
Abstract
All aerobic organisms are exposed to oxidative stress during their lifetime and are required to respond appropriately for maintenance of their survival and homeostasis. Sustained exposure to oxidative stress has devastating effects in organisms, and, not surprisingly, oxidative stress has been implicated in numerous human diseases. Therefore, an understanding of how mammals respond to oxidative stress is crucial both biologically and clinically. Intracellular signaling pathways, which are activated in response to excessive oxygen radicals, play essential roles in overcoming oxidative stress. The mitogen-activated protein kinase (MAPK) signaling pathways are involved in diverse physiological processes, and are critical for induction of oxidative stress responses. In this review, we will discuss the physiological roles of MAPKs in oxidative stress, the upstream signaling pathways leading to MAPK activation, their regulation, and the MAPK downstream substrates, with a focus on mammalian systems. Antioxid. Redox Signal. 15, 205–218.
Introduction
The MAPK Cascade
The MAPK cascades are intracellular signaling pathways comprising three components, the MAPK, the MAPK kinase (MAPKK or MAP2K), and the MAPKK kinase (MAPKKK or MAP3K), whose functions and structures have been conserved throughout evolution from unicellular organisms, such as yeast, to complex organisms, including mammals (140). MAPKs are activated by numerous cellular stresses and ligand–receptor bindings. They have crucial roles in regulating a number of key cellular processes, such as cell proliferation, survival, differentiation, and death, and have been implicated in various human diseases. The MAPK cascade is initiated by activation of the MAP3K, which is induced by either direct self-activation or upstream signaling. Subsequently, the MAP3K phosphorylates and activates the MAP2K, which in turn phosphorylates and activates the MAPK (Fig. 1). The MAPK phosphorylates serine and threonine residues of substrate proteins, including other protein kinases, transcription factors, phospholipases, and cytoskeletal proteins, which regulate various cellular activities. For all three components of the MAPK cascade, there are several different kinases, and currently 21 MAP3K, 7 MAP2K, and 11 MAPK genes have been identified. The MAP3Ks are activated by various signals, and function as signaling hubs, integrating these signals into activation of specific MAP2K/MAPK signaling pathways (Fig. 2).

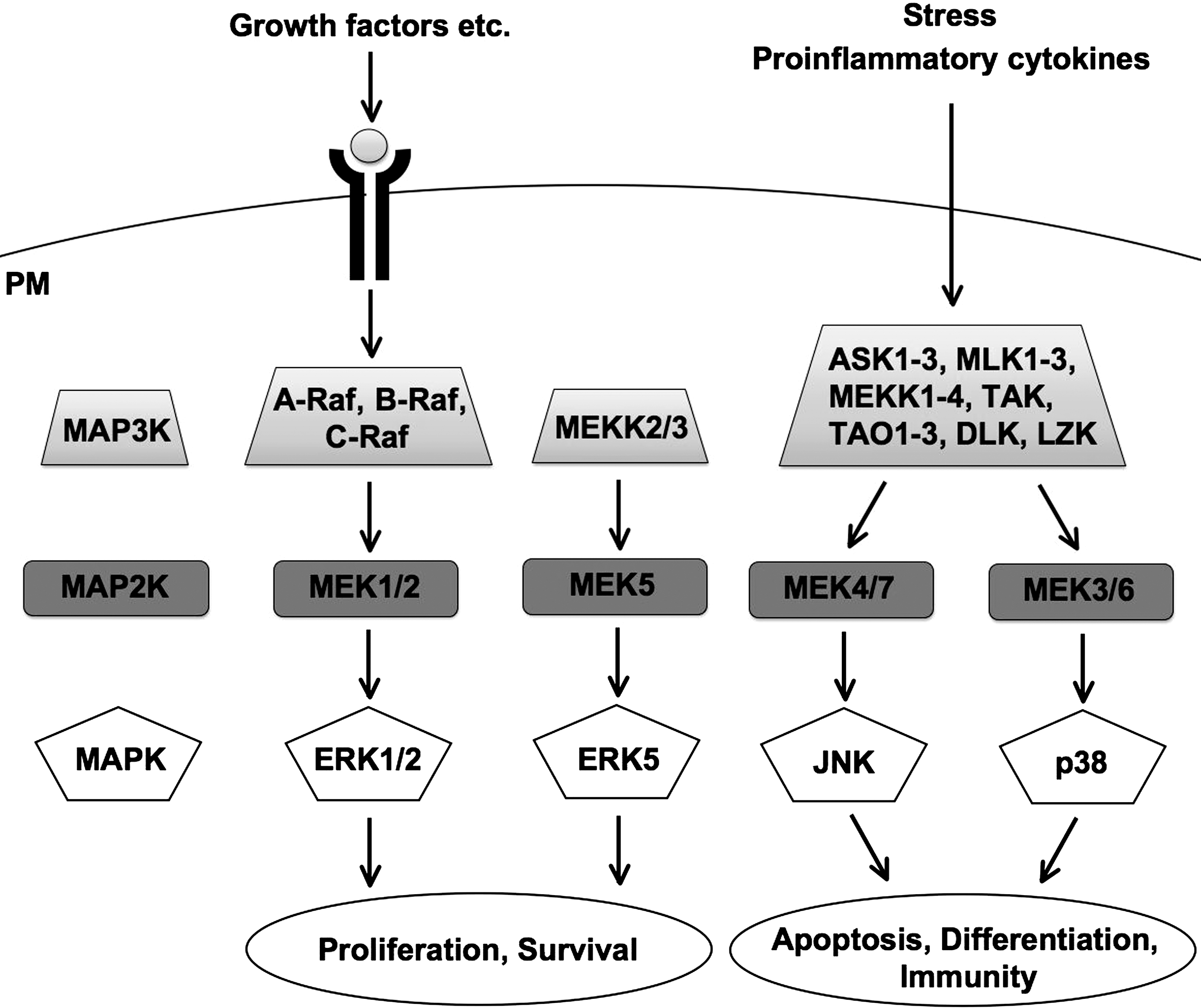
The response of the MAPK cascade to a specific stimulus is often very complex. One stimulus can activate several different MAP3Ks, and each MAP3K can be activated by several different kinds of stimuli. This means that the response to one stimulus is mediated by several different MAP3Ks and that each MAP3K is usually involved in several different cellular responses. Activation of each MAP3K is integrated into a unique pattern of MAPK activation and substrate phosphorylation, leading to a specific cellular response to the stimulus. The MAPK cascade shows further complexity, since it is extensively regulated by various factors, such as kinases and phosphatases, and there is extensive crosstalk between different MAPK and other signaling pathways. The duration and strength of MAPK activation are also important in determining the specific cellular response. This remarkable complexity ensures appropriate cellular responses to a specific stimulus.
H2O2 activates several MAPK signaling pathways, all of which mediate different and specific cellular responses. All of the four conventional MAPK families are activated by H2O2, namely, extracellular signal-regulated kinase 1 (ERK1) and ERK2 (ERK1/2) (91), ERK5/big MAP kinase 1 (BMK1) (2), c-Jun N-terminal kinase (JNK) (85), and p38 (118). The atypical MAPKs, such as ERK3 and ERK4, have not been reported to be activated by H2O2 and will not be considered further in this review.
Extracellular Signal-Regulated Kinases 1 and 2
ERK1 and ERK2 (ERK1/2) were the first MAPKs to be identified and have been extensively characterized due to their diverse physiological roles and critical involvement in a number of diseases. As the name implies (extracellular signal-regulated kinases), activation of the canonical ERK pathway is initiated by ligand binding to receptor tyrosine kinases at the plasma membrane, such as the EGF receptor, leading to activation of the small G protein Ras. Ras in turn activates the MAP3K Raf, which phosphorylates and activates MAP2Ks, MEK1 and MEK2, followed by phosphorylation and activation of ERK1/2. Finally, ERK1/2 phosphorylate a large number of substrates leading to an appropriate cellular response. Several key components of other signaling pathways have also been shown to activate ERK1/2, including other receptor tyrosine kinases (39), Src kinases (143, 153), calcium (14), protein kinase C (PKC) (12), and G protein-coupled receptors (59).
Physiological roles of ERK1/2 in oxidative stress
ERK1/2 have diverse physiological roles, but are mostly known for their important involvement in stimulating cell survival and cell cycle progression. ERK1/2 are usually activated by growth factors or cytokines, but can also be activated by some stresses. ROS is a well-known potent activator of ERK1/2. Several different kinds of ROS can activate ERK1/2 in several cell lines (91), suggesting that ERK1/2 play important roles in oxidative stress responses in diverse tissues. Consistently with their known pro-survival role, ERK1/2 have been shown to have proliferative and protective effects on cells exposed to oxidative stress. Low and adequate concentration of H2O2 is mitogenic (11), which is partly due to activation of ERK1/2. ERK1/2 have also been shown to promote survival and have antiapoptotic effects on cells injured by exposure to oxidative stress (42), and to protect against oxidative stress-induced aging (146). ERK1/2 was thought to only promote proliferation and survival, but recently, it has emerged that ERK1/2 also have pro-apoptotic roles. H2O2-induced apoptosis has been reported to be dependent on ERK1/2 in opossum kidney and human glioma cells (77, 79). Whether ERK1/2 promote cell survival or death is probably in large part cell type specific.
ROS-regulated signaling pathways that activate ERK1/2
H2O2 does not directly activate ERK1/2, but instead induces the activation of upstream signaling components. MEK1/2 are required for ERK1/2 activation, since the treatment of cells with MEK1/2 inhibitors blocks H2O2-induced ERK1/2 activation (77, 79). Several major signaling pathways are activated by ROS, finally leading to the activation of ERK1/2 (Fig. 3). EGF receptor is usually activated by EGF ligand binding, causing receptor dimerization and phosphorylation. The EGF receptor-associated protein SOS functions as a guanine nucleotide exchange factor for Ras, thereby activating it and leading to ERK1/2 activation. H2O2 induces phosphorylation and activation of the EGF receptor independently of EGF ligand (153). In a similar way, platelet-derived growth factor receptor is also activated by H2O2 and induces the activation of ERK1/2 (69). Ras itself can also be a target of ROS. Nitric oxide (NO) has been shown to activate Ras through S-nitrosylation of a cysteine residue of Ras (Cys118) (72). In addition, other oxidants, such as H2O2, S-nitrosoglutathione, diamide, glutathione disulfide, and cystamine, modify reactive cysteines in Ras, and thereby regulate the activity of ERK1/2 (86). Certain members of the Src kinase family have also been shown to be activated by H2O2. H2O2-induced activation of Src leads to the activation of ERK1/2 through different pathways. H2O2-induced activation of EGF receptor has been shown to depend on Src, since a Src inhibitor protein phosphatase (PP)1 inhibits H2O2-induced phosphorylation and activation of EGF receptor and ERK1/2 (153). Src also directly activates C-Raf by phosphorylation of residues Tyr340/341, which is inhibited by treatment with ROS scavengers (143). Fyn, another Src kinase family member, activates JAK2 upon H2O2 stimulation, which induces ERK1/2 activation via the Shc-Ras signaling pathway (1).

Through the activation of several different signaling pathways, H2O2 increases the intracellular concentration of calcium, which is a known activator of ERK1/2 (6). H2O2 induces phosphorylation and activation of phospholipase C (PLC)-γ, which causes cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol and inositol triphosphate (IP3) (124). IP3 induces release of calcium from the endoplasmic reticulum. This PLC-γ activation is further enhanced by Src, which has been shown to phosphorylate and activate PLC-γ in a H2O2-dependent manner (136). ROS also increases the intracellular concentration of calcium by causing flux through calcium channels (70). Several signaling pathways activated by calcium lead to ERK1/2 activation. Calcium and diacylglycerol activate PKC (156), and the increase of intracellular calcium concentration also activates CaM kinases (27, 122) and PYK2 (81), resulting in ERK1/2 activation.
Another mechanism of ERK1/2 activation by ROS is through the regulation of protein phosphatases. H2O2 inhibits several protein phosphatases, such as protein tyrosine phosphatases (PTPs) (116, 139), by the modification of catalytic cysteine residues. H2O2 stimulation inhibited several PTPs, including SHP-1, CD45, and hePTP, and overexpression of all three PTPs caused a reduction in ERK1/2 activation, indicating that ROS-induced inhibition of PTPs leads to ERK1/2 activation (78).
ERK1/2 substrates
All signaling pathways leading to ERK1/2 phosphorylation induce conformational changes in ERK1/2, which allows substrate binding and ERK1/2 translocation to the nucleus, enabling activation of nuclear substrates. ERK1/2 have at least around 160 substrates [see (148) for a detailed review on ERK1/2 substrates] and this number is likely to increase. Since ERK1/2 also activate other kinases, such as ribosomal S6 kinases (RSKs), which have their own specific substrates, the ERK signaling cascade probably involves hundreds of proteins. Approximately half of ERK1/2 substrates are nuclear, whereas the other half are located in the cytosol, the plasma membrane, and cellular organelles. ERK1/2 substrates share a consensus motif, Pro-X-Ser/Thr-Pro, where the Ser/Thr residue is phosphorylated by ERK1/2. The phosphorylation domains of ERK1/2 substrates do not provide sufficient binding affinity to ERK1/2 or substrate specificity, so to ensure this, ERK1/2 substrates usually also have an ERK1/2 binding domain, such as a D- or DEF-domain. Phosphorylation by ERK1/2 induces conformational changes in substrates, which have distinct effects, such as either activating or inhibiting their functions, regulating intracellular localization, or altering protein–protein interactions. In addition, ERK1/2 also regulate substrates in a kinase activity-independent manner. For example, ERK1/2 directly interact with and activate topoisomerase IIα by induction of its conformational change (123). Well-characterized ERK1/2 substrates include transcription factors such as Elk1, c-Fos, and c-Myc, and kinases such as RSKs and MSK1/2 (148). All ERK1/2 substrates are potentially phosphorylated by oxidative stress, but which ERK1/2 substrates are actually phosphorylated under oxidative stress remains poorly characterized. However, two known substrates are AP-1 and Nrf2. The NADPH oxidase gene has been reported to be induced through ERK1/2-dependent AP-1 activation, which is triggered by angiotensin II (Ang II) and TNFα (87). Even though it is not clear whether Nrf2 is a direct target of ERK1/2, it seems to be an important downstream target of the ERK1/2 signaling pathway. Under stable conditions, Nrf2 interacts with the E3 ligase Keap1, which mediates Nrf2 degradation (55, 92). Exposure to oxidative stress induces Nrf2 dissociation from Keap1 and Nrf2 translocation to the nucleus where it upregulates key antioxidant and detoxifying proteins (49, 103). All MAPKs, especially ERK1/2, have been shown to regulate Nrf2; however, the details of how ERK1/2 and other MAPKs regulate Nrf2 remain unknown. The ERK1/2 signaling pathway has been shown to stabilize Nrf2 (105) and to be required for Nrf2 translocation to the nucleus (155), but the molecular mechanism is not clear.
ERK5 (BMK1)
ERK5, also known as BMK1, is a more recently identified MAPK, and is not as well characterized as ERK1/2, JNK, and p38. ERK5 was cloned in a yeast two-hybrid screen using MEK5 as a bait (151) and was found to be ubiquitously expressed, being especially abundant in heart and muscle. The ERK5 signaling pathway is initiated by the MAP3Ks, MEKK2 and MEKK3. These MAP3Ks phosphorylate and activate a MAP2K, MEK5, which in turn phosphorylates ERK5 on threonine and tyrosine residues, thereby activating it.
Physiological roles and signaling pathways of ERK5 in oxidative stress
ERK5 has similar roles to ERK1/2 and is involved in diverse physiological processes, including cell proliferation (65), survival, and differentiation (13). The ERK5 signaling pathway is activated by various factors, such as cellular stress (3), G protein-coupled receptors (89), and growth factors (65). H2O2 strongly activates ERK5 in several different cell lines (2). In H2O2-exposed PC12 cells, inhibition of the ERK5 signaling pathway by MEK5 inhibitor treatment enhanced cell death, suggesting that ERK5 has protective effects on H2O2-induced apoptosis (126).
Several different pathways lead to oxidative stress-induced ERK5 activation (Fig. 4). Src inhibitors and a kinase negative mutant of Src inhibited H2O2-induced activation of ERK5, suggesting that the Src pathway activates ERK5 as well as ERK1/2 in response to oxidative stress (3, 126). Activation of ERK5 induced by H2O2 was shown to be calcium dependent in several cell lines (2), implicating that the ROS–calcium signaling pathway (discussed for ERK1/2) is also involved in ERK5 activation. This suggests that CaM kinases and PYK2 could be potential activators of ERK5. However, PKC, which is also activated by elevated intracellular calcium levels, was shown to inhibit ERK5 in PC12 cells (33), in contrast to ERK1/2. Another redox-sensitive signaling cascade activating ERK5 is mediated by Ang II. Stimulation of Ang II receptor was found to activate ERK5 by inducing NADPH oxidase-dependent ROS production, partially through the transactivation of insulin-like growth factor-1 receptor (IGF-R1) and EGF receptor. Ang II-induced activation of ERK5 was ROS dependent, since treatment with ROS scavengers inhibited ERK5 activation (132).

Several types of proteins, such as phosphatases and kinases, are known to regulate the ERK5 pathway, but their roles in ROS-mediated regulation of ERK5 are not clear. The serine/threonine phosphatases PP1 and PP2A were shown to regulate ERK5 activity in cells exposed to H2O2. Treatment with PP1 and PP2A inhibitors, such as okadaic acid and calyculin, inhibited H2O2-induced activation of ERK5 in HeLa and PC12 cells, suggesting that PP1 or PP2A, or both are required for H2O2-induced activation of ERK5; however, the mechanism of how they mediate this remains unknown (33).
ERK5 substrates
ERK5 substrates are not as well characterized as for other MAPKs, and only around 10 have so far been identified. ERK5 have both nuclear and cytosolic substrates, and translocates to the nucleus upon its activation. The best characterized ERK5 substrates are the transcription factors MEF2A, MEF2C, and MEF2D, whose transcriptional activities are increased by ERK5-dependent phosphorylation (8, 63, 64). Other ERK5 substrates include transcription factors [c-Myc, Sap1, FOXO3a, PPAR-γ1 (5, 26, 61, 135)], kinases [p90RSK, SGK (46, 115)], and Bcl-2 family proteins [Bad (135)]. Phosphorylation by ERK5 has different effects on these substrates. Generally, the activity of proliferative proteins is enhanced, such as c-Myc and SGK, whereas that of pro-apoptotic proteins is inhibited, such as Bad and FOXO3a (135).
Among them, only MEF2C has been shown to be regulated by oxidative stress-activated ERK5 (126). Treatment of PC12 cells with H2O2 induced ERK5 activation, which correlated with enhanced MEF2C binding affinity to DNA. Inhibition of ERK5 activity increased H2O2-induced apoptosis and decreased MEF2C binding affinity to DNA, suggesting that MEF2C phosphorylated by ERK5 have antiapoptotic functions in oxidative stress-exposed cells.
JNK and p38
JNK and p38 are classified together as stress-responsive kinases, which are involved in numerous physiological processes. Since JNK and p38 often have similar physiological roles and are in many cases activated by similar signaling pathways, they will partly be discussed together in this review.
JNKs were identified as stress-activated kinases in cells treated with protein synthesis inhibitors (22, 71). The JNKs comprise three proteins, JNK1/2/3, encoded by three genes, which can be alternatively spliced generating at least 10 isoforms. JNK1 and JNK2 are ubiquitously expressed, whereas JNK3 is highly expressed in brain. The JNK signaling pathway is initiated by activation of several MAP3Ks, including apoptosis signal-regulating kinase (ASK)1/2, MEKK1/2/3/4, mixed lineage kinase (MLK)1/2/3, DLK, and TAK1, which phosphorylate and activate the MAP2Ks, MEK4/7. MEK4/7 phosphorylate JNK on critical threonine and tyrosine residues leading to JNK activation.
p38 was identified in a screen as a target molecule of anti-inflammatory drugs (43, 76). The p38 kinases consist of four proteins, p38α/β/γ/δ. p38α is ubiquitously expressed, whereas p38β, in general, shows low expression and its role in MAPK signaling is unclear. p38γ and p38δ are expressed in specific tissues. p38γ is predominantly expressed in muscle, whereas p38δ is expressed in salivary, pituitary, and adrenal glands, and they both probably have specialized functions. In the literature, p38 mostly refers to p38α. In a similar way to JNK, the p38 signaling pathway is initiated by activation of several MAP3Ks, including ASK1/2, MEKK1, MLK3, and TAK1, which phosphorylate and activate the MAP2Ks, MEK3/6 (41, 50, 95, 128, 129). MEK3/6 phosphorylate and activate p38 (23, 95).
JNK and p38 are activated by a variety of stressors, such as osmotic stress (32, 43), UV irradiation (22, 58), heavy metals (80), and chemotherapeutic drugs (29). Although JNK and p38 are mostly known for being activated by stressors, several receptors have also been shown to activate JNK and p38. Proinflammatory cytokine receptors, especially TNFα receptor and Fas, strongly activate JNK and p38 (30, 60, 73, 138), and growth factor receptors have also been implicated in JNK and p38 activation (40, 44).
Physiological roles of JNK and p38 in oxidative stress
Exposure of cells to excessive stress is potentially very harmful to multicellular organisms, and an effective defense to this is that the inflicted cells undergo apoptosis. JNK and p38 have critical roles in stress-induced cell death and are in many cases required for stress-induced apoptosis. Treatment with JNK and p38-specific inhibitors or expression of dominant negative JNK and p38 mutants suppresses apoptosis induced by various types of stress (130). Especially, the sustained activation of JNK and p38 is important for their ability to induce apoptosis (88). Beside important roles of JNK and p38 in apoptosis, they are also involved in several other physiological processes, such as immunity and cell differentiation (17, 25). JNK and p38 were thought to oppose the proliferative functions of ERK1/2 (144), but recently, JNK and p38 have also been shown to have proliferative effects. It has been reported that p38 has proliferative roles in retinal pigment epithelial cells exposed to oxidative stress (112), and that JNK phosphorylates Nrf2, thereby enhancing its transcriptional activity and inhibiting oxidative stress-induced apoptosis (109).
JNK and p38 are both strongly activated by H2O2, and are critical mediators of oxidative stress-induced apoptosis. These JNK and p38 signaling pathways are initiated and regulated by the upstream MAP3Ks. Among various MAP3Ks, ASK1/2, MEKK1, and MLK3 have been reported to be closely associated with oxidative stress. ASK1/2 activate both JNK and p38, whereas MEKK1 and MLK3 preferentially activate JNK in response to oxidative stress (Fig. 5). In the next section, ASK family proteins will be discussed in detail, since they are critically involved in oxidative stress responses, determining whether or not cells commit to apoptosis.

ROS-regulated signaling pathways of ASK family proteins
The ASK family consists of two members, ASK1/2, which are activated by ROS and mediate downstream activation of JNK and p38, leading to apoptosis (50, 121, 128). The first member to be identified was ASK1, which is ubiquitously expressed in various tissues. H2O2-induced cell death was strongly inhibited in ASK1−/− cells (131), providing direct evidence for the requirement of ASK1 in oxidative stress-induced apoptosis. ASK1 interacts with the antioxidant protein thioredoxin (Trx). This ASK1-Trx complex functions as a redox sensor that initiates the activation of JNK and p38 when cells are exposed to excessive oxidative stress. Under normal conditions, the reduced forms of Cys-32 and −35 in the redox active site of Trx mediate noncovalent binding to the N-terminus of ASK1 and suppress ASK1 kinase activity by inhibiting N-terminal oligomerization between ASK1 molecules (121). When cells are exposed to H2O2, the formation of an intramolecular disulfide bond between Cys-32 and −35 of Trx is induced and this mediates Trx dissociation from ASK1. After this, TRAF2/6 are recruited to ASK1, facilitating ASK1 N-terminus interaction and autophosphorylation of Thr845 (106) (Fig. 6). Activated ASK1 directly phosphorylates and activates MKK4/7 and MKK3/6, leading to JNK and p38 activation. In addition, under H2O2 stimulation, internal cysteine residues of ASK1 are oxidized and form intermolecular disulfide bonds between different ASK1 molecules, which are required for the downstream activation of JNK and execution of apoptosis (101). By its thiol reductase activity, Trx reduces these internal disulfide bonds in ASK1, thereby inactivating it (100).
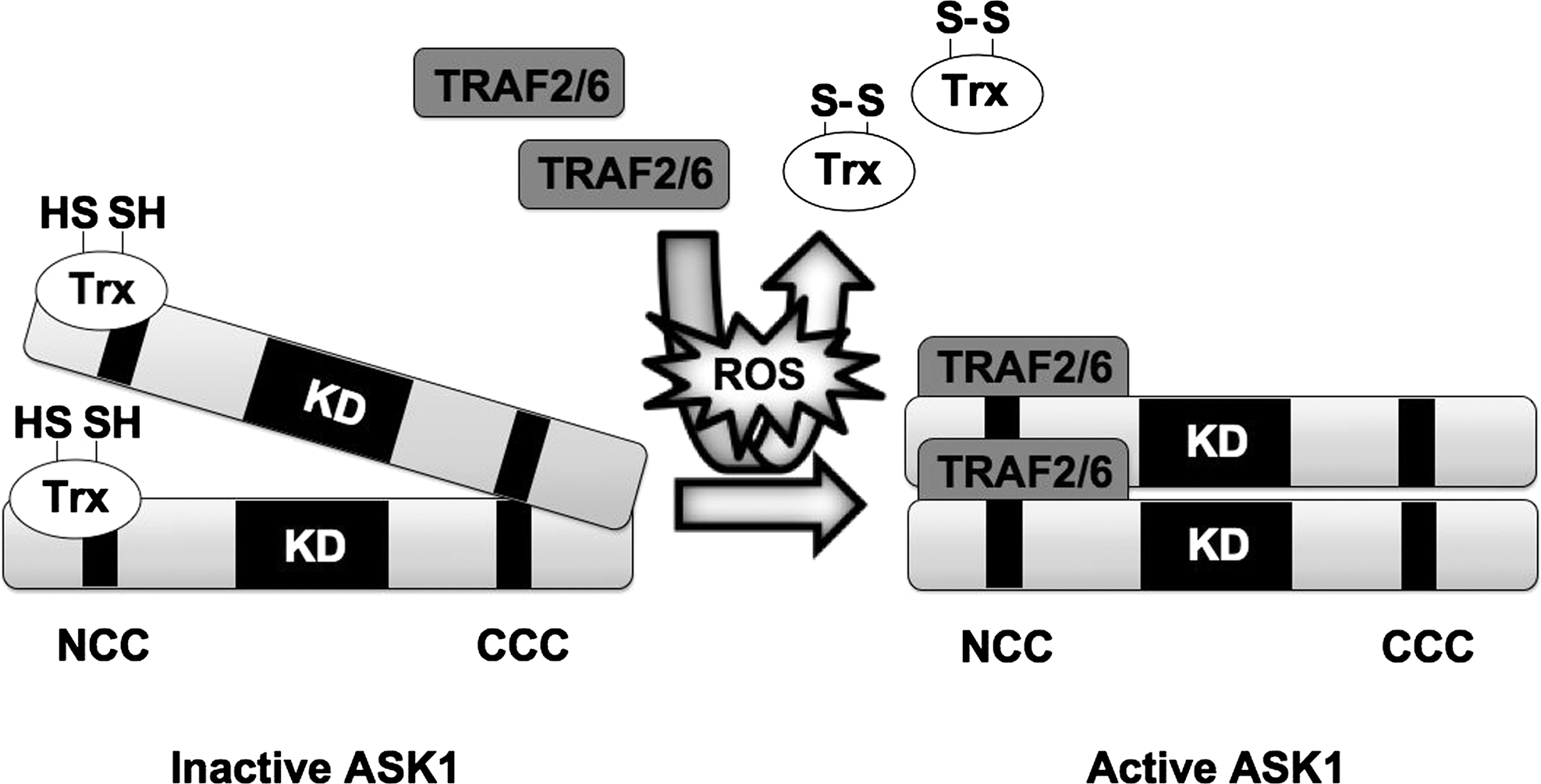
H2O2-dependent activation of ASK1 also plays important roles in receptor signaling. H2O2 has dual functions both upstream and downstream of cytokine and growth factor receptors. It has been shown that ROS induces the activation of several receptors independently of ligand binding (110), and that the stimulation of several cytokine and growth factor receptors generates ROS (84, 107), where it functions as a second messenger in downstream signaling pathways. ASK1 has been shown to be activated by the activation of several receptors in a ROS-dependent manner.
TNFα is a well-known proinflammatory cytokine that has crucial roles in inflammation, proliferation, and cell death. TNFα binds to the TNFα receptor and initiates several intracellular pathways, including the ASK1-JNK/p38 pathway. The activation of TNFα receptor induces ROS production (38), and ROS scavengers inhibit TNFα-induced activation of JNK and p38 (45, 125), suggesting that ROS production is required for TNFα-induced activation of JNK and p38. ASK1 is also required for this activation and apoptosis, since TNFα-induced activation of JNK and p38 and apoptosis are reduced in ASK1−/− cells (131). Upon TNFα stimulation, ASK1 is activated in a similar way to oxidative stress, by ROS-dependent dissociation of Trx from ASK1 and subsequent association of TRAF2 (82, 108).
Toll-like receptors (TLRs) play key roles in innate immunity and are activated by broadly shared pathogenic markers such as pathogen-associated molecular patterns (PAMPs). Lipopolysaccharide (LPS) is one of the PAMPs that bind to and activate TLR4. Activation of TLR4 leads to association of TRAF6 and other adaptor proteins, triggering intracellular signaling cascades, including JNK and p38 pathways (147). LPS ligation to TLR4 was found to induce ROS production, which activated the TRAF6-ASK1-p38 signaling pathway (90). Treatment with ROS scavengers inhibited LPS-induced activation of ASK1 and p38, and prevented association of TRAF6 with ASK1. Probably, LPS-induced ROS production activates ASK1 through the dissociation of Trx from ASK1 followed by the recruitment of TRAF6.
ASK2, a member of the ASK family, has been shown to be a functional binding partner of ASK1, and is also critically involved in oxidative stress-induced apoptosis. ASK2 is highly expressed in tissues that are exposed to the external environment, such as skin, lung, and gastrointestinal tract (54). In contrast to ASK1, ASK2 is intrinsically unstable, and the stability of ASK2 depends on heteromeric complex formation with ASK1 (128). Interestingly, although ASK2 stability depends on ASK1 expression, ASK1 kinase activity is not important for ASK2 activation. When ASK2 was overexpressed with a kinase negative mutant of ASK1, ASK2 was autophosphorylated and induced the activation of p38 and JNK. H2O2 was found to induce activation of ASK2, and ASK2 was required for oxidative stress-induced JNK activation in B16 melanoma cells and apoptosis. ROS generated by carcinogenic chemicals also induced ASK2-dependent apoptosis in keratinocytes (54).
ROS-regulated signaling pathways of other kinases that activate JNK
JNK can also be activated by other oxidative stress-activated pathways. As mentioned before, oxidative stress induces the activation of Ras, which leads to activation of Raf and subsequently ERK1/2. However, oxidative stress-activated Ras have also been shown to activate the MAP3K MEKK1. Ras-activated MEKK1 phosphorylates MEK4/7, leading to JNK activation (56). The MEKK1-JNK pathway cooperated with and positively regulated the nuclear factor kappa B signaling pathway, suggesting that, in this case, JNK has proliferative roles. However, MEKK1 can also be negatively regulated by oxidative stress. Menadione-induced oxidative stress inhibited MEKK1 activity by inducing glutathionylation of a crucial residue in the ATP binding domain of MEKK1, Cys1238, thereby probably inhibiting ATP binding (18). Another MAP3K activated by ROS is MLK3. Treatment with genipin, an antidiabetes drug, induced ROS production via NADPH oxidase, and activated the MLK3-JNK pathway (48). Transfection with a dominant negative mutant of MLK3 inhibited genipin-induced JNK activation, indicating that MLK3 is required for the genipin-ROS-JNK pathway. A JNK inhibitor suppressed genipin-induced cell death, suggesting that the MLK3-JNK pathway is pro-apoptotic. PKC also has several roles in the activation of JNK. TPA-induced ROS production was found to depend on PKC, and in PKC-deficient cells, the activation of MEKK1 and JNK was abolished (20). PKC also has more direct functions in oxidative stress-induced JNK activation. Ethanol-induced ROS production caused PKC activation, leading to PKC-dependent degradation of MAPK phosphatase 1. This resulted in sustained activation of JNK, which promoted apoptosis in hepatocytes (90).
Regulation of ASK1-JNK/p38 signaling pathway in oxidative stress
The ROS-ASK1-JNK/p38 signaling axis is extensively regulated by different types of proteins, such as kinases and phosphatases. Especially, ASK1 is strictly regulated, perhaps due to its critical role as a redox–sensor.
Autophosphorylation of ASK1 at Thr845 is critical for ASK1 activation, and several phosphatases have been shown to dephosphorylate ASK1 at this residue, thereby negatively regulating the ASK1-JNK/p38 signaling pathway. The threonine/serine phosphatases, PP5 and PP2Cɛ, negatively regulate ASK1 activation. PP5 recognizes H2O2-activated ASK1 and, in a negative feedback mechanism, dephosphorylates Thr845, inhibiting ASK1 activation in response to H2O2 (96). PP2Cɛ was found to inhibit basal activity of ASK1. Under basal conditions, PP2Cɛ interacted endogenously with ASK1 and retained ASK1 in its inactive form by dephosphorylation of Thr845. Exposure to H2O2 induced the dissociation of PP2Cɛ from ASK1, thereby increasing autophosphorylation and activity of ASK1 (120).
ASK1 activity is also regulated by phosphorylation of other residues than Thr845. The 14-3-3 protein family is composed of highly conserved phospho-threonine/phospho-serine binding proteins that are involved in regulation of diverse intracellular signaling pathways (97). Under stable conditions, 14-3-3 proteins bind to ASK1 by recognizing phosphorylated Ser966, and thereby inhibiting ASK1 kinase activity (150). H2O2-induced dephosphorylation of ASK1 at Ser966 correlated with the dissociation of 14-3-3 proteins from ASK1, resulting in enhanced ASK1 activity (37). PP2B was found to be responsible for dephosphorylation of ASK1 at Ser966 and subsequent dissociation of 14-3-3 proteins (83). 14-3-3 Proteins can also dissociate from ASK1 in other ways. Recently, Ste20/oxidant stress response kinase (SOK) 1 has been shown to phosphorylate 14-3-3ζ at Ser58 in an oxidative stress-dependent manner, thereby inducing the dissociation of 14-3-3ζ from ASK1 (152). Phosphoglycerate mutases (PGAMs) are involved in glycolysis, but recently, it was shown that PGAM family member, PGAM5, regulates ASK1 activity. PGAM5 interacted endogenously with ASK1 and dephosphorylated ASK1 at inhibitory phosphorylation sites, leading to ASK1 activation (127). The Akt/PKB family consists of threonine/serine kinases that promote cellular proliferation and survival by inactivating pro-apoptotic proteins, such as Bax and Bad (21, 34), via their direct phosphorylation. Akt was shown to interact with ASK1 and induced phosphorylation of ASK1 at Ser83 (68). The phosphorylation of Ser83 decreased oxidative stress-induced activation of ASK1 and apoptosis.
ASK1 is also regulated by other types of proteins. It has been reported that DJ-1 is involved in the progression of early onset familial Parkinson's disease, and has cytoprotective effects, which in part are due to its antioxidant activity. DJ-1 was found to interact directly with ASK1 and negatively regulate ASK1 activation. Overexpression of DJ-1 inhibited H2O2-induced activation of ASK1 by preventing ASK1 homo-oligomerization (94). Further, DJ-1 has been shown to block dissociation of Trx from ASK1, thereby retaining ASK1 in its inactive configuration (51). Recently, the deubiquitinating enzyme USP9X was found to be a critical regulator of ASK1 activation during oxidative stress (102). USP9X interacted with ASK1 in a H2O2-dependent manner and prevented ubiquitin-mediated degradation of ASK1. In USP9X−/− cells, H2O2-induced JNK activation and apoptosis were reduced, suggesting that USP9X enhances the apoptotic response of ASK1 by inhibiting its ubiquitin-dependent degradation.
Other components of the ASK1-JNK/p38 pathway are also regulated by oxidative stress. Several proteins have been shown to bind to and regulate p38 and JNK. The antioxidant protein peroxiredoxin I was shown to be required for p38 activation in H2O2-treated macrophages (16). Another antioxidant protein, glutathione S-transferase Pi, was found to bind to JNK and inhibit its activity under normal conditions (4). H2O2 induced the dissociation of glutathione S-transferase Pi from JNK, allowing JNK activation. As mentioned earlier, sustained activation of JNK is required for stress-induced apoptosis. JNK-inactivating phosphatases were found to be inhibited by ROS, resulting in sustained JNK activation and cell death (62). Ferritin heavy chain, an iron storage factor, was found to inhibit TNFα-induced ROS production, thereby blocking sustained activation of JNK and cell death (111). Similar to ERK1/2, JNK and p38 pathways are also regulated by H2O2-induced inhibition of PTPs, since overexpression of PTPs inhibits JNK and p38 activation by H2O2 stimulation (78).
JNK substrates
JNK has at least around 60 substrates (9), and just as for ERK1/2, approximately half of the substrates are nuclear, whereas the other half are located in the cytosol. In fact, many of the details discussed in the section of ERK1/2 substrates also apply to JNK, including an identical phosphorylation consensus motif. However, the binding domain of JNK substrates to JNK is different from that of ERK1/2 substrates, and is termed JNK binding domain. The effect of JNK phosphorylation on substrates is also similar to ERK1/2 effects. Theoretically, all JNK substrates are candidates for JNK phosphorylation induced by oxidative stress, and in contrast to ERK1/2, several substrates have been shown to be phosphorylated by oxidative stress-activated JNK (Fig. 7).
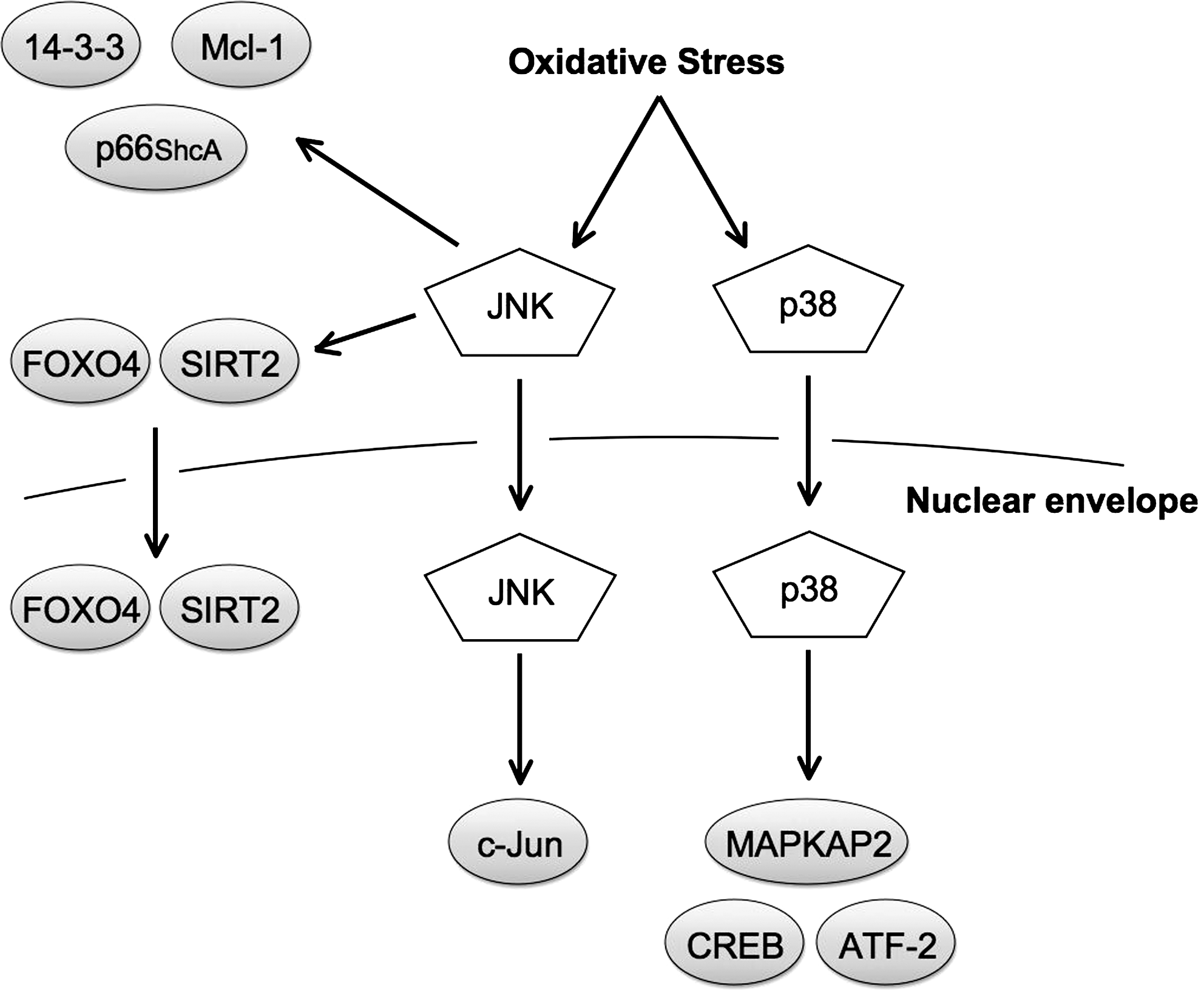
JNK has been shown to regulate several pro-apoptotic and anti-apoptotic proteins in oxidative stress. c-Jun is one of the best characterized substrates of JNK. c-Jun functions as an transcription factor, which regulates cell proliferation and death, and forms the AP-1 complex together with other transcription factors, such as c-Fos and ATF-2. c-Jun has been shown to be involved in oxidative stress-induced cell death. In hepatocytes exposed to menadione-induced oxidative stress, JNK activation and cell death were increased. Adenoviral expression of a dominant negative mutant of c-Jun blocked menadione-induced apoptosis, suggesting that c-Jun is required for oxidative stress-induced apoptosis in hepatocytes (19). JNK regulates the pro-apoptotic adaptor protein p66ShcA, which has important roles in oxidative stress-induced cell death. Mice lacking p66ShcA showed decreased sensitivity to oxidative stressors, such as H2O2 and UV irradiation, suggesting that p66ShcA is required for oxidative stress-induced cell death (93). Further, pro-apoptotic signals induced ROS production in the mitochondria, which depended on p66ShcA (36). Phosphorylation of p66ShcA at Ser36 was shown to be critical for oxidative stress-induced cell death, and JNK was identified as the responsible kinase for Ser36 phosphorylation in response to UV irradiation (75). c-Abl is a pro-apoptotic tyrosine kinase, which is indirectly regulated by JNK. Nuclear c-Abl is activated by diverse genotoxic agents and induces apoptosis. Under stable conditions, c-Abl is localized in the cytosol and binds to 14-3-3 proteins. Oxidative stress-activated JNK phosphorylates 14-3-3 proteins, which are dissociated from c-Abl, allowing c-Abl translocation to the nucleus (149). JNK also regulates anti-apoptotic proteins. Mcl-1, an anti-apoptotic member of the Bcl-2 family, was found to be phosphorylated on Ser121 and Thr163 by JNK in H2O2-stimulated cells (53). Transfection with an unphoshorylatable Mcl-1 mutant decreased oxidative stress-induced apoptosis compared to Mcl-1 wild-type, indicating that JNK-mediated phosphorylation inhibits Mcl-1 anti-apoptotic activity.
As discussed earlier, under some conditions, JNK also has anti-apoptotic roles in response to oxidative stress. SIRT1 is a nicotinamide adenine dinucleotide-dependent deacetylase that is involved in cellular protection against stress and apoptosis. SIRT1 deacetylates a number of nuclear substrates, including histones, FOXOs, PGC-1α, and p53. JNK was shown to interact with and induce phosphorylation of SIRT1 in an oxidative stress-dependent manner (104). JNK-mediated phosphorylation induced SIRT1 translocation to the nucleus and increased its enzymatic activity. This result partly accounts for the anti-apoptotic response of JNK. The transcription factors of the FOXO protein family have also been implicated in cellular protection against oxidative stress, by the upregulation of antioxidants such as MnSOD and catalase. H2O2-activated JNK was found to phosphorylate FOXO4 on Thr447 and Thr451 and facilitate FOXO4 translocation to the nucleus and an increase in transcriptional activity, thereby enhancing the protective response to oxidative stress (28). Similar to ERK1/2, JNK seems to regulate Nrf2 activity. Expression of JNK increased transcriptional activity of Nrf2, which was inhibited by overexpression of a JNK dominant negative mutant (66). JNK was also found to induce translocation of Nrf2 to the nucleus (145). In addition, JNK phosphorylated c-Jun interacts with Nrf2, which induced transcription of several antioxidant genes (57, 134).
p38 substrates
p38 has numerous and diverse substrates, and the regulatory mechanisms of p38 substrates share most of the characteristics discussed in the section of ERK1/2 and JNK substrates. p38 substrates specifically activated by oxidative stress are poorly characterized, but both pro- and antiapoptotic proteins have been identified (Fig. 7).
The transcription factors CREB and ATF-2 are among the best characterized substrates of p38, and both have been shown to be phosphorylated by p38 under oxidative stress conditions. Cadmium-induced oxidative stress resulted in sustained activation of p38 and phosphorylation of CREB and ATF-2 (119). p38 inhibitors blocked phosphorylation of CREB and ATF-2 and cadmium-induced cell death, suggesting that p38 mediates oxidative stress-induced apoptosis in part by the activation of CREB and ATF-2. Another well-characterized substrate of p38 is the kinase MAPKAPK2. Treatment of neonatal rat ventricular myocytes with H2O2 induced MAPKAPK2 phosphorylation. Activated MAPKAPK2 subsequently phosphorylated the small heat shock protein HSP25/27, which may be important for cardioprotection. These effects were blocked by p38 inhibitors, suggesting a protective effect of p38 on myocytes exposed to oxidative stress (15). The p38 pathway also regulates Nrf2. p38 has been reported to be required for Nrf2 binding to promoters by an unknown mechanism (154). However, p38 has also been shown to inhibit Nrf2 activity by inducing formation of the Nrf2 and Keap1 complex (67), thereby inhibiting Nrf2 nuclear translocation. Whether p38 activates or inhibits Nrf2 is probably both cell type and cell context specific.
Conclusion
ROS is produced by numerous chemicals and stresses and plays roles in diverse pathologies. Especially in cancer cells, ROS seems to be a key player in mediating tumor development by diverse mechanisms. Recently, MAPKs were shown to have crucial roles in inhibiting the oncogenic potential of ROS by inducing apoptosis (24). This emphasizes the importance of understanding the effects of ROS and their link to MAPKs in both normal and pathological conditions.
The big question is how cells determine the appropriate response to a given ROS stimuli. H2O2 functions as a proliferatory signal, a second messenger, or as a signal for apoptosis by the activation of different MAPKs. When cells are exposed to H2O2, often all MAPKs are activated and send conflicting signals to determine cell fate. This necessitates strict regulation to ensure that inappropriate responses are avoided. Low concentrations and local cellular distribution of ROS mediates its role as a second messenger, whereas high concentrations of ROS through out the cell mediate its role as a death signal. Perhaps by keeping the ROS levels low and local, the cell ensures that the ROS-induced apoptotic stimuli by activation of p38 and JNK are not sufficient to initiate the apoptotic program. As ROS levels increase, a certain threshold is reached, and apoptosis is induced. In the end, what determines the cell response is the availability of MAPK substrates, and this is largely cell type specific. Which substrates are specifically activated by ROS in different cells remains poorly characterized and will be an important future area of research.
Footnotes
Acknowledgments
We thank all the members of Cell Signaling Laboratory for their critical comments. This work was supported by grants-in aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation, and Global Center of Excellence Program.