
Abstract
Zn2+ has emerged as a major regulator of neuronal physiology, as well as an important signaling agent in neural injury. The intracellular concentration of this metal is tightly regulated through the actions of Zn2+ transporters and the thiol-rich metal binding protein metallothionein, closely linking the redox status of the cell to cellular availability of Zn2+. Accordingly, oxidative and nitrosative stress during ischemic injury leads to an accumulation of neuronal free Zn2+ and the activation of several downstream cell death processes. While this Zn2+ rise is an established signaling event in neuronal cell death, recent evidence suggests that a transient, sublethal accumulation of free Zn2+ can also play a critical role in neuroprotective pathways activated during ischemic preconditioning. Thus, redox-sensitive proteins, like metallothioneins, may play a critical role in determining neuronal cell fate by regulating the localization and concentration of intracellular free Zn2+. Antioxid. Redox Signal. 15, 2249–2263.
Introduction
Neuronal tolerance to lethal ischemic cell death can be conferred by sublethal preconditioning stimuli, which activate endogenous survival pathways that limit or resist subsequent lethal injury (51). Increasing evidence suggests that preconditioning stimuli induce the sublethal activation of cell death factors that trigger survival pathways, which, in turn, prevent subsequent lethal injurious signaling (32). Along these lines, a novel role for Zn2+ as an important, early signal in the initiation of survival pathways critical to neuronal tolerance has emerged (5, 65). The results from these studies offer insights into endogenous mechanisms that protect neurons in the face of lethal cellular injury. In this review, we discuss the roles for Zn2+ in cell physiology, neuronal cell death pathways, and neuronal tolerance, focusing on the intersection of Zn2+-dependent pathways with redox-sensitive proteins and other damage-associated molecular patterns (DAMPs).
Physiological Roles of Zinc
Zinc is the second most prevalent trace element in the body and is very abundant in the brain (overall content of ∼150 μM) with highest concentrations in forebrain regions, including the hippocampus, amygdala, and cortex (127). Since its discovery in the brain almost 55 years ago, Zn2+ has been shown to play critical roles in protein structure, enzymatic activity, and gene regulation (29). Zn2+ can associate with over 300 enzymes, where it can interact strongly with electronegative sulfur, nitrogen, and oxygen moieties in multiple coordination geometries, serving catalytic and structural roles in maintaining active peptide conformations (23). In addition to metalloenzymes, Zn2+ is most known for its ability to bind and stabilize proteins involved in gene regulation in domains called zinc fingers, zinc clusters, and zinc twists (122). Because of the critical role Zn2+ plays in protein structure and function, it is not surprising that chronic dietary Zn2+ deficiency manifests in multisystemic dysfunction, including growth failure, skin changes, delayed wound healing, immune system impairment, neurosensory disorders, and lack of sexual development (101).
Besides regulating the structure and function of proteins, brain Zn2+ may also be important in modulation of neuronal activity (29). Approximately 10% of neuronal Zn2+ is selectively stored in presynaptic vesicles of certain glutamatergic neuronal terminals by the neuronal-specific zinc transporter 3 (ZnT3). Because of this, much attention has been given to determining whether synaptic Zn2+ release is activity dependent (96). Activity-dependent Zn2+ release was elegantly and clearly demonstrated by Qian and Noebels, who used a membrane-impermeant form of the Zn2+-selective indicator FluoZin-3 to show extracellular Zn2+ in mossy-fiber synapses of wild-type, but not ZnT3 knock-out mice after individual action potentials (102, 103). Zn2+ released from presynaptic neurons has several potential postsynaptic targets. Zn2+ inhibits N-methyl-D-aspartate (NMDA) and gamma-aminobutyric acid receptor subtypes, and potentiates glycine receptors (96, 110). In addition to modulating ionotropic pathways, synaptic Zn2+ has also been reported to transactivate receptor tyrosine kinase b (42) and regulate metabotropic activity through the activation of a recently described Zn2+ receptor, ZnR (10). Of interest, the molecular identity of the ZnR has been proposed to be the orphan G protein-linked receptor GPR39 (10). However, since the exact magnitude and time course of synaptic Zn2+ remain unclear (47), the full range of physiological roles for vesicular Zn2+ has yet to be fully described.
A critical new role of Zn2+ is its emergence as an intracellular signal. Yamasaki and colleagues recently found evidence for intracellular Zn2+ release in mast cells within minutes of extracellular stimulation of the high-affinity immunoglobulin E receptor 1 (FcɛR1), a phenomenon they termed a “zinc wave” (129). Using confocal fluorescent imaging, the authors found that the Zn2+ wave originated near the endoplasmic reticulum and other perinuclear areas (129). Rapid Ca2+ influx and mitogen-activated protein/extracellular signal-regulated kinase (MEK) activity were required for the Zn2+ rise, although neither the Ca2+ ionophore ionomycin nor an MEK activator was sufficient to elicit a Zn2+ wave (129). Zn2+ chelation decreased FcɛR1-induced interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) mRNA expression, while increasing free Zn2+ in mast cells with pyrithione/Zn2+ prolonged mRNA expression (129). Similarly, mitogen-activated protein kinase (MAPK) activation was decreased with Zn2+ chelation and prolonged with Zn2+ supplementation after FcɛR1 stimulation (129). A likely role for such intracellular Zn2+ signals is the modulation of protein phosphorylation. While increased cellular Zn2+ can activate many kinase pathways, including extracellular signal-regulated kinase and p38 (135), these studies suggest a target upstream in the signaling cascade rather than direct effect of Zn2+ on these kinases. For the most part, however, activation of Zn2+-dependent kinase cascades have been examined within the context of large exogenous Zn2+ exposure, sometimes leading to cellular toxicity. On the other hand, physiologically relevant nanomolar concentrations of Zn2+ have been shown to effectively inhibit protein tyrosine phosphatase (PTP) activity (38). These studies were performed with a truncated form of the phosphatase containing only the catalytic domain, suggesting that the inhibitory action of Zn2+ is through direct interaction with this highly conserved domain of PTPs (37). Thus, a major physiological role for Zn2+ may be the modulation of cell signaling cascades, especially those involving protein phosphorylation.
Regulation of cellular zinc
Zn2+ does not passively cross biological membranes and thus requires specialized mechanisms for its uptake and release. Dietary Zn2+ is absorbed from the gastrointestinal tract by a ZnT, ZIP4, located on the apical membrane of enterocytes, whereas another ZnT, ZnT1, is involved in zinc efflux from the enterocyte into the circulation (70). Mutations in the ZIP4 gene cause Acrodermatits enteropathica, a rare, lethal autosomal recessive inherited human zinc deficiency (59, 125). Once in the circulation, Zn2+ is bound to albumin and other plasma proteins facilitating transport to tissues, including the brain. Despite relatively high Zn2+ content in the brain, extracellular Zn2+ concentrations are normally below 500 nM and the intracellular levels of free Zn2+ are generally 1 nM or less (127). Intracellular free Zn2+ is tightly regulated though the opposing actions of the solute-linked carrier 39 (SLC39) family of transporters (also known as Zrt- and Irt-like proteins or Zips) and the SLC30 transporter family (also known as ZnTs) (110). Zip and ZnT proteins appear to have opposing roles in Zn2+ homeostasis, as Zips increase cytoplasmic Zn2+, whereas ZnTs promote Zn2+ efflux from the cytoplasm into intracellular compartments or across the plasma membrane (70). Detailed mechanisms and energetics of transport by these proteins are not well characterized, but are believed to be mediated by facilitated diffusion, secondary active transport, or symporters (70). Recent work investigating the activity of the ZnT5 transporter, which is localized in the Golgi apparatus and mediates Zn2+ transport into this compartment, found that Zn2+ transport is catalyzed via Zn2+/H+ exchange and is powered by the vesicular H+ gradient (88). Further, using a bacterial Zn2+ transporter YiiP as a homology model, this study identified Asp 599 and His 451 as two residues on the C-terminal region of ZnT5 that are critically responsible for ZnT5-mediated Zn2+ transport and sequestration to the Golgi (88). This work represents an important step toward determining the transport mechanisms involved in Zn2+ homeostasis.
Metallothioneins (MTs) also play a major role in regulating intracellular Zn2+ homeostasis by binding, releasing, and distributing Zn2+ (Fig. 1). MTs are a family of nonenzymatic, cysteine-rich, metal-binding polypeptides (61–68 amino acids) found throughout mammalian tissues (6). In the mammalian adult central nervous system, three major MT isoforms are expressed. The precise cellular localization of MT I, II, and III is still a matter of debate due to the lack of isoform-specific antibodies leading to conflicting results between in vivo and in vitro studies and between mRNA and protein expression assays (41). The general consensus, however, is that MT I and MT II are the predominant astrocytic isoforms (where they are expressed over seven-fold higher than in neurons), whereas MT III is predominantly expressed in neurons (41). MT III was first identified during investigations of putative mechanisms underlying Alzheimer's disease neuropathology. Abundant MT III expression, which was then identified as growth inhibitory factor due to its ability to inhibit neurotrophic factors and neurite outgrowth, was found in normal brain astrocytes, but not in Alzheimer's disease brains (121). However, since then, the association between MT III and Alzheimer's disease has not fully developed (19). Instead, MTs are thought to play a major role in heavy metal detoxification and Zn2+ homeostasis. Expression of MT I and II is highly inducible in response to a range of stimuli in addition to Zn2+ and Cd2+, including hormones, cytokines, oxidative agents, inflammation, and ischemia (2). Because of this, MTs have been implicated in mediating a more general cellular response to injury (128). Exciting recent reports have shown that MT can be secreted by astrocytes and internalized by neurons after brain injury, promoting neuronal cell survival (25). Thus, MTs play diverse roles in regulating cellular Zn2+ homeostasis, heavy metal detoxification, and intercellular response to injury.
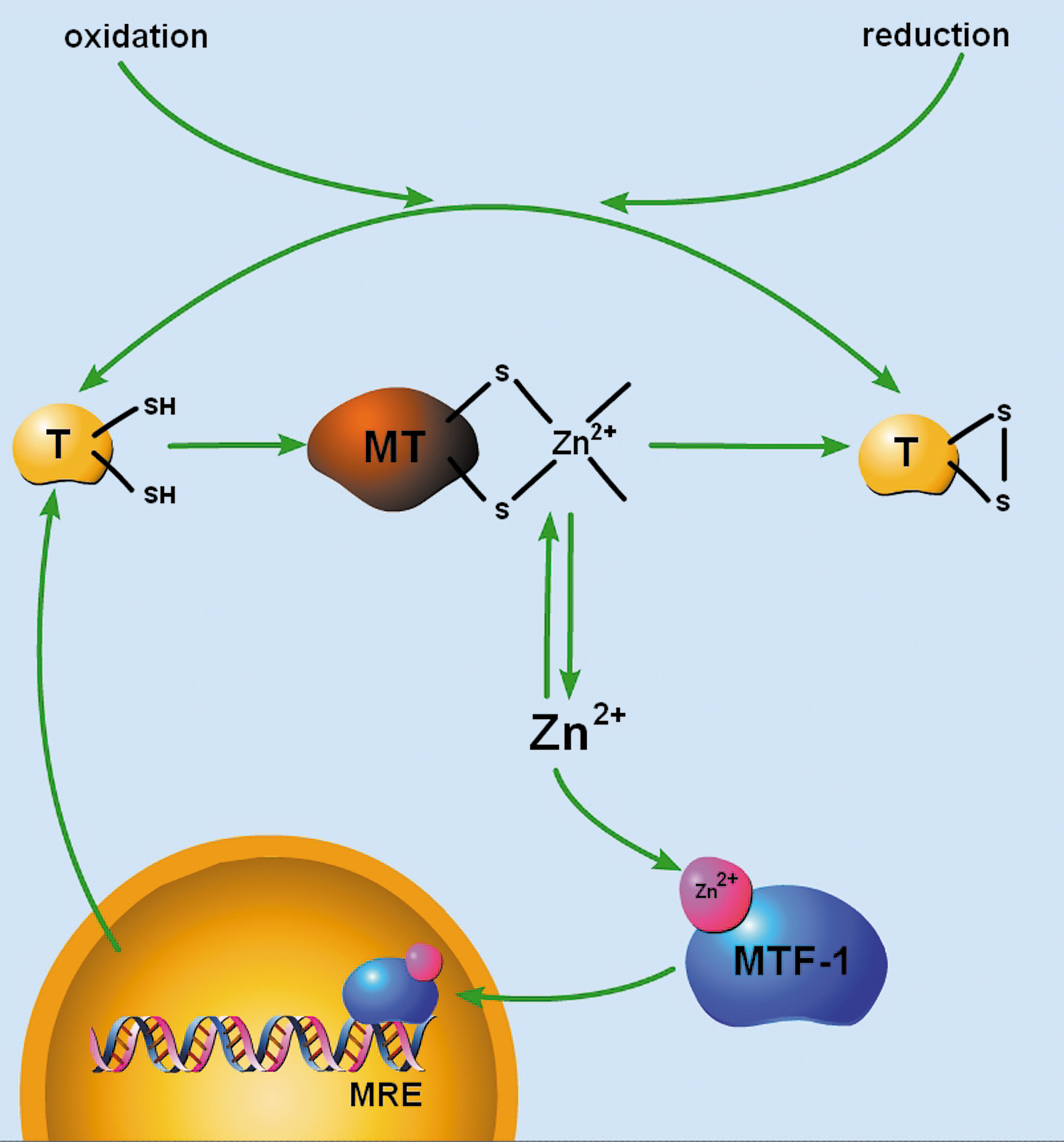
MTs are structurally composed of two globular metal-binding domains (α and β) containing a total of 20 sulfur-donating cysteine residues, allowing them to bind up to seven Zn2+ ions in Zn3S9 and Zn4S11 configurations or up to seven Cd2+ ions or 12 Cu2+ ions (73). These cysteine residues in MTs occur in unique Cys-X-Cys, Cys-X-Y-Cys, and Cys-Cys motifs (45). For MTs to buffer cellular-free Zn2+, these high-affinity ligands for Zn2+ must normally not be saturated with metal. Indeed, biochemical analysis of the association of Zn2+ with the apoprotein thionein revealed three classes of binding affinities that vary by several orders of magnitude. Four Zn2+ ions were bound tightly (log K d = 11.8), making the thionein a strong chelating agent; one Zn2+ was relatively weakly bound (log K d = 7.7), making MT a potential Zn2+ donor, and the remaining two Zn2+ ions were with intermediately bound (log K d ∼10) (57). This suggests that MT molecules are not saturated with seven Zn2+ ions under normal physiological conditions and can actively participate in cellular Zn2+ buffering and distribution (57). In addition, differential fluorescent labeling of the MT cysteine clusters from rat kidney, liver, and brain tissue extracts showed that tissues contain almost as much of the apo-protein thionein as metal-bound MT (130). Thus, due to the presence of Zn2+-lacking species along with apo-thionein, MT III serves a significant role in buffering neuronal free Zn2+. Further, the unique Zn2+ coordination environment of MT allows the redox environment of the cell to ultimately dictate overall cellular Zn2+ availability.
Zinc-regulated gene expression
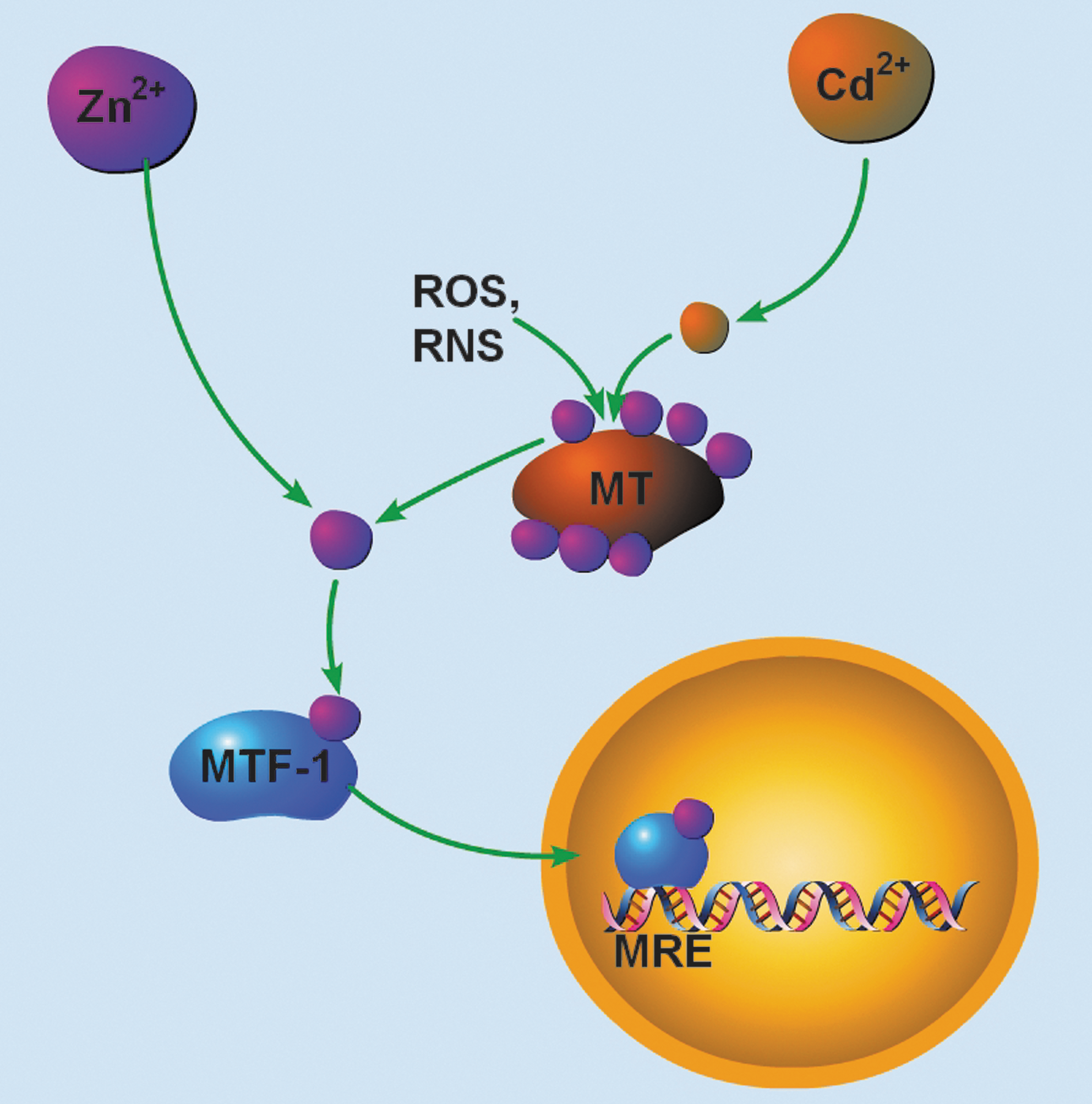
A major role for physiologic Zn2+ is the regulation of the proteins that control the intracellular concentration of the metal (Fig. 2). The inducible MT isoforms, MT I and MT II, are regulated in a coordinated manner through the activation of gene transcription (39). The promoter regions of MT I/II genes contain several cis-acting DNA elements, including metal response elements (MRE), glucocorticoid response elements (GREs), antioxidant response elements (ARE), and IL-6/signal transducers and activator of transcription responsive elements (IL-6 RE) that can bind trans-acting transcription factors involved in the regulation of both constitutive and inducible MT expression (39). MRE transcription factor 1 (MTF-1) is the predominant gene regulatory protein mediating constitutive as well as metal- and oxidative stress-induced MT I/II expression (2). MTF-1 is a 72.5-kDa, ubiquitous zinc-finger transcription factor in the Cys2His2 family that binds to DNA sequence motifs, known as MREs, at the consensus site TGCRCNC (3). MRE sequences are present in five, nonidentical copies (MREa-MREe) in the 5′ flanking region of the MT I/II gene (3). MTF-1-mediated MRE activation sufficiently upregulates MT I/II in response to Zn2+ or Cd2+, oxidative stress, and hypoxia (2, 83). After Zn2+, acute hypoxia, or oxidative stress induced by H2O2 or tert-butylhydroquinone, MTF-1 translocates from the cytoplasm to the nucleus and acquires tight DNA-binding ability (2). However, after exposure to other heavy metals, most notably Cd2+, the DNA-binding ability of MTF-1 is inhibited in cell-free DNA binding assays, despite induction of MTF-1-dependent MT gene transcription in vivo (3). This paradox was solved by demonstrating that transcriptional activation after Cd2+ requires Zn2+-saturated MT (134). Cd2+, which binds to MTs with higher affinity than Zn2+, can displace Zn2+ from the metal-binding protein, making Zn2+ available for MTF-1 activation (134). Activation of gene transcription also requires the occupancy of the Zn2+ fingers with Zn2+ and the interaction among three distinct transcriptional activation domains in the C-terminal region of MTF-1 (3). While most of the evidence characterizing Zn2+-regulated gene expression has been performed studying MT gene activation, changes in ZnT1 transporter expression can also be induced by Zn2+ or Cd2+ (2). Constitutive and metal-induced activation of the ZnT1 gene also requires MTF-1 (62). While the organization of the promoter regions of all MT genes is conserved, the MT III gene was found to contain a “silencing element” in the 5′ flanking regions upstream from the first cluster of putative MREs (20), eliminating Zn2+-regulation and making MT III a constitutively expressed protein in neurons. Close inspection of the silencing element of the rat MT III promoter reveals that it contains a quadruplicate CTG sequence ∼400 base-pairs upstream of the translation start site (20). CTG-repeat elements have been shown to act as repressors on a number of heterologous promoters independent of its orientation and proximity to the gene, and thus may account for the silencing effect on the promoter region of the MT III gene (20). Besides MT I/II and ZnT1, many other MRE-containing MTF-1 target genes have been identified, including γ-glutamylcysteine synthetase (a key enzyme in glutathione synthesis), α-fetoprotein, and transforming growth factor β-1 (68).
Role of Zinc in Neuronal Cell Death
Translocation of vesicular zinc
Unregulated increases of free intracellular Zn2+ lead to cell death. Using TSQ (N-[6-methoxy-8-quinolyl]-P-toluenesulfonamide) and acid fuschin to simultaneously stain the presence of Zn2+ and degenerating neurons, respectively, Tonder et al. suggested a translocation of Zn2+ from mossy fibers terminals to degenerating dentate hilar neurons 2–24 h after 20 min of cerebral ischemia (118). This type of observation, coupled with reports suggesting that the amount of Zn2+ stored in synaptic vesicles (∼300 μM) was sufficient to kill neurons (132), led to what is generally known as the Zn2+ translocation hypothesis. This hypothesis proposes that the extracellular release of vesicular Zn2+ and its uptake by postsynaptic neurons is responsible for the toxic accumulation of this metal in neurons after ischemia and other neurological disorders (28). In support of this model, Koh et al. demonstrated that intraventricular injection of the membrane-impermeant Zn2+ chelator, ethylenediaminetetraacetic acid saturated with Ca2+ (CaEDTA), blocked the accumulation of neuronal Zn2+ and prevented neuronal death after transient global cerebral ischemia (53). While Zn2+ was previously known to be toxic to neurons when applied exogenously (24), these reports were first to implicate endogenous accumulation of Zn2+ in ischemic neuronal death.
Extracellular Zn2+ can lead to toxic intracellular Zn2+ accumulations by entering through voltage-sensitive Ca2+ channels (VSCC), NMDA receptors, or Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)/kainate receptor channels (Ca2+-A/K) (110). Of these, Ca2+-A/K channels exhibit the highest Zn2+ permeability (111) and are thought to be the primary route of Zn2+ entry after ischemia (131). Indeed, in hippocampal slices, ischemia-induced Zn2+ accumulation and subsequent neurodegeneration can be attenuated with a Ca2+-A/K channel inhibitor but not NMDA receptor or VSCC channel antagonists (131). Hippocampal pyramidal neurons, which are especially sensitive to ischemic injury, express Ca2+-A/K channels on distal dendritic branches (87). Unlike all other heterotetrameric AMPA receptors, Ca2+-A/K channels lack the GluR2 subunit (60). GluR2 subunits contain an arginine (R) in its pore-forming domain due to RNA editing of a genomically encrypted glutamine (Q) codon (60). The arginine is functionally dominant because it dictates gating kinetics, channel conductance, channel assembly, and, importantly, Ca2+ permeability (35). Edited GluR2(R) subunits form Ca2+-impermeable channels, whereas unedited GluR2(Q) channels are Ca2+ permeable (60). Generation of mice with unedited GluR2(Q) exhibit fulminant seizures and death by 3 weeks of age (14). GluR2 expression can be modulated by ischemic injury, as ischemia has been shown to induce GluR2 downregulation (34) and disrupt Q/R editing (98). This modulation of GluR2 subunits render AMPA channels Ca2+ permeable, allowing entry of toxic Ca2+ and Zn2+ during ischemia (91).
Regulation of Ca2+-A/K channel composition after ischemic injury may also involve Zn2+-dependent signaling. In an in vivo model of stroke, application of the cell-impermeant Zn2+ chelator CaEDTA 30 min before global ischemia attenuated the ischemic downregulation of GluR2, the delayed rise in neuronal Zn2+, and neuronal death (16). Ischemic downregulation of GluR2 occurs via the Zn2+-dependent expression of the nine Zn2+-finger transcription factor restrictive element-1 silencing transcription factor (REST-1), which suppresses neuronal-specific targets, including GluR2 (16, 17). Application of CaEDTA 48–60 h after global ischemia, presumably after GluR2-lacking A/K channels are already expressed, attenuated only the late rise in neuronal Zn2+ and cell death (16).
In spite of the aforementioned work, several key observations have shown that Zn2+ translocation cannot fully account for the accumulation of free Zn2+ in dying neurons after ischemia. For instance, Zn2+ accumulation in neurons after injury is observed in areas that are not highly innervated by glutamate- and Zn2+-releasing fibers, such as thalamic neurons (53, 61). Intracellular sources of Zn2+ must play a major role in lethal Zn2+ accumulation in these brain regions. More importantly, if presynaptic Zn2+ were the only source of toxic Zn2+ contributing to ischemic injury, then animals without presynapic Zn2+ would not be susceptible to Zn2+-dependent toxicity. However, mice lacking the ZnT3, which show no histochemically reactive Zn2+ in presynaptic vesicles, still undergo significant Zn2+ accumulation in degenerating CA1 and CA3 neurons after kainate-induced seizures (64). This suggests that lethal neuronal injury can induce intracellular Zn2+ accumulation and neurodegeneration in the absence of presynaptic vesicular Zn2+. In initial studies, CaEDTA nearly completely abolished Zn2+ accumulation and neuronal cell death, suggesting an extracellular source for Zn2+ (53). However, CaEDTA has since been shown to deplete intracellular Zn2+ compartments, including synaptic vesicles (30). CaEDTA may trap extracellular Zn2+, giving rise to a steep Zn2+ gradient across the plasma membrane resulting in the removal of Zn2+ from the neuronal cytoplasm. As such, accumulation of intracellular Zn2+ after neuronal injury may not be solely dependent on translocation of the metal from presynaptic neurons. Intracellular sources of Zn2+ may therefore play a substantial and critical role in neurodegenerative processes.
Redox-dependent liberation of intracellular zinc
As Zn2+ accumulation can occur in degenerating neurons even in the absence of vesicular Zn2+, liberation of Zn2+ from intracellular stores may be a significant mechanism of cell injury, especially in brain regions with little or no synaptic Zn2+. Vallee and colleagues suggest that Zn2+ binding and release in cells are dynamic processes that are dependent on the intracellular redox status (Fig. 1) (44). Of the Zn2+ metalloproteins, only those with Zn2+/sulfur coordination environments are susceptible to oxidative mobilization of Zn2+ during injury (72). These coordination environments allow for tight binding of Zn2+ and for the mobilization of redox-inert Zn2+ (9) by biological oxidants. Of the thiol-Zn2+ metalloproteins in neurons, MT III is a likely source of injury-induced mobilized Zn2+. Despite binding Zn2+ with high thermodynamic stability (K d = 1.4 × 10−13 M for human MT, pH 7.0), the redox potential for MT is very negative (−366 mV), causing even mild cellular oxidants to release Zn2+ from the metal-binding protein (73). In HT-29 cells, it was found that as the intracellular redox potential became more oxidizing, the buffering capacity of MT was lowered, leading to a pronounced elevation in free Zn2+ (55). Liberation of Zn2+ from MT has been demonstrated in a variety of systems, including cell-free assays (73), as well as in cortical neurons (1, 136), where it is an event critical to initiation of oxidant-induced neuronal apoptosis (1). In mouse hepatoma (Hepa) cells, oxidants activate MRE-binding by MTF-1 (2). Further, nitric oxide (NO) and subsequent peroxynitrite (ONOO−) formation, two powerful cellular oxidants contributing to ischemic nitrosative stress, also liberate neuronal Zn2+ from intracellular stores triggering neurodegeneration (13, 136). NO-induced Zn2+ liberation in pulmonary artery endothelial cells was shown to effectively activate MTF-1 and transcription of MT (113). NO has been shown to more readily release Zn2+ from MT III than other MT isoforms due to the unique presence of consensus motifs for catalytic nitrosylation and the preferential reactivity of S-nitrosothiols with MT III through transnitrosylation, allowing direct transfer of NO between sulfhydryl groups that result in sequential release of Zn2+ ions (21). Other redox signals, including superoxide/peroxide, selenium compounds, and NADPH, have also been shown to induce the release Zn2+ from MT (71, 107). Thus, liberation of Zn2+ from intracellular stores, especially neuronal MT III, may be a significant source of the Zn2+ rise after ischemia. Depending on the availability of unoccupied Zn2+ binding sites as well as the intracellular redox environment, MT III can serve as an important sink, as well as a source, for neuronal-free Zn2+.
Zinc-dependent signaling in neuronal death
Accumulation of neuronal Zn2+, likely mediated by a combination of Zn2+ translocation from presynaptic vesicles and Zn2+ liberation from intracellular stores, can trigger subsequent neurodegenerative signaling after ischemia (Fig. 3). Zn2+-induced cell death involves several serial and parallel signaling events, has features of both apoptosis and necrosis (50), and is likely mediated by oxidative and nitrosative stress (48). The diversity of cell death signaling attributed to Zn2+ may depend on the intensity of Zn2+ exposure, as brief exposure to high concentrations of Zn2+ lead to signs of necrotic, caspase-independent cell death, whereas longer exposures to lower Zn2+ concentrations trigger apoptotic, caspase-dependent cascades (48). In addition, accumulation of Zn2+ after ischemia may trigger divergent signaling mechanisms along multiple temporal profiles. While a delayed rise in neuronal Zn2+ in degenerating neurons after ischemia has been well described (53), recent reports suggest that an early Zn2+ rise after ischemia onset also contributes to ischemic injury (79, 114). Mechanisms involved in Zn2+-dependent ischemic neuronal death are incompletely understood, and may resemble the complexity in Ca2+-mediated cell death pathways (22). Diverse Zn2+-dependent cell death signaling events have been described, including regulation of Zn2+-dependent transcription factors, induction of p75 neurotrophin receptor-mediated pathways, and activation of kinases and poly-ADP-ribose polymerase (PARP-1) (110). Recent work has also illuminated a previously unknown link between Zn2+ and the K+/Cl− cotransporter 2 (KCC2) after oxygen–glucose deprivation (40). Here, we highlight some recent examples of neuronal cell death pathways that critically involve an accumulation of free Zn2+.
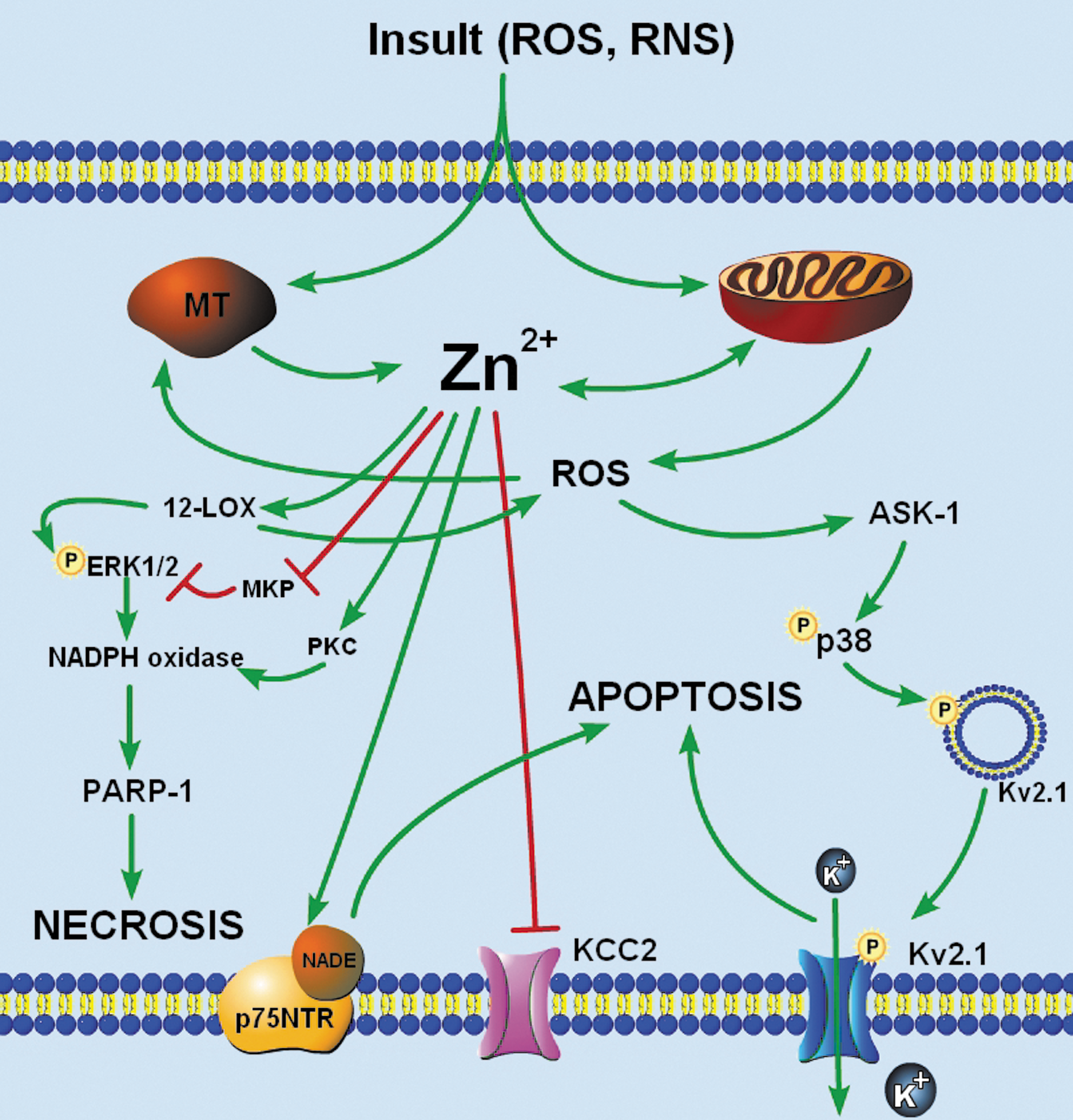
Role of Zn2+ in Kv2.1-facilitated apoptosis
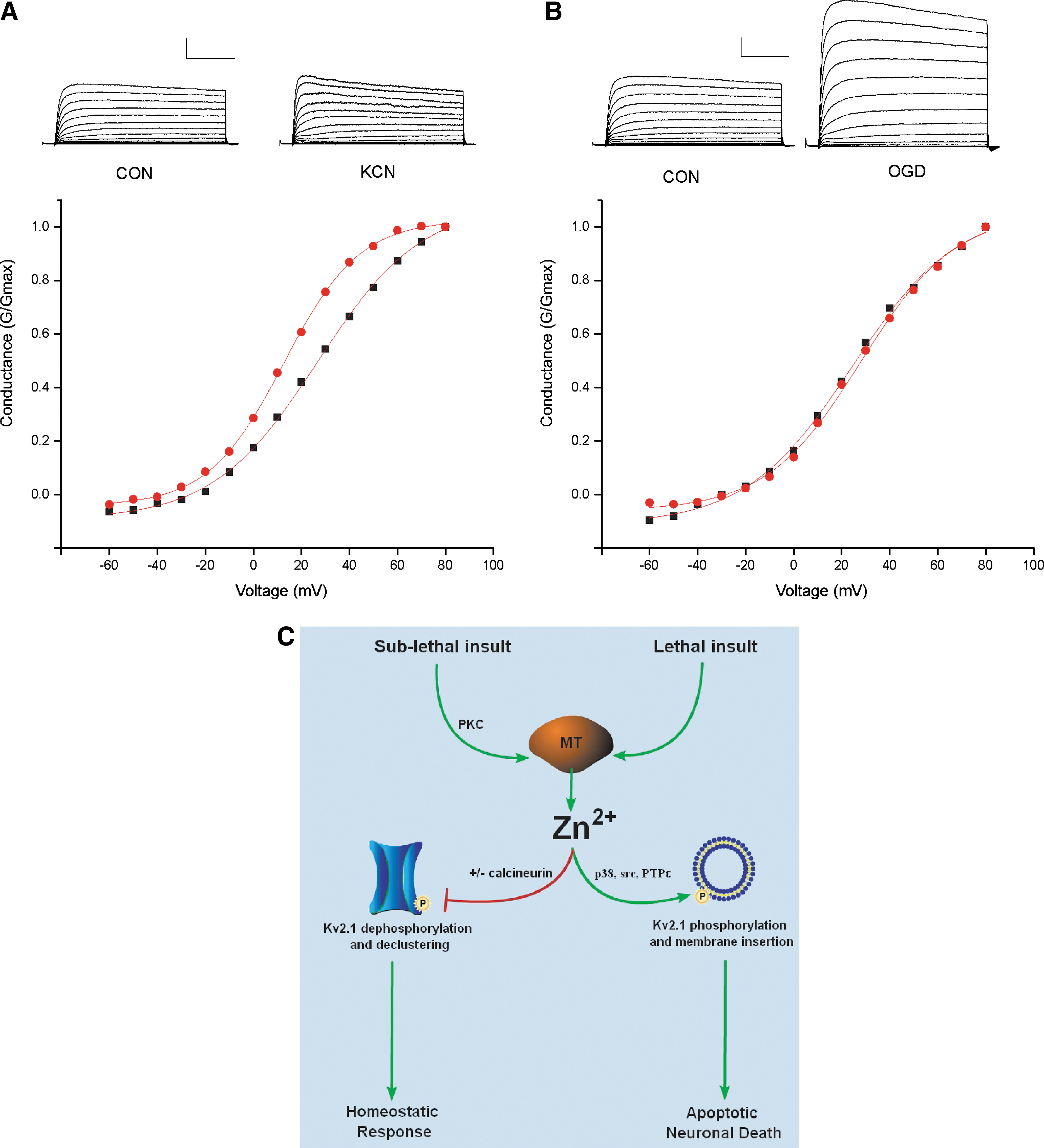
A well-characterized caspase-dependent apoptotic pathway has been critically linked to oxidant-induced Zn2+ liberation. The enhancement of voltage-gated K+ channel (Kv) activity, resulting in cellular K+ efflux, is a critical step in many apoptotic programs (12). Apoptotic K+ current enhancement leads to a decrease in the concentration of this cation in the cytoplasm (133), which serves as a permissive apoptotic signal (12), as pro-apoptotic factors are activated most efficiently at reduced K+ concentrations (43). In our laboratory, Kv2.1 was identified as the critical mediator of K+ efflux during neuronal apoptosis (92). Kv2.1 is the major component of delayed-rectifying K+ current in cortical neurons (82), and exists in large, highly phosphorylated clusters on the somatic surface and proximal dendrites of cortical neurons (109). Stimulation of a caspase-dependent neuronal apoptotic cascade by oxidative injury triggers the liberation of intracellular Zn2+, leading to p38 MAPK-dependent phosphorylation and insertion of new Kv2.1-encoded channels (Fig. 3) (77, 93, 105). Recent molecular studies have shown that, in addition to p38 phosphorylation, the apoptotic program requires Kv2.1 to be phosphorylated on a N-terminal tyrosine (Y124) residue (104), a target of Src kinase and cytoplasmic PTP-epsilon (PTPɛ). Zn2+ plays a critical role in Kv2.1-mediated apoptosis by coordinating the activation of p38 MAPK and Src kinase, along with the concomitant inhibition of cytoplasmic PTPɛ, thereby enabling the completion of the apoptotic program (104).
Role of Zn2+ in immune cell/DAMP-mediated neuronal death
Microglia are resident immune cells of the central nervous system that can serve several beneficial functions in neuronal cellular maintenance and innate immunity. Analogous to peripheral macrophages, microglia monitor the brain parenchyma through expression of diverse membrane receptors that can identify a wide range of signals (11). Integral to the innate immune response are constitutively expressed pattern recognition receptors that identify and bind DAMPs and pathogen-associated molecular patterns (PAMPs). Indeed, microglia have been shown to express many of the well-recognized pattern recognition receptors, including toll-like receptors 1–9 (90), scavenger receptors, and the receptor for advanced glycation endproducts (11). However, the microglial response to signals in innate immunity can differ from that elicited by neuronal damage.
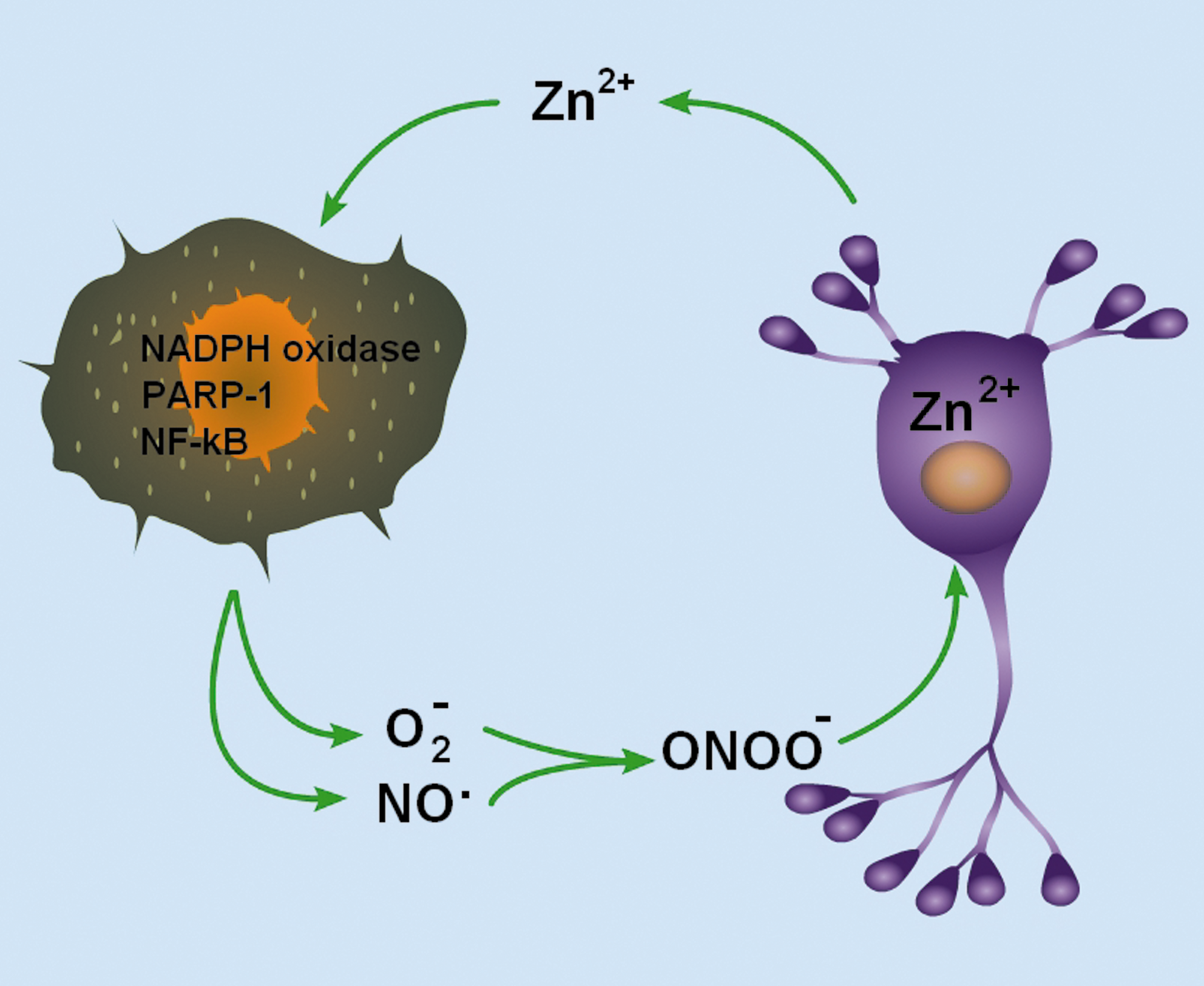
When stimulated during brain injury, the overactivation and dysregulation of microglia can compound neuronal damage by releasing neurotoxic cytokines, matrix metalloproteinases, and NO and superoxide (O− 2), leading to the formation of the powerful cellular oxidant, peroxynitrite (ONOO−) (66). Since ONOO− can traverse membrane lipids faster than its decomposition pathways (74), it is possible that activated microglia may trigger oxidant-induced Zn2+-dependent neuronal cell death. Indeed, in a coculture model, lipopolysaccharide (LPS)-activated microglia triggered an apocyanin- and 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrinato iron(III) chloride-dependent accumulation of neuronal free Zn2+ (52). Further, microglia-derived reactive oxygen and nitrogen species (ROS and RNS) triggered the well-characterized apoptotic signal-regulating kinase-1 (ASK1)/p38-dependent, Kv2.1-mediated K+ current surge (52). When these studies were repeated in neurons overexpressing MT III, thereby chelating free Zn2+, the K+ current surge and neuronal cell death were markedly reduced (52). Thus, microglial activation can lead to a neuronal free Zn2+ rise and Zn2+-dependent neuronal cell death. Of note, peroxynitrite is also responsible for the LPS-induced degeneration of developing oligodendrocytes in coculture experiments (66). However, when astrocytes were present along with oligodendrocytes and microglia, the LPS-induced microglia-dependent toxicity to developing oligodendrocytes was mediated by a mechanism involving TNF-α signaling (67). Thus, in addition to redox-triggered signaling, microglia-mediated toxicity is also dependent on a number of immunologically relevant cytokines that can activate conserved DAMPs and PAMPs.
Microglia can stimulate an accumulation of free Zn2+ in target neurons, and the metal itself can trigger microglial activation. After exogenous Zn2+ exposure, cultured mouse microglia transformed into the activated morphology, increased NO production and F4/80 expression, and altered cytokine release (46). Zn2+-induced microglial activation was dependent on NADPH oxidase and PARP-1 activation (46). Importantly, intracerebroventricular injection of Ca2+-EDTA prevented microglial activation in an in vivo model of cerebral ischemia (46). Thus, a positive feedback model emerges in which ischemic injury leads to microglial activation causing the release of cellular oxidants and other DAMPs, which lead to the liberation of neuronal free Zn2+ (Fig. 4). In addition to activating cell death signaling pathways, the accumulation of free Zn2+ may overwhelm homeostatic mechanisms and lead to Zn2+ release, further activating microglia. Other DAMPs may also contribute to microglia-mediated neurodegeneration after stroke. For example, the prototypical DAMP, high-mobility group box 1 (HMGB1) is elevated in the serum of stroke patients (81), may be secreted from neurons triggering microglial activation (49), and contributes significantly to neurodegeneration after ischemia (69, 81). Thus, further studies are needed to better understand the precise mechanisms by which immune cells and inflammatory signaling contributes to neurodegeneration after acute neuronal injury, as well as the relative roles of Zn2+ in these processes.
Zinc in Neuronal Preconditioning
Preconditioning refers to the activation of endogenous adaptive processes by sublethal stimuli, which in turn can increase cellular tolerance to subsequent, lethal injury (86). Tolerance to lethal ischemic injury was first described by preconditioning cardiac myocytes with brief sublethal ischemic episodes (84). This phenomenon was later shown to also be present in the brain (51). A variety of stress stimuli known to cause ischemic brain injury when utilized at sublethal levels have been reported to trigger neuronal tolerance, including ischemia, hypoxia, metabolic inhibitors, excitotoxins, and inflammatory cytokines (32). Thus, the ultimate fate of neurons may not depend on which molecules are activated after a particular insult, but rather the extent and duration of their activity.
Cell death pathways trigger tolerance
Recent evidence suggests that pro-survival mechanisms conferring neuroprotection in ischemic preconditioning may involve sublethal activation of cell death pathways. For example, antioxidants and protease inhibitors, which limit cell death after lethal stimuli, can paradoxically increase vulnerability to subsequent lethal injury when administered during the preconditioning period (76). Similarly, autophagy, a lysosomal degradation process of the cell's own components, has been shown to be protective after a preconditioning stimulus, but may contribute to cell death after lethal insults (97). Caspase-3, an apoptosis executioner cysteine protease, has also been implicated in neuronal tolerance (Fig. 5). Our group first demonstrated that preconditioning led to caspase-3 cleavage in vivo and that caspase-3 activity was required for neuronal tolerance in vitro (76). Pro-caspase cleavage and caspase enzymatic activity reach maximum levels 6 h after a preconditioning stimulus and are dependent on ROS generation and ATP-sensitive potassium channel (KATP) channel opening (31, 76). Although widespread caspase-3 activation characterizes many cell death processes, the relatively modest preconditioning-induced caspase-3 activation is held in check by sequestration with caspase-binding proteins, including the constitutively active heat shock protein 70 (HSP70) homolog HSC70 (76) and the pro-survival inhibitor-of-apoptosis (IAP) family member cIAP (116). The depletion of the free pool of HSC70 leads to increased synthesis of HSP70, which is observed 24 h after preconditioning. HSP70 is then able to buffer lethal cellular signaling processes, including caspase-3 generation (76). Importantly, inhibiting the activation of caspase-3 dramatically attenuates the neuroprotective effect of preconditioning (76).
Aside from upregulating survival proteins in preconditioned neurons, caspase-3 has also been shown to target PARP-1 (31). PARP-1, which accounts for >80% of nuclear PARP activity, facilitates DNA repair by mediating the enzymatic transfer of ADP-ribose groups from NAD+ to form branched ADP-ribose polymers on acceptor proteins in the vicinity of DNA strand breaks or kinks (26). However, extensive PARP-1 activation, which occurs in ischemia (124), depletes NAD and ATP, leading to cellular energy failure and cell death (36). Caspase-3 can irreversibly cleave the catalytic site of PARP-1 from its DNA binding domain, effectively inactivating the polymerase (63). Accordingly, preconditioning stimulates caspase-dependent PARP-1 cleavage, attenuating PARP-1 activity, and protecting neurons from subsequent PARP-1-mediated cell death (31). Caspase inhibition during preconditioning blocked PARP-1 cleavage and severely diminished the neuronal tolerance (31). Thus, the killer protease caspase-3, when activated to sublethal levels, is a critical mediator of neuroprotection in preconditioned neurons.
Zinc accumulation in preconditioned neurons
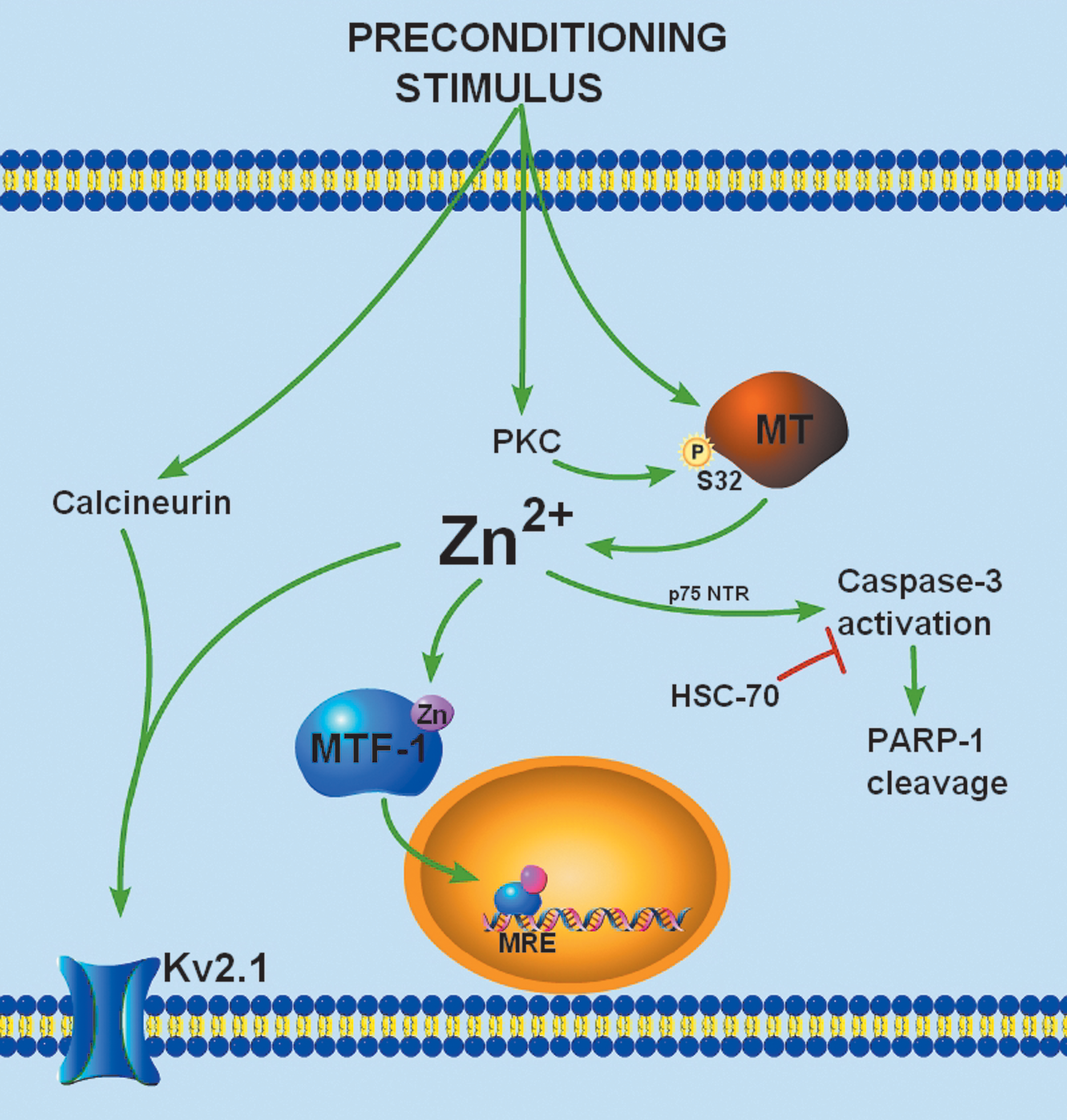
Although preconditioning-induced caspase activation and some of its downstream targets have been characterized, little is known about proximal signaling events that contribute to sublethal caspase activation. Until recently, the intracellular accumulation of free Zn2+ has been largely identified as a characteristic of degenerating neurons. Using in vivo and in vitro systems, two recent reports have shown that a sublethal stimulus led to an accumulation of free Zn2+ in preconditioned neurons (5, 65). The free Zn2+ rise occurred immediately after the preconditioning stimulus, subsided 24 h after the sublethal stimulus, and was required for neuronal tolerance (5, 65). The increase in neuronal free Zn2+ activated caspase-3-dependent PARP-1 cleavage, HSP70 upregulation, and Zn2+-regulated gene transcription conferring delayed neuronal tolerance (Fig. 5) (5, 65). In addition, the rise in free Zn2+ led to modulation of Kv2.1 channel activity and localization (4), which may be an important immediate response to ischemic injury (80). Together, these studies have described a critical, novel role for an accumulation of neuronal free Zn2+ in the activation of cell survival signaling pathways.
Source of zinc rise in preconditioning
While oxidative- and nitrosative-stress mediated liberation of neuronal Zn2+ has been previously described as a characteristic of cell death (1, 13), a similar mechanism may account for the Zn2+ signal in preconditioned neurons. Indeed, sublethal oxidative signaling has been shown to be required for tolerance in models of preconditioning (76). In preconditioned neurons, sublethal ROS generation can modify redox-sensitive protein kinases, phosphatases, or redox-sensitive transcription factors, such as nuclear factor-κB and activator protein-1, modulating gene expression and conferring neuronal tolerance (100). Similarly, NO generation, triggered by the influx of Ca2+ through NMDA receptors, has been shown to play a central role in preconditioning signal transduction by activating downstream kinases that ultimately modulate gene transcription (33). Thus, multiple redox signals known to trigger Zn2+ release from MT are activated in preconditioned neurons. The predominant source of the preconditioning-induced Zn2+ rise is indeed intracellular, as the low-affinity extracellular Zn2+ chelator tricine and the AMPA receptor blocker 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) had little effect on preconditioning-induced increase in Zn2+ fluorescence (5). Our laboratory found evidence for a highly conserved, putative protein kinase C (PKC) phosphorylation site on MT at serine 32 (S32) that is critically involved in Zn2+ regulated gene expression and in conferring neuronal tolerance (5), providing a novel role for PKC in modulating the MT/Zn2+ interaction to facilitate Zn2+ release from the metal-binding protein. Thus, it is likely that the preconditioning-induced Zn2+ signal is mediated by the redox modulation of the MT/Zn2+ interaction. Further, signaling kinases can serve as regulating factors in neuronal Zn2+ homeostasis.
Preconditioning triggers zinc-regulated gene expression
One of the hallmark characteristics of delayed neuronal tolerance is its dependence on de novo protein synthesis elicited by the preconditioning stimulus (8). However, the genomic response to preconditioning may not simply involve an immediate activation of quiescent survival genes. Instead, DNA oligonucleotide microarray analysis has shown that a large number of genes may be repressed after preconditioning (112). Nevertheless, protein synthesis inhibition by cyclohexamide before, or during preconditioning blocks neuronal tolerance in vivo and in vitro (76), suggesting a major role for gene activation and protein synthesis in neuroprotection. Changes in gene expression/repression may occur along several temporal profiles. Some genes are altered within minutes or hours (e.g., adenosine A2a receptor and vascular endothelial growth factor), whereas others are affected days after the preconditioning stimulus (e.g., calbindin and serine/threonine protein kinases) (117). Thus, the highly regulated modulation of gene expression in preconditioned neurons is an important cellular response that may ultimately confer neuronal tolerance.
We found evidence suggesting that a preconditioning-induced Zn2+ rise triggers the activation of processes that can prevent subsequent toxic Zn2+ accumulations (5). Matsushita and colleagues (75) reported that exogenous ZnCl2 pretreatment reduced neuronal death after in vivo global ischemia when Zn2+ was administered 24 and 48 h, but not 1 h, before ischemia. In light of the requirement of new protein synthesis in neuronal tolerance, and that proteins involved in buffering-free Zn2+ are regulated at the level of transcription, it is likely that preconditioning triggers the upregulation of proteins involved in maintaining cellular Zn2+ homeostasis. To this end, our group found that preconditioning activates MRE-dependent transcription in cortical neurons, suggesting that Zn2+-regulated gene expression can be stimulated in preconditioned cells. Thus, the Zn2+-induced upregulation of Zn2+-regulated proteins, such as MT I/II and ZnT1, may play a role in neuronal tolerance. Indeed, upregulation of MT I and II have been shown in multiple models of preconditioning (18, 27, 117). A two-fold induction of MT I/II mRNA can be detected as early as 3 h after preconditioning, peaking at 12 h with a 6-fold change in expression, and returning to baseline levels 24 h after preconditioning (18, 27). The induction of MT by application of transition metals protects the heart against oxidative damage (108) and cortical cells against irradiation damage (15). Moreover, mice overexpressing MT I have reduced lesion volume and sensorimotor deficits after in vivo ischemia compared with wild-type mice (123). On the other hand, mice lacking MT I/II suffer worse outcomes than wild-type mice after a range of CNS injuries, including focal cryolesion, experimental autoimmune encephalomyelitis (an experimental model of multiple sclerosis), motor neuron disease, and stroke (99). Finally, MT I/II double-knockout mice had approximately three-fold larger infarcts and significantly worse neurological outcome than wild-type mice after transient ischemia (119). Thus, MT plays an important neuroprotective role in response to cellular injury.
Despite the abundance of data describing the upregulation of MTs in limiting neuronal injury, the inducible isoforms MT I and II are predominantly expressed in astrocytes (54). Thus, if MT I/II buffer lethal neuronal free Zn2+ in models of neuronal tolerance, then brain astrocytes may respond to preconditioning by upregulating MT I/II and secreting it into the extracellular space. In fact, accumulating evidence has supported the injury-induced secretion of astrocytic MT, the presence of MT in the extracellular space, and a potential receptor that may mediate MT internalization in neurons (25). In response to brain injury, extracellular MT has been shown to promote axonal regeneration and neurite outgrowth (25). It is also conceivable that, in addition to its effects on neuronal proliferation, secreted MT could also chelate Zn2+ released into the extracellular space from injured neurons. By chelating extracellular Zn2+, MT may be able to indirectly reduce intracellular Zn2+, analogous to the previously reported effects of CaEDTA (30). In addition, the internalization of astrocytic MT by neurons (25) may buffer lethal intracellular Zn2+ accumulations. These various hypotheses, however, require additional experimental studies to be validated.
Another likely target of preconditioning-induced Zn2+-regulated gene expression is the plasma membrane Zn2+ efflux transporter, ZnT1. ZnT1 is expressed by neurons (120) and can be induced by Zn2+ in an MTF-1-dependent pathway (62). ZnT1 facilitates Zn2+ efflux (95, but also see 89), and may actually be more efficient than MTs in rapidly regulating intracellular accumulation of Zn2+ (94). Using in situ hybridization, ZnT1 mRNA expression has been shown to be upregulated in gerbil CA1 pyramidal neurons 12 h after transient forebrain ischemia (120). However, not all changes in ZnT1 mRNA have been realized as proportional changes in protein levels (78, 120), indicating possible post-translational regulation of ZnT1 protein expression. It is also unclear whether blocking ZnT1-mediated Zn2+ efflux after ischemia exacerbates neuronal injury. Unlike MT, changes in ZnT1 mRNA and protein expression in preconditioned neurons have not been extensively investigated. However, a conceptually similar investigation has been performed in astrocytes (85). In these cells, sublethal exogenous Zn2+ exposure induced ZnT1 protein expression and conferred resistance to subsequent lethal Zn2+ toxicity (85). Further, in nonpreconditioned astrocytes, heterologous expression of ZnT1 reduced toxic intracellular Zn2+ accumulations and induced Zn2+-tolerance (85).
Zinc mediates Kv2.1-dependent homeostatic response after sublethal ischemia
Ischemia leads to profound changes in neuronal excitability, manifested as an early phase of cellular hyperpolarization and depression of neural activity, followed by a second phase of dramatic enhancement of excitability (58). Changes in metabolic state or intracellular Ca2+ concentration after ischemia can modulate a variety of K+ channels, including KATP channels, Ca2+-activated large conductance K+ channels, and delayed rectifier voltage-dependent K+ channels (7, 80, 106). Recent evidence has shown that sublethal ischemic injury is associated with a protein-phosphatase 2B (PP2B or calcineurin)-dependent dephosphorylation of existing Kv2.1 channels, which is accompanied by a dispersal of somatodendritic Kv2.1 clusters and hyperpolarizing shifts in voltage-dependency (80). The latter has been proposed to limit neuronal excitability and thus prevent widespread excitotoxic cell death (115). We have observed that chelation of Zn2+ during sublethal ischemia blocks the hyperpolarizing shifts in the activation kinetics and prevents Kv2.1 de-clustering (Fig. 6) (4). As ischemia-induced Zn2+ accumulation is not blocked by a calcineurin inhibitor, nor is calcineurin activity altered by Zn2+, the Zn2+ rise and calcineurin activation may act in concert to modulate Kv2.1 channels (4).
Concluding Remarks
Currently, the most effective therapy for ischemic stroke is timely reperfusion. Intravenous administration of human recombinant tissue plasminogen activator within 3 h of ischemic stroke onset can restore blood flow before major brain damage occurs (126). However, thrombolytic drugs must be used with caution, as lethal intracranial bleeding may result. In light of the serious side effects and limited time window of efficacy, it is imperative that alternative therapeutic strategies for ischemic stroke be developed. Over a decade, experimental evidence has described a central role for a toxic accumulation of intracellular Zn2+ in ischemic neuronal death. In several animal models of cerebral ischemia, Zn2+ chelation has consistently been shown to reduce neuronal death (16, 53). However, emerging in vivo and in vitro studies suggest that a sublethal increase in neuronal free Zn2+ may also trigger pathways that limit cell injury after injury and confer long-term tolerance (5, 65). The mechanisms accounting for the free Zn2+ accumulation and downstream signaling pathways mediating neuroprotection may provide novel therapeutic targets for regulating intracellular Zn2+ signals, and ultimately cell survival. This new, neuroprotective role for increases in free Zn2+ could aid in the development of future therapies for ischemic stroke.
Footnotes
Acknowledgments
This work was supported by NIH grant R01 NS043277 to E.A. M.A.A. was supported by American Heart Association Pre-doctoral Fellowship 0715176U.