodes 69 different F-box proteins (317), each with its own substrate binding preferences. Additional variability of the CRL complexes is derived from incorporation of different cullin proteins, of which there are at least 8 in humans.
A group of RING-related E3 proteins is defined by the so-called U-box domain proteins. The U-box was first identified in the budding yeast E4 enzyme, Ufd2, and is essentially an E2-binding domain that structurally resembles a RING domain. One of the best-studied members of the U-box family is C-terminus Hsp70 interacting protein (CHIP), which together with the proteasome cofactor, BAG-1, orchestrates the ubiquitylation and transfer of misfolded Hsp70 clients to the 26S proteasome for degradation (8, 216). CHIP associates with Hsp70-type chaperones, and accordingly most of CHIP's known substrates are misfolded proteins. Here the chaperone serves as a specificity factor, recognizing the misfolded protein substrate. Thus, by outsourcing substrate recognition to Hsp70, CHIP utilizes a pre-existing cellular system for selectively targeting damaged or misfolded proteins for proteasomal degradation.
The E3 called E6AP is the founding member of the HECT E3 family. E6AP is a well-characterized enzyme, and is best known for its role in degradation of p53 in cells infected with oncogenic strains of human papillomavirus (296). In the infected cells, the viral protein E6 associates with E6AP, which then gains the ability to bind and ubiquitylate the host cell p53 protein, resulting in p53 degradation, viral DNA replication, inhibition of apoptosis, and cancer (296).
After addition of the first ubiquitin moiety to the substrate protein, E3 and E4 enzymes catalyze the elongation of the growing ubiquitin chain by ubiquitylating a lysine residue in the substrate-bound ubiquitin unit. Since ubiquitin contains seven lysine residues (K6, K11, K27, K29, K33, K48, and K63), the ubiquitin chains may adopt various conformations, depending on which lysine residue is utilized during chain elongation (185, 266). Recently, linear chains, where the ubiquitin moieties are linked by peptide bonds between the N- and C-termini, forming head-to-tail fusion proteins, were ascribed a function in NF-κB signaling (350). In addition, ubiquitin chains may be homotypic or heterotypic (81, 185); that is, the chain may be linked via the same lysine residue throughout the chain, or the chain may contain more than one linkage type or even be branched. Homotypic ubiquitin chains, linked through K48, are the preferred signal for protein degradation via the UPS (270, 348). In addition, the chain length may vary, but chains are normally found to contain 3–5 ubiquitin units. In relation to proteasomal protein degradation, the length of K48-linked ubiquitin chains has a significant influence on the affinity to the 26S proteasome (270) and thus on the degradation kinetics. Longer ubiquitin chains are recognized more efficiently than shorter chains, and normally the chains must contain at least 4 ubiquitin units to be efficiently recognized by the 26S proteasome (78, 348, 379). Finally, the ubiquitin signal is not always composed of a ubiquitin chain. It can also be conjugated to the target protein as either a single moiety in a process termed monoubiquitylation, or as several single ubiquitin moieties by a process termed multiple monoubiquitylation.
The various ubiquitin signals function in diverse cellular pathways (Fig. 2). For instance, monoubiquitylation is important for transcriptional regulation (183), whereas K63-linked and linear ubiquitin chains are important for DNA repair (320) and NF-κB signaling (350), respectively. However, in many cases the function of specific ubiquitin modifications is still unknown.
B. Deubiquitylation
Like most other regulatory post-translational modifications, ubiquitylation is a reversible process. The human genome encodes ∼100 different putative deubiquitylating enzymes (DUBs). The DUBs fall into two classes of proteases based on their mechanism of catalysis: cysteine proteases and metalloproteases (285). All DUBs may potentially deubiquitylate the target protein back into its unmodified form (186, 285) and thus ultimately rescue a protein from degradation (200, 387) (Fig. 4) or terminate a signaling event. For instance, the human DUB called Usp29 was recently found to bind and cleave polyubiquitin chains from p53 in response to oxidative stress (212). This, in turn, leads to stabilization of p53 and apoptosis (212).
Another example of a DUB that may counter protein degradation is the proteasome-associated DUB, called Usp14 in humans. Treating cells with a Usp14 inhibitor enhances degradation of several proteasome substrates, including oxidized proteins (200). However, other proteasome-associated DUBs do not deubiquitylate the substrate until the proteasome is committed to degrade it, and in these cases the DUBs may function to promote protein degradation (200, 230). DUBs also serve other purposes in the cell. Since ubiquitin is expressed as a linear fusion protein with either multiple copies of itself or with one ubiquitin unit fused to the N-terminus of certain small ribosomal proteins, a key function of DUBs is to generate free ubiquitin by specific cleavage of the ubiquitin precursors (186). In addition, DUBs are involved in maintenance of ubiquitin homeostasis. For efficient protein degradation and to allow cells to swiftly respond to stress conditions, it is essential that cells contain large amounts of free ubiquitin that can be rapidly conjugated to target proteins (138). For this purpose, proteasome-associated DUBs ensure that ubiquitin is released before substrate translocation (137). Thus, ubiquitin is not degraded along with its substrate, but instead recycled (186, 201) (Fig. 4). If ubiquitin levels are low, cells experience the so-called ubiquitin stress, which in yeast leads to induction of the proteasome-associated DUB Ubp6. Subsequently, proteasomes become more efficient at recycling ubiquitin and maintaining the ubiquitin pool (137).
In general, DUBs are highly specific. For instance, some DUBs discriminate between different ubiquitin linkage types (186). Each of the seven lysine residues in ubiquitin is located in a unique sequence context, which may be important for recognition by DUBs. Finally, yet another layer of specificity is achieved by protein interaction domains within the DUBs that bind specific substrates or substrate adaptors (285).
Some data suggest that E3s and DUBs often work in pairs, similar to kinases and phosphatases. For instance, the proteasome-associated E3 and DUB, respectively, called Hul5 and Ubp6 in budding yeast, appear to counteract each other (63). A more extreme example is the protein A20, which combines E3 ligase and DUB activity in the same polypeptide. This attribute allows A20 to edit the ubiquitylation status of the NF-κB signaling component RIP1. Hence, removal of K63-linked ubiquitin chains on RIP1 will reduce signaling, and replacing them with K48-linked chains will lead to RIP1 degradation and thus terminate signaling (150).
III. The Proteasome
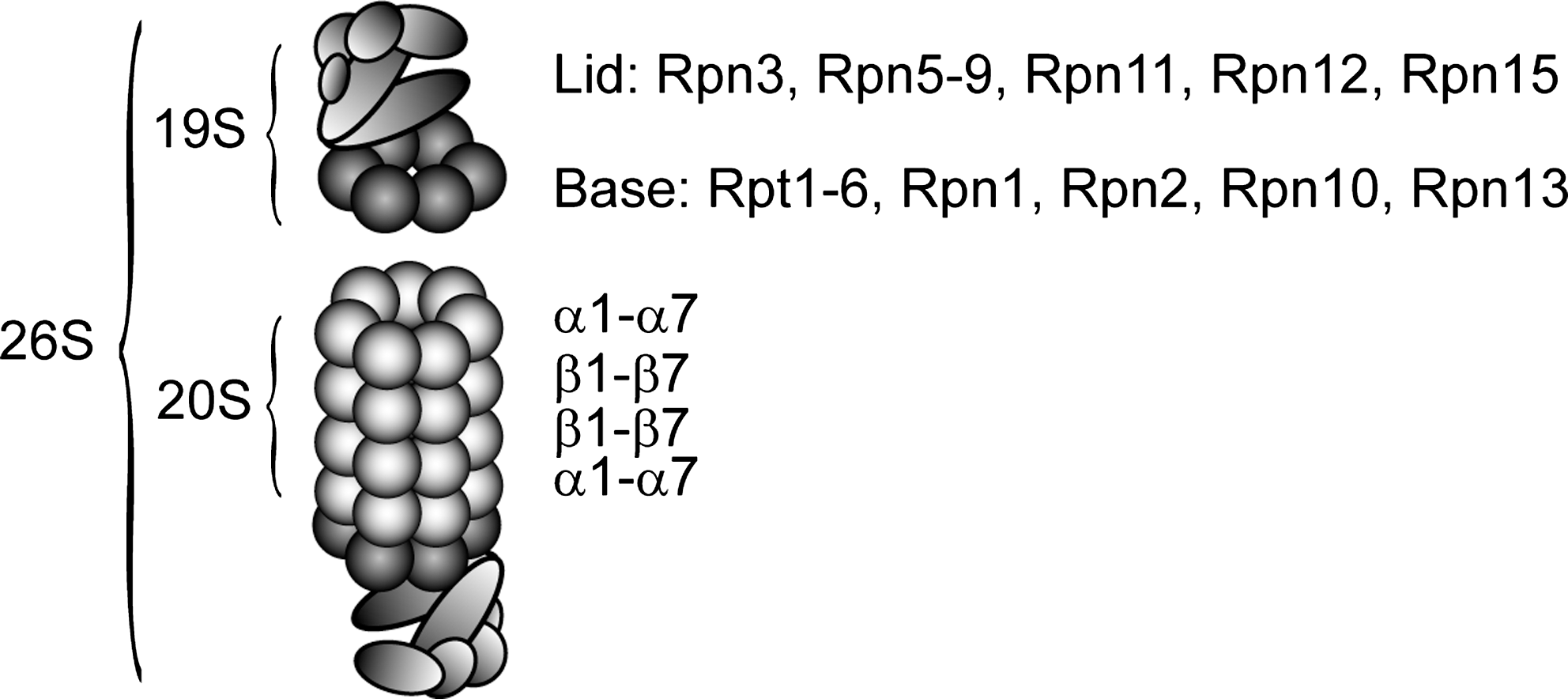
The 26S proteasome is a stable particle (193) that is composed of two subcomplexes: the proteolytically active 20S core particle and 19S regulatory complexes that bind to one or both ends of the 20S proteasome (Fig. 5) (99). In mammalian cells, the 26S proteasome is distributed throughout the cytoplasm and nucleus (31). However, in yeast the 26S proteasomes are primarily found along the nuclear periphery (90), and this localization is certainly important since protein degradation is impaired in mutants that mislocalize proteasomes (33, 124, 343).
Since the nomenclature of mammalian proteasome subunits is confusing, we use the simpler and more logical yeast nomenclature also for mammalian proteins, unless otherwise stated.
A. The 20S proteasome
The structure of the 20S catalytic core has been solved and shows a complex that is built from 28 subunits, arranged as four stacked heptameric rings, forming a cylindrical structure enclosing a central chamber and two antechambers (Fig. 6) (127). The two identical outer rings contain seven different α subunits (α1–α7) and the two identical inner rings each contains seven different β subunits (β1–β7), leading to an overall α1–7β1–7β1–7α1–7 structure (Fig. 5) (127). Three of the β subunits (β1, β2, and β5) are threonine-type proteases that expose their active sites toward a central chamber inside the 20S cylinder (127). The β1 subunit preferentially cleaves after acidic residues, β2 displays trypsin-like activity and cleaves after basic residues, whereas β5 displays chymotrypsin-like activity and cleaves after hydrophobic residues (62, 127). Normally, the outer α-rings are closed, but upon binding of the 19S regulatory complex, a central pore is formed that allows substrate access to the proteolytic sites within (Fig. 7) (126, 375).


The assembly process of the 20S proteasome is a highly orchestrated process that relies on assistance from chaperones and specialized proteasome maturation proteins (227). Of these the best characterized example is the protein called Ump1 in yeast and POMP in humans, which after mediating proteasome assembly is degraded by the nascent particle (227). Consistent with a role of the 20S and 26S proteasome in degradation of oxidized proteins that we describe below, overexpression of Ump1 in yeast (323) and POMP in fibroblasts (51) protects cells from oxidative stress.
B. The 19S regulatory complex
The 19S regulatory complex is an asymmetric particle composed of about 19 different subunits (99). So far, no high-resolution structural information on the 19S particle is available, but elegant electron microscopy and biochemical studies on 26S proteasomes, 19S particles, and related protein complexes have revealed important information regarding the overall structure and subunit topology of the complex (89, 102, 103, 248, 351). By introducing mutations or treatment with high concentrations of salt, the 19S complex disassembles into two subcomplexes, termed the base and the lid (Fig. 5) (118). The base contains six different ATPase subunits (Rpt1–Rpt6) and 4 non-ATPase subunits. The ATPase subunits belong to the family of ATPases associated with various activities (AAA), and like most members of this family, the Rpt1-6 subunits form a ring (102). The C-termini of some ATPase subunits bind to pockets between the 20S proteasome α subunits and thereby accomplish opening of the pore in the 20S particle (Fig. 7) (318, 346). However, even upon opening, the entry pore to the 20S particle is still too narrow to allow access of a fully folded protein substrate although peptide chains and loops may enter (126). Accordingly, the ATPase subunits also function in unfolding and translocation of the protein substrates into the 20S cylinder (29, 335).
The more distal lid subcomplex is composed of nine subunits and covers the base (Fig. 5). It functions in deubiquitylation of incoming substrates (361) and resembles the COP9 signalosome (CSN) and the translation initiation complex eIF3 (118). Accordingly, both the CSN and eIF3, are also, at least in part, connected with intracellular protein degradation via the UPS. For instance, the best understood biochemical function of CSN is regulating the activity of cullin-RING E3 ligases (219). The compositionally more divergent eIF3 functions as a scaffold during translation initiation, but also forms larger complexes with the proteasome and other components of the UPS (258, 305). However, we shall not discuss the CSN and eIF3 further here.
Another essential function of the 19S particle relates to binding of ubiquitylated substrates (99). This task is accomplished by several 19S subunits and certain extrinsic proteasome cofactors (78, 157, 196, 297, 379). So far, three proteasome subunits (Rpt5, Rpn10, and Rpn13) have been found to form specific noncovalent interactions with polyubiquitylated substrates (78, 157, 196, 297). Intriguingly, these subunits all belong to the base subcomplex of the 19S particle, and unlike most other proteasome subunits, Rpn10 and Rpn13 are not essential for viability in yeast (157, 379). The substrate-binding proteasome cofactors, called Rad23 and Dsk2 in yeast and HHR23 and ubiquilin/protein linking IAP with cytoskeleton (PLIC) in humans, respectively, all contain an N-terminal ubiquitin-like (UBL) domain and one or two C-terminal ubiquitin-associated (UBA) domains (142, 154). Since the UBL domains associate with the 26S proteasome (295, 379) and the UBA domains associate with ubiquitin chains (379), these so-called UBL/UBA proteins or substrate shuttle proteins may recruit substrates to the 26S proteasome (88, 99, 141, 362, 379). Surprisingly, like the Rpn10 and Rpn13 subunits, Rad23 and Dsk2 are also not essential for viability in yeast. However, in combination the lack of Rpn10, Rad23, and Dsk2 is lethal in yeast (88, 379), revealing that for efficient substrate recognition the 26S proteasome relies on an intrinsic affinity for ubiquitin-conjugated substrates via specific 19S subunits, and functionally overlapping extrinsic ubiquitin-binding cofactors (141).
C. Other proteasome activators
In lower eukaryotes and under normal growth conditions in mammalian cells, the 26S proteasome, as described above, is the protease responsible for the majority of intracellular protein degradation. However, proteasomes are modular in their composition, and changes in the proteolytic requirements can induce specialized subunits and proteasome activators (99).
In addition to the 19S regulatory complex, several other proteasome activators are known (327). The best characterized examples are PA28αβ and PA28γ. However, since none of these complexes contain subunits with ubiquitin-binding capabilities, they are predicted to be involved mainly in ubiquitin-independent degradation (99). In addition, these other activators do not possess ATPase activity, suggesting that PA28αβ and PA28γ substrates are primarily peptides or proteins that contain unstructured regions so that the active substrate unfolding by the 19S complex ATPases can be bypassed (99).
Like the 19S regulatory complex, the other 20S proteasome activators associate with the peripheral α-rings of the 20S particle, but their association with the 20S proteasome is not mutually exclusive. Thus, hybrid proteasomes containing a 19S particle on one side of the 20S particle and a different activator on the other side can form (146). These hybrid proteasomes constitute a significant fraction of the total proteasome pool in mammalian cells (339), suggesting that they are biologically relevant.
The proteasome activator PA28αβ (also known as REGαβ and the 11S regulator) is only found in vertebrates. It is a heteromeric complex composed of PA28α and PA28β subunits arranged in a heptameric ring-shaped complex (99, 313). The expression of PA28αβ is induced by interferon and the complex is required for efficient presentation of some antigens on major histocompatibility complex (MHC) class I molecules (313). Upon binding to the 20S cylinder, the pore to the central proteolytic chamber is opened, which may allow easier access of unfolded substrates and more efficient product release, but association also changes the cleavage specificity of the 20S proteasome (313, 375).
PA28γ (also known as REGγ) structurally resembles PA28αβ. However, unlike PA28αβ it is found in most higher eukaryotes and it is not induced by interferon. PA28γ is predominantly a nuclear protein, and accordingly it plays a role in ubiquitin-independent degradation of nuclear proteins, like the steroid hormone receptor SRC-3 (209) and certain cyclin-dependent kinase inhibitors (46).
D. The immunoproteasome
An additional level of proteasomal variation is achieved through replacement of specific subunits. In response to interferon-α, interferon-β, and interferon-γ, as well as upon virus infection (302), the three proteolytically active 20S proteasome subunits, β1, β2, and β5, are each replaced with the induced β1i, β2i, and β5i subunits (99), forming the induced 20S proteasome, also known as the immunoproteasome (Fig. 8) (99). Expression of the immunoproteasome is not restricted to cells of the immune system (79, 315). Like the standard proteasomes, the immunoproteasomes localize to the cytosol, but are also associated with the ER membrane (31). The induced β subunits are also proteolytically active, but the immunoproteasome particles display altered cleavage specificity that may produce peptide products that are better suited for presentation on MHC class I molecules (99). Recently, immunoproteasomes have been linked to degradation of oxidized proteins during inflammatory responses and they are therefore discussed in more detail later.
Also, the so-called mixed proteasomes where only some of the constitutive β subunits are replaced by immunosubunits have been described (180). However, as of yet the functional significance of these particles remains unknown.
IV. Degradation of Misfolded Secretory Proteins
Many mammalian genes encode proteins that are secreted and must therefore pass the ER. Although the environment in the ER is specialized to allow protein folding, some proteins may still misfold and accumulate inside the lumen. Such proteins are eliminated by the ER-associated degradation (ERAD) pathway (278), which, since the ER is devoid of a ubiquitin system, instead relies on the cytosolic UPS.
ERAD is a multistep process that commences in the ER lumen (Fig. 9). Here, the misfolded proteins are first recognized by general ER-resident molecular chaperones such as BiP and protein disulfide isomerase (PDI) (242). Misfolded glycoproteins are recognized by the sugar-binding chaperones calnexin and calreticulin, before mannosidase I trims a single mannose unit from the oligosaccharide. This, in turn, allows the ERAD-relevant lectin EDEM to associate with the glycoprotein and target it for ERAD (155, 163, 241, 254). After recognition, the ERAD substrates are shuttled to the cytosol in a process termed retrotranslocation by a mechanism and a channel, which remain unknown (278). However, upon reaching the cytosol, ERAD substrates are conjugated to ubiquitin by ERAD E3 ligases such as Hrd1, gp78, and CHIP (278, 389, 390). After ubiquitylation, the AAA-type ATPase, called p97 or valosin-containing protein (VCP) in mammals and Cdc48 in yeast, powers the extraction of the ERAD substrates. However, p97 does not work alone, but is dependent on various cofactors (221), like the Ufd1/Npl4 heterodimer that binds the ubiquitylated substrates (221, 238, 286, 299). Finally, the ubiquitylated ERAD substrate is delivered to the 26S proteasome and degraded (Fig. 9). Importantly, for several ERAD substrates, the retrotranslocation process is coupled to proteasomal degradation (222), suggesting that the individual steps in the ERAD process are tightly connected.
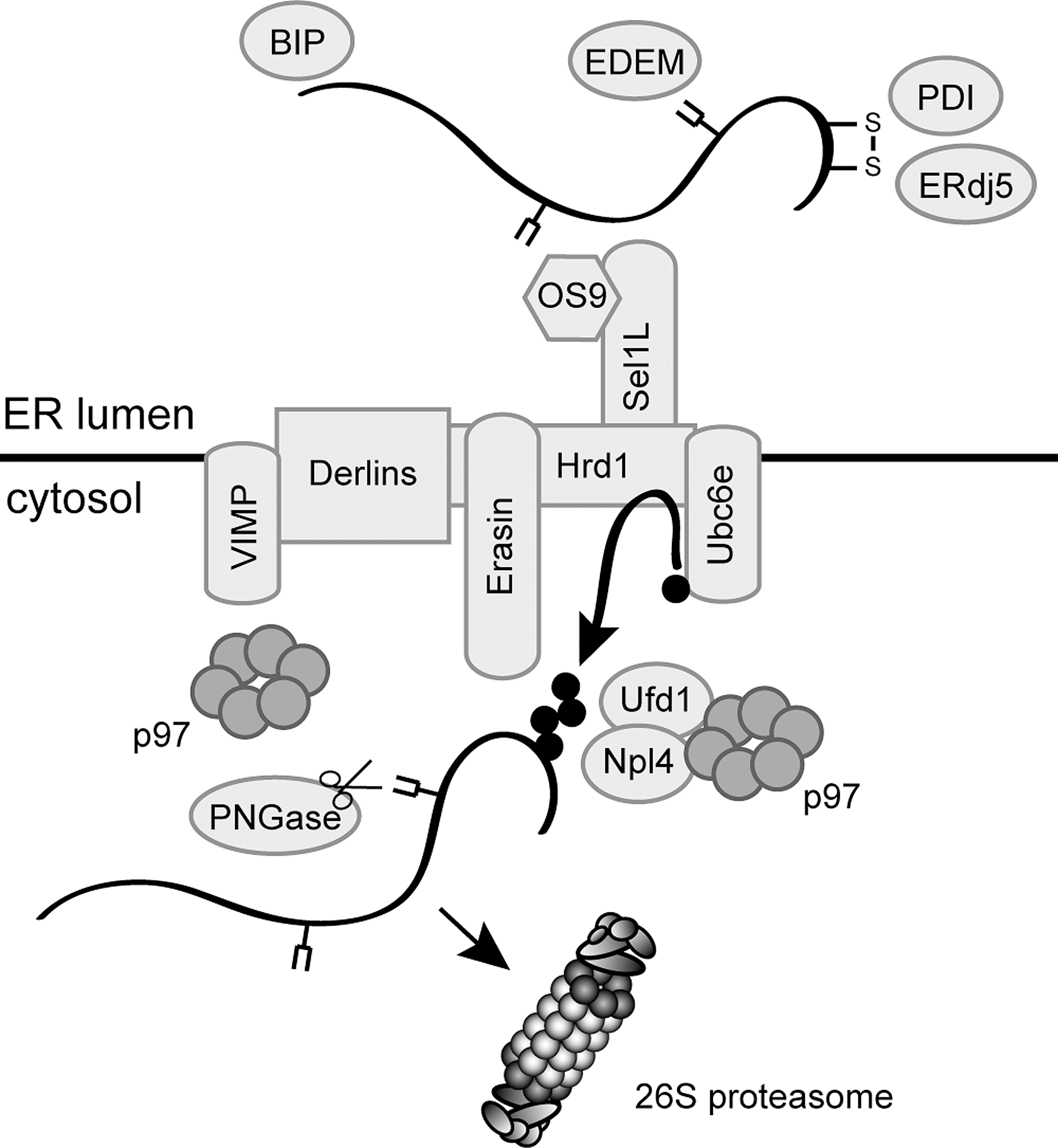
Since some ERAD substrates contain bulky carbohydrates, efficient translocation into the narrow openings of the 20S proteasome core requires that these glycans are removed before degradation. Deglycosylation is catalyzed by the peptide:N-glycanase (PNGase) that associates directly with both p97 and the substrate-binding proteasome cofactor Rad23 (206, 207). Certainly, more tightly folded proteins that contain disulfide bonds may pose a problem for translocation into the 20S particle (373). However, as to where the reduction of disulfide bonds in ERAD substrates occurs, the answer is not clear. The ER luminal ERdj5 and PDI play a role in ERAD and may reduce some ERAD substrates already inside the ER lumen (355). In the case of short-lived immunoglobin heavy chains, reduction of interchain disulfides has been shown to precede retrotranslocation (94). Considering that glycosylated ERAD substrates can be efficiently retrotranslocated and that such carbohydrates are probably much larger than the structures induced by disulfide bonds, the dimensions of the channel will probably also accommodate some oxidized proteins and the reduction of disulfide bonds might therefore not be a prerequisite for retrotranslocation of all substrates. Accordingly, the Caenorhabditis elegans ortholog of PNGase contains an active thioredoxin domain (171, 336), indicating that disulfide bond reduction and deglycosylation are closely linked and that some substrates may require cytosolic oxidoreductases to catalyze the reduction of disulfide bonds before degradation. In mammalian cells the proteasome-associated thioredoxin, Txnl1, which we describe in detail later, is a candidate enzyme to provide this activity. However, so far no ERAD defects have been reported upon knockdown of Txnl1 (5). Another protein that may be involved in reduction of ERAD substrates containing disulfide bonds is the ER transmembrane p97 cofactor called VIMP or SelS (390). The bulk of VIMP faces the cytosol and since VIMP is a selenoprotein it might be redox active.
V. Protein Aggregation and Aggresomes
As mentioned, a crucial role for the UPS is protein quality control whereby cytosolic, nuclear, or ER-derived proteins with abnormal structures due to, for example, genetic mutations or oxidative damage, are selectively degraded (8, 112, 119). Some misfolded proteins, however, are beyond salvation by chaperones and are also not destroyed by the UPS. Instead, these form insoluble aggregates reminiscent of the insoluble protein deposits observed in conformational diseases such as Parkinson's disease (188). Experimentally, aggregate formation can be induced by inhibition of the proteasome or by protein overexpression. During these conditions the aggregated proteins merge into a single large structure termed the aggresome (169, 381). Aggresome formation depends on p97 (382) and on dynein-driven transport of smaller aggregates along microtubules. At the centrosome these aggregates are deposited in the aggresome (111, 169, 381). Certainly, misfolded proteins may form various types of aggregates, but for the purpose of this review we shall in the following focus on aggresomes.
Upon aggresome formation, the vimentin intermediate filament cytoskeleton often rearranges, resulting in a cage of vimentin surrounding the aggresome (169). The aggresome also contains chaperones, including Hsp70 and Hsp90, and UPS components such as proteasomes and ubiquitin (92, 378, 381), that even under basal conditions may also concentrate at the centrosome (378). Although the UPS degrades misfolded proteins and therefore fights protein aggregation, once aggresomes are formed they are probably more efficiently cleared by autophagy (104, 188). The function of aggresomes is still a matter of some debate. Certainly, the presence of aggregated proteins may negatively affect cellular homeostasis in several ways and inhibit the UPS (21). It is possible that aggregated proteins directly trap and inhibit the proteasome, that is, proteasomes choking on aggregated substrates, but it has also been suggested that the inhibitory effect of aggresomes on protein degradation could be the result of trapping UPS components at the centrosome. However, data suggest that in vivo, protein aggregation is a rather specific process, so unrelated misfolded proteins only seldom coaggregate (280), but this might not be true for all types of protein aggregates. Certainly, aggresomes and other proteinaceous aggregates could also provide a safe environment for storage of the aggregated proteins and may thus constitute a beneficial self-defense mechanism (188). An example of a protein that accumulates in aggresomes is the cystic fibrosis transmembrane conductance regulator (CFTR). Recently, it was shown that expression of defective CFTR results in upregulation of ROS, defects in the autophagy system, and a decreased clearance of aggresomes (215).
The so-called aggresome-like structures (ALIS) were observed in several mammalian cell types in response to oxidative stress and starvation (337). Although ALIS are spherical and ubiquitin-rich and resemble aggresomes, they are transient structures that do not localize to the centrosome (202). Also ALIS are not caged in vimentin and can be formed in the presence of microtubule destabilizing drugs (202). The formation of ALIS depends on protein synthesis and is increased by puromycin, suggesting that ALIS primarily contain misfolded nascent proteins (337).
VI. The Role of the Proteasome in Degradation of Oxidized Proteins
The importance of efficient clearance of damaged proteins by degradation was appreciated early on and appointed as a defense mechanism during oxidative stress. Degradation relieves the threat of oxidized proteins forming toxic aggregates, and the amino acid products, released in the course of degradation, may also become oxidized and therefore function as ROS scavengers that protect more important cellular components from oxidation (68, 70, 139).
A remarkable correlation between protein damage by oxidation and an increase in proteolysis has been described by various groups already before characterization of the proteasome (49, 66, 67, 69, 203). In the mid-1980s Rivett isolated different proteolytic systems from liver, including a large protease complex, which showed increased activity toward the oxidized model substrate, glutamine synthetase from bacteria (287). Independently, the Davies group purified a protease complex, named macroxyproteinase, which was responsible for the degradation of oxidatively damaged proteins (260, 262). Later, it became clear that these proteases were identical to the particle named the 20S proteasome (9).
Although other proteases have also been connected with the clearance of oxidized proteins, about 70%–80% of turnover of oxidized protein has been attributed to the proteasome (262). In addition, proteasome depletion by either immunoprecipitation or by knockdown of subunit α1 led to a decline in proteolysis of bulk proteins and oxidatively damaged model substrates (129, 130). Another indication came with the development of specific proteasome inhibitors, with which the dependency of proteasome activity in degradation of oxidized proteins could also be shown (316). Hence, proteasome activity is clearly important for proteolysis of oxidized proteins and for the oxidative stress response. However, as to the individual roles of the 20S and 26S proteasomes and the involvement of the ubiquitin system, there is some controversy. In the sections below we summarize results on both 20S proteasomes and 26S proteasomes. This is then followed by a discussion on recent findings which reveal that immunoproteasomes protect cells from oxidative stress.
A. The role of the 20S proteasome in degradation of oxidized proteins
In general, ubiquitin- and ATP-dependent protein degradation by the 26S proteasome is the predominant mechanism for protein degradation in all eukaryotic cells (117). However, in some cases, proteins are degraded by the proteasome in a ubiquitin-independent manner. The classical example is the degradation of ornithine decarboxylase, a key enzyme in polyamine biosynthesis. Its degradation depends on the protein called antizyme, which mediates its proteasomal targeting (244). Several other proteins have been reported to be degraded by the proteasome in a 19S complex- and ubiquitin-independent manner. Especially, intrinsically disordered proteins are prone to ubiquitin-independent degradation (211). Recently, it was suggested that the free 20S proteasome may account for the degradation of 20% of all proteasome-dependent degradation (17), but several nonubiquitylated substrates are also degraded by the 26S proteasome with the same or a better efficiency (17). Hence, more studies are needed to confirm this surprisingly high figure. Still, in vitro most unfolded proteins, including proteins that have been oxidatively damaged and therefore in part become unstructured, can be degraded by purified 20S proteasomes (98), whereas more severely oxidized proteins form aggregates and are therefore not likely to be suitable as proteasome substrates (68). In addition, aggregated proteins may inhibit proteasome function, and in this manner oxidative stress may indirectly affect proteolysis (30, 188, 277).
As we describe later, the 26S proteasome plays an important role in clearing the cell of oxidatively damaged proteins. However, results suggest that in addition to the 26S proteasome, the free 20S particle also degrades oxidized proteins in a process that is independent of both ubiquitin conjugation and ATP hydrolysis (13, 67, 68, 71 –73).
With purified proteasomes or cell extracts, ATP may even be slightly inhibitory for the degradation (129, 261, 262). Also, ATP deprivation of intact red blood cells as well as ATP depletion in whole cell extracts (93) from different sources such as red blood cells (70), hepatocytes (131, 287), skeletal muscle (113), or heart tissue (332) has been shown to have no effect on the observed enhanced turnover rate triggered either by oxidizing conditions or on the turnover of oxidized model substrates. Modification of some oxidatively damaged proteins with ubiquitin to signal degradation might not be a prerequisite for degradation either. Studies using mammalian cells expressing a temperature sensitive E1 mutant showed that the cells are able to efficiently clear some oxidatively damaged proteins (312). Further, in vitro studies with ubiquitin conjugating systems revealed that increasing oxidative damage of model proteins reduces the ubiquitylation efficiency (312). These data pointed to a role of the free 20S proteasome in degradation of oxidatively damaged proteins, and indeed in vitro studies with purified 20S proteasomes showed that this complex was sufficient to degrade various oxidized model proteins, including glutamine synthetase, hemoglobin, SOD, and ferritin (68, 128, 262, 287). Since protein oxidation leads to partial protein unfolding and subsequently to exposure of hydrophobic patches (44), a possible mechanistic explanation for these observations revolves around these hydrophobic patches having some affinity for the α subunits of the 20S proteasome, and direct binding is believed to promote opening of the 20S proteasome gate and degradation (179, 261). Supporting this model are observations that some naturally unstructured proteins (i.e., intrinsically disordered proteins) such as p21Cip1 may bind directly to the 20S proteasome α subunits and become degraded (211, 353). However, many proteins, including p21Cip1, that have been found to be degraded by the free 20S proteasome are degraded by the 26S proteasome with the same or a better efficiency (17, 211).
Other studies also link the 20S proteasome to degradation of oxidized proteins in vivo. Thus, whereas siRNA-mediated knockdown of 19S particle subunit Rpt2 in mammalian cells only results in a partial stabilization of oxidized proteins, knockdown of the 20S core subunit β5 has a much stronger effect (271). Since the siRNA-mediated knockdowns might not be equally efficient and since 20S proteasome subunits are also components of the 26S proteasome, one should be careful with such comparisons. However, observations in budding yeast also support the role of free 20S proteasomes in degradation of oxidized proteins. Hence, deletion of RPN9 leads to a defect in 26S proteasome assembly, but oxidized proteins are degraded more efficiently in rpn9 mutants than in wild-type cells (159).
In some cases 20S proteasome-mediated degradation of oxidized proteins may depend on other proteasome cofactors. For instance, in the nucleus, 20S proteasome activity may be more resistant to aging and to oxidative stress (236, 283). This effect appears to be connected to the enzyme PARP-1, which has been reported to be a positive regulator of 20S proteasome activity (236, 283). However, mechanistic details concerning this activation are lacking.
Calmodulin is one of the most intensely studied proteins during oxidation induced protein degradation. Calmodulin is the primary Ca2+ signal transducer in eukaryotes and belongs to the family of EF-hand domain Ca2+ sensors (48). Upon Ca2+ binding, calmodulin undergoes significant conformational changes, exposing hydrophobic areas, which are rich in methionine residues (48). With age, calmodulin signaling is reduced (110), and this loss of functionality may be attributed to age-induced oxidation of its methionine residues to methionine sulfoxides. However, the methionine residues appear to differ in their susceptibility to oxidation (321).
Oxidized calmodulin can be degraded by both intact 26S proteasomes and by 20S proteasomes associated with the molecular chaperone Hsp90 (98, 342, 377). Although calmodulin can be ubiquitylated, the degradation of calmodulin appears to be independent of ubiquitin (342).
Recognition of the oxidized form of calmodulin by the proteasome cannot solely be explained by the exposure of hydrophobic areas since Ca2+ binding to native calmodulin also leads to an increase in surface hydrophobicity, but in this state calmodulin is actually resistant to degradation (98, 342). Recently, the oxidation of specific methionine residues within calmodulin was suggested to trigger proteasomal degradation (14, 292). This is interesting since it implies that there are special mechanisms present in the cell that recognize defined oxidation patterns, besides hydrophobic areas and misfolding. Thus, one proposal is that oxidation of certain methionine residues in calmodulin to methionine sulfoxide leads to untangling of an otherwise cryptic sequence, distantly resembling PEST-degron (14), which is a sequence motif found in short-lived proteins that is connected with rapid protein turnover. Since oxidation of certain methionine residues may also inhibit calmodulin binding to specific interaction partners, it seems that calmodulin function is regulated at two stages during oxidative stress: firstly on the level of target activation and secondly by degradation (321).
B. The role of the 26S proteasome in degradation of oxidized proteins
Although oxidatively damaged proteins can be degraded in the absence of ATP and ubiquitin by free 20S proteasomes in vitro, the ATP-dependent UPS plays an important role in this process in vivo. Early on, experiments with cell extracts proved that ATP-dependent proteolysis was required for efficient protein degradation induced by oxidative stress (120).
Since then, several other reports have tied ubiquitin-dependent protein degradation with oxidative stress. During cellular oxidation, the amount of oxidized ubiquitylated protein is increased (308). This could of course be a consequence of oxidative modifications of already ubiquitylated proteins, but could also be due to oxidized proteins that have been ubiquitylated and are awaiting degradation. More recently, ubiquitin conjugates isolated from oxidatively stressed mammalian cells were found to be highly enriched in oxidatively modified proteins and blocking the UPS rendered cells more susceptible to oxidative stress-induced cytotoxicity (87). Compromising ubiquitin-dependent protein degradation by expression of mutant ubiquitin renders cells more susceptible to oxidative stress, but has no effect on 20S proteasome activity (87). Further, the iron regulatory protein 2 is found to be specifically ubiquitylated by the E3 ubiquitin ligase HOIL-1 after oxidative damage, pointing to an involvement of the 26S proteasome in degradation of oxidized protein (161, 386). Also, Hsp70, which via the proteasome cofactor BAG-1 and the E3 CHIP plays a role in ubiquitylation of misfolded proteins (8), has been proposed to play a role in degradation of oxidized Hsp90 clients (274). More recently, the mammalian F-box protein Fbxw7β, which is localized in the ER-membrane, was shown to protect cells from oxidative stress (228). However, no oxidized Fbxw7β substrates are currently known.
The importance of the 26S proteasome in degrading oxidized proteins was also recently demonstrated through studies on the DUB called Usp14 in mammalian cells and Ubp6 in yeast. Usp14 associates with the base of the 19S regulatory complex, and when adding a Usp14 inhibitor to mammalian cells, the degradation of oxidized proteins was accelerated and the cells were more resistant to oxidative stress (199).
Recently, elegant studies, performed in budding yeast, showed that the degradation of newly synthesized proteins is enhanced two- to threefold by oxidative stress, induced by either deletion of the gene encoding SOD, or by treatment with paraquat or cadmium. In contrast, the degradation rates of long-lived proteins were unaffected (234). The degradation of these oxidatively damaged proteins was ubiquitin and ATP dependent, since the E2 enzymes Ubc4 and Ubc5, the 26S proteasome ubiquitin-binding subunit Rpn10, and p97/Cdc48 in complex with Ufd1 and Npl4 were all required for the degradation (234). When any of these components were missing, the oxidatively damaged proteins accumulated in intracellular aggregates and though it remains to be identified, the responsible E3 enzyme is therefore probably related to mammalian CHIP or other E3s that are specific for misfolded proteins. Although p97 in complex with Ufd1 and Npl4 is connected with degradation via the ERAD pathway (278), the degradation of the oxidatively damaged proteins did not rely on the ERAD E2 Ubc7 (234) and these proteins are therefore not degraded via the ERAD pathway.
Considering that the majority of yeast proteins are long lived, the finding that only the degradation of newly synthesized proteins was affected by oxidative stress is surprising. In addition, it was shown that the degradation was only enhanced for about 1 h (234). This indicates that, although susceptible to oxidative modification, the long lived proteins, which have already attained a native conformation when oxidative stress is applied, are not rapidly degraded, but instead probably repaired by cellular thioredoxins, glutaredoxins, and other enzymes with reducing properties (23). However, some of the nascent proteins proceed through a fragile period, lasting up to 1 h, where oxidative modification leads to misfolding to an extent that they are not repaired, but rather eliminated by proteolysis. Since most proteins attain their native structure in seconds or minutes, the nascent proteins, which are prone to oxidation-induced degradation, are likely to be components of larger protein complexes, or require specific cofactors or are perhaps slow to reach their final destination in specific compartments and therefore undergo a longer maturation period.
Interestingly, recent evidence has coupled protein synthesis and protein degradation together in a more direct manner. The translation elongation factor eEF1A has, in addition to its function in recruiting codon-specific aminoacyl-tRNAs to the ribosome, also been shown to possess chaperone activity (217) and to target nascent misfolded proteins for degradation (45, 54). Biochemical and genetic studies have shown that eEF1A binds ubiquitylated proteins and the proteasome subunit Rpt1, and can suppress the growth defect of a rad23Δrpn10Δ double mutant (45, 54). Hence, eEF1A is well suited to detect and promote the degradation of aberrant proteins during translation and may therefore indirectly show specificity for oxidatively damaged nascent proteins. However, whether eEF1A mutants are sensitive to oxidative stress and display a reduced clearance of oxidatively damaged nascent proteins remains to be shown.
In general, proteins that become oxidatively damaged or modified are believed to be degraded more rapidly. However, recently an example of oxidation-induced stabilization was reported. Under normal cellular conditions the SUMO protease SENP3 is ubiquitylated by CHIP and rapidly degraded by the 26S proteasome, but in response to oxidative stress SENP3 is oxidized at specific cysteine residues near its N-terminus. This in turn recruits the molecular chaperone Hsp90, which then protects SENP3 from CHIP-mediated ubiquitylation and degradation (338). This is interesting not only because it provides another link between stress signaling and sumoylation, but also because it suggests that there are other examples of such redox-regulated ubiquitylation events.
VII. The Immunoproteasome Protects Cells from Inflammation-Induced Oxidative Stress
In addition to the standard β subunits, alternative, catalytically active β subunits (β1i, β2i, and β5i), are incorporated into nascent proteasome complexes upon induction with interferon (302) (Fig. 8). Two of these subunits, β1i and β5i, are encoded within the MHC class II region, which led to the terms immunosubunit and immunoproteasome (3). Together, these findings connected immunoproteasome function with antigen presentation, whereby these special 20S proteasomes are responsible for the generation of peptides, derived from pathogens or cellular proteins, that are subsequently presented to cytotoxic T-cells by MHC class I molecules on the cell surface (391).
In response to proinflammatory cytokines, typically produced early upon an infection by the innate immune system, cells rapidly shift to immunoproteasome formation, which is a strongly accelerated and transient response (144). In addition, these cytokines also induce the synthesis of the proteasome activator PA28αβ that also influences proteasomal antigen processing and presentation on MHC class I (80, 125).
Newly translated polypeptides are thought to represent a main source of proteasomal substrates, from which peptides designed for MHC class I cell surface presentation are generated (284, 298). Such polyubiquitylated nascent proteins are also termed defective ribosomal products (DRiPs) (392, 393). In contrast with earlier claims (298), these DRiPs may not be particularly abundant under normal cellular conditions (356). In addition, the type of defect in the DRiPs remained unclear and there existed no explanation on how DRiP levels can be adapted to changing immunological requirements in terms of peptide supply.
Immunoproteasomes exhibit altered peptidase activities and cleavage site preferences, resulting in more efficient liberation of many MHC class I epitopes (182, 194). Expression of immunoproteasomes as a consequence of infections and concomitant interferon production has been shown to exert protective effects against bacterial infection. In particular, immunoproteasomes seem to be important for triggering an effective early cytotoxic T-cell response (76). Studies, using mice lacking a specific immunosubunit, revealed that immunoproteasomes can influence immunodominance hierarchies of antiviral CD8+ T-cells by generating different amounts of a specific epitope. However, in some cases the lack of fully functional immunoproteasomes may also influence the available T-cell repertoire, in that certain viral epitopes or self-epitopes will not be efficiently generated while others are (76, 349).
Despite these positive examples for immunoproteasome function in the immune response, a number of studies reported only subtle effects of immunoproteasomes on the immune system and the specific immune response. Thus, the lack of fully functional immunoproteasomes as in β1i−/− and β5i−/− mice revealed either no or only negligible effects on the priming of CD8+ cytotoxic T lymphocytes or on the hierarchy of known T-cell epitopes. Moreover, in pathogen-challenged tissues also noninfected cells induce immunoproteasome formation in response to cytokines although these cells have no need for improved antigen processing (391). Based on these results, it was suggested that immunoproteasomes are not generally important for an antiviral immune response and that their true cellular function may lie elsewhere (250, 391).
Until today the function of immunoproteasomes was almost exclusively acknowledged in connection with antigen processing and MHC class I antigen presentation. However, recently immunoproteasome deficiency was also linked with cell stress (158). In addition to induction by cytokines or infections, immunosubunits have been reported to be induced by insults like heat stress (34), arsenite (399), nitric oxide (190), or in the course of neurodegenerative diseases (276). Thus, the expression of immunoproteasomes may be initiated in different damage scenarios, supporting the view of a more general physiological function of immunoproteasomes apart from antigen processing.
It was recently observed that interferons upregulate protein ubiquitylation and the translation machinery, resulting in a transient accumulation of newly synthesized K48-linked polyubiquitin conjugates (Fig. 10), which essentially require immunoproteasomes for their efficient degradation (301). Notably, proinflammatory cytokines like interferons are also potent triggers for the endogenous formation of ROS that cause oxidative stress (265, 294, 372). As mentioned, nascent proteins are particularly sensitive to oxidative stress (234), causing their partial unfolding and their tendency to aggregate. Thus, oxidative stress in combination with increased sensitivity of newly translated proteins to oxidant damage results in cytokine-dependent enhanced formation of DRiPs, which are subsequently degraded by immunoproteasomes. Thereby, increased DRiP levels in response to interferons also meet the increased substrate demand of the MHC class I antigen presentation machinery. Therefore, under cytokine-induced oxidative stress, the DRiPs do indeed represent defective nascent proteins (301). Interestingly, the requirement of immunoproteasomes in generating MHC class I ligands from oxidized proteins has already been suggested in the so-called protein oxidation and immunoproteasome or PrOxI hypothesis (345). However, it was shown that DRiPs are ubiquitylated even in response to inflammation and thus require 20S immunoproteasomes associated with 19S regulatory particles, forming 26S immunoproteasomes, for their degradation (301). The correlation between oxidative damage and ubiquitin conjugation was further established using the antioxidant, sulphoraphane, which impaired the interferon-induced accumulation of both polyubiquitylated protein conjugates and oxidatively damaged proteins (301).
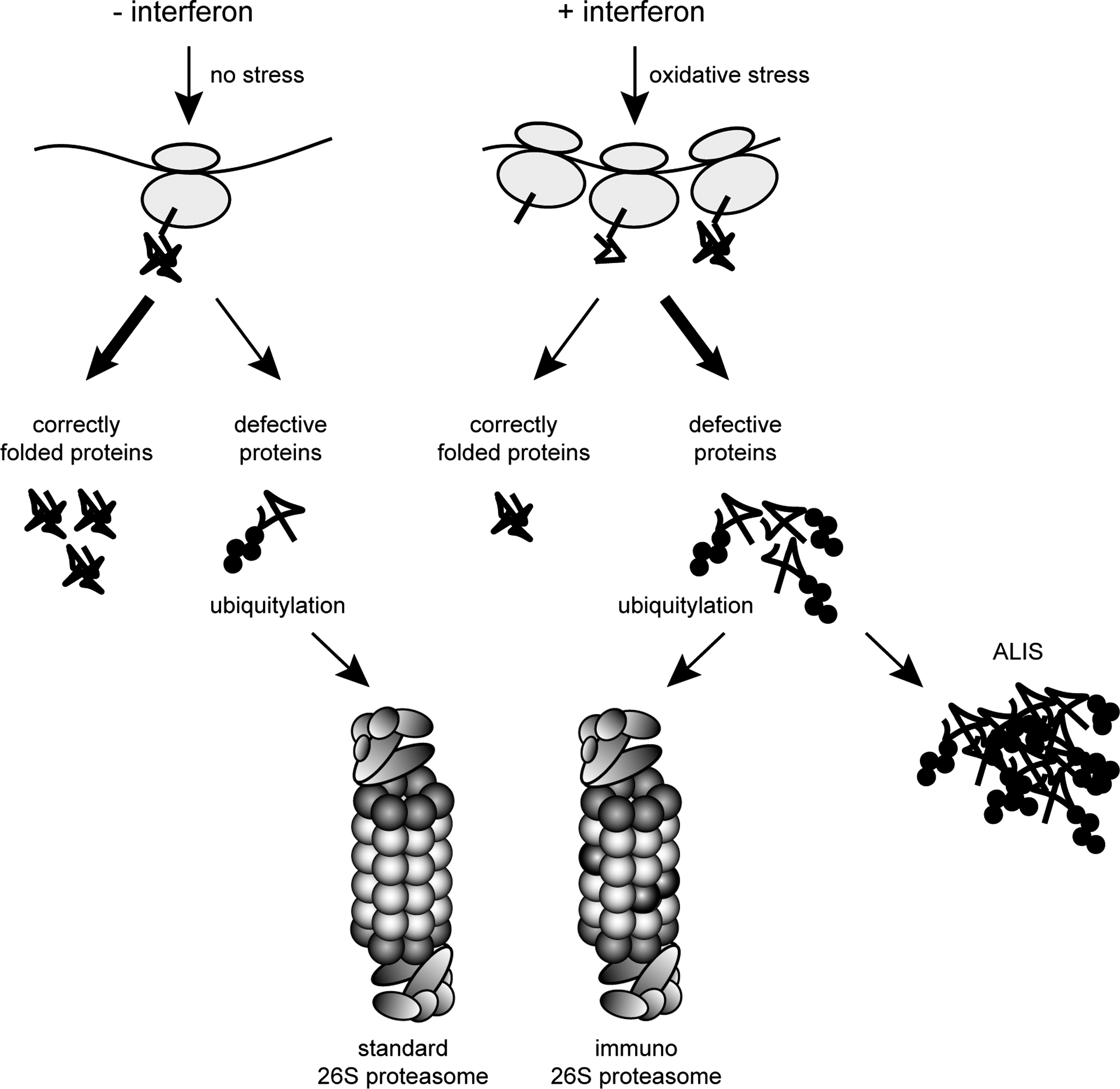
Immunoproteasome-deficient cells and tissues accumulate polyubiquitin conjugates, oxidatively damaged proteins, and form ALIS-type inclusions upon an interferon challenge (301). As mentioned, such ALIS are ubiquitin-rich cytosolic inclusions, which are formed in response to severe cellular stresses such as heat shock, proteasome inhibition, infection, starvation, and oxidative stress. Inflammatory responses, in turn, often provoke oxidative stress in cells by induction of endogenous ROS production (202, 337). Consequently, the accumulation of oxidatively damaged proteins and ALIS represents a severe physiological stress condition, which essentially requires immunoproteasome function to clear and protect cells from the accumulation of protein aggregates (Fig. 10). This was shown to be due to the enhanced capacity of 26S immunoproteasomes for accelerated degradation of nascent oxidant-damaged and polyubiquitylated proteins. Immunoproteasome deficiency results in the accumulation of intracellular protein aggregates, which in turn causes a higher susceptibility to apoptosis (301). More importantly, immunoproteasome-deficient transgenic mice suffer severely from impaired survival and clearance rates upon infection (53, 160, 333) and from the consequences of encephalomyelitis (301). Therefore, the primary physiological function of immunoproteasomes lies in the maintenance of protein homeostasis of cells under interferon-induced oxidative stress conditions. Thus, they are important for the protection of cells against potentially toxic effects of ALIS, which in the long term may result in neurodegenerative diseases. These data crucially expand the concept of immunoproteasome function by demonstrating that accelerated immunoproteasome formation is pivotal for the maintenance of protein homeostasis and the preservation of viability in cells and tissues exposed to inflammatory insults. Therefore, the present paradigm of an exclusive function of immunoproteasomes in antigen processing should be reconsidered.
VIII. Regulating Proteasome Expression
Coordinated regulation of the UPS is crucial for the cell to adjust the capacity of its intracellular protein turnover to specific cellular or environmental requirements. Although gene expression is an essential prerequisite for proteasome formation, the regulation of proteasomal gene expression has been neglected for a long time. The first indication of a coordinated control of proteasomal gene expression in eukaryotes came from the yeast system. In the budding yeast Saccharomyces cerevisiae, genes encoding proteasomal subunits are preceded by a common upstream activating cis-element, the so-called proteasome-associated control element (PACE), which serves as a target sequence for the transcription factor Rpn4 that activates proteasomal gene expression (223). The extremely short-lived Rpn4 protein is not only a transcriptional activator for the 26S proteasome, but also a substrate for the 26S proteasome. Such a regulatory mechanism yields a negative feedback circuit in yeast: the same protein that induces proteasome formation is destroyed by the newly formed proteasomes (384).
Rpn4 is strictly required to control balanced levels of proteasomal subunits and thereby for balanced intracellular proteolysis in yeast. Transcriptional profiling revealed a concerted Rpn4-dependent upregulation of all proteasomal subunits upon treatment with proteasome inhibitors, suggesting that Rpn4 is responsible for the ability of the cell to compensate for impaired proteasome activity (101). Over the last 10 years it has been demonstrated by various groups that Rpn4 represents an important stress-responsive mediator, which is critical for cell survival under various stress conditions (135, 137, 237, 259, 370). The activation of Rpn4 is achieved by binding of transducing transcription factors to the promoter of RPN4, like heat-shock transcription factor 1, multidrug resistance-related transcription factors Pdr1 and Pdr3, and Yap1, a transcription factor that plays an important role in response to oxidation and DNA damage (135, 259). Hence, by Yap1-mediated induction of Rpn4, oxidative stress indirectly results in increased proteasome synthesis. In addition, Rpn4 can be directly activated by proteasome inhibition or by the unfolded protein response (UPR) (237). Rpn4, in turn, induces a large number of stress-responsive genes, including the proteasome genes, genes encoding proteasome-associated proteins, genes connected to the ubiquitylation machinery, DNA repair, and other cellular processes (370).
In higher eukaryotes no homologs of Rpn4 or similarities to transcriptional control elements like PACE could be identified, though proteasome abundance is also controlled by negative feed-back loops in Drosophila and man (218, 235). Induction of proteasome gene expression in response to proteasome inhibitions in Drosophila is dependent on the 5′ untranslated regions of the mRNAs (218). However, the factor mediating this induction remains to be identified.
Besides the induction of immunosubunits by an immunological challenge, it was shown that treatment of cells with nontoxic doses of proteasome inhibitors induces a transient and concerted upregulation of all mammalian 26S proteasome subunit mRNAs. Inhibitor-induced proteasome gene activation results in enhanced protein synthesis and subsequent de novo formation of proteasome complexes (235). Thus, mammalian cells can compensate for the impairment of proteasomal activity by upregulating standard 26S proteasome subunits. In conclusion, the abundance of proteasomes in mammalian cells is regulated at the transcriptional level, although the transcription factor until recently remained unknown.
For the mammalian system, it has been reported that antioxidants enhance mouse proteasome expression through the Keap1-Nrf2 signaling pathway, thus establishing a link between the antioxidative response and the upregulation of the standard 26S proteasomes (195). Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) belongs, like p45-NF-E2, TCF11/Nrf1 and Nrf3, to the cap ’n’ collar–basic leucine zipper (CNC-bZIP) family of transcription factors. The CNC-bZIP motif comprises a basic DNA-binding domain and a leucine zipper dimerization domain. Members of the CNC-bZIP family are key components of the oxidant stress response. The transcription factors bind to antioxidant-response elements (AREs) in the promoters of stress defense genes. AREs are typical promoter elements of many detoxification and antioxidant enzymes, like glutathione-S-transferases and enzymes involved in removal of oxidatively damaged proteins such as proteasomes (108, 289).
Kelch-like erythroid-cell-derived ECH-associated protein (Keap1) functions as a redox sensor that normally anchors the Nrf2 transcription factor within the cytoplasm at the cytoskeleton, targeting it for ubiquitylation and degradation by the 26S proteasome (195, 352). Keap1 functions here as a substrate adaptor protein for the Cullin3 containing CRL-type E3-ligase complex. During oxidative stress, Nrf2 is released from the Keap1 CRL-complex, resulting in the stabilization and activation of Nrf2 (Fig. 11) (232, 396). Accordingly, proteasome inhibitors have been found to elevate the amount of glutathione (171), probably via stabilization of Nrf2 that drives production of γ-glutamylcysteine synthetase, a rate-limiting enzyme in glutathione synthesis.
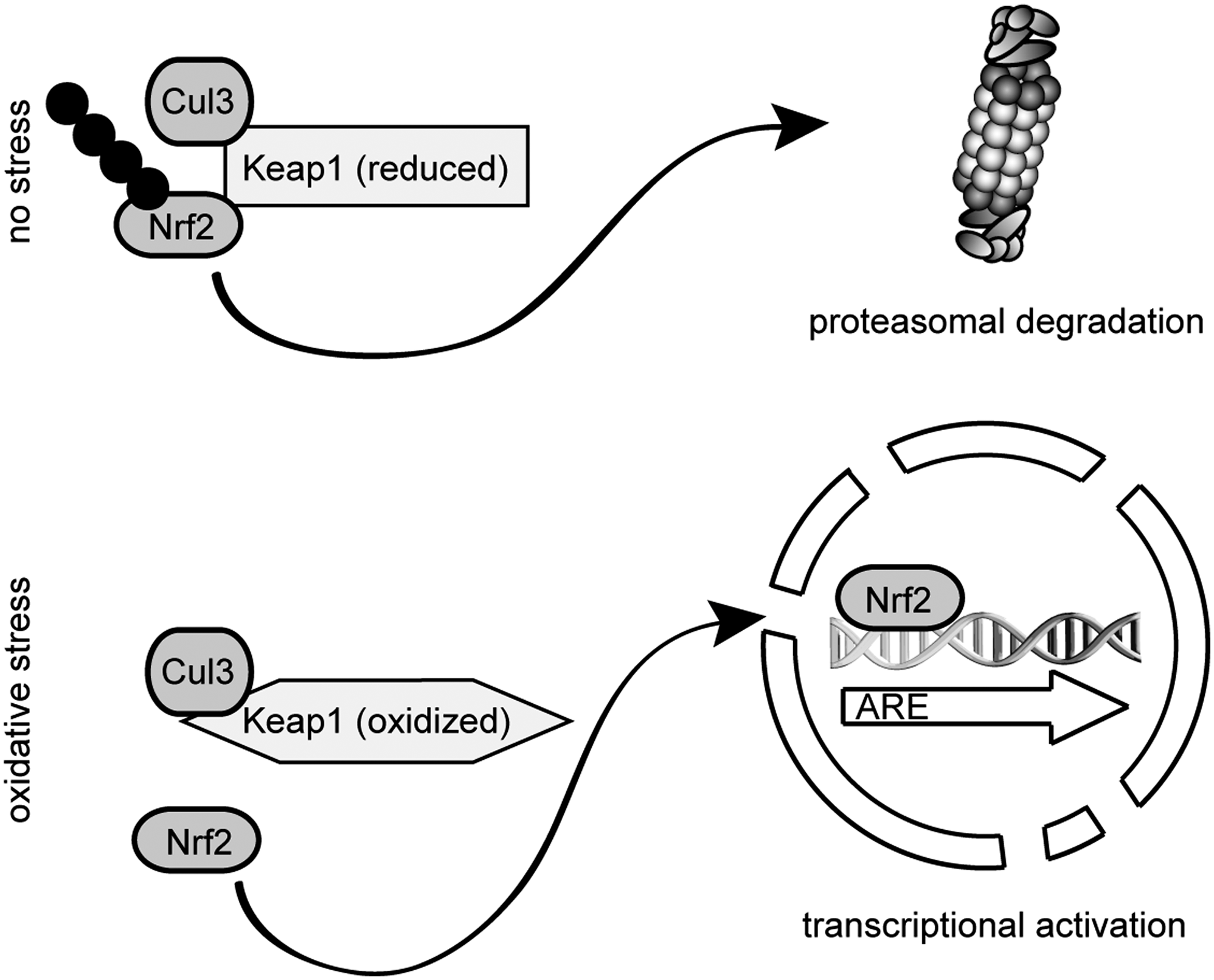
While the knockout of Nrf2 leads to a higher sensitivity to chemical-induced DNA damage, oxidative stress, and higher susceptibility toward cancer development, high constitutive levels of Nrf2 are observed in many tumors or cancer cell lines, conferring resistance to chemotherapeutics (174). Nrf2 regulates expression of a variety of genes involved in xenobiotic metabolism, antioxidant defense, stress response, as well as proteasomal degradation. This collectively constitutes a cellular defense against carcinogens (174).
It was shown before that TCF11/Nrf1, like Nrf2, induces transcription of cytoprotective genes (156, 246). Knockout of the NFE2L1 gene, coding for TCF11/Nrf1, results in embryonic lethality (42). A conditional knockout in adult mice induces hepatic cancer (385). Nrf1 has been described to be primarily a membrane-bound protein, located in the ER membrane (368, 398). In humans, Nrf1 also occurs in a longer isoform known as transcription factor 11 (TCF11), which contains an additional exon and harbors a potential nuclear export signal (156, 166, 385). In the nucleus, TCF11/Nrf1 and Nrf2 hetero-dimerize with other bZIP transcription factors, for example, small Mafs, then bind to the AREs and increase the transcription of antioxidant defense genes (116, 167, 245, 360).
Since most studies on the interference of proteolytic capacity of proteasomes have focused on apoptosis or diseases (65), the capacity of cells to respond to a reduced proteasomal activity, induced by nontoxic doses of proteasome inhibitors, by proteasome gene expression was until recently not clear. Also, the therapeutic potential of proteasome inhibitors as drugs in inflammatory and hyperproliferative diseases is increasingly realized. Several studies have reported that Nrf2 can be activated by proteasome inhibition (84, 85, 192, 293, 374). Moreover, Nrf2 is discussed as an important factor that confers apoptosis protection in tumor cells through induction of proteasome levels (6). It was therefore assumed that the ARE-Nrf2-Keap1 pathway is responsible for the expression of proteasome genes in general.
Recently, the transcription factor TCF11 was identified as the key regulator for 26S proteasome formation in human cells (279, 329). Proteasome inhibitors trigger the accumulation of oxidant-damaged proteins and promote the nuclear translocation of TCF11, permitting activation of proteasome gene expression by binding to AREs in their promoter regions. In addition to genes encoding standard proteasome subunits and the proteasome maturation protein POMP, more than 46 UPS-related genes were upregulated by TCF11 in response to the proteasome inhibitor epoxomicin (329). Thus, in response to proteasome inhibition and the resulting proteotoxic stress (64), the cell appears to increase the level of UPS enzymes involved in ubiquitin and protein homeostasis. Vice versa, the cytokine-inducible immunosubunits and PA28αβ genes were downregulated or remained unchanged upon proteasome inhibition (235). Inflammatory cytokines induce ROS production and thus promote oxidative stress, whereas the antioxidative defense machinery regulated by Nrf2 or TCF11 counteracts cytokine-mediated oxidative damage (290). In fact, TCF11 additionally functions as a repressor of the radical-producing enzyme, interferon-inducible nitric-oxide synthase, via tumor growth factor β signaling, supporting the above conclusions (22). Moreover, indirect antioxidants like sulforaphane, which detoxify radicals by induction of phase II enzymes via Nrf2, counteract oxidant-damage of proteins during cytokine responses (301). Under these conditions the damage of proteins is sufficiently low for clearance by standard proteasomes.
In light of these results, the use of proteasome inhibitors as therapeutic agents for treatment of cancer can result in tumor resistance to proteasome inhibition by upregulation of proteasomes (1, 109) through TCF11-dependent gene activation. Otherwise it has been shown that cancer cells are more sensitive to proteasome inhibitors than healthy cells, maybe due to a restricted activation of TCF11 (1, 151). Under noninducing conditions, TCF11 resides in the ER membrane. ER-resident proteins are degraded by the ERAD system (152, 359), and indeed TCF11 is rapidly degraded by ERAD, involving direct ubiquitylation by the ER-membrane resident E3 ligase Hrd1 together with the extraction capacity of p97 under noninducing conditions (Fig. 12). Application of chemical proteasome inhibitors results in nuclear translocation and stabilization of TCF11. Thus, human proteasome-dependent protein degradation is regulated by a transcriptional control loop involving the ERAD system to counteract proteotoxic stress, caused by proteasome inhibition (Fig. 12) (329). A coordinated induction of proteasome subunits by TCF11 and their assembly into active proteasome complexes is a critical mechanism of protection against proteotoxic stress. Therefore, in a manner remarkably parallel to that in yeast, proteasome-dependent proteolysis in higher eukaryotes is autoregulated by degradation of the same regulators of gene expression that control their synthesis.
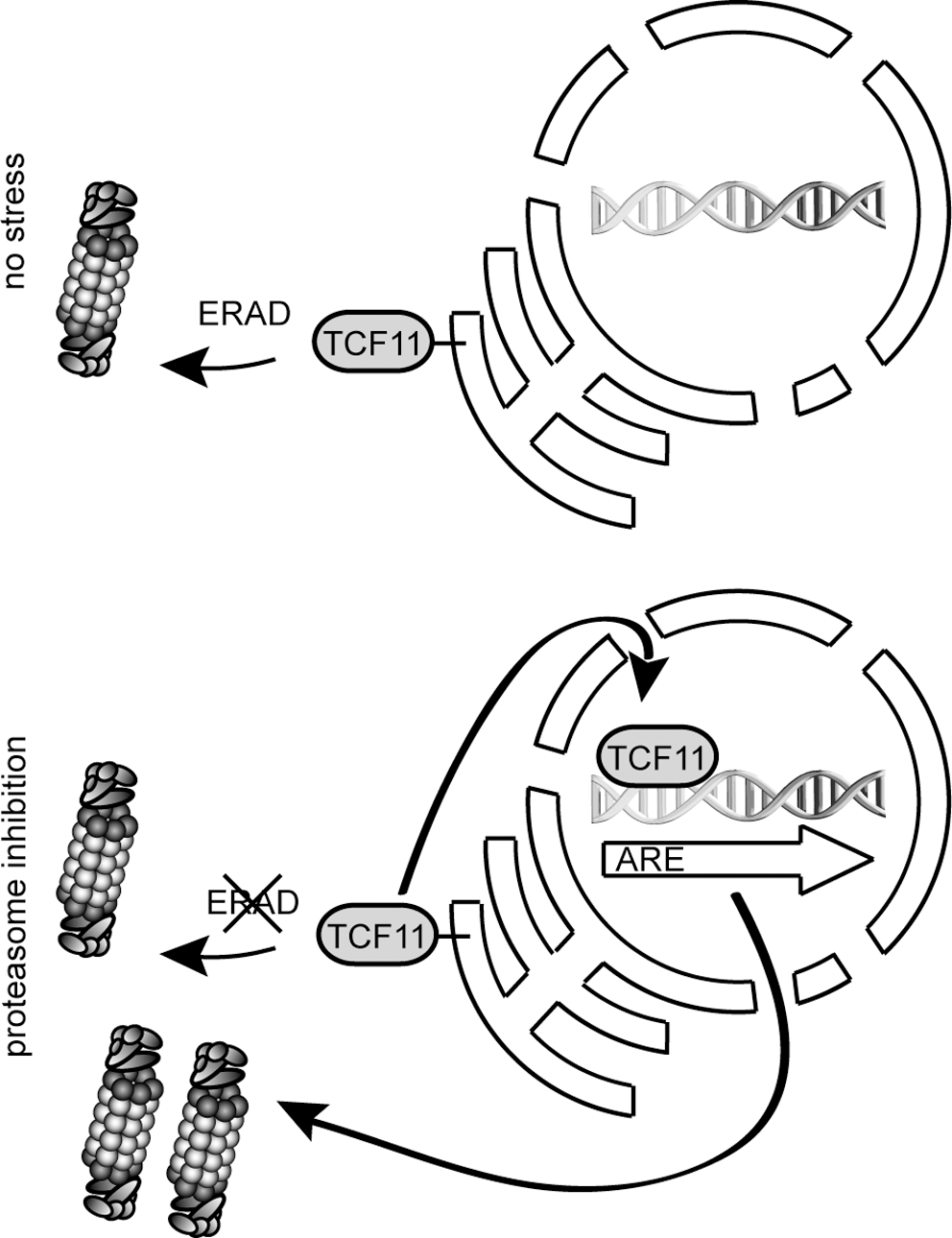
IX. Oxidative Stress and the Ubiquitylation System
It is generally accepted that since protein degradation is increased during stress, the cellular demand for ubiquitin is also increased and cells therefore induce synthesis of ubiquitin in response to stress conditions (65, 100). In yeast, the stress-inducible polyubiquitin gene, UBI4, has been shown to be upregulated in response to oxidative stress (47). Similarly, the mouse ubiquitin genes Ubb and Ubc are both upregulated upon oxidative stress (291). However, oxidative stress has also been found to have adverse effects on the ubiquitylation process.
Upon oxidation, the sulfhydryl group in a cysteine residue (Cys-SH) can form a disulfide bond to another cysteine residue in proteins or in glutathione. Alternatively, the addition of molecular oxygen to the sulfhydryl can lead to cysteine sulfenic acid (one oxygen) (75), cysteine sulfinic acid (two oxygens), or even cysteine sulfonic acid (three oxygens). Whereas sulfenic acid modifications are reversible, oxidation to cysteine sulfonic acid is irreversible. The reduction of cysteine sulfinic acid is so far only described for peroxiredoxins (26). Also, methionine residues are susceptible to oxidation. The resulting methionine sulfoxides are readily reduced by methionine sulfoxide reductase (326).
S-glutathionylation is a common reversible protein modification that is observed in response to oxidative stress (139) as a means of protecting proteins from other more destructive oxidation reactions, such as formation of cysteine sulfonic acid. During oxidative stress, the ratio of the major cellular redox buffer system, reduced and oxidized glutathione (GSH/GSSG), decreases dramatically (139, 357). This change in the intracellular redox environment is also reflected by alterations of the protein thiol status (139) and is connected to changes in protein activity.
When looking at the central enzymes in the ubiquitin pathway, the DUBs and the E1, E2, and E3 enzymes, one finds that most contain active site cysteine residues, which are prone to oxidation and may become modified by, for example, S-glutathionylation. Accordingly, treatment with the oxidizing agent diamide leads to a decrease in the level of ubiquitin-loaded E1 and E2 enzymes, lower levels of ubiquitin-protein conjugates, and retarded ability to form ubiquitin-protein conjugates and E1 and E2 ubiquitin thioesters de novo (162, 253, 309). However, the effect is transient and when the intracellular redox conditions are restored to normal, the ubiquitin system is also re-established to full functionality (162, 253, 309), and, curiously, mild oxidative stress increases ubiquitin-conjugation activity (307).
The transient inhibition of the ubiquitin-conjugating enzymes, caused by reversible S-glutathionylation, can serve adaptive functions in the cell. The E1 and E2 enzymes may be protected from permanent oxidative damage by S-glutathionylation. Subsequently, deglutathionylation leads to rapid re-activation when the oxidative stress has lifted. The temporary incapacitation of the UPS may also serve to protect the many proteins that are only affected by small conformational changes in response to oxidation. These can thus be salvaged when the stress period has ended (139). By disabling the predominant mechanism for protein degradation during oxidative stress, the cell retains the possibility to quickly return to a normal state. However, this is dependent on the stress period not being too long or too severe. One should note, however, that the inhibition of the ubiquitylation system in response to oxidative stress is not easily reconcilable with the observed increase in ubiquitin-protein conjugates during oxidation (308). Hence, more detailed studies are required before such controversies can be resolved.
X. Oxidation-Induced Changes in the 26S Proteasome
Oxidative stress causes an elevated degradation of oxidized proteins by the proteasome (70, 93, 229). However, the 26S proteasome is also itself subject to modulation during oxidative stress (282, 283). Unlike the 20S particle and the proteasome activator PA28αβ, which are surprisingly resistant to oxidation (331), the 19S regulatory particle is prone to oxidative damage (275, 283).
A decline in 26S proteasome activity was detected at H2O2 concentrations between 0.4 and 1 mM (281), and although the induction of oxidative damage in liver epithelial cells in this concentration range did not affect cell viability, these H2O2 concentrations are probably not physiologically relevant. Under extreme conditions, such as the oxidative burst during pathogen invasion or in the aqueous humor surrounding the ocular lens, the concentration of H2O2 maximally reaches only 0.1 mM (86), where 26S proteasomes should still be perfectly active (198). However, recently it was reported that signaling can lead to inactivation of peroxiredoxin which in turn allows H2O2 to accumulate around membranes (397). Hence, in some rare cases unexpectedly high local concentrations of H2O2 might build up. Due to the instability of H2O2, one should in general be cautious when assessing experimental data. Thus, when adding H2O2 to cells, the actual H2O2 concentration will probably quickly drop to unknown levels and is therefore not readily comparable with the H2O2 concentrations observed in vivo. However, since no effect on 26S proteasome activity was observed by treating proteasomes with 0.4 mM H2O2 or less (283), it is unlikely that the maximal naturally occurring concentration of 0.1 mM (86) affects 26S proteasome activity. Accordingly, the UPS is functional even during atherogenesis despite a significant increase in oxidative stress (149).
Under normal conditions the 26S proteasome is a rather stable particle (193). However, upon oxidation, the 26S proteasome may dissociate into free 20S and 19S particles (371), which in turn may lead to accumulation of ubiquitylated proteins (371). Recently, the oxidation-triggered dissociation of the budding yeast 26S proteasome was shown to depend on the proteasome-interacting protein Ecm29 (371). Since yeast mutants lacking Ecm29 are more sensitive to oxidative stress, this suggests that Ecm29 functions as a redox switch that, upon oxidation, induces 26S proteasome disassembly, leading to higher amounts of 20S proteasome particles and better cellular recovery from oxidative stress (371). Accordingly, yeast mutants, which are defective in 26S proteasome assembly, show an increased degradation of oxidized proteins (159). Interestingly, Ecm29 has also been suggested to serve as an adaptor for coupling 26S proteasomes to specific cellular compartments, such as secretory compartments and to the centrosome (122, 123), but whether these functions are connected with oxidative stress is still unknown. The details of how Ecm29 function to dissociate 26S proteasomes during oxidative stress are also still unknown, but oxidation-induced protein modification of Ecm29 could play a role.
During oxidative stress, 26S proteasomes are also more directly affected by modifications such as carbonylation, glycation, ubiquitylation, as well as binding to lipid peroxidation products and glutathione (Fig. 13) (37, 73, 74, 255, 401). However, the functional relevance of these modifications is unknown.
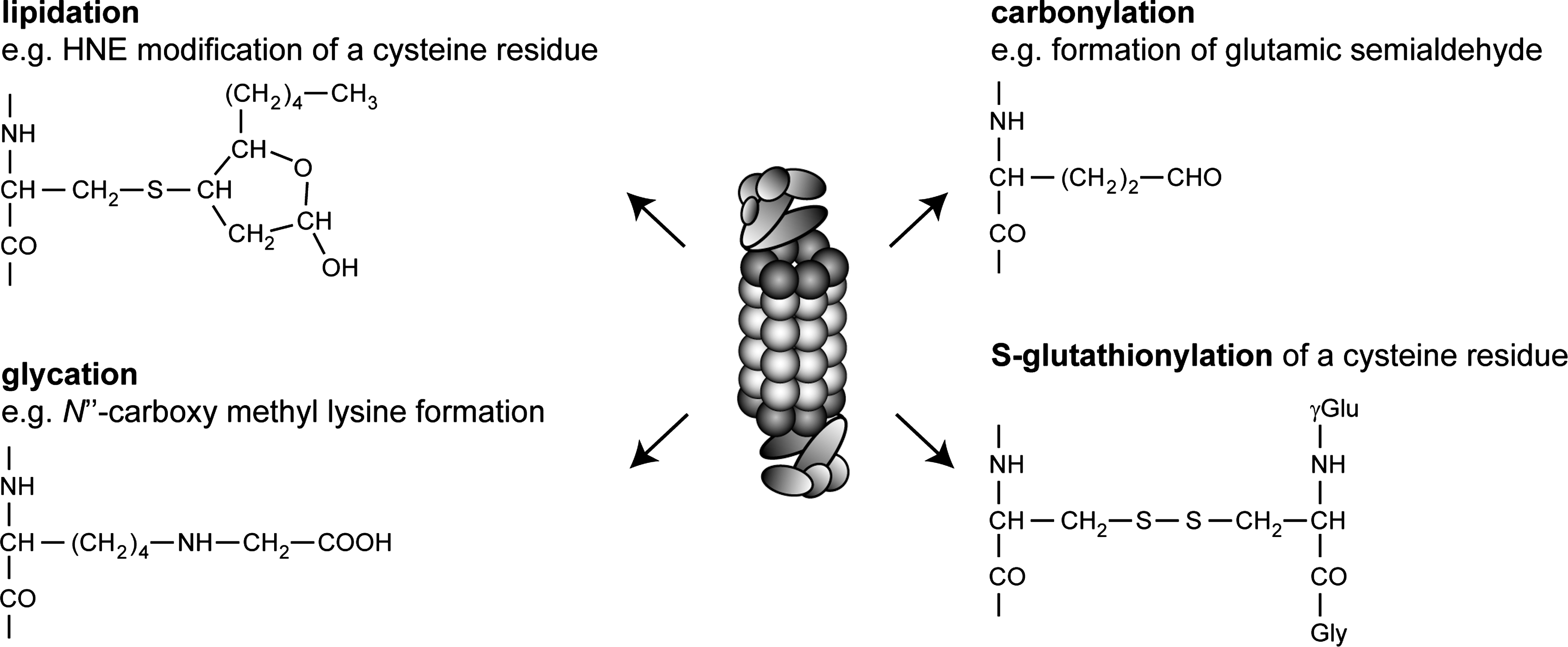
Carbonylation is a well-recognized marker of protein oxidation (204, 251, 383), but a basal level of carbonylation of 20S proteasomes has been reported (32). Induction of oxidative damage leads primarily to the introduction of carbonyl groups in the α2, α4, α6, and β3 subunits in purified mammalian 20S proteasomes (2, 401).
Under oxidizing conditions, certain amino acid residues may react with sugar carbonyls, such as glyoxal, thus forming glycation products. These advanced glycation end products (AGEs), such as N′′-carboxymethyl-lysine, lead to protein damage (347). With age, the proteasome subunits showing increased glycation were α1, α2, β1, β5i, and β7 as well as the 19S subunits Rpt1, Rpt2, and Rpn11 (2, 37).
Another secondary protein modification is caused by 4-hydroxy-2-nonenal (HNE). HNE is a prominent product of lipid peroxidation after oxidative cell damage (354). This highly cytotoxic component is known to react with both DNA and proteins (264, 354). Exposed lysine, histidine and cysteine residues are especially prone to HNE modification, which may lead to intra- and intermolecular cross-linking, which result in protein inactivation and aggregation. Proteasome inhibition after oxidative stress and during aging has been linked to HNE addition (32, 255). After inducing oxidative damage, HNE-modified proteasomes can be observed after a few hours, but full recovery is in place within 48 h (255). This time span correlates nicely with the observed decline and recovery of proteasome activity (255).
Some data suggest that the inhibitory effect of HNE on proteasome activity is tissue specific. Thus, purified 20S proteasomes from rat heart showed a HNE-dependent decline in the trypsin-like activity, whereas proteasomes derived from liver were mainly inhibited in their chymotrypsin-like activity (32, 95). The hydrolytic inactivation appears to be an indirect effect of HNE-modified proteasomal α subunits and proteolytic inactive β subunits, since hardly any HNE was found to be attached to the enzymatic β subunits (32, 37, 95). The active β subunits appear to only become modified with age or when proteasomes are treated with high concentrations of HNE (95). Thus, not all subunits have the same susceptibility toward HNE addition. Typical subunits found to be modified both in vivo and in vitro include all α subunits with the exception of α3. Other subunits, such as β1, β5, β7, and Rpt4, have only been observed to be modified in vivo, whereas subunits α3, β1i, β3, β4, and β6 are only modified in vitro (15, 16, 32, 36, 95, 97). Intriguingly, interaction of the molecular chaperone Hsp90 with 20S proteasomes seems to protect certain subunits from HNE modification (58, 59, 97).
Oxidation-induced differences in proteasome activity are also observed between purified proteasomes and proteasomes in crude cell extracts, indicating that other factors than direct HNE addition to proteasomal subunits play a role. For instance, oxidative damage in rat heart extract causes a decrease of all three hydrolytic activities, whereas only the trypsin-like activity of purified HNE-modified 20S proteasome from heart was decreased (32). One possible explanation is that accumulated protein aggregates or HNE-linked proteins inhibit proteasome function (107). However, it is likely that also other oxidative proteasome modifications are involved.
In addition, proteasomal activity was found to be inhibited by S-glutathionylation (73, 74, 400). In mammals, S-glutathionylation of the 19S complex subunits Rpn1 and Rpn2 correlates with an inhibition in the chymotrypsin-like activity (400). However, this S-glutathionylation does not affect the stability or integrity of the 26S particle. In yeast, S-glutathionylation of the proteasome has also been observed to inhibit the proteolytic activity (74). In general, proteins that are stably S-glutathionylated are very rare (139). However, low levels of S-glutathionylation are observed in proteasomes even under normal growth conditions (314), yet the physiological significance of this observation is still unknown. Removal of glutathione from the 20S protease in vitro by treatment with, for example, dithiothreitol restores the proteolytic activity completely (314). However, within an intact cell, deglutathionylation of the proteasome is probably achieved via enzymatic catalysis. In yeast, the major glutaredoxin, Grx2, may be involved in the release of glutathione from 20S proteasomes (314). The cytosolic yeast thioredoxins Trx1 and Trx2 interact with the proteasome (132) and may also catalyze deglutathionylation of the 20S proteasome in vitro (314). In mammalian cells and in fission yeast, the 26S proteasome stably associates with the thioredoxin Txnl1, described in detail below (5, 380). However, Txnl1 does not appear to support glutathione liberation from the 26S proteasome (4).
Considering the importance of the proteasome in clearing the cell of oxidatively damaged proteins, it appears counterproductive that proteasome activity is partially inactivated by glutathione during oxidative stress. However, as with the ubiquitylating enzymes and DUBs, the reversible modification of proteasome subunits with glutathione also protects the proteasome from irreversible damage. Thus, after induction of glutaredoxins and thioredoxins by the antioxidant defense, the proteasome will be rapidly reduced and fully regain its enzymatic activity to further mitigate the oxidative stress.
Importantly, oxidative stress has also been found to affect proteasome activity indirectly through phosphorylation. Hence, the tyrosine kinase c-Abl is activated by oxidative stress and γ-irradiation and phosphorylates tyrosine 153 in the α4 20S proteasome subunit, leading to a decrease in proteasome activity (190). This mechanism may allow c-Abl to regulate cell cycle arrest in response to γ-irradiation and may serve a regulatory function during oxidative stress, for example, by protecting salvageable oxidized proteins from premature degradation.
XI. Proteasome-Associated Oxidoreductases
Recently, human 26S proteasomes were found to associate directly with the thioredoxin called Txnl1 or Trp32 in humans and Txl1 in the fission yeast Schizosaccharomyces pombe (4, 5, 380). Like proteasome subunits, Txnl1 is induced by proteasome inhibition (5, 218) in a TCF11-dependent manner (329). However, unlike most other proteasome cofactors, Txnl1 associates with the 26S proteasome in near stoichiometric amounts by a direct interaction between the Txnl1 C-terminal proteasome-interacting thioredoxin (PITH) domain and 19S regulatory subunits near the substrate binding site (5, 380). In vitro Txnl1 displays a robust thioredoxin activity, and since Txnl1 is found to be fully reduced in vivo, it most likely catalyzes protein disulfide reduction (5, 380). Potential substrates for a proteasome-associated thioredoxin, like Txnl1, include the proteasome itself, proteasome substrates and proteasome cofactors (Fig. 14). Although the PA28 complex has been shown to contain a disulfide bond (380), no proteasome subunits have been isolated as Txnl1 substrates, and while the proteasome clearly prefers to degrade reduced proteins (4, 373), no natural proteasome substrates have yet been identified as Txnl1 substrates. However, substrate capture experiments, using active site Txnl1 mutants, did reveal eEF1A as a Txnl1 substrate (5), indicating that perhaps during oxidative stress, Txnl1 keeps eEF1A reduced to allow disposal of misfolded nascent proteins.
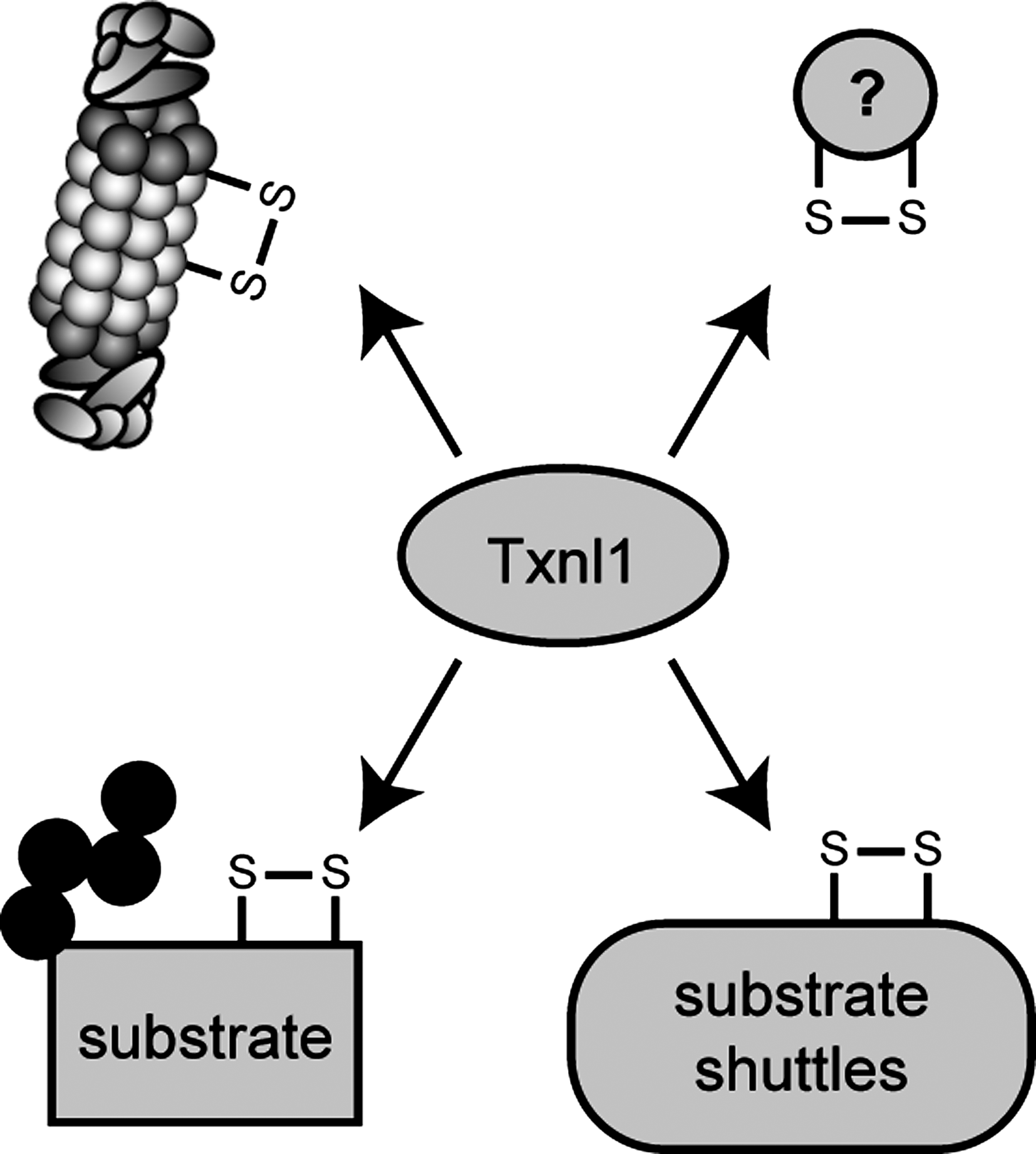
Surprisingly, no obvious phenotype was observed when Txnl1 was knocked out in mice and fission yeast (4, 380) and in fission yeast Txnl1 does not display any strong synthetic defects with Trx1, the only other cytosolic thioredoxin (4). This suggests that, unlike most other thioredoxins, Txnl1/Txl1 is a fairly specific enzyme that only targets a subset of oxidized proteins. Neither Txnl1-deficient mouse cells nor a txl1Δ fission yeast mutant was sensitive to oxidative stress (380), and also the degradation of bulk protein and model ERAD and cytoplasmic substrates were unaffected by loss of Txnl1 (5). However, genetic screening of a fission yeast txl1Δ mutant did reveal a synthetic growth defect in combination with a cut8Δ mutant (4, 380). In yeast and Drosophila, Cut8 tethers 26S proteasomes to the nuclear rim (343). So far, no Cut8 ortholog has been found in mammals. In the cut8Δ mutant, proteasomes mislocalize and become evenly distributed throughout the cytosol (343). Presumably, this mislocalization affects some activity that is worsened by additional loss of Txl1 in a txl1Δcut8Δ double mutant. However, since expression of a Txl1 active site mutant can rescue the growth defect of the txl1Δcut8Δ double mutant (4, 380), the genetic interaction between txl1Δ and cut8Δ is not connected with Txl1 thioredoxin activity, but probably rather linked to some structural instability of the 26S proteasome when Cut8 and Txl1 are both missing. Accordingly, Txnl1 has been suggested to play a role in proteasome assembly since it is associated with nascent 26S proteasomes early in the assembly process (147). However, the lack of any obvious phenotype in mouse and fission yeast knockouts argues against this hypothesis.
Although Txnl1 is conserved in most eukaryotes, no ortholog is present in budding yeast, implying that the other cytoplasmic thioredoxins, Trx1 and Trx2, perhaps maintain Txnl1 function in this organism. Indeed, both Trx1 and Trx2 have been found to associate with 26S proteasomes in budding yeast (132), and may here function in deglutathionylation of proteasome subunits (74, 400). However, in fission yeast Txnl1 does not appear to share this function (4).
Txnl1 is not the only example of a proteasome-associated redox active enzyme. Recently, the degradation of certain transcription factors was shown to be regulated by a flavin-dependent and proteasome-associated redox switch. The human flavin-dependent NADPH:quinone reductase, NQO1, interacts with the 20S proteasome and under reducing conditions recruits the transcription factors p53 and p73 (10), thereby probably protecting them against proteasomal degradation (11). The budding yeast NQO1 ortholog Lot6 is also associated with 20S proteasomes (319) and, like its human counterpart, Lot6 binds and protects the transcription factor Cin5 from degradation (Fig. 15) (319).
XII. Arsenite Regulation of Proteasome Composition
Proteasomes are subject to regulation by a variety of stress conditions, but in relation to redox conditions arsenite, As(III), induced stress is especially relevant. Exposure to As(III) triggers the redox defense mechanisms and causes accumulation of ROS that modify protein structures and cause protein misfolding (310). In turn, this promotes activation of the heat-shock response and integrated stress response (168, 205), which induce expression of molecular chaperones, proteasome components, and other stress-relieving enzymes. Intriguingly, As(III) exposure induces Txnl1 degradation by the 26S proteasome (380). Under normal conditions, Txnl1 is a stable protein in mammalian cells (5, 380), but upon treatment with As(III) the half-life of Txnl1 is reduced to about 1 h (380). This observation is somewhat counter intuitive, since one would expect Txnl1 to provide the 26S proteasome with redox capabilities that are important during As(III)-induced oxidative stress.
In acute promyelocytic leukemia, the promyelocytic leukemia protein (PML) is fused to the retinoic acid receptor and the disease can be effectively treated by arsenic administration. Like Txnl1, the PML protein has been shown to be degraded in an As(III)-dependent manner (344). In the case of PML, arsenic triggers intermolecular disulfide formation (164). The disulfide-linked PML multimers are then modified with chains of the small UBL modifiers SUMO-2 and -3 (164). This in turn recruits the SUMO-binding E3 ubiquitin ligase RNF4 that ubiquitylates PML and targets it for degradation (Fig. 16) (344). Txnl1 has also been reported to be conjugated to SUMO (133), and RNF4 might therefore also be responsible for As(III)-induced ubiquitylation and degradation of Txnl1.
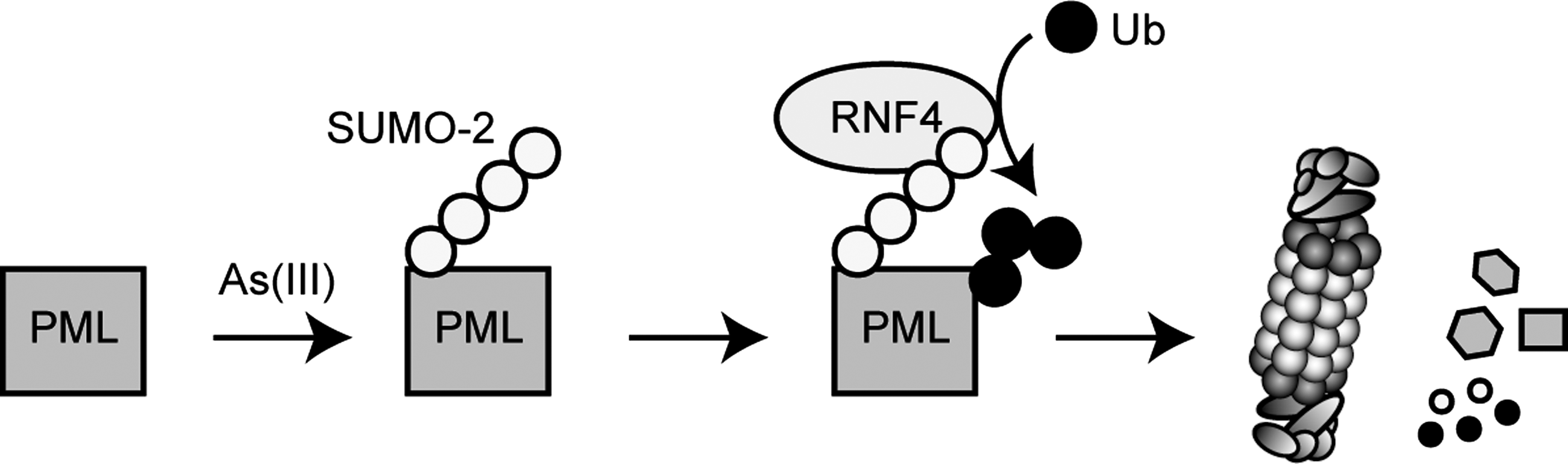
Exposure to As(III) also leads to other changes in the 26S proteasome composition. In response to As(III)-induced protein misfolding, transcription of the AIRAP gene is strongly upregulated and AIRAP is incorporated into pre-existing 19S regulatory particles (328, 395). Upon incorporation, AIRAP stimulates proteasomal cleavage of peptide substrates (328, 395), suggesting that AIRAP adjusts proteasome activity to counter the proteotoxic stress provoked by As(III). Accordingly, AIRAP knockout mouse cells and C. elegans lacking AIRAP accumulate polyubiquitylated proteins and are sensitive to stress (328, 395). The worms also exhibit a shortened lifespan (395).
Intriguingly, mammals have a second AIRAP-like (AIRAPL) gene that also encodes a proteasome-associated protein with the ability to stimulate peptide cleavage. However, unlike AIRAP, the larger AIRAPL is constitutively expressed, but As(III) exposure stimulates its proteasome-binding capabilities (395). Both AIRAP and AIRAPL associate with the 26S proteasome via their N-terminal AN1 Zn-finger domains (328, 395), but an important difference between AIRAP and AIRAPL is the presence of two ubiquitin-interaction motifs (UIMs) in the C-terminus of AIRAPL that are lacking in AIRAP. This combination of proteasome- and ubiquitin-binding domains in the same protein resembles the UBL/UBA proteins Rad23 and Dsk2, and suggests that AIRAPL may also play a role in recruiting ubiquitylated substrates to the 26S proteasome in response to As(III) exposure.
XIII. Proteasome Function in Mitochondrial Maintenance
In response to stress, cells do not only utilize the UPS to mitigate damage and increase survival, but also rely on autophagy (175). The ubiquitin- and proteasome-binding protein p62 (also named sequestosome-1) is a central autophagy component that shuttles ubiquitylated proteins to the proteasome (300). Since p62 also associates directly with LC3, a well-characterized autophagic marker, p62, links polyubiquitylated proteins with autophagy (176). During defects in the autophagic system, p62 is upregulated and aggregates in polyubiquitin-containing cytoplasmic inclusions (376). Recently, it was shown that accumulation of p62, due to a defect in autophagy, causes oxidative stress (226). Intriguingly, p62 associates directly with the Nrf2 regulating CRL-type E3, Keap1, and sequesters Keap1 into aggregates, resulting in the inhibition of Keap1-mediated Nrf2 ubiquitylation and subsequent proteasomal degradation (198). Thus, autophagy deficiency also activates the Nrf2 pathway by a redox-independent mechanism.
Mitochondria are the major source of ROS in cells. The UPS plays a role in mitochondrial fission and fusion events (35, 56), but along with autophagy, also clears damaged mitochondria from the cell.
Recently, it was reported that the UPS and autophagy cooperate in mitochondrial maintenance during cell quiescence, but not in proliferating cells (338). During quiescence, the proteasome is required for normal mitochondrial function, and inactivation of the proteasome by mutation results in accumulation of ROS and antioxidant proteins (338). Since mitochondria are the primary source of ROS production in cells, autophagy of mitochondria prevents lethal accumulation of ROS and promotes longevity. Accordingly, double mutants, where both the proteasome and the autophagy system are compromised, invoke hyperaccumulation of ROS and lethality (338).
So far, only a few mitochondrial proteins were found to be degraded by the UPS (12, 213, 224, 340), but since these substrates were all found recently, others are bound to exist and the UPS could therefore provide a common method for degrading specific mitochondrial proteins. The detailed mechanism of this mitochondrial-associated degradation is still unclear and many of the components involved have yet to be identified and characterized. However, again the p97 ATPase is involved (340), suggesting that this system may function in an ERAD-like manner. Interestingly, an E3, called Parkin, that is involved in heritable forms of Parkinson's disease, appears to play a major role in both UPS-mediated clearance of mitochondrial proteins and in autophagy. Hence, the ubiquitin and autophagy systems also converge on mitochondrial regulation in Parkinson's disease (see below).
XIV. The UPS and Aging
The role of the UPS in aging has been reviewed extensively (40, 52, 243, 273), and we shall only discuss it briefly here.
It has long been recognized that ROS and protein oxidation contribute to aging (140, 256, 324). Thus, generally aging cells contain more oxidized proteins (173, 324), and transgenic mice overexpressing thioredoxin have an extended lifespan (240). Some results indicate that when cells age, their ability to maintain protein degradation is also compromised (50, 173). This effect may be caused by direct oxidation-induced modification of UPS components (40, 52, 243, 273). In aging cells oxidized and misfolded proteins accumulate (363), and this may in turn lead to protein aggregation and various diseases, including several neurodegenerative diseases (28) and muscle maintenance disorders (7). Due to light-induced oxidation, retinal cells are prone to oxidative stress and aging, accordingly oxidative stress and proteasome function also converge during macular degeneration (272). Moreover, aging cells often contain a protein and lipid aggregate called lipofuscin (173). These structures are distinct from aggresomes and some data indicate they inhibit the proteasome (283). Although some of the accumulated proteins, particularly oxidized proteins, may be degraded in a ubiquitin-independent manner, the increased steady state level of these proteins is still thought to inhibit the UPS during cell aging and senescence (30). Accordingly, proteasome-associated proteins and E3 ligases have been reported to affect longevity in C. elegans (115, 395) and long living species such as molerats appear to have a more active UPS than mice (267).
Intriguingly, recent studies have shown that the UPS is not affected by aging in the brain of transgenic UPS-reporter mice (60). However, studies in C. elegans have shown an age-related impairment of the UPS in C. elegans dorsorectal ganglion, but not in muscle cells (136). Hence, further studies on the UPS and oxidative stress are required before we may fully comprehend the importance of these systems during aging.
XV. Proteasome Inhibitors and Cancer
In 2003 the U.S. Food and Drug Administration approved the proteasome inhibitor bortezomib (Velcade™), previously known as PS-341, for treatment of multiple myeloma (306), but the drug also shows clear efficacy against other hematological malignancies, including Waldenstrom's macroglobulinaemia and mantle cell lymphoma. However, no, or only limited, effects on solid tumors have been reported in human trials so far (231). Bortezomib is a reversible peptide boronate that binds the β5 subunit and thereby inhibits the chymotrypsin-like activity of the 20S proteasome (178). More recently, two other proteasome inhibitors, carfilzomib and NPI-0052, entered clinical trials (330). These new clinically relevant inhibitors are not related to bortezomib, and notably NPI-0052 targets all three proteolytic activities of the 20S proteasome.
Since the proteasome degrades most intracellular proteins and is therefore involved in a series of cellular processes, it was surprising that proteasome inhibitors are not more toxic and are relatively specific for myeloma cells. However, the 26S proteasome is an abundant protease constituting about 1% of the protein in mammalian cells (145, 341) and therapeutic doses of bortezomib only decrease protein degradation by about 40% (177), which may explain that no dramatic toxicity is observed. Today, the specificity of bortezomib for myeloma cells is primarily ascribed to its inhibitory effect on NF-κB signaling and ERAD (231). In most cells and certainly in cancers, NF-κB is a major inhibitor of apoptosis. In myelomas, NF-κB plays an important role in transcribing genes encoding important growth factors such as IL-6, but also for production of angiogenic factors like VEGF (231). In addition, since myeloma is a cancer of plasma cells, these cells produce large amounts of immunoglobins. If the heavy and light chains of the immunoglobins are synthesized in unbalanced amounts, the surplus needs to be degraded. In addition, a fraction of these immunoglobins will be misfolded, and myeloma cells are therefore probably active in degrading these proteins via the ERAD pathway. Accordingly, plasma cells display symptoms of ER stress and the UPR transcription factor XBP-1 is essential for differentiation of B lymphocytes into plasma cells (41). Proteasome inhibitors block ERAD, which leads to accumulation of misfolded proteins and activation of the UPR and if the misfolded proteins persist due to proteasome inhibition, apoptosis is induced (165, 252). Since formation of disulfide bonds leads to production of ROS it is also reasonable to assume that plasma cells experience some level of oxidative stress. Accordingly, decreasing intracellular glutathione through buthionine sulfoximine treatment enhances bortezomib toxicity, whereas antioxidants protect myeloma cells from bortezomib-mediated cell death (247). Since ubiquitin-protein conjugates are more abundant in lymphocytes from chronic leukemia patients than in lymphocytes from healthy donors (225), it appears that transformed cells rely more heavily on the UPS than do untransformed cells.
In relation to oxidative stress, generation of ROS has also been implicated in cell death induced by proteasome inhibitors (43). Indeed, proteasome inhibitors induce ROS (106, 210, 239, 268). In lung cancer cells proteasome inhibitors cause an increase in superoxide levels leading to oxidative stress, which is followed by apoptosis (210). Addition of the antioxidant Tiron blocked the bortezomib-induced ROS production and apoptosis (210), suggesting that ROS generation plays a critical role in the initiation of the bortezomib-induced apoptosis. Later this view was challenged since a series of antioxidants failed to protect cells from bortezomib-induced cell death and some antioxidants like vitamin C may bind and inactivate the inhibitor directly (402). However, in hematological cancer models, the ability of bortezomib to induce ROS and antioxidants to provide cytoprotection appears to be conserved (43, 247).
It is possible that ROS are produced in response to protein aggregation, but since ROS production is connected to mitochondrial events during apoptosis, ROS production could also be triggered by stabilization of the proapoptotic BCL-2 family members. However, recently induction of ROS was observed at low doses of proteasome inhibitors that did not trigger apoptosis (25), suggesting that generation of ROS occurs independently of apoptosis.
Looking ahead, drugs that target E3s and DUBs are likely to provide more specific alternatives to proteasome inhibitors in cancer treatment. Several pharmaceutical companies are already pursuing these ideas in the hope of developing novel therapeutics not only for cancer, but also for inflammatory diseases and neurodegenerative disorders (19).
XVI. The UPS in Neurodegenerative Diseases
A growing body of evidence indicates that various neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and polyglutamine (polyQ) diseases have a common pathological feature, namely, aggregation of misfolded proteins in the affected brain regions. It therefore appears that dysfunction of the protein quality control plays a central role in the etiology of these diseases (220). Since misfolded proteins are discarded by either autophagy or via the 26S proteasome (8), malfunctions in the UPS will profoundly affect the protein quality control system and is therefore connected with a series of such proteinopathies. However, again the ubiquitin and autophagy systems appear to be connected and perhaps play overlapping roles. For instance, knockout mice lacking the essential autophagy component Atg7 display massive neuronal loss in the cerebral and cerebellar cortices, resulting in behavioral defects, movement reduction, and death within 28 weeks of birth (187). Notably, polyubiquitylated proteins accumulated as inclusion bodies in Atg7-deficient neurons, but no obvious alteration in proteasome function was observed (187).
Alzheimer's disease is the most common cause of dementia, characterized by a cognitive decline and memory impairment (277). A characteristic feature of Alzheimer's disease is the accumulation of amyloid β-peptide (Aβ) and hyperphosphorylated tau protein in the brain (303). Tau is a microtubule binding protein that promotes microtubule assembly, whereas Aβ peptides are natural metabolic products of about 40 amino acids, but both Aβ peptides and hyperphosphorylated tau may spontaneously aggregate. The generation of Aβ is maintained by presenilin, the catalytic core of γ-secretase. Under certain conditions, presenilin may also form aggregates. The proteasomal cofactor and substrate shuttle protein, PLIC/ubiquilin-1, controls protein aggregation and deposition of aggregated proteins (134), and mutations in the gene encoding PLIC/ubiquilin-1 are genetically associated with Alzheimer's disease (24). Brain protein is more oxidized in Alzheimer's disease, and experimental models have shown that oxidative damage precedes the pathological changes observed in Alzheimer's disease (249). The Aβ peptide is a potent generator of both ROS and reactive nitrogen species (57, 148) and is considered a prime initiator of this damage. Oxidative inactivation of proteasomes has also been observed in Alzheimer's disease (39), and accordingly proteasome subunits were found to contain carbonyls and to be HNE modified (39). The main risk factor for Alzheimer's disease is age (277). Aging is associated with increased oxidative stress, impaired protein-folding in the ER, and perhaps deficient proteasome- and autophagy-mediated clearance of damaged proteins, all factors that accelerate the accumulation of amyloid proteins in Alzheimer's disease (214). A vicious circle is formed since Aβ oligomers inhibit proteasome function that further facilitates the build-up of tau and accumulation of Aβ in aggregates (277).
Proteins with abnormally long glutamine expansions (polyQ), such as the Huntington's disease protein, Huntingtin, are especially prone to misfold and form intracellular aggregates (367). Certainly, oxidative stress is implicated in the etiology of Huntington's disease and antioxidant compounds have shown some promise in mouse models for Huntington's disease (96, 322, 323). However, the details regarding the connection between oxidative stress and Huntington's disease are still rather unclear. Recently, some elegant studies using transgenic mice, carrying reporters for the activity of the UPS, demonstrated a global impairment of the UPS by polyQ proteins in vivo (257). However, the inhibition of the ubiquitin system occurred only transiently upon expression of mutant Huntingtin and restoration correlated with appearance of protein aggregates. This indicates that aggregation shields the ubiquitin system from mutant Huntingtin. Accordingly, the recovery of the UPS was abrogated when aggregate formation was prevented through administration of aggregation inhibitors (257).
Like for most other neurodegenerative diseases, mitochondrial dysfunction and oxidative stress are connected with Parkinson's disease (172, 388). Oxidative stress causes α-synuclein aggregation (143), which in turn impairs proteasome function (233). Although most cases of Parkinson's disease are sporadic, some 15 genes have been linked to rare familial forms of the disease (20). Of these genes at least three, Parkin (PARK2), UchL1 (PARK5), and FBXO7 (PARK15), play a role in the ubiquitin system. UchL1 is a DUB, whereas Parkin and FBXO7 are E3 ligases. Of these, Parkin is the best characterized protein. Parkin contains two RING-finger modules and displays E3 ligase activity in vitro (311), and because protein aggregation is a common pathological feature of Parkinson's disease, previous models of Parkin function have suggested that mutations in parkin/PARK2 lead to accumulation of toxic proteins (170). However, Parkin also collaborates with two other Parkinson's disease proteins, PINK1 (PARK6) and DJ-1 (PARK7), to regulate mitochondrial function (82). Upon an oxidative insult, mitochondria may become depolarized. Recently, Parkin, in conjunction with PINK1, was shown to mediate selective autophagy of depolarized mitochondria (114), suggesting that mutant Parkin contributes to Parkinson's disease by causing a failure to trigger the removal of dysfunctional mitochondria. PINK1 spans the outer mitochondrial membrane and recruits Parkin upon mitochondrial depolarization (114, 366). When reaching the mitochondrial membrane, Parkin catalyzes the ubiquitylation of the voltage-dependent anion channel 1 (VDAC1), which in turn recruits p62 and initiates mitochondrial autophagy (Fig. 17) (114). Importantly, these results are supported by genetic evidence from Drosophila, where aberrant mitochondria were observed in Parkin and PINK1 mutants (263). More recently, Parkin and PINK1 were also connected with ubiquitylation of mitofusins (340). Here, ubiquitylation appears to prevent damaged mitochondria from fusing with undamaged mitochondria, thus promoting mitochondrial quality control.
XVII. Conclusions
From the examples given above, it is obvious that the fields of oxidative stress, redox signaling, and proteasomal degradation are connected at many levels. Thus, during the past 20 years or so, many examples of proteasome-dependent degradation of oxidized proteins have been uncovered. At the interface between the fields of oxidative stress and protein degradation, it is clear that there is some controversy among scientists as to the relative importance of the 20S proteasome (ATP- and ubiquitin-independent) and the 26S proteasome (ATP- and ubiquitin-dependent) in degradation of oxidized proteins. Thus, an important question that remains to be answered is to what extent and under which conditions do ubiquitin-dependent and -independent degradation of oxidized proteins prevail? It is possible that the two systems coexist and deal with different subsets of proteins. For instance, the 26S proteasome could degrade ubiquitylated proteins that have only been moderately damaged, whereas the 20S proteasome degrades more severely damaged proteins that have also become unstructured to a point where they resemble disordered proteins. More detailed studies on Ecm29-dependent dissociation of 26S proteasomes in response to oxidative stress, and perhaps other forms of stress, may cast some light on this matter. In addition, characterizing the E2s, E3s, and DUBs that play a role in ubiquitin-dependent degradation of oxidized proteins will be fundamental. The recent report showing that inhibition of the proteasome-associated DUB Usp14 increases the degradation of oxidized proteins (199) is one example, which may also offer a novel strategy to reduce the levels of aberrant proteins during aging and disease. Work in yeast link the E2s Ubc4 and Ubc5 with degradation of oxidized proteins (234), but are there any E3 ubiquitin-protein ligases that are specific for oxidized proteins, or are the oxidized proteins recognized simply by being misfolded?
Here we have focused primarily on the role of ubiquitin in redox regulation. However, other UBL modifiers also play a role during oxidative stress. Recently, the ubiquitin-related modifier Urm1 was found to be conjugated to a range of proteins in response to oxidative stress in both yeast and mammalian cells (358); however, Urm1 conjugation is probably not connected with protein degradation. Oxidized PML is degraded via the UPS after modification with chains of SUMO-2 and SUMO-3 (344). How many other proteins are targeted via these pathways? Also, is it true that the proteins that are most susceptible to oxidation-induced degradation are components of, for example, larger protein complexes, and is it possible, perhaps using a proteomics approach, to isolate and identify these proteins? Such a resource would be very useful for more detailed studies on the role of the UPS in degradation of oxidized proteins and also allow for testing 26S proteasomes in meaningful in vitro studies using relevant ubiquitylated substrates and purified 26S proteasomes.
In regard to aging, more in vivo and genetic studies are required before we may understand the connection to the UPS more clearly. Also, for the aging and oxidation-induced modification of proteasome subunits, tools such as antibodies specific for modified proteasome subunits (121) would be valuable for more detailed studies.
The connection between cellular redox control and the UPS is further consolidated by the identification of Txnl1 as a proteasome-associated thioredoxin. However, the function of Txnl1 is still rather enigmatic and further in-depth studies are needed to provide us with the detailed mechanism on the function of Txnl1 and other proteasome-associated redox active enzymes.
In relation to ERAD and redox regulation, an important question that remains to be answered is whether disulfide reduction of ERAD substrates only occurs inside the ER lumen or if cytosolic ERAD and/or redox components are also involved?
The recent research development revealing that proteasomes are induced by oxidative stress and that immunoproteasomes protect cells from inflammation-induced oxidative stress manifests that the UPS has to be classified as a direct component of the oxidative defense machinery. Herein, the UPS sorts out irreversibly damaged proteins to maintain protein homeostasis, which has important implications for the protection from neurodegeneration and for the general vitality of cells. However, the exact mechanism of activation of TCF11 is an open question, just like whether TCF11 can be activated by stimuli other than proteasome inhibition. Why should 26S immunoproteasomes be better than standard 26S proteasomes for efficient turnover of polyubiquitylated proteins? Is this a matter of structural changes due to incorporation of immunosubunits? All these open questions demand further detailed studies to expand our understanding of the role of UPS within the defense against oxidative stress.
Footnotes
Acknowledgments
We thank Dr. Klavs B. Hendil and Dr. Jakob R. Winther for helpful discussions and the reviewers for their constructive critique and comments on the article. We apologize to those authors whose work we were not able to cite due to space constraints. The work of R.H.-P. is supported by grants from the Lundbeck Foundation, The Novo Nordisk Foundation, the Danish Council for Independent Research, the Danish Natural Science Research Council, and the Harboe Foundation. E.K. is supported by grants from the Deutsche Forschungsgemeinschaft.