
Abstract
Most antigenic peptides are generated by proteasomes in the cytosol and are transported by the transporter associated with antigen processing (TAP) into the endoplasmic reticulum, where they bind with nascent major histocompatibilitiy complex class I molecule (MHC-I). Although the overall process of peptide–MHC-I complex assembly is well studied, the mechanism by which free peptides are delivered from TAP to MHC-I is unknown. In this study, we investigated the possible role of protein disulfide isomerase (PDI) as a peptide carrier between TAP and MHC-I. Analysis of PDI–peptide complexes reconstituted in vitro showed that PDI exhibits some degree of specificity for peptides corresponding to antigenic ligands of various human leukocyte antigen (HLA) alleles. Mutations of either anchor residues of the peptide ligand or the peptide-binding site of PDI inhibited the PDI–peptide interaction. The PDI–peptide interaction increased under reducing conditions, whereas binding of the peptide to PDI decreased under oxidizing conditions. TAP-associated PDI was predominantly present in the reduced form, whereas the MHC-I-associated PDI was present in the oxidized form. Further, upon binding of optimal peptides, PDI was released from TAP and sequentially associated with HLA-A2.1. Our data revealed a redox-regulated chaperone function of PDI in delivering antigenic peptides from TAP to MHC-I. Antioxid. Redox Signal. 15, 621–633.
Introduction
Optimization of MHC-I-bound peptide is the most crucial process for enhancing MHC-I structural stability and thereby presenting a strong signal to the immune system. Because peptide binding of individual MHC-I is selectively controlled by allele-specific peptide-binding motifs, most peptides in the ER are presumably suboptimal for binding to MHC-I (41). Quantitative analysis shows that only 0.05% of peptides are available for MHC-I loading (27, 29, 40). Additionally, many peptides confront cytosolic peptidases before they are translocated across the ER membrane (43). Excess peptides that cannot be loaded onto MHC-I are exported from the ER into the cytosol through the Sec61p channel (22). To overcome these constraints, cells utilize the cellular machinery to improve the stability and specificity of peptides both in the cytosol and in the ER. TRic and Hsp90, well-known cytosolic chaperones, protect both antigenic peptides and their precursors from cytosolic amino-peptidase activity and ensure their safe delivery to the TAP heterodimer (24). The TAP heterodimer has a relatively broad spectrum of substrate specificity for peptides transported into the ER according to their lengths and sequences (28). Similarly, in the ER, the “editing” mechanism of the tapasin-ERp57 heterodimer plays a crucial role in regulating optimal peptide loading onto MHC-I (19, 59). Further, ER-aminopeptidase-1 (ERAP1/ERAAP) trims the peptide precursors to a size suitable for MHC-I binding (47, 49, 61).
Some ER proteins, including calreticulin, gp96, ERp72, grp170 and PDI, are thought to be associated with antigenic peptides translocated into the ER by TAP, but their functions in MHC-I antigen presentation are unclear (26, 31, 52, 53). For example, gp96 associates with peptides and induces MHC-I antigen presentation via the endocytic pathway in antigen presenting cells (4, 5). However, the putative role of gp96 as a peptide carrier in the ER has been received with some skepticism because reduced gp96 expression does not influence the level of MHC-I on the cell surface (42).
PDI is a classical member of the ER oxidoreductase family (13) and catalyzes the oxidation, reduction and isomerization of disulfide bonds in newly synthesized or misfolded proteins (48). PDI consists of four distinct domains (a-b-b′-a′) with a C-terminal KDEL ER retention sequence. The a and a′ domains contain a characteristic CXXC motif that catalyzes disulfide bond formation. The b′ domain is the principal binding site for small peptides containing 10–15 amino acids (12, 18, 21, 23). The b domain has no catalytic activity but may be involved in substrate binding (8). Our previous study has shown that PDI facilitates optimal peptide loading by regulating the oxidation state of the disulfide bond in the MHC-I peptide-binding cleft (36). Interestingly, ectopic expression of a PDI-F258W/I272A mutant, in which the catalytic site is intact but the peptide-binding activity of the b′ domain is destroyed (39), failed to restore optimal peptide loading of MHC-I in endogenous PDI-depleted cells (36). These observations suggest that the peptide-chaperoning activity of the b′ domain of PDI is also essential for optimal peptide loading. However, we were unable to separate this potential additional function of the b′ domain from the catalytic function of PDI in the previous study because of apparent crosstalk between these two functions in a cellular context. To overcome this limitation and to exclusively test the role of the peptide-chaperoning activity of PDI in antigen processing, we utilized in vitro reconstitution of the PDI–peptide complex and conducted an in vivo analysis. We report that PDI can serve as a peptide carrier for delivering peptides from TAP to MHC-I.
Materials and Methods
Cell lines and antibodies
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Hyclone), penicillin (50 U/ml), and streptomycin (50 μg/ml). The human B lymphoblastoid cell line, 721.220, stably expressing HLA-A2.1 was generated by infection with a retroviral system and maintained in RPMI-1640 medium (Hyclone) with 0.5 mg/ml of G418 (Calbiochem). The mAb BB7.2 recognizes HLA-A2.1 (34). We raised rabbit polyclonal antibodies against the recombinant PDI protein, tapasin (C-terminus region including the KDEL sequence as an epitope), and TAP1 (nucleotide-binding domain as an epitope). We purchased antibodies for ERp57 (Santa Cruz Biotechnology), MHC class I (Santa Cruz Biotechnology), and Calreticulin (Stressgen).
Site-directed mutagenesis
Isoleucine-to-alanine (I272A) and phenylalanine-to-tryptophan (F258W) replacement mutations within the b′ domain of PDI and cysteine-to-serine (C36, C39, C357, and C360) mutations in the a and a′ domains of PDI were made by site-directed mutagenesis with Pfu DNA Polymerase (Stratagene).
Protein expression and purification
For bacterial recombinant protein expression, cDNA corresponding to mature polypeptide (except for the signal sequence) of human PDI wild-type or PDI point mutants were cloned into the recombinant protein expression vector pET28a (Novagen). This vector was transformed into Escherichia coli strain BL21 (DE3) (Novagen). Cells were grown to an OD600 of approximately 0.5 in LB medium with 1 mg/ml kanamycin at 37°C. Expression of recombinant PDI wild-type and point mutants was induced by 1 mM isopropyl-β-
Peptides
Peptides were synthesized by Fmoc solid phase peptide synthesis (SPPS) using ASP48S (Peptron, Inc.) and purified by reverse-phase high-performance liquid chromatography (HPLC) using a Vydac Everest C18 column (250 mm×22 mm, 10 μm). Elution was carried out with a water–acetonitrile linear gradient (10%–75% [v/v] of acetonitrile) containing 0.1% (v/v) trifluoroacetic acid. Molecular weights of the purified peptides were confirmed using liquid chromatography/mass spectrometry (LC/MS; Agilent HP1100 series; Agilent). Peptides were biotinylated or conjugated with fluorescein isothiocyanate (FITC) at the N-terminus for detection and contained a lysine residue for crosslinking at position 4.
Crosslinking
Crosslinking was performed using the homobifunctional crosslinker DSG (Pierce). Wild-type PDI and PDI mutants were incubated with the biotinylated peptide in phosphate-buffered saline (PBS) for 10 min on ice followed by further incubation with added DSG (a final concentration of 10 mM) for 60 min on ice. The crosslinking reaction was stopped by boiling the samples in a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. The sample contents were separated by 8% SDS-PAGE and subjected to an immunoblot assay. The biotinylated peptide–protein complex was probed with horseradish peroxidase (HRP)–streptavidin (Pierce). Triton X-100 (Sigma–Aldrich) and β-estradiol (Sigma–Aldrich) were used for characterization of the PDI–peptide interaction. β-Estradiol is a known inhibitor of PDI, targeting the b′ domain and decreasing the thermostability of MHC-I (36, 57).
Metabolic labeling and reimmunoprecipitation
For analysis of the redox state of PDI in association with TAP or HLA-A2.1, 721.220-A2.1 cells (5×106) were starved for 40 min in medium lacking methionine/cysteine and labeled with 0.1 mCi/ml [35S]methionine/cysteine (NEN) for 20 min. Cells were lysed using 1% digitonin (Calbiochem) in lysis buffer [25 mM HEPES, 100 mM NaCl, 10 mM CaCl2, 5 mM MgCl2 (pH 7.6), and 10 mM N-ethylmaleimide supplemented with protease inhibitors] for 30 min on ice. After preclearing cell lysates with protein G-Sepharose (GE Healthcare UK Ltd.), primary antibodies for TAP or HLA-A2.1 and protein G-Sepharose were added to the supernatant and incubated on ice with rotation for 2 h. The beads were washed three times with 0.1% digitonin in lysis buffer. Proteins were eluted from the beads by boiling the samples in reimmunoprecipitation buffer (1% NP-40, 1.5% SDS, and 10 mM N-ethylmaleimide in PBS), and the eluted proteins were diluted into 1% NP-40 in PBS and reprecipitated with the anti-PDI antibody. Precipitated PDI was eluted by reducing or nonreducing SDS sample buffer and separated by 12% SDS-PAGE. The gels were dried, exposed to BAS film for 14 h, and analyzed using the Phosphor Imaging System BAS-2500 (Fuji Film).
Coimmunoprecipitation and immunoblot analysis
Cells or microsomes were lysed in 1% digitonin in buffer containing 25 mM HEPES, 100 mM NaCl, 10 mM CaCl2, and 5 mM MgCl2 (pH 7.6) supplemented with protease inhibitors. Lysates were precleared using protein G-Sepharose for 1 h on ice. For coimmunoprecipitation, samples were incubated with the appropriate antibodies for 2 h on ice before protein G-Sepharose beads were added. Beads were washed four times with 0.1% digitonin and bound proteins were eluted by boiling the samples in SDS sample buffer. Proteins were separated by 12% SDS-PAGE, transferred to an immobilon-P membrane (Millipore), blocked with 5% skim milk in PBS containing 0.1% Tween 20 for 2 h, and probed with the appropriate antibodies overnight at 4°C. Membranes were washed three times with PBS containing 0.1% Tween 20 and incubated with HRP-conjugated streptavidin (Pierce) for 1 h at 4°C. Immunoblots were visualized using an ECL detection reagent (Pierce).
Microsome purification
Microsomes from cells expressing HLA-A2.1 were prepared and purified as previously described (35). Cells expressing HLA-A2.1 were washed once with PBS and harvested by centrifugation. Cells were resuspended to a concentration of 2×108 cells/ml in a cavitation buffer containing 0.25 M sucrose, 25 mM potassium acetate, 5 mM magnesium acetate, 0.5 mM calcium acetate, 50 mM Tris–HCl (pH 7.4), 5 μg/ml leupeptin and 0.25 mM PMSF. Then, cells were homogenized by 16 passages through a 26-gauge needle. Unbroken cells and nuclei were removed by centrifugation at 1500 g for 10 min at 4°C. The resulting supernatants were ultracentrifuged at 100,000 g for 1 h at 4°C. Membrane pellets were resuspended in the cavitation buffer, snap-frozen in liquid nitrogen and stored at −80°C until use.
Peptide binding assay and UV crosslinking
Biotinylated peptides ILDKFPVTV (high-affinity ligand for HLA-A2.1) and IEDKFPVTD (low-affinity ligand for HLA-A2.1) were conjugated to the photoreactive crosslinker N-5-azido-2-nitrobenzoyloxysuccinimide (ANB-NOS; Pierce) as described previously (36). For the peptide-binding assay, reporter peptides were mixed with 15 μl of microsomes (concentration of 60 A280/ml) in a total volume of 50 μl of RM buffer (250 mM sucrose, 50 mM triethanolamine–HCl, 50 mM potassium acetate, 2 mM magnesium acetate, 1 mM DTT, and 10 mM ATP). The mixture was incubated for 30 min at 26°C in a flat-bottom 96-well tissue culture plate. Samples were maintained on ice during a 3-min exposure to shortwave (365 nm) UV irradiation. After centrifugation, membranes were washed once with cold RM buffer, lysed with 1% digitonin, and the crosslinked proteins were immunoprecipitated with primary antibodies. Precipitates were separated by 12% SDS-PAGE followed by detection using immunoblot. Peptide translocation was determined after incubating microsomes with FITC-conjugated reporter peptides under the same experimental conditions. Microsomal membranes were recovered by centrifugation at 75,000 g for 10 min through a 0.5 M sucrose cushion in cold RM buffer. After washing with cold RM buffer twice, the membrane pellet was directly dissolved in sample buffer. Samples contents were separated using tricine/SDS-PAGE and probed with HRP–streptavidin. Relative densities of the peptide bands were determined using an imaging densitometer (GS-700; Bio-Rad) and Multianalyst densitometer software (Bio-Rad).
Results
PDI specifically interacts with peptides through its b′ domain
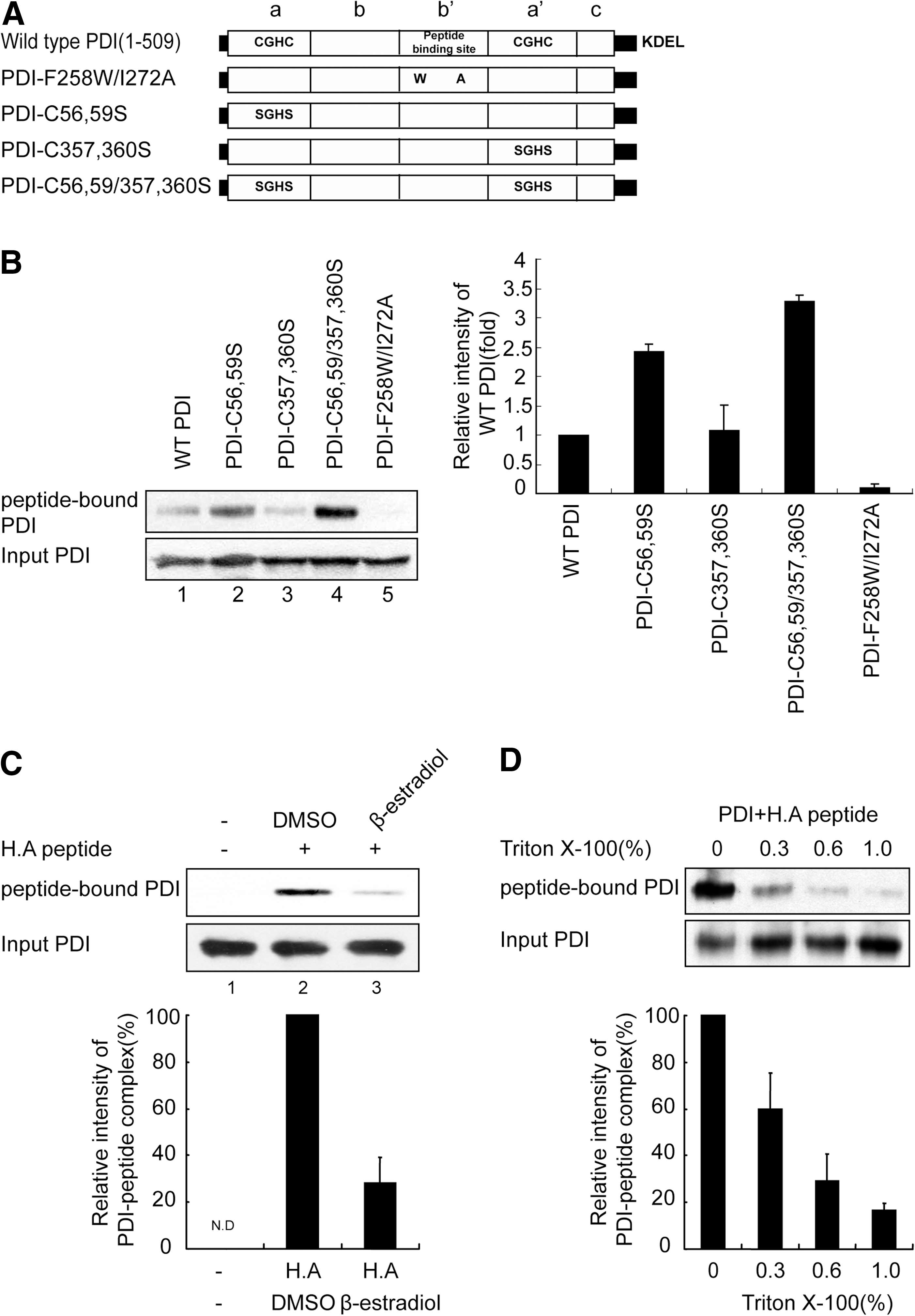
We previously showed that the b′ domain of PDI, a binding site for substrates such as small peptides that are 10–15 amino acids in length (21), is also involved in optimal peptide loading to MHC-I (36), suggesting that PDI may serve as a peptide carrier in antigen processing. To test this hypothesis, we first determined whether PDI binds antigenic peptides. We constructed several PDI proteins with point mutations in the two active site domains (PDI-C56, 59S; PDI-C357, 360S; and PDI-C56, 59/357, 360S) or in the b′ substrate-binding domain (PDI-F258W/I272A) and purified the recombinant proteins (Fig. 1A and Supplementary Fig. S1A; Supplementary Data are available online at
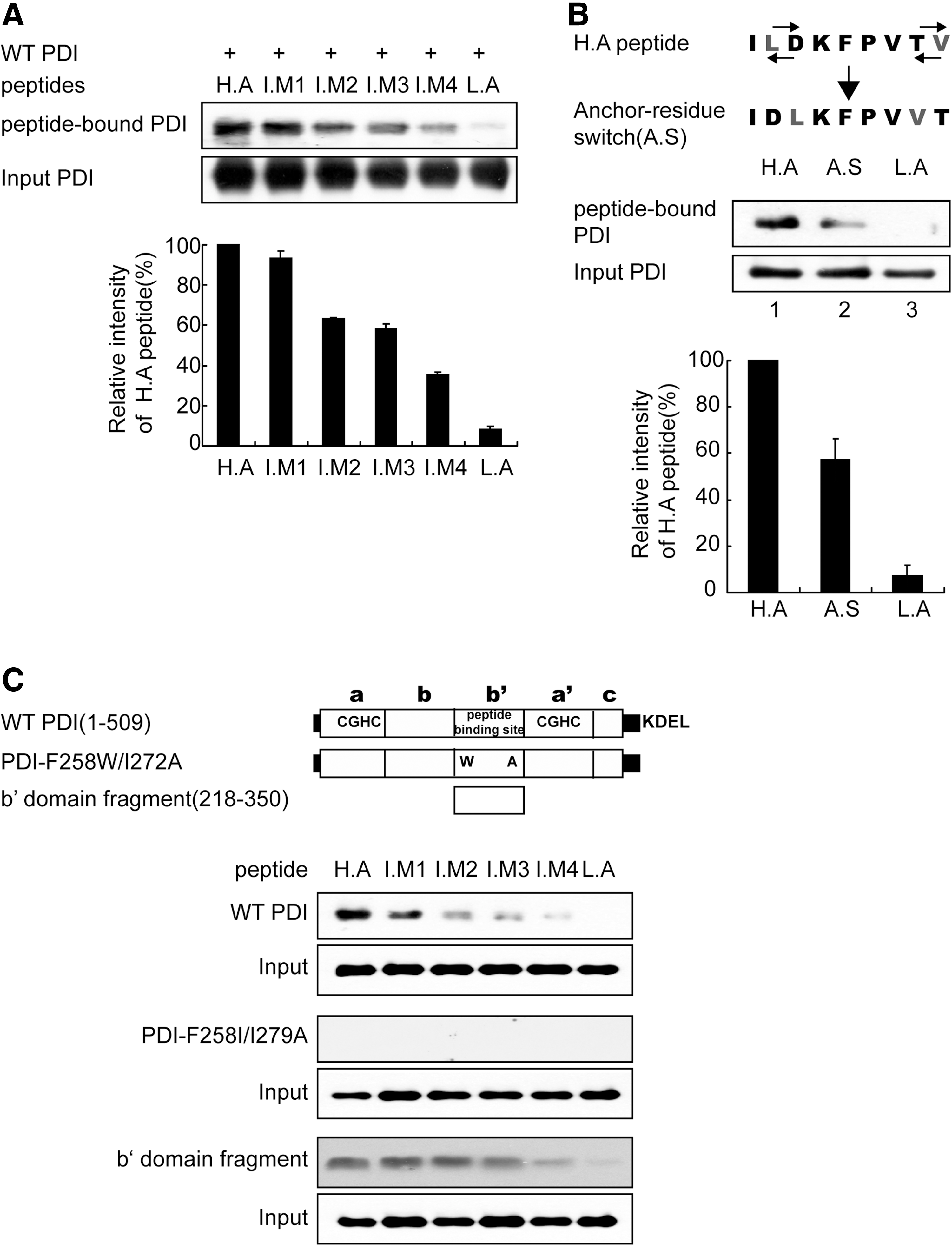
ILDKFPVTV represents high-affinity peptide for HLA-A2.1 and its anchor residues at the P2 and P9 positions are indicated by bold letters. Mutations on anchor residues to reduce the binding affinity for HLA-A2.1 are underlined in bold letters. All peptides possessed a lysine residue for crosslinking with homobifunctional crosslinker disuccinimidyl glutarate or photoreactive-crosslinker N-5-azido-2-nitrobenzoyloxysuccinimide (ANB-NOS).
Representative of binding affinity of each peptide to HLA-A2.1. The affinity of each peptide was estimated from a previous study (10).
Sum of hydrophobicity of amino acids in each peptide with the Eisenberg scale (54).
To examine the interaction between PDI derivatives and peptides, PDI derivatives were incubated in the presence of ILDKFPVTL peptide (H.A peptide, a high-affinity binder to HLA-A2.1) and were chemically crosslinked with disuccinimidyl glutarate (DSG) followed by immunoblotting with HRP–streptavidin for detection of the biotinylated peptide–PDI complex. An apparent size of 60 kDa that corresponded to the size of the PDI–peptide complex was detected for wild-type PDI (Fig. 1B, lane 1), indicating that PDI can bind to the ligand of HLA-A2.1 as well as to relatively long peptide substrates (21). Interestingly, active site mutants displayed differential affinities for peptide, and the C56, 59/357, 360S double mutant exhibited the highest degree of association (Fig. 1B, lanes 2–4). Because mutation of the active site cysteine mimics reduction of the active site, this result suggests that PDI interaction with peptides is regulated by the redox state of PDI. No interaction was observed for the F258W/I272A peptide-binding site mutant (Fig. 1B, lane 5), confirming the previous findings (36, 39) that the b′ domain of PDI is crucial for small peptide binding. This result was further confirmed by the observation that β-estradiol, a selective inhibitor that targets the b′ domain of PDI (57), inhibited the association of wild-type PDI with peptide (Fig. 1C). Interaction of PDI with peptide decreased in the presence of Triton X-100 in a concentration-dependent manner (Fig. 1D). Virtually no PDI–peptide complex remained in 1% Triton X-100, suggesting that PDI associates with antigenic peptide primarily through a hydrophobic interaction. Because the peptide-MHC-I interaction is generally preserved in 1% Triton X-100 (60), the PDI–peptide interaction appears weaker than the MHC–peptide interaction. Next, we estimated the affinity of the PDI–peptide interaction using a competition assay in conjunction with a gel retardation assay (37). PDI was preincubated with FITC-conjugated H.A peptide at 4°C for 1 h. After incubation, varying amounts of cold H.A peptide competitor were added and the mixture was directly subjected to native-PAGE. PDI-FITC-H.A peptide complex was visualized and quantified using a fluorimager. We roughly estimated the IC50 value for PDI–peptide interaction as ∼7 μM (Supplementary Fig. S2). The affinity of peptide for PDI appears lower than the affinity of peptide for MHC-I (∼0.4 μM) (50) or TAP (∼1.2 μM) (14), suggesting that the transfer of PDI-bound peptide to MHC-I is energetically favorable.
PDI displays selectivity toward antigenic peptide
Next, we tested whether PDI exhibits a preference for peptides with MHC-I binding motifs. Wild-type PDI was incubated with various peptides biotinylated at their N-terminus for 10 min, and the reaction mixture was subjected to crosslinking followed by immunoblotting. Surprisingly, PDI displayed differential affinity for each peptide, and the increase in PDI–peptide affinity was proportional to the increase in peptide affinity to HLA-A2.1 (Fig. 2A). The sequential order of affinity of the designed peptides to HLA-A2.1 coincided with the total hydrophobicity of each peptide (Table 1) (54). Peptide binding to PDI was dependent on peptide hydrophobicity (Fig. 1D); these differences in peptide hydrophobicity might have resulted in our data. To rule out this possibility, we switched positions of anchor residues in the H.A peptide, keeping the total hydrophobicity of the anchor-residues switched (A.S) peptide unchanged. Interestingly, the interaction between the anchor-residues of the switched peptide (A.S) and PDI significantly decreased (Fig. 2B, compare lanes 1 and 2). This result suggested that peptide anchor motifs affect its interaction with PDI in a similar manner as the peptide–HLA-A2.1 interaction. To extend and generalize these findings, we tested more peptides for their interaction with PDI. Four peptides were previously identified as CTL epitopes specific for the influenza A virus and possess differential specificity toward the HLA allele supertype (2). These viral peptides were named as A1 (VSDG
Specific interaction of PDI with peptides depends on the PDI redox state
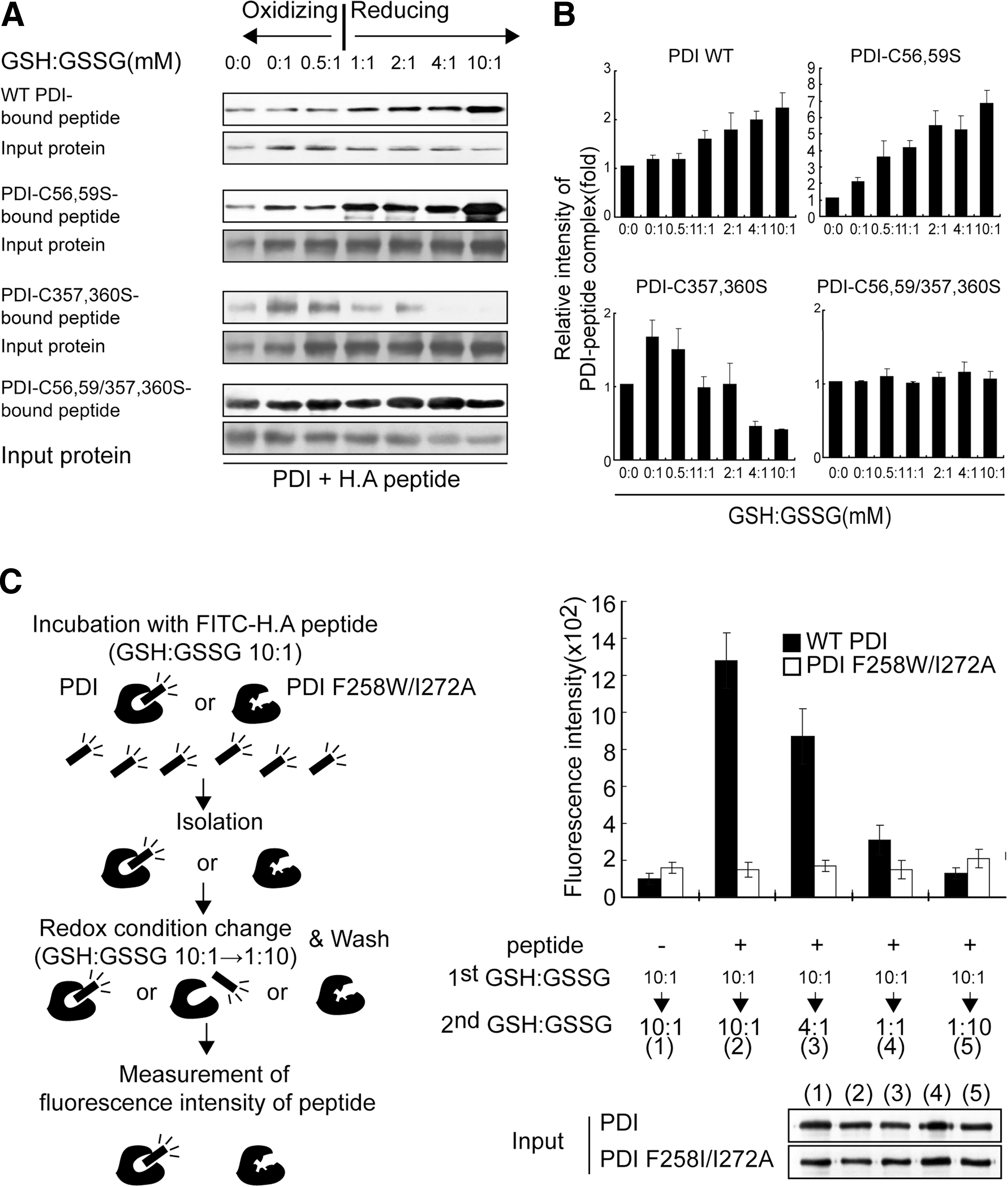
Although the b′ domain is a substrate binding site, other domains are also known to influence substrate binding (20, 21). Thus, we investigated whether the redox state of the PDI a and/or a′ domain affects its peptide-binding ability. Wild-type PDI or its mutants were incubated with H.A peptide in redox buffer containing varying ratios of oxidized glutathione (GSSG) and reduced glutathione (GSH), followed by crosslinking and immunoblotting. Interestingly, PDI variants displayed different associations with H.A peptide. Wild-type PDI and PDI-C56, 59S mutant showed increased interaction with peptide under reduced conditions (Fig. 3A, first and third panels; Fig. 3B). However, the PDI-C357, 360S mutant exhibited decreased interaction with peptide under the same reducing conditions (Fig. 3A, fifth panel; Fig. 3B). Regardless of redox conditions, PDI-C56, 59S/C357, 360S double mutant, in which both catalytic sites are mutated, was able to evenly bind peptides (Fig. 3A, seventh panel; Fig. 3B). To monitor the redox state of PDI under varying redox buffer conditions, aliquots of reaction mixture were precipitated by TCA and chemically modified with malPEG, and then analyzed using nonreducing SDS-PAGE. Wild-type PDI gradually shifted from the oxidized to the reduced form as the GSH/GSSG ratio increased (Supplementary Fig. S4, first panel). Interestingly, PDI-C56, 59S mutant maintained reduced state over all range of redox buffer conditions (Supplementary Fig. S4, second panel). Reduction of PDI-C357, 360S much higher GSH/GSSG ratio when compared with wild-type PDI (Supplementary Fig. S4, third panel). As expected, the redox state of PDI-C56, 59/357, 360S mutant remained unchanged in all buffer conditions. We performed a similar experiment under oxidizing conditions. The PDI–peptide interaction was inversely proportional to the oxidizing power (Supplementary Fig. S5). To exclude the possibility that GSSG, as small peptide, competed with H.A peptide for interaction with PDI and thereby produced an experimental artifact, we examined the interaction of H.A peptide with PDI-C56, 59/357, 360S mutant in which all active cysteine residues are mutated but the b′ substrate binding domain is intact. Consistent with the result shown in Figure 3A, the interaction of peptide with this mutant was independent of GSSG concentration under oxidizing condition (Supplementary Fig. S5). These results suggest that the interaction between PDI and peptide is regulated by the redox state of PDI. To further examine this possibility, we preincubated PDI with FITC-conjugated H.A peptide under reducing conditions (GSH:GSSG=10:1) for 10 min and isolated the PDI–peptide complex. The isolated PDI–peptide complex was subjected to a second incubation under various oxidizing conditions (GSH:GSSG=10:1∼1:10), and the fluorescence intensity of PDI-bound peptides was determined (Fig. 3C, left panel). Consistent with the observation above (Fig. 3A, B and Supplementary Fig. S5), release of peptide from PDI was facilitated under oxidized conditions (Fig. 3C, right panel). On the basis of these results, we concluded that the differential redox potential of each catalytic site in the a and a′ domains enable PDI to bind and release peptides.
Interaction of endogenous PDI with peptide
To extend these in vitro observations to cells, the interaction of endogenous PDI with peptide was analyzed using microsomes isolated from HeLa cells. Following preincubation with a photoreactive crosslinker, biotinylated-peptides were mixed with microsomes. After UV-crosslinking, microsomes were washed and lysed using 1% NP-40; PDI was precipitated using the indicated antibodies, and the immunoblot was performed using streptavidin-HRP. Consistent with in vitro observations (Fig. 2A), the binding affinity of endogenous PDI for peptides coincided with that of HLA-A2.1 for peptides (Fig. 4A). To gain insight into the putative relay network of peptides in the PLC, we examined the interaction of H.A peptide with PLC components. Only three proteins, TAP, MHC-I and PDI, displayed specific interaction with the H.A peptide (Fig. 4B). Interestingly, we did not detect an interaction between H.A peptide and ERp57 despite the structural and functional similarities between ERp57 and PDI.
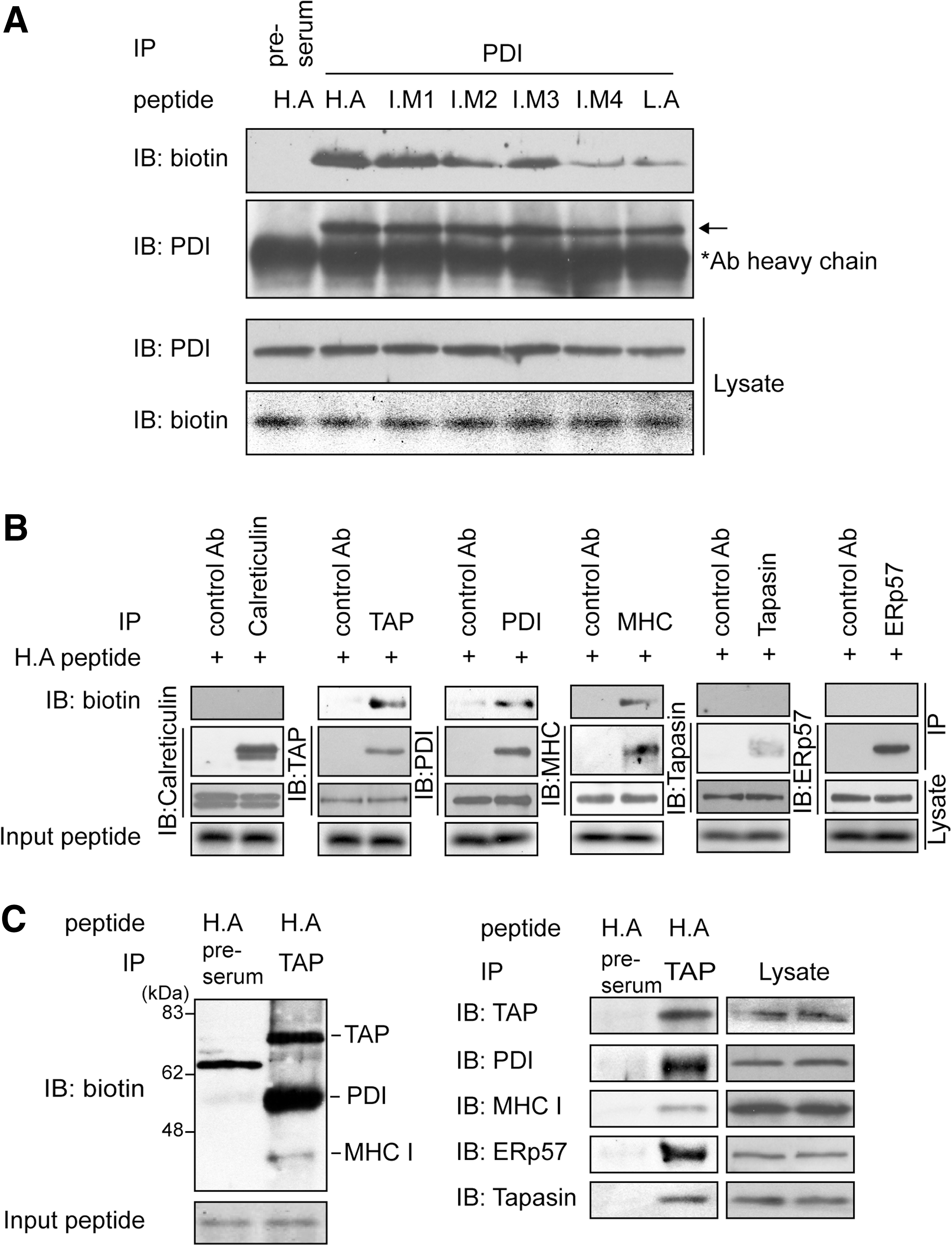
Then, we confirmed whether the specific interaction of peptides with TAP, MHC-I, and PDI occurs within the PLC. Following incubation of microsomes with biotinylated H.A peptide and UV-crosslinking, microsomal lysates were coimmunoprecipitated with anti-TAP1 serum followed by immunoblot for peptide. Biotin blotting revealed three distinct bands corresponding to the molecular weights of TAP, PDI, and MHC-I (Fig. 4C, left panel, second lane). Stripping and reblotting of the membrane with the respective antibodies verified their identities as TAP, PDI, and MHC-I (Fig. 4C, right panel, second lane). Notably, a predominant protein species bound to H.A peptide was PDI. This result is consistent with a previous study that identified PDI as the dominant peptide acceptor in the ER (26). Other PLC components, ERp57 and tapasin, were also present in TAP immunoprecipitates, as shown by stripping and reblotting the same membrane using anti-ERp57 and antitapasin antibodies (Fig. 4C, right panel). Our results suggest that PDI plays a major role in delivering peptides from TAP to MHC-I.
Peptide-dependent sequential association of PDI with TAP and HLA-A2.1
Cytosolic peptides are transported into the ER through TAP and loaded onto MHC-I with the assistance of PLC. However, a detailed understanding of the overall flow of peptide from TAP to MHC-I remains elusive; this is partly because of the complicated architecture of PLC components. In particular, tapasin plays several structural and functional roles in the process of peptide loading, which include physically bridging MHC-I to TAP (46) and peptide editing by cooperation with ERp57, its disulfide-linked partner (19, 59). Therefore, to exclude the effect of tapasin and its related functions, we used human tapasin-deficient 721.220 cells stably expressing HLA-A2.1, a tapasin-independent allele (35). We reasoned that if PDI transfers peptides from TAP to MHC-I, the transient association of PDI with TAP and MHC-I would be detectable. Thus, we first investigated the interaction of PDI with TAP or HLA-A2.1 at steady state. Coimmunoprecipitation of digitonin lysate of 721.220-HLA-A2.1 with anti-TAP or BB7.2 mAb specific for HLA-A2.1 revealed that PDI directly interacts with TAP or HLA-A2.,1 respectively (Supplementary Fig. S6A, B). Since no tapasin was present to physically bridge TAP to MHC-I, the interaction of PDI with TAP or HLA-A2.1 appears to be direct. Consistent with this observation, Raghaven and colleagues have recently reported an interaction between PDI and TAP in a tapasin-independent manner (44). Next, we examined the interaction between PDI and TAP or HLA-A2.1 in the presence of high- (ILDKFPVTV) or low-affinity peptide (IEDKFPVTD). Microsomes prepared from 721.221-HLA-A2.1 cells were incubated with peptides and were lysed with digitonin and coimmunoprecipitated with anti-TAP or BB7.2 mAb specific for HLA-A2.1. Interestingly, in the presence of high-affinity peptide, the interaction between PDI and TAP decreased in a dose-dependent manner (Fig. 5A, E), whereas the interaction between PDI and HLA-A2.1 increased in a dose-dependent manner (Fig. 5B, E). By contrast, the supply of exogenous low-affinity peptides influenced interactions between PDI and TAP or HLA-A2.1, but to a much smaller extent compared with the effects observed in the presence of high-affinity peptides (Fig. 5C, D, F). These results suggest that PDI is released from TAP upon binding TAP-transported peptides and sequentially associates with MHC-I for peptide transfer.
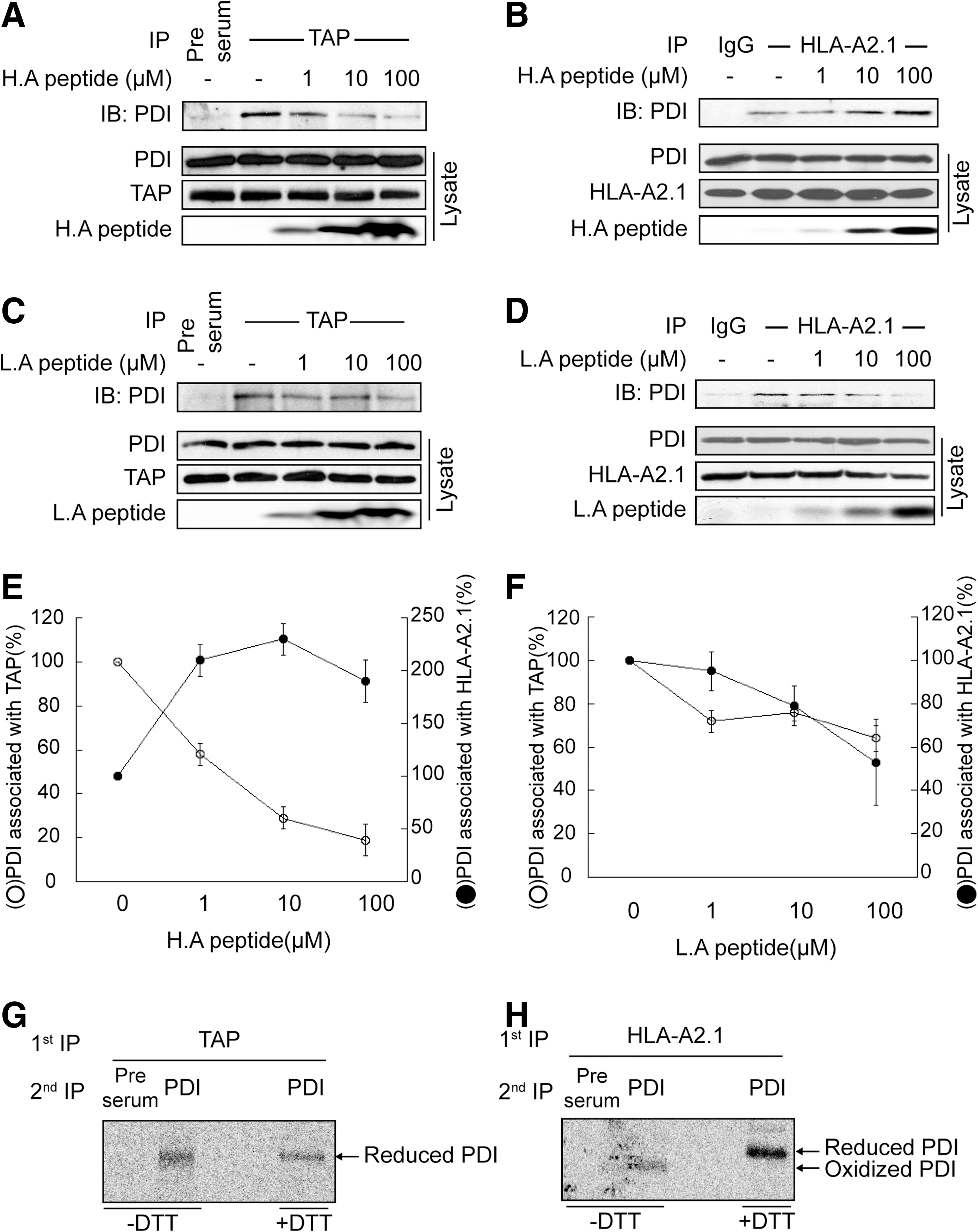
Since our in vitro data suggest that the redox state of PDI influences association and dissociation between PDI and peptide (Fig. 3), we investigated how the redox state of PDI affects its association with TAP or HLA-A2.1. The redox state of TAP- or HLA-A2.1-associated PDI was analyzed using metabolically labeled cell lysate and nonreducing SDS-PAGE. Analysis by serial immunoprecipitation using anti-TAP and anti-PDI antibodies revealed that TAP-associated PDI exhibited a reduced form (Fig. 5G). In contrast, most of the HLA-A2.1-associated PDI was present in the oxidized form (Fig. 5H).
Discussion
Although numerous studies have been performed to characterize the components and functions of antigen presentation machinery, most have mainly focused on quality control during different stages of the MHC-I protein, such as MHC-I folding, assembly, and peptide loading (38). Detailed information regarding the quality control mechanism for MHC-I binding to a partner, a peptide ligand, is currently unavailable. One challenging questions that remains to be addressed is how peptides are delivered from TAP to MHC-I in the ER. It is unlikely that TAP-translocated peptides reach MHC-I by simple diffusion because of the activities of various peptidases (45) and low abundance of optimal peptides with correct binding motifs in the ER (22, 27, 40). On the basis of the observations that ER-resident chaperones, including ERp72, gp96, and calnexin, can bind peptides (25, 26, 52, 53), it has been suggested that these chaperones may serve as peptide carriers in the ER. Although PDI is a known dominant receptor for TAP-translocated peptides (26, 52), its physiological role as peptide chaperone in the ER lumen is unknown.
In this study, we clarified the distinct role of PDI as a peptide carrier using an in vitro peptide-binding analysis system. Our data showed that PDI has several important characteristics required for delivering peptides from TAP to MHC-I. First, PDI has a peptide binding profile similar to that of MHC-I, albeit with lower affinity (Fig. 2 and Supplementary Fig. S2). Not only systematic variants of model peptides specific for HLA-A2.1 but also several well known influenza A virus epitopes for various HLA allele supertypes (HLA-A1, A3, B44, and B27) can bind to PDI. Because the nonionic detergent, Triton X-100, nearly abolished the interaction between PDI with peptide, hydrophobic contacts appear to dominate the primary binding interaction between PDI and peptide in the b′ domain. Generally, some antigenic peptide is generated by the proteasome and some by peptidase in the cytosol/ER; the peptide C-terminus is thought to primarily be processed by the proteasome. Most peptides specific for various HLA alleles share the hydrophobic amino acid residue at the C-terminus (51). Switching the C-terminal amino acid with the adjacent nonhydrophobic amino acid in high-affinity peptide prevented its interaction with PDI (Fig. 2B). This result implies that the nature of the peptide C-terminal amino acid may be important for binding onto “the binding pocket” of PDI. The affinity of peptide for PDI (IC50: ∼7 μM) is lower than the affinity of peptide for MHC-I (∼0.4 μM) (50), which makes it thermodynamically favorable to deliver peptides from PDI to MHC-I. Although the affinity of peptide for TAP (∼1.2 μM) (14) is higher than the affinity of peptide for PDI (IC50: ∼7 μM), the thermodynamics of peptide flow from TAP to PDI are different from that of peptide flow from PDI to MHC-I. TAP binds to a peptide via its cytosolic nucleotide-binding domain (33), whereas peptide binding to PDI occurs in the ER lumen where the biochemical environment may be very different from that of the cytosol.
Similarly, cytosolic heat shock protein 70 and gp96, the ER homolog of heat shock protein 90, can bind to antigenic peptides in the cytosol and the ER. Both chaperones appear to have peptide-binding pockets and preferences for peptides that can bind to MHC-I (7, 17, 30, 32). Further, gp96 is known for its ability to elicit an immune response against its chaperoned antigenic peptides (1, 5). Therefore, it was proposed that gp96 is a peptide carrier for MHC-I, although the detailed mechanism remains unknown. However, analysis of gp96-bound peptides showed that peptide sequences only partially match with those of peptides presented by MHC-I. Moreover, only 0.1–0.4% of gp96 is occupied with peptides (9), and peptide binding to gp96 is irreversible (3). Although it seems clear that gp96 contributes to antigen presentation in some way, these results do not support the role of gp96 as a peptide carrier.
Second, “peptide carriers” are expected to possess a regulatory mechanism for peptide binding as well as peptide release. Our data suggest that the association and release of peptides are dependent on a thiol-based redox-switch of PDI. Under reducing conditions, the interaction of wild-type PDI and peptide increased and TAP-associated PDI was predominantly in the reduced form. However, under oxidizing conditions, the interaction of PDI with peptide decreased and HLA-A2.1 associated PDI exhibited a partially oxidized form (Figs. 3 and 5G, H). These results support a role for PDI in delivering peptides from TAP to MHC-I. This redox-switched regulation of protein–protein interaction has been reported for several chaperone activities (15, 56). For example, Tsai and colleagues showed that a change of redox state in the PDI catalytic domains can regulate its affinity for substrates, mediated by a conformational change (56). Interestingly, we found that the redox state of each catalytic domain differentially influences interaction of PDI with peptide (Fig. 3A). The a domain mutant (PDI-C56, 59S), which mimics a reduced domain, exhibited increased affinity for peptide under increasingly reduced conditions (Fig. 3). In contrast, the a′ domain mutant (PDI-C357, 360S) displayed the opposite phenotype under the same conditions. Our data showed that the reduction rates of the two catalytic sites in the a and a′ domains are different from each other in various reducing condition (Supplementary Fig. S4). In accordance with this observation, it was previously shown that PDI displays asymmetry in the oxidation rate of its two catalytic sites because of the difference in the architecture of the a and a′ domains in the context of full-length PDI (23). In particular, PDI-C56,59/357,360S, in which all active sites are mutated, displayed strong affinity to peptide. Considering that PDI-C56,59/357,360S mimics a fully reduced PDI, fully reduced PDI represents an open conformer for the most efficient peptide binding, and conformational changes induced by differential redox regulation underlies the molecular mechanism for peptide binding and release by PDI. The ER maintains a relatively oxidizing environment at a GSH/GSSG ratio of 1:1 to 3:1 (16). Additionally, it is generally accepted that redox potential is not evenly distributed throughout the ER lumen. Thus, we speculate that the redox microenvironment around TAP favors reduction of a domain of PDI, whereas the redox microenvironment around MHC-I promotes oxidation of the a′ domain.
Finally, we showed that upon supply of high-affinity peptides, the interaction of PDI with TAP decreased in a dose-dependent manner, whereas its interaction with MHC-I increased. Given that a high-affinity peptide promoted faster dissociation of PDI from TAP and its subsequent association with MHC-I than a low-affinity peptide (Fig. 5), the binding of high-affinity peptide to PDI likely induces a conformational change of PDI, thereby altering the interaction of PDI with TAP or MHC-I. These results provide another line of evidence in favor of a sequential peptide transfer pathway by PDI from TAP to MHC-I.
In summary, our data indicate that PDI utilizes catalytic activity coordinated with peptide binding activity for selective peptide transport to MHC-I. On the basis of our findings, we propose a model for PDI-mediated delivery of peptide from TAP to MHC-I (Fig. 6). The reduced form of PDI binds to TAP before binding with peptides. Upon binding to TAP-translocated peptides, peptide-bound PDI is released from TAP and sequentially associates with MHC-I; this is followed by delivery of the peptide to MHC-I.

Footnotes
Acknowledgments
This work was supported by the National Creative Research Initiative Program of MOST/KOSEF. C.O., K.K., and J.R. were recipients of the BK21 fellowship.
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.