
Abstract
Two principal pathways exist by which cells can undergo apoptotic death, known as the extrinsic and the intrinsic pathways. Binding of a ligand to a death receptor activates the extrinsic pathway. In the intrinsic pathway, an apoptotic stimulus, such as neurotrophin withdrawal or exposure to a toxin, causes a proapoptotic member of the Bcl-2 family of proteins, such as Bax, to permeabilize the outer mitochondrial membrane. This allows redistribution of cytochrome c from the mitochondrial intermembrane space into the cytoplasm, where it causes activation of caspase proteases and, subsequently, cell death. A dramatic increase occurs in mitochondria-derived reactive oxygen species (ROS) during the apoptotic death of sympathetic, cerebellar granule, and cortical neurons. These ROS lie downstream of Bax in each cell type. Here I review possible mechanisms by which Bax causes increased ROS during neuronal apoptosis. I also discuss evidence that these ROS are an important part of the apoptotic cascade in these cells. Finally, I discuss evidence that suggests that neurotrophins prevent release of cytochrome c in neurons through activation of an antioxidant pathway. Antioxid. Redox Signal. 14, 1437–1448.
Introduction
Apoptosis serves its most important physiological role in the nervous system during neurogenesis, when the apoptotic death of a large number of excess neurons matches the number of innervating neurons to the size of their targets and thus sculpts the developing nervous system. About 50% of the neurons produced during neuronal development die by this process before birth or shortly thereafter (66). Acquisition of a sufficient quantity of a neurotrophic substance supplied by target or other tissues is a major determinant of which neurons escape the period of developmental death and live into the adulthood of the organism. Those neurons that do not obtain a sufficient quantity of neurotrophin execute the apoptotic program, and phagocytic cells rapidly remove them from the developing organism. The neurotrophic hypothesis posits that larger target tissues can supply more neurotrophic substances than can smaller targets and thus can prevent more cell death during development than can the smaller targets. The result is greater innervation of larger targets in adult animals. Neuronal death with apoptotic features also occurs inappropriately in many neuropathologies [for example, in neurodegenerative diseases such as Alzheimer disease (28, 54, 57, 93) and in neurotrauma and stroke (10, 20)]. Apoptosis may also be involved in the aging process in the nervous system and other organ systems (26, 68).
Increased levels of reactive oxygen species (ROS) occur in neurons and many other types of cells undergoing apoptotic death (8, 30, 72, 73, 76, 78, 81). Elevated production of ROS occurs both in neurons dying during developmental apoptosis and in neurons dying in pathologic states in which apoptotic features are observed (1, 10, 20, 28, 54, 57, 93). It has long been known that externally generated ROS can induce apoptosis (78). However, it was uncertain whether the ROS produced internally in apoptotic cells are important for death or if they are epiphenomena that have little or nothing to do with the actual apoptotic process (43, 47). Recent evidence clarifies this issue and suggests that the elevated ROS in apoptotic cells create a cellular pro-oxidant state that is a necessary component of apoptotic death.
The subject of this review is the role of ROS in regulating the intrinsic apoptotic cascade in neurons. Because most of the work aimed at understanding the role of ROS in neuronal apoptosis has been done in developmental models, I focus on these systems.
The Molecular Machinery of Apoptosis: The Extrinsic and Intrinsic Pathways
During the development of the nematode, Caenorhabditis elegans, 131 cells normally die by apoptosis. Certain mutations in the gene, ced-3, prevent the death of most of these cells (22). This gene encodes a cysteine protease that has the unusual feature of cleaving proteins after an aspartate residue. About 15 mammalian homologues of ced-3 have been identified and termed caspases (3). By attacking critical cellular substrates, caspases act as the executioners of apoptotic death in many cell types, including neurons (17, 77, 93). Two other genes, ced-4 and ced-9, are also central to developmental death in C. elegans (22). The ced-4 gene product is proapoptotic, whereas that of ced-9 is antiapoptotic (91). Many mammalian homologues of ced-9 exist, some antiapoptotic and some proapoptotic. Members of this family of proteins, known as the Bcl-2 family, include Bcl-2, Bcl-xL, Bcl-xS, Bax, Bad, and Bak (53). Bcl-2, Bcl-xL, and other family members have antiapoptotic effects, whereas Bcl-xS, Bax, Bad, and Bak are proapoptotic (11, 48, 53).
Another group of proteins belonging to this family are known as BH3-only (Bcl-2 homology 3) proteins, as they are missing the BH domains 1, 2, and 4 that are found in full-length Bcl-2 or Bcl-xL. The BH3 domain forms an amphipathic α helix that is required for cell killing, and it mediates interactions with other Bcl-2 family members. BH3-only proteins bind to and inhibit the antiapoptotic proteins Bcl-2, Bcl-xL, and Mcl-1 and, in some cases, directly activate the multidomain proapoptotic proteins Bax and Bak. In some types of cells, including neurons, BH3-only proteins are upregulated during apoptosis and augment the apoptotic process (11, 71). The mammalian homologue of ced-4 is known as apaf-1 (apoptosis protease activating factor-1) (89, 97). Like ced-4, the protein product of apaf-1 (Apaf-1), has a proapoptotic effect. Also, as with ced-4, Apaf-1 exerts its proapoptotic effect by activating caspase proteases.
Mammalian caspases are activated and cause cell death through two different signaling cascades known as the extrinsic and intrinsic pathways (Fig. 1).
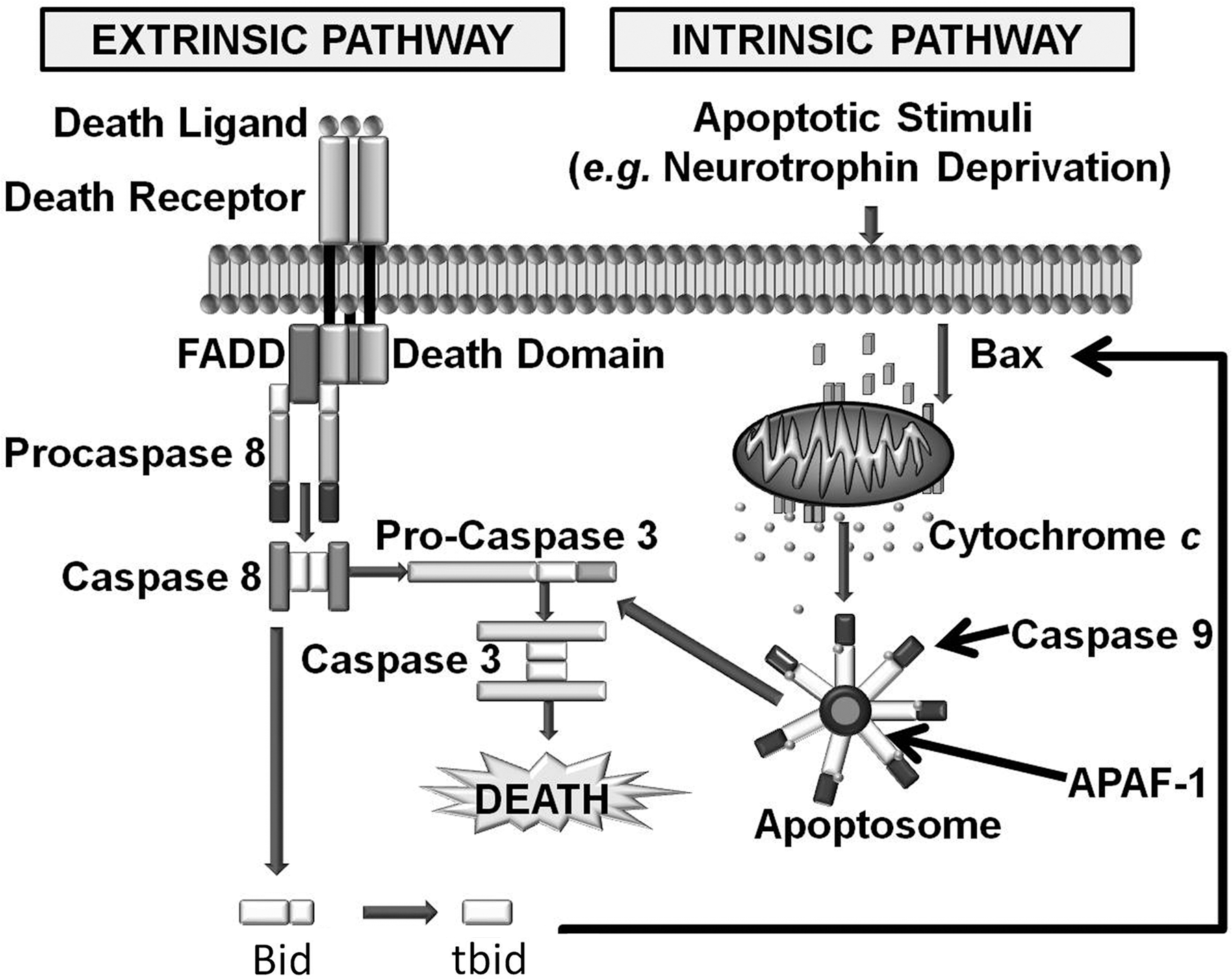
Activation of the extrinsic pathway occurs when a proapoptotic ligand such as FasL binds onto a cell-surface death receptor. This binding causes the adaptor protein FADD (Fas-associated protein with death domain) and the initiator caspases 8 or 10 to form a complex known as DISC (death-inducing signaling complex). The formation of this complex activates the caspases (97). These caspases in turn cleave and activate downstream effector caspases (3, 6, or 7) that cleave many cellular substrates, directly causing cell death.
In contrast, the intrinsic pathway involves a cascade of molecular events occurring entirely within cells. Plasma-membrane death receptors are not involved. The intrinsic pathway is activated by many different stimuli including DNA damage, toxins, trophic factor deprivation, ionizing radiation, and other cellular stresses (Fig. 1). The intrinsic pathway appears to be the major route to apoptotic death during neuronal development, whereas the extrinsic pathway is of importance in several neuronal pathologies (31, 71, 86).
Mitochondria are central to the intrinsic apoptotic pathway (11, 18, 35). In addition to their roles in adenosine triphosphate (ATP) production by oxidative phosphorylation and in intermediary metabolism, mitochondria function to induce or regulate the intrinsic apoptotic pathway by releasing a number of proapoptotic molecules into the cytoplasm. The most important of these molecules is cytochrome c. In healthy cells, the mature form of this protein is sequestered in the space between the inner and the outer mitochondrial membranes (IMM and OMM), where it functions to shuttle electrons from respiratory complex III (bc1 complex) to complex IV (cytochrome oxidase) in the electron-transport chain (64). On receipt of an apoptotic signal, a proapoptotic member of the Bcl-2 family, such as Bax, oligomerizes and inserts into the OMM, permeabilizing it and allowing cytochrome c to redistribute into the cytoplasm. Once in the cytoplasm, cytochrome c binds onto Apaf-1, causing seven Apaf-1 molecules to oligomerize. Each Apaf-1 molecule recruits a molecule of the initiator caspase, caspase 9. The cytochrome c–induced formation of this structure, known as an apoptosome, brings together and activates seven caspase 9 molecules (2, 97). As with caspase 8 in the extrinsic pathway, caspase 9 then cleaves and activates downstream effector caspases such as caspases 3 and 7 that cleave many protein substrates and cause cellular death. Although cytochrome c appears to be the most important mitochondria-sequestered proapoptotic protein, several other proteins released from mitochondria have proapoptotic functions (19, 56, 79). Preeminent among these are apoptosis-inducing factor (AIF) and Smac. The former is involved in DNA fragmentation, whereas the latter relieves inhibition of caspases by their endogenous inhibitors, the inhibitors of apoptosis proteins (IAPs). The extrinsic pathway can also augment the intrinsic pathway by cleaving the BH3-only member of the Bcl-2 family, Bid, to form tBid. tBid can then activate the intrinsic pathway by permeabilizing the OMM through activation of Bax (16). Many cytoplasmic proteins are also implicated in regulation of the intrinsic pathway (13).
Production and Clearance of ROS in Neurons and Other Cell Types
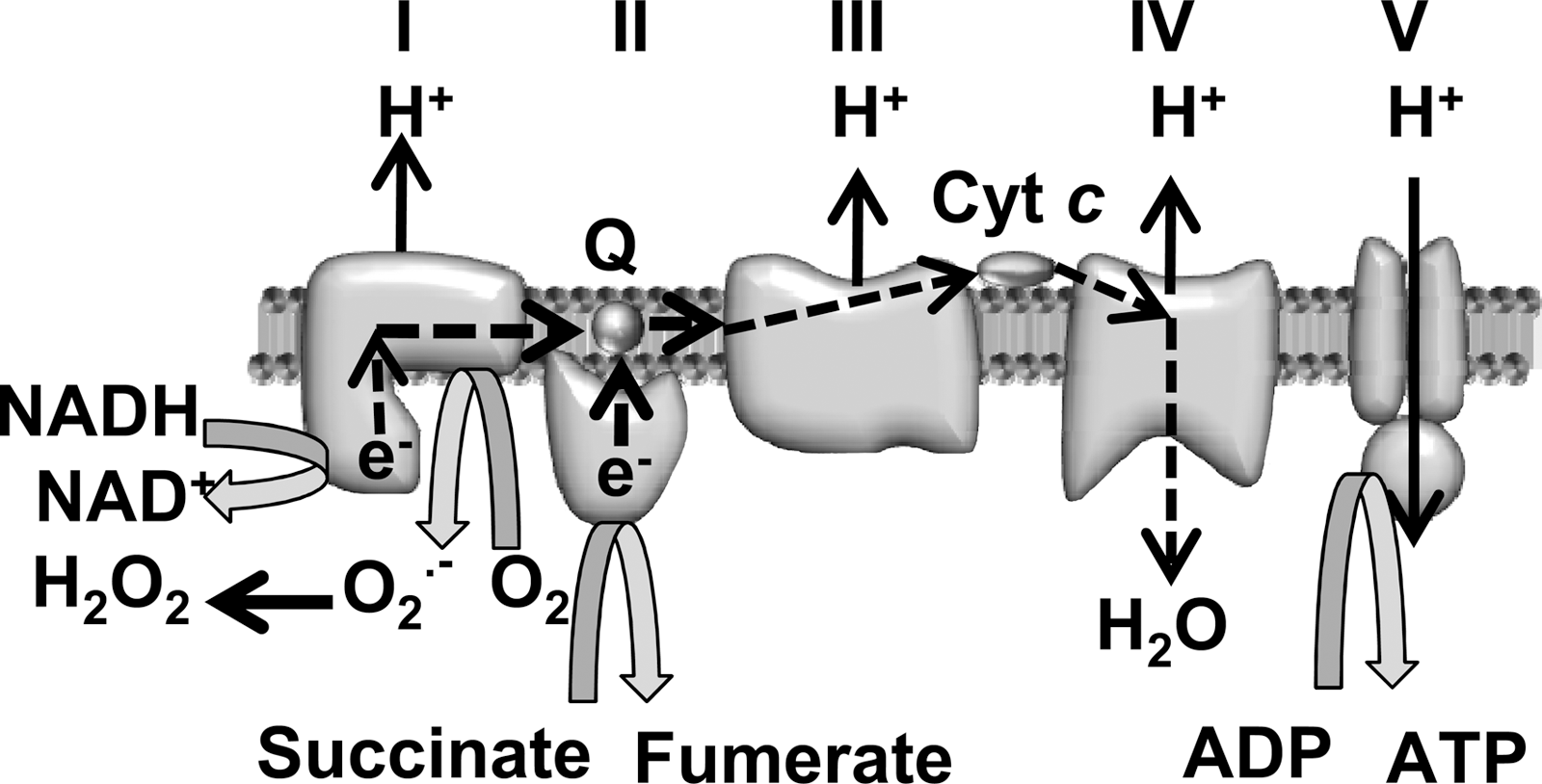
ROS are species of molecular oxygen that are more reactive than diatomic oxygen (O2) (32). They comprise one group of a larger class of reactive molecules collectively known as reactive species (RS). ROS include both free radicals, molecules that have at least one unpaired electron, and non–free radical forms. Living organisms produce several types of ROS. Important free radical ROS produced by biologic tissues include superoxide (O2 ·−), hydroxyl radicals (OH.), peroxyl radicals (RO2 .), and alkoxyl radicals (RO.). The most important non–free radical ROS produced biologically is H2O2. A primary source of cellular O2 ·− is the mitochondrial electron-transport chain, in which a significant fraction of O2 consumed undergoes one-electron reduction to O2 ·− by electrons leaked from the respiratory complexes (Fig. 2) (4, 58). Another potential source for ROS in many cells, including neurons, are NADPH oxidases that can convert O2 to O2 ·− (6, 80). The O2 ·− produced by either mitochondria or oxidases is rapidly converted to H2O2 by the dismutation reaction catalyzed by superoxide dismutase (SOD) enzymes. The H2O2 can transform into other ROS (e.g., OH.). However, most H2O2 is converted to H2O by oxidation of the tripeptide glutathione (GSH; Fig. 2) via a reaction catalyzed by the enzyme glutathione peroxidase (GPx). The enzyme glutathione reductase then catalyzes the reduction of oxidized glutathione (GSSG) to create more GSH by using reducing equivalents obtained from NADPH. In this way, GSH neutralizes the potentially damaging effects of H2O2. The enzyme catalase also detoxifies H2O2. However, in neurons, this is a minor antioxidant pathway.

ROS in Neuronal Apoptosis
Most of the work aimed at understanding the molecular mechanisms underlying apoptotic neuronal death has been done with cultures of sympathetic neurons deprived of nerve growth factor (NGF), cultures of repolarized/serum-starved cerebellar granule neurons, and cultures of toxin-treated cortical neurons (38, 43
–47, 61, 90). All of these neurons die with most of the features common to many types of cells undergoing apoptosis (15, 17, 18, 61). Greenlund et al. (30) were among the first to demonstrate increased ROS levels during the apoptotic death of neurons. They used the redox-sensitive dye 5 (and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate bis(acetoxymethyl) ester (75) in conjunction with confocal microscopy to demonstrate that rat sympathetic neurons in cell culture enter a transient prooxidant state that peaks about 3 h after depriving them of NGF, their developmentally required neurotrophic factor. This ROS increase occurs long before any of the cells is committed to die, as apoptosis does not begin in any of the NGF-deprived cells until about 18 h after withdrawal. Expression of human SOD in these cells slowed their death after NGF withdrawal, suggesting that the ROS burst was an important part of the apoptotic cascade in them. They also found that downregulation of SOD with an antisense expression vector made sympathetic neurons more susceptible to death caused by NGF withdrawal. By using a similar redox-sensitive dye and confocal microscopy, my laboratory later demonstrated that, in addition to this early transient burst of ROS in NGF-deprived rat sympathetic neurons, a later ROS increase also occurs (43). This delayed elevation of cellular ROS begins about 12 h after NGF withdrawal and continues throughout the apoptotic process (Fig. 3). Concurrent with the decline of the early transient ROS increase, GSH levels also increase. Buthionine sulfoximine (BSO), an inhibitor of glutathionine synthesis, blocks the increase in GSH concentration and prevents the transience of the early ROS increase. In BSO-treated neurons deprived of NGF, ROS continue to increase before reaching a peak at about 18 h after NGF withdrawal and remains at this level throughout the rest of the apoptotic process. These findings suggest that the cells continue to produce ROS but that these ROS are rapidly removed by increased activity in the glutathione redox-cycling pathway. Surprisingly, NGF-deprived mouse sympathetic neurons exhibit only the later increase in ROS levels and not the early one (Fig. 3). Therefore, the same types of neurons from even two relatively closely related species can exhibit disparate patterns of ROS production and clearance during apoptosis caused by the same apoptotic stimulus. In both cases, the antioxidants N-acetyl-
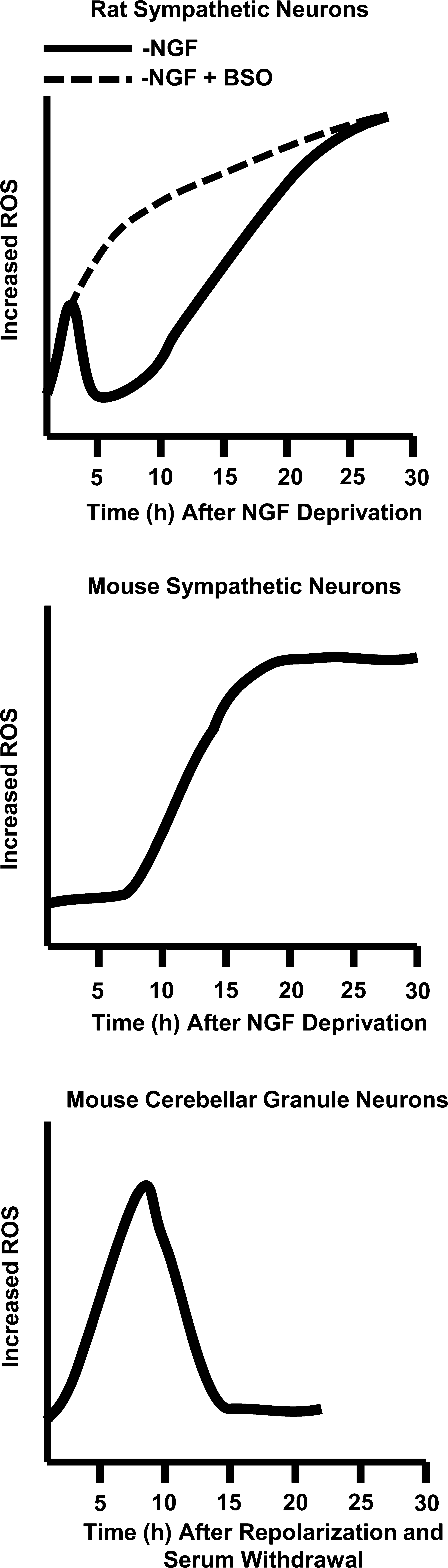
Cerebellar granule cells and several other types of neurons can be maintained alive in cell culture in medium containing elevated concentrations of potassium (27, 61). The high potassium chronically depolarizes their plasma membrane potentials and induces a low-level, sustained activation of plasma membrane calcium channels. These channels support a calcium influx that gives rise to a small sustained increase of intracellular free calcium concentration that blocks apoptosis by an undetermined mechanism (27). When switched from a depolarizing medium (25 mM K+)-containing serum to one containing 5 mM K+ without serum, the plasma membrane potential of cerebellar granule neurons repolarizes, shutting calcium channels and returning the calcium influx and intracellular calcium concentration to baseline levels (61). ROS increase, and apoptosis begins in these cells soon after the repolarization. These ROS reach a peak level by 9 h after the switch and then decline to baseline levels by 15 h (Fig. 3) (47). Antioxidants inhibit the death of these cells, suggesting a proapoptotic role for the ROS (76). Increased levels of ROS have been reported during the apoptotic death of a number of other types of neurons as well (12, 81).
Data that my laboratory published and results from other laboratories suggest that most or all of the ROS in apoptotic and nonapoptotic sympathetic neurons derive from production and dismutation of O2 ·− produced by electrons leaking from the mitochondrial electron-transport chain (21, 43, 47). Electrons derived from NADH and succinate generated by the TCA cycle in the mitochondrial matrix normally transfer from respiratory complexes I and II to ubiquinone (coenzyme Q), which then transfers them to complex III. They are then carried to complex IV by cytochrome c. As electrons pass through the respiratory complexes, their energy is dissipated and is used to translocate protons from the mitochondrial matrix into the intermembrane space of the mitochondria. These protons then cross the IMM back into the matrix through the F1.FO–ATP synthase (respiratory complex V), where the energy stored in the electrochemical proton gradient across the IMM is used to produce ATP from ADP. Leakage of electrons from the respiratory chain to form O2 ·− from O2 is thought to be able to occur in only the first three respiratory complexes, but primarily at complex I (Fig. 4) (55). The mitochondrial uncoupler, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), completely blocks the ROS increase measured by CM-H2DCFDA in NGF-deprived rat and mouse sympathetic neurons (21, 43, 47). Dissipating the proton gradient across the IMM with FCCP leads to a faster transit time for electrons traveling through the electron-transport chain, less electron stalling, and, as a result, fewer electrons leaking from the chain to form O2 ·−. Suppression of ROS by an uncoupler is strong evidence that the electrons contributing to the ROS derive from mitochondria.
Additional evidence for a mitochondrial origin for the increased ROS during neuronal apoptosis comes from experiments with the new mitochondria-targeted redox-sensitive dye MitoSOX Red (39, 74, 94, 95). MitoSOX consists of hydroethidine linked by a hexyl carbon chain to a triphenylphosphonium group. The triphenylphosphonium cation targets the molecule to the mitochondrial matrix because of the high membrane potential across the inner mitochondrial membrane (74). Oxidation of the hydroethidine moiety by O2 ·− generates 2-hydroxyethidium, which becomes intensely fluorescent on intercalation into mitochondrial DNA (74, 95). Exposing the dye to ultraviolet or near-ultraviolet light excites the O2 ·−/MitoSOX product but not the oxidation products of MitoSOX generated by other ROS, including H2O2 and ROS downstream of H2O2. MitoSOX colocalizes with mitochondria in sympathetic and cerebellar granule neurons in cell culture. Exposure of mouse sympathetic neurons to culture medium containing menadione, a compound that generates O2 ·− intracellularly by mediating transfer of electrons from NADH or NADPH to O2, increases MitoSOX fluorescence (82). H2O2 at a concentration that causes a large increase in CM-H2DCFDA fluorescence intensity has no effect (46), indicating that MitoSOX is an effective tool for investigating production of O2 ·− by the mitochondria of these neurons, without significant interference from H2O2-associated ROS. Mitochondria-localized MitoSOX fluorescence increases in NGF-deprived mouse sympathetic neurons in parallel with the increase in the ROS detected by CM-H2DCFDA. FCCP blocks this increase. Together, the data strongly suggest that the mitochondria of neurons undergoing apoptotic death increase production of O2 ·− and that the ROS detected by CM-H2DCFDA lie downstream of this increase.
The Role of Bax and Caspases in the ROS Increase During Neuronal Apoptosis
Bax is central to the apoptotic death of many types of neurons in vivo (86) and in vitro, including NGF-deprived rat and mouse sympathetic neurons, repolarized/serum-starved cerebellar granule neurons, and cortical neurons treated with excitotoxins in cell culture (14, 61, 90). Bcl-2 and other antiapoptotic members of the Bcl-2 family reside primarily in the OMM (62, 63). The antiapoptotic function of these proteins seems to be primarily to cause retention of cytochrome c and other apoptogenic factors within the mitochondrial intermembrane space (11,13). In healthy cells, most Bax is located in the cytoplasm or is loosely associated with the cytoplasmic side of the OMM. During apoptosis, Bax oligomerizes and inserts into the OMM, permeabilizing it and causing cytochrome c to be released into the cytoplasm, where it activates the intrinsic apoptotic cascade (70, 71, 88). A great deal of effort has gone into investigating how Bax induces the release of cytochrome c from mitochondria, but the exact mechanism remains undetermined (11, 60). Bax can form pores in liposomes that are large enough to allow passage of cytochrome c (13). However, it is unclear whether it can form similar channels in the OMM (11). It is possible that Bax pores in the OMM are the main exit route for cytochrome c redistribution into the cytoplasm. However, considering the current literature, it also is possible that Bax causes release, at least in part, through activation of other OMM channels (13). It is thought that much of the cytochrome c associates with the cytoplasmic side of the IMM and that this association must be broken for cytochrome c in mitochondria to exit the intermembrane space, even if the OMM is permeabilized. Evidence exists that this may occur through peroxidation of the IMM lipid, cardiolipin (37, 67). We presented evidence that such peroxidation occurs in rat sympathetic neurons deprived of NGF (45).
Withdrawal of NGF from sympathetic neurons in culture causes cessation of somatic growth and neurite outgrowth. It also causes a steep decline in many metabolic parameters (15). Similar effects occur in repolarized/serum-starved cerebellar granule neurons in culture (61). These events transpire at about the same rate in neurons taken from Bax-null mice as in those taken from wild-type mice, indicating that Bax is not required for the trophic effects. However, neurons taken from Bax-null animals do not die after withdrawal of trophic support. For as long as cultures can be maintained, these cells will live, despite receiving an apoptotic signal (14, 18). Additionally, ROS levels do not increase in either Bax-null, NGF-deprived sympathetic neurons or repolarized/serum-starved cerebellar granule cells. Treatment of cortical neurons in culture with the kinase inhibitor staurosporin induces them to undergo an apoptotic death that is blocked in Bax-null cells. Bax deletion also prevents increased ROS in them. In each of these cell types, the ROS block is complete. Therefore, Bax lies upstream of all increased ROS production occurring during the apoptotic death of these three representative types of neurons.
Several possible mechanisms exist by which Bax could cause increased production of O2 ·− by the mitochondria of apoptotic neurons (Fig. 5). First, Bax-induced release of cytochrome c from the mitochondrial intermembrane space may cause increased production of O2 ·− by blocking the flow of electrons through the electron-transport chain secondary to depleting the chain of cytochrome c. Such a block can augment leakage of electrons from carriers upstream of cytochrome c (8, 25). Second, Bax could increase O2 ·− production by releasing cytochrome c and activating caspases that then damage mitochondrial respiratory complexes. The damaged complexes could leak more electrons, augmenting O2 ·− production. Consistent with the latter mechanism, the broad-spectrum caspase inhibitors boc-aspartyl-(OMe)-fluoromethylketone (BAF) or zVAD greatly attenuate but do not prevent increased ROS in NGF-deprived sympathetic neurons, suggesting that much of the ROS is caspase dependent (43, 47). Sympathetic neurons from caspase 3–null animals do not die when deprived of NGF, indicating that it is the primary effector caspase in these cells (87). Caspase 3 activity increases mitochondrial ROS production in some cell lines by cleaving the NDUFS1 subunit of the respiratory complex I (72, 73). This subunit and its consensus caspase 3 cleavage site are ubiquitously expressed in mammalian tissues. Whether cleavage of the NDUFS1 subunit or other components of the respiratory chain occurs in neurons or other primary cells and the relevance of such cleavage to ROS production has not yet been determined. The suppression of ROS by caspase inhibitors is consistent with this possibility.

Additionally, we have found that NGF-deprived sympathetic neurons from caspase 3–null mice exhibit a much smaller increase in ROS compared with wild-type cells, consistent with caspase attack on respiratory substrates in the cells (data not shown). The rate of cytochrome c exit was not affected in the caspase 3–knockout cells. Therefore, it appears that the bulk of increased ROS, at least in apoptotic sympathetic neurons, arises from caspase activity, most likely as a result of caspase cleavage of respiratory complex subunits.
Finally, Bax might influence mitochondrial ROS production by a variety of mechanisms independent of cytochrome c release or caspase activation. BAF does not inhibit cytochrome c redistribution in NGF-deprived sympathetic neurons (47). Maximal cytochrome c loss occurs by 48 h after NGF withdrawal. In unreported data, we found that bax+/+ and bax+/− neurons deprived of NGF for 24 to 48 h and maintained alive with BAF had identical levels of cytochrome c remaining in their mitochondria. The bax+/+ neurons had slightly higher ROS levels than did the bax+/− neurons. By using the sensitive Caspase-Glo assay, we found that caspase 3/7 activity was completely blocked in these cells by BAF. It is likely that no other effector caspases are expressed in these neurons (18, 87). Therefore, Bax affects mitochondrial O2 ·− production in them by a mechanism in addition to that caused by cytochrome c redistribution and caspase activation. One possible mechanism is Bax-induced mitochondrial biogenesis. Bax influences mitochondrial fission/fusion (40, 41), a process that could, ostensibly, increase total cellular mitochondrial volume and, thereby, elevate ROS.
Another possible mechanism is a Bax-induced increase in mitochondrial metabolism so that more electrons are available to the respiratory chain. In support of such a mechanism, the related antiapoptotic protein, Bcl-2, increases levels of reduced cellular pyridine nucleotides in cells (e.g., NADH) (50 –52). A similar effect of Bax could explain some of its effect on O2 ·− production. It also is possible that Bax has direct or indirect effects on respiratory complexes that cause increased electron leakage. Zimmerman et al. (95) presented evidence that, in cerebellar granule cells, Bcl-2 exerts an antioxidant effect on mitochondria by binding GSH and that the BH3 domains of proapoptotic proteins like Bax can displace this GSH. They also showed that BH3 mimetics displace a mitochondrial pool of GSH. Such displacement could, ostensibly, decrease removal of H2O2, causing oxidative stress, and might be one mechanism by which Bax exerts a prooxidant effect. However, this finding cannot explain how Bax increases O2 ·− production by mitochondria.
Taken together, the available data suggest several possible mechanisms by which Bax increases ROS during neuronal apoptosis.
The Role of ROS in Cytochrome c Release During Neuronal Apoptosis
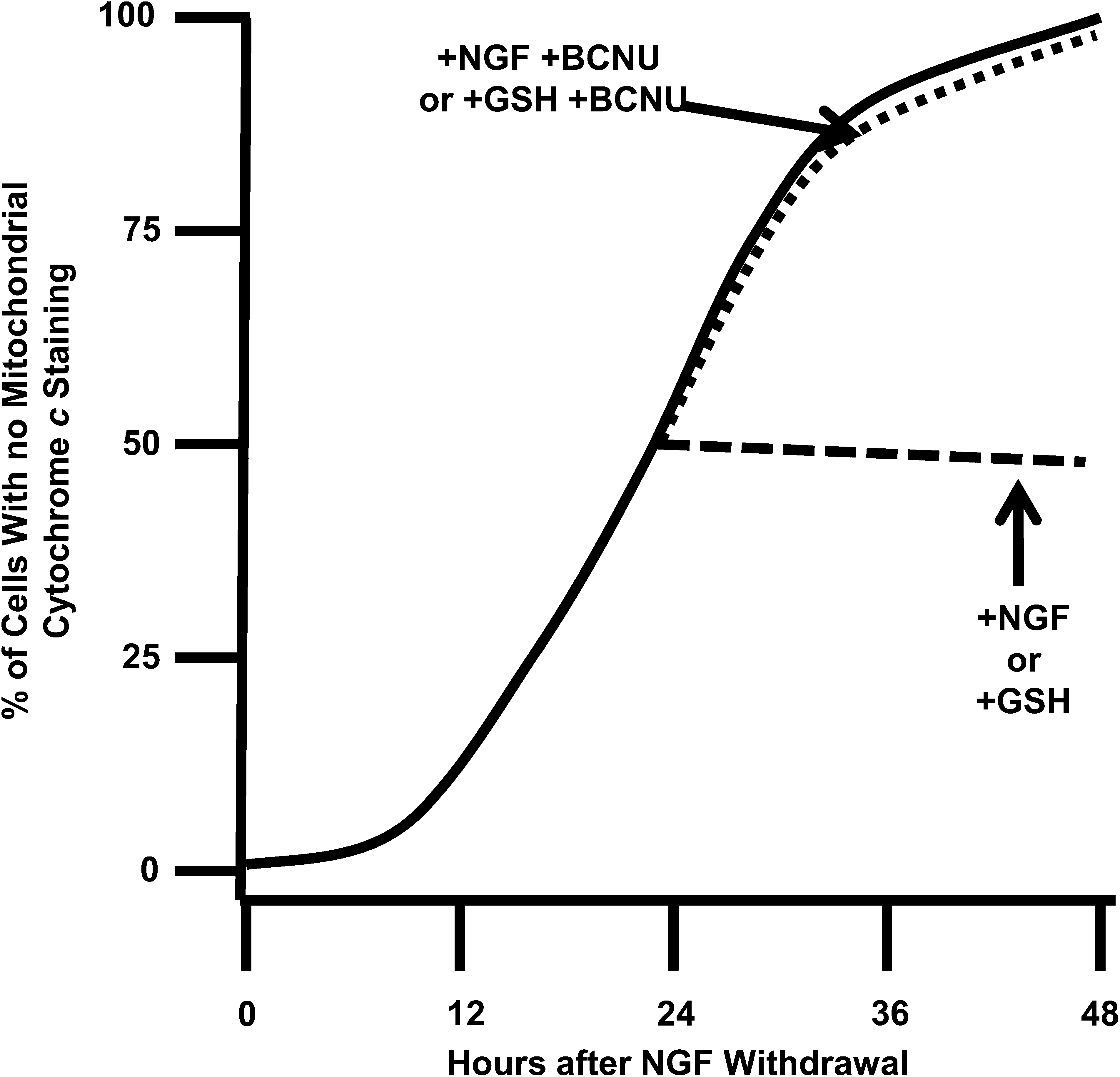
Dugan et al. (21) showed that readdition of NGF to NGF-deprived cultures of rat sympathetic neurons rapidly suppresses the elevated ROS levels in them. We found a similar effect in mouse sympathetic neurons (46). NGF readdition to mouse cultures deprived of NGF for 24 h suppresses CM-H2DCFDA intensity by ∼80% within 20 min of exposure. This suppression continues for at least 2 h. In contrast to the rapid and potent suppressant effect that NGF readdition has on CM-H2DCFDA intensity in NGF-deprived neurons, reexposure of cells deprived of NGF for 24 h to NGF does not suppress MitoSOX fluorescence in them for at least 6 h after the addition. These findings suggest that mitochondria in NGF-deprived sympathetic neurons continue to generate O2 ·− but not H2O2 after the readdition of NGF. The explanation for the disparate effects of NGF on the two different types of ROS appears to be that NGF readdition rapidly activates antioxidant mechanisms for removing the H2O2 from the cells. When the neurons are treated with N,N′-bis(2-chloroethyl)-N-nitrosourea (BCNU) (5), an inhibitor of glutathione reductase, the enzyme necessary for reducing GSSG to GSH, NGF no longer suppresses the increased ROS. This finding suggests that NGF activates the glutathione redox-cycling pathway, the primary means for removing H2O2 from these cells (46). A possible mechanism underlying this activation is phosphorylation and activation of GPx1, the most abundant of the approximately five GPxs in mammalian cells, by the Src family of tyrosine kinases c-Abl and Arg (9). GPx1 is found in most cells, including neurons. Both c-Abl and Arg are involved in the cellular response to oxidative stress, are widely expressed, and associate with the high-affinity NGF receptor TrkA (92). Therefore, it is possible that NGF activates glutathione redox cycling and suppresses ROS, at least in part, by causing phosphorylation and activation of GPx1.
NGF may also modulate cellular antioxidant status through its effect on glucose uptake. Hexokinase catalyzes the conversion of glucose to glucose-6-phosphate (G6PD) in the first step of glycolysis. G6PD + NADH can then enter the pentose phosphate pathway rather than the glycolytic pathway by conversion to 6-phosphogluconolactone + NADPH in a reaction catalyzed by G6PD dehydrogenase. The pool of NADPH produced is the primary source of reducing equivalents for the conversion of GSSG to GSH. Withdrawing NGF from sympathetic neurons in cell culture causes glucose uptake to decrease to about 20% of control within 6 h (14). A decrease in available glucose in NGF-deprived neurons or a decrease in the activity of either of the two relevant enzymes could, ostensibly, result in fewer NADPH reducing equivalents being available for the reduction of GSSG to GSH. Readdition of NGF increases glucose uptake and could increase NADPH concentration (15).
The membrane-permeant antioxidants N-acetyl-

ROS have been reported to cause cytochrome c release in several types of cells (37, 43, 47, 59). However, Jekabson and Nicholls (36) reported that no correlation exists between ROS at the individual cell level and apoptosis in dying cerebellar granule neurons in culture. These cells were scored as being apoptotic if they stained for annexin V, a marker of apoptotic cells. They suggested that this finding is consistent with a model whereby the increased ROS may provide a permissive environment for cytochrome c redistribution but that is not instructive for release. However, it is difficult to reconcile our findings in sympathetic neurons with the permissive hypothesis. H2O2 concentrations that cause an increase in CM-H2DCFDA similar to that seen in NGF-deprived cells induce release. It is possible that the lack of correlation between annexin V staining and ROS could merely be that the ROS burst is fast, brief, and the cells are then dead and can no longer produce ROS.
ROS, JNK, and Autocatalytic Processing of Bax
c-Jun N-terminal kinase (JNK) becomes activated in NGF-deprived sympathetic neurons and repolarized/serum-starved cerebellar granule cells soon after they receive an apoptotic stimulus (23, 61). Oxidative stress can activate JNK. However, JNK activates to the same extent and over the same time course in Bax-null neurons as in wild-type cells, even though ROS levels remain very low in the knockout cells. Therefore, in these cells, oxidative stress does not lie upstream of JNK activation. Harris and Johnson (34) demonstrated that the BH3-only proteins Dp5 and Bim are upregulated in both sympathetic neurons and cerebellar granule cells during apoptosis. They also demonstrated that the death-promoting activity of these proteins in these cells requires Bax. Inhibition of JNK activity blocked much of this upregulation, suggesting that JNK lies upstream of much of the induction. Inhibition of JNK also supports long-term survival in NGF-deprived cells (33). JNK has been reported to cause Bax translocation to mitochondria in COS-1 cells through phosphorylation of 14-3-3 proteins that anchor it in the cytoplasm. The phosphorylation causes Bax to dissociate from these proteins (84). However, such a mechanism seems unlikely to have much of a role in Bax translocation during the apoptotic death of sympathetic and cerebellar granule neurons as JNK is fully activated soon after receipt of an apoptotic stimulus in these cells but many hours before cytochrome c is released (23). Because both JNK and ROS appear to be important for apoptotic neuronal death, JNK must lie either upstream of the ROS increase or affect death through a parallel pathway. One possibility is that the upregulated BH3-only proteins cause increased association of Bax with mitochondria and that this association then leads to elevated ROS.
In many cell types, including neurons, cytochrome c redistributes into the cytoplasm from most or all mitochondria in a cell over a period of just a few minutes (29, 47). This rapid release implies the existence of a signaling mechanism that coordinates near-simultaneous release of cytochrome c from many mitochondria. An increase in intracellular Ca2+ has been suggested to signal rapid release in some cells (7). However, cytoplasmic Ca2+ does not increase during the period of cytochrome c redistribution in NGF-deprived dorsal root ganglia neurons (83) or NGF-deprived sympathetic neurons (our unreported data). Upregulation of BH3-family proteins are one possible candidate for activation of Bax and cytochrome c release. However, it is unclear how the induction of these proteins could trigger rapid coordinate release from most mitochondria in a cell over a period of a few minutes. Also, it is not clear why ROS would be important to this release. Based on our data and those of others, I propose that the increased H2O2 is the principal messenger molecule that coordinates this release (Fig. 8). Nie et al. (65) demonstrated that H2O2 can cause oligomerization and activation of Bax, apparently by oxidizing the conserved cysteine 62 residue. This finding, in combination with the prooxidant effects of Bax, raises the possibility of a Bax/ROS-induced autocatalytic cycle in which Bax increases O2
·− production and
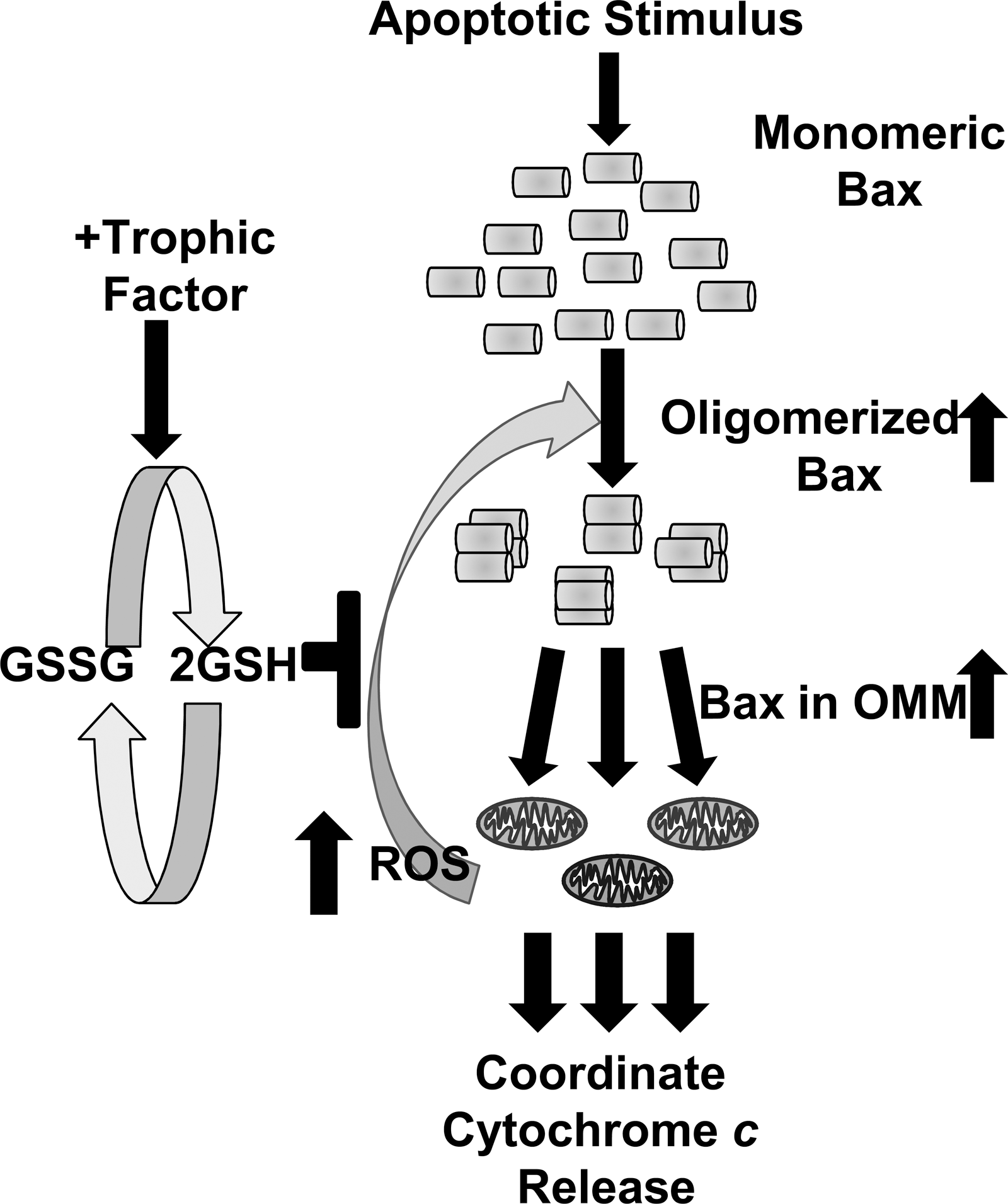
The Role of ROS in Priming Neuronal Caspase Activity
In addition to its apparent role in causing cytochrome c release, ROS also have an additional function during neuronal apoptosis (49, 85). Vaughn and Deshmukh (85) demonstrated that the ability of cytochrome c to induce apoptosis in sympathetic neurons depends on its redox state. In nonapoptotic neurons, cytochrome c is maintained in a reduced state by GSH generated via the pentose phosphate pathway. The reduced cytochrome c is ineffective at causing apoptotic death. Suppression of the pentose phosphate pathway or GSH synthesis with inhibitors placed the cells in a prooxidant state, oxidizing the cytochrome c. The oxidized cytochrome c effectively induces death. The Bax-dependent increase of ROS and the decline of glutathione redox cycling in NGF-deprived neurons mentioned earlier may contribute to this oxidation.
Conclusions
ROS concentrations increase during the apoptotic death of many types of cells, including neurons. Available data suggest that these ROS are important regulators of the intrinsic apoptotic cascade in neurons. Specifically, they appear to be necessary for induction of cytochrome c release from mitochondria. The proapoptotic protein Bax lies upstream of the elevated production of ROS during neuronal apoptosis. Bax appears to increase ROS in part through activation of caspases secondary to Bax-induced redistribution of cytochrome c. It is likely that the activated caspases increase ROS in neurons, as in other cell types, by cleaving mitochondrial respiratory complexes. Bax may also contribute to ROS production by depleting cytochrome c from the mitochondrial electron-transport chain or by a variety of other mechanisms that may affect leakage of electrons from the chain. I hypothesize that ROS coordinates rapid release of cytochrome c from most mitochondria in a neuron by an autocatalytic mechanism involving Bax oxidation. Additionally, ROS primes neuronal caspase activity by oxidizing cytochrome c, an event that appears to be a necessary step for cytochrome c to initialize apoptosis.
Footnotes
Acknowledgments
This work was supported by NIH grant RO1NS37110. I thank Rebecca A. Kirkland for critical reading of the manuscript.