
Abstract
Morphine increases the production of nitric oxide (NO) via the phosphoinositide 3-kinase/Akt/neural nitric oxide synthase (nNOS) pathway. Subsequently, NO enhances N-methyl-D-aspartate receptor (NMDAR)/calmodulin-dependent protein kinase II (CaMKII) cascade, diminishing the strength of morphine-activated Mu-opioid receptor (MOR) signaling. During this process, NO signaling is restricted by the association of nNOS to the MOR.
Introduction
A series of G protein-coupled receptors (GPCRs) activate protein kinase C (PKC) and Src to enhance NMDAR-regulated calcium currents (21, 30). The MOR to regulate NMDAR activity requires PKC, Src, and other signaling proteins as well (36, 38). One of these proteins, the histidine triad nucleotide binding protein 1 (HINT1), binds to the MOR C terminus (CT) and to the Rz subfamily of regulators of G protein signaling, RGSZ1 and RGSZ2 (2, 18, 37). Upon activation of MOR, this HINT1-RGSZ complex recruits other proteins to the MOR environment, such as the inactive form of neural-specific PKCγ (36). This translocation raises the PKCγ activation threshold by reducing its sensitivity to local concentrations of diacylglycerol (DAG) (36), and it probably prevents the early potentiation of NMDAR, for example, before the strength of morphine signaling would require such control. Since the initial report associating PKC with MOR-dependent enhancement of NMDAR function (8), other elements have been seen to bridge the gap between these receptors. In this respect neural nitric oxide synthase (nNOS) exerts a well-established negative influence on MOR function (13, 22). This regulation is attributed to the NMDAR/nNOS cascade (20, 36), mainly because NMDAR activation or nitric oxide (NO) donors reduce the analgesic potency of morphine and accelerate the development of opioid tolerance (22). However, the nNOS that drives the MOR to NMDAR connection appears essentially to be recruited by MOR activity (39). Thus, the MOR-activated phosphoinositide 3-kinase (PI3K)/Akt cascade activates nNOS, carrying signals from the MOR toward the NMDAR. The PI3K/Akt/nNOS pathway acts upstream of PKC/Src and it plays a decisive role in the potentiation of NMDAR currents (39).
Innovation
Pharmacological, electrophysiological, and behavioral studies have demonstrated a relevant role of the glutamate-binding N-methyl-D-aspartate receptor (NMDAR) in the genesis and/or maintenance of chronic/persistent pain states. In these conditions, mu-opioid receptor (MOR)-activating opioids do not provide efficacious relief and this complicates their clinical use to treat persistent neuropathic pain. The relationship between MORs and NMDARs is bidirectional, and tolerance to morphine develops as a consequence of MOR-induced potentiation of NMDAR activity via nitric oxide (NO)-mediated release of zinc, zinc-mediated recruitment of PKCγ, and the subsequent PKC-mediated activation of Src. Our study shows that morphine regulates neural nitric oxide synthase (nNOS) by a push–pull mechanism. It promotes nNOS activation via the MOR/phosphoinositide 3-kinase (PI3K)/Akt pathway and it also diminishes NO production, fomenting the protecting association of nNOS with the MOR apart from the activating effects of Akt. This regulation requires binding of the nNOS N-terminal PDZ domain to the regulator of G protein signaling Z2 (RGSZ2) PDZ binding motifs that lie upstream of the RGS box. The MOR bears the histidine triad nucleotide binding protein 1 (HINT1)-RGSZ2 complex in its C-terminus, where nNOS-RGSZ2 interaction reduces NO signaling. Impairment of RGSZ2 expression results in the overactivation of nNOS/NO-regulated NMDAR/calmodulin-dependent protein kinase II (CaMKII) cascade. This provokes a fast and lasting tolerance to morphine, comparable to the attenuation of opioid efficacy observed in states of persistent pain. Thus, RGSZ2 and NO emerge as potential targets to treatment of neural diseases in which abnormally strong NMDAR activity depresses that of the associated MOR.
As observed for PKCγ in terms of the MOR to NMDAR connection (36), the morphine-induced potentiation of nNOS is counterbalanced by its translocation to the HINT1-RGSZ2 complex (39). Indeed, free zinc ions released by nNOS/NO facilitate the binding of PKCγ to HINT1, the cysteine-rich domain (CRD) in the PKCγ regulatory region physically associating with the histidine residues in the HINT1 protein. The association of nNOS with the MOR also requires the HINT1-RGSZ2 complex, although little is known about how this process occurs. Since nNOS lacks a CRD, the MOR-associated RGSZ2 protein rather than HINT1 could support this negative regulation. There is evidence indicating that RGSZ2 interacts with the MOR. The RGSZ2 gene lies close to the MOR gene, suggesting that they may be coordinately expressed (25, 44), and, indeed, the MOR and RGSZ2 genes are coregulated after voluntary oral morphine consumption and/or in the morphine-quinine two-bottle choice paradigm (11). Thus, RGSZ2 has been implicated in the development of morphine tolerance (35) and impaired RGSZ2 expression augments morphine analgesic activity, which is rapidly followed by sustained tolerance to its effects (15).
Significantly, nNOS is associated with the postsynaptic membrane through a PDZ domain at its N terminus (5), a region that is absent from the endothelial and inducible isoforms of the enzyme. This domain binds to the second PDZ domain of PSD-95 and couples nNOS to the NMDAR-mediated permeation of calcium fluxes that promote the formation of calcium-calmodulin (Ca2+-CaM). Here we have examined how the PDZ domain affects the control of morphine-activated nNOS by RGSZ2 in PAG synaptosomes. We found that the nNOS PDZ domain binds to the PDZ-binding motifs upstream of the RGSZ2 RGS domain, and that this binding prevents Akt from enhancing the activity of the enzyme. In RGSZ2-deficient mice, the nNOS/NMDAR/calmodulin-dependent protein kinase II (CaMKII) pathway is excessively activated, and morphine antinociception is greatly reduced. These results reveal a critical role for the HINT1-RGSZ2 complex in the control of MOR-generated NO signaling.
Results
The N terminal PDZ domain of nNOS binds to the RGSZ2 PDZ domain binding motifs
Activation of PAG MOR is coupled to the Akt/nNOS/NO pathway (39), and after icv administration of 10 nmol morphine, increased cysteine S-nitrosylation of a series of MOR-associated proteins was detected ex vivo. However, no such modification was observed for the 60 kDa MOR itself (Fig. 1A).

Using antibodies characterized previously in mouse brain synaptosomes, we were able to detect the MOR, RGSZ2, and associated proteins. In PAG synaptosomes, the immunoprecipitated MOR was recognized by antibodies directed against different regions of the MOR sequence (Supplementary Fig. S1; Supplementary Data are available online at
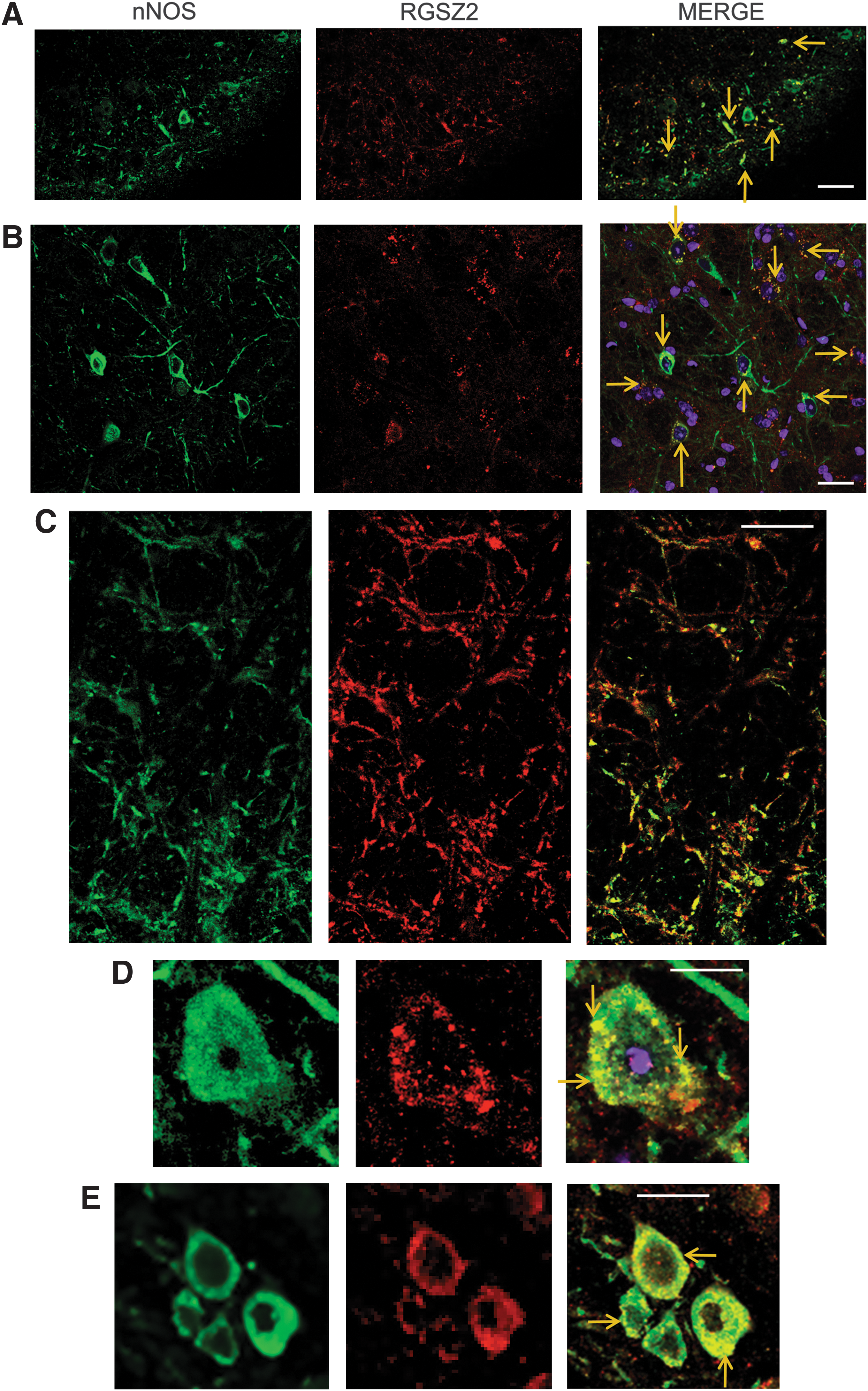
Immunohistochemical analysis revealed that RGSZ2 is widely expressed in PAG neurons, whereas those producing nNOS were much less abundant in this region. Importantly, the RGSZ2 protein colocalized with the nNOS enzyme, a generator of NO. Moreover, RGSZ2 immunoreactivity was distributed in dense spots along the plasma membrane, a pattern that is compatible with a regulatory role for RGSZ2 on GPCR function (Fig. 2). Eukaryotic Linear Motif (ELM:

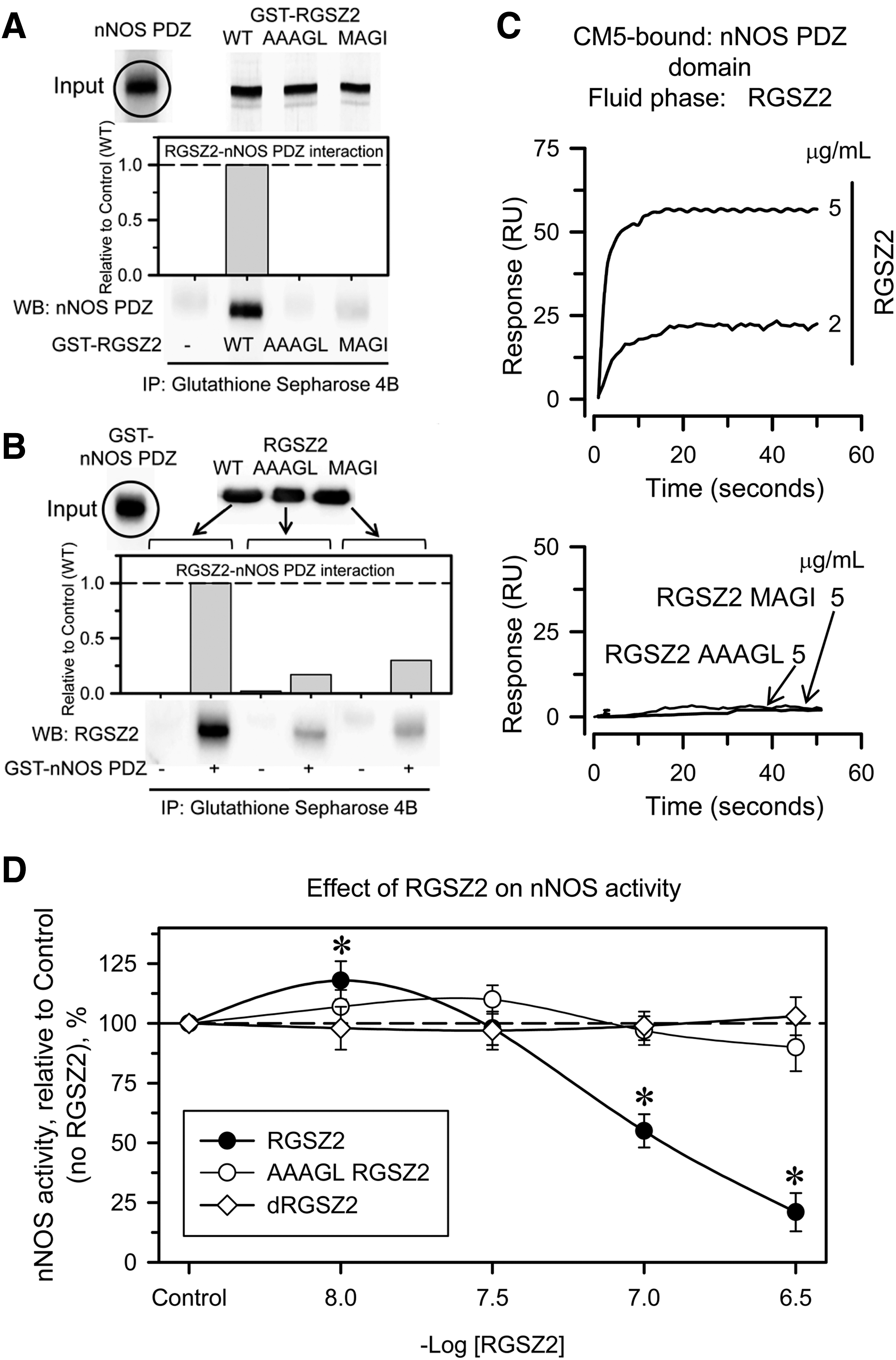
In in vitro pull-down assays and through Surface Plasmon Resonance analysis, we also observed a direct physical interaction between recombinant murine RGSZ2 and the canonical nNOS 15–98 PDZ domain. This interaction was lost in mutated RGSZ2 E62A+S63G or RGSZ2 D76A+E77A+V78G (Fig. 4A–C). In vitro, wild-type (WT) RGSZ2 had a biphasic effect on nNOS activity. At concentrations below those of nNOS (50 nM), RGSZ2 moderately increased its enzymatic activity, whereas when the RGSZ2 concentration was greater or equal to that of nNOS its activity was greatly reduced (Fig. 4D). Thus, the N terminal PDZ domain of nNOS binds to internal peptide sequences of RGSZ2 (PDZ binding motifs), an interaction that mostly impairs nNOS activity.
RGSZ2 prevents morphine from producing rapid and long-lasting analgesic tolerance
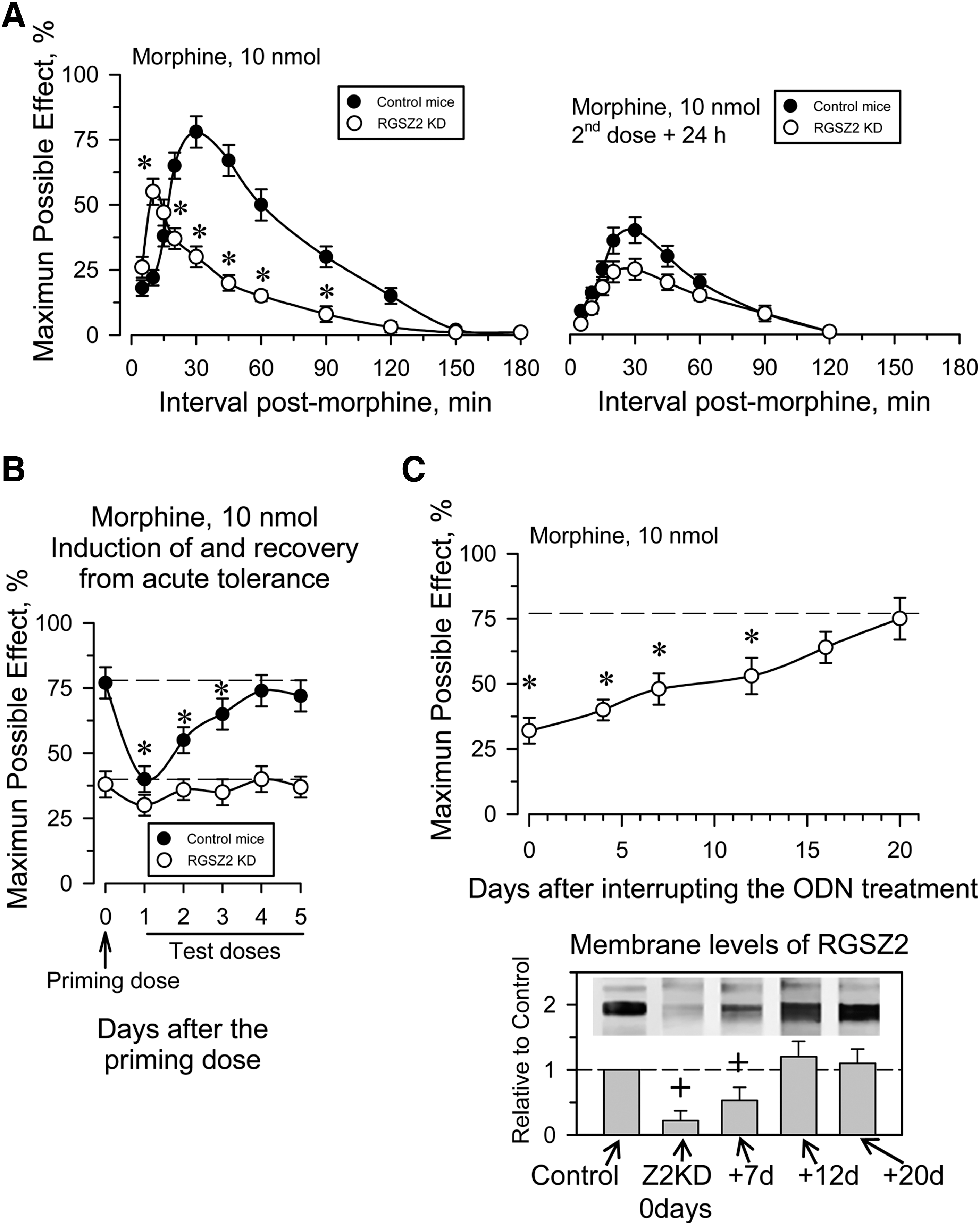
Neural RGSZ2 is a sumoylated and glycosylated protein, and while RGSZ2 knockout (KO) mice are available, previously characterized synthetic end-capped phosphorothioate antisense oligodeoxynucleotides (ODNs) were used to silence the expression of the RGSZ2 protein (15). This procedure reduces the MOR-associated RGSZ2 by about 50%–70% (Supplementary Fig. S4), without affecting the expression of RGSZ1, G proteins or MOR (15). A single icv dose of about 10 nmol morphine per mouse is suitable to promote MOR desensitization, whereas smaller doses (e.g., 3 nmol morphine) only produce moderate single-dose tolerance [see e.g., Garzón et al. (15)]. As reported previously in RGSZ2-deficient mice, 10 nmol morphine elicits a stronger analgesic effect during the early postopioid interval, followed by a rapid desensitization during the time-course of morphine analgesia (see Fig. 5A). However, the analgesia evoked by a second and identical dose of morphine 24 h later was clearly diminished in both RGSZ2-deficient and WT mice. Indeed, in RGSZ2-knockdown mice, the second dose of morphine lacked the initial sharp response and the peak effect was now observed closer to the 30 min postopioid interval. Control mice desensitized by administration of 10 nmol morphine recovered the morphine analgesic response 48 h later, reaching baseline levels after 4–5 days. However, in the absence of RGSZ2 the analgesic response remained dampened over this period (Fig. 5B). In a parallel group of mice that received ODN injections on 5 days there was a gradual recovery of the protein when the treatment was suspended, as well as recovery of the analgesic response to morphine that was complete after ∼2 weeks (Fig. 5C).
RGSZ2 reduces the impact of morphine-activated nNOS/NO on NMDAR-CaMKII pathway
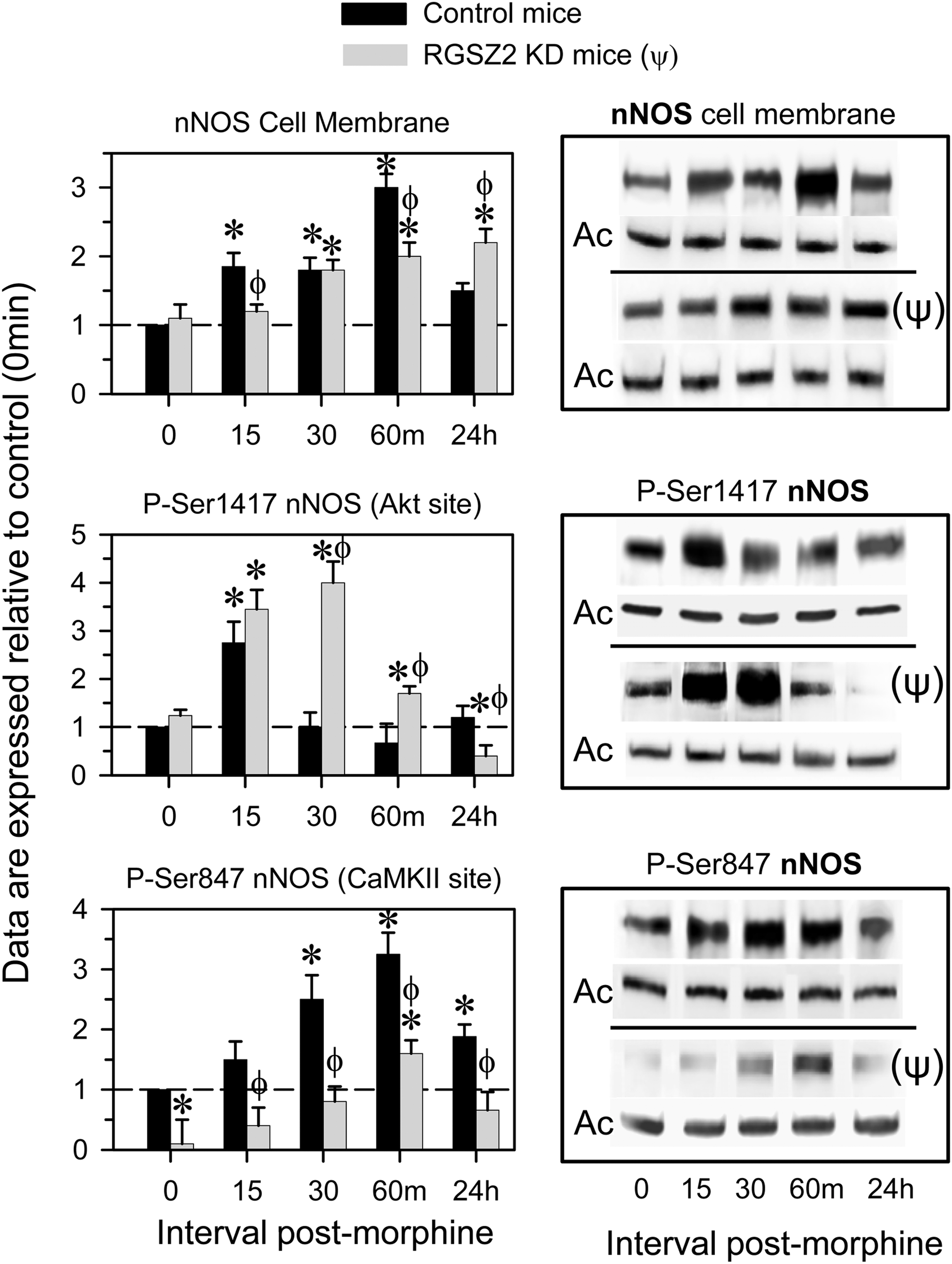
Before icv morphine injections of RGSZ2-deficient mice, the membrane levels of nNOS and of its activating Akt-mediated Ser1417 phosphorylation were almost identical to that of control mice. By contrast, CaMKII-mediated Ser847 phosphorylation was clearly weaker. In RGSZ2-deficient mice morphine increased the membrane levels of nNOS and of its activating Ser1417 phosphorylation for longer than in control mice, in conjunction with reduced nNOS inactivating Ser847 phosphorylation (Fig. 6). Thus, silencing the MOR-associated RGSZ2 protein weakened the MOR's negative influence on nNOS activity.
Morphine did not increase the presence of the MOR in the cytosolic fraction of PAG synaptosomes in either control or RGSZ2-deficient mice (Fig. 7A). The experimental reduction of RGSZ2 levels brought about the hyper-phosphorylation of the MOR. Nonetheless, the serine 375 that has been linked to GRK2- and arrestin-mediated internalization of the MOR was not phosphorylated (42). Notably, the NMDAR-CaMKII pathway was strongly activated, as evident through the phosphorylation of the NMDAR subunits and of CaMKII (Fig. 7B, C).
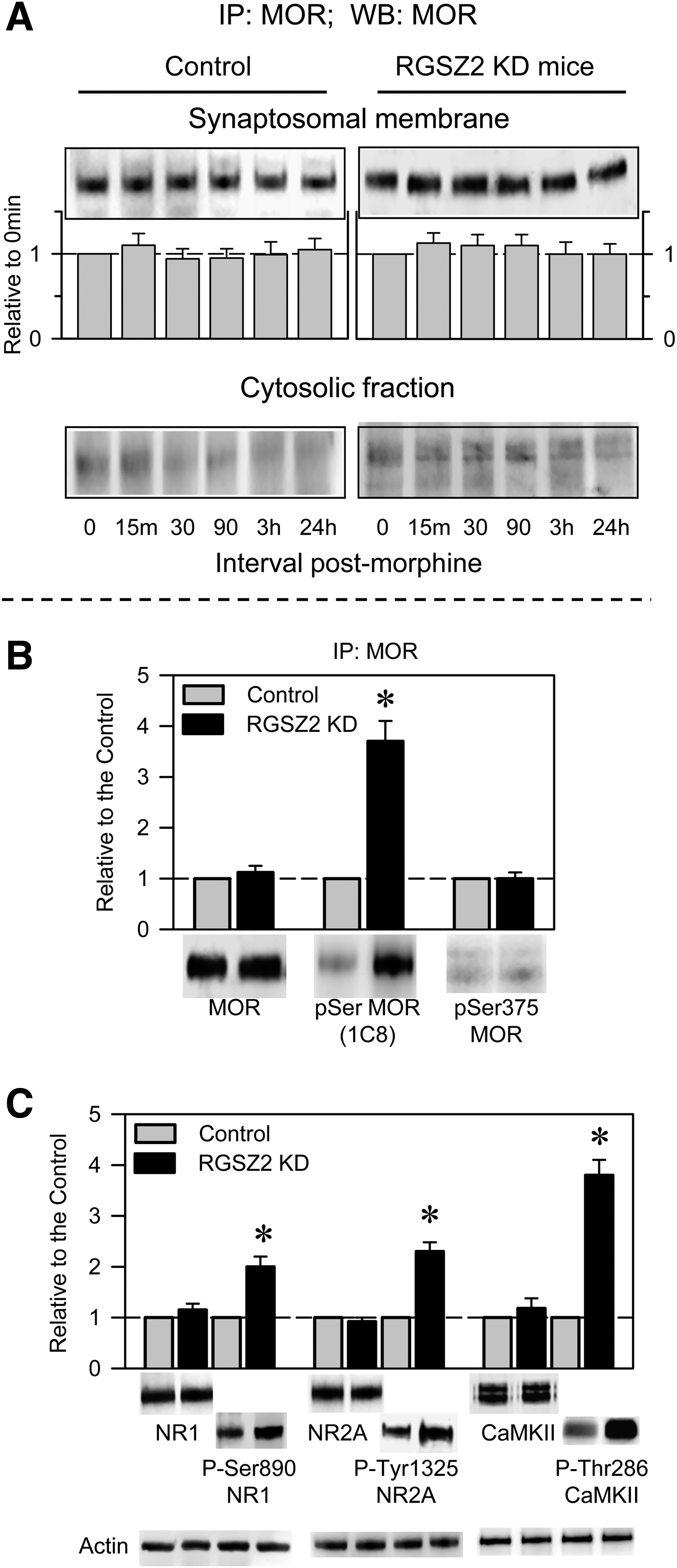
In the absence of RGSZ2, disruption of the nNOS/PKC/NMDAR/CaMKII pathway restores the function of the MOR and rescues morphine analgesia
Those changes brought about by RGSZ2 knockdown in the PAG MOR-NMDAR connection were dampened by in vivo inhibition of nNOS, PKC, CaMKII, or by antagonism of the glutamate-driven NMDA receptor. Therefore, MOR serine phosphorylation, CaMKII Thr286 autophosphorylation, as well as the phosphorylation of Tyr416 nonreceptor tyrosine kinase (Src/Fyn) and Tyr1325 NMDAR2A all diminished within few minutes of treating the mice with the corresponding compounds (Fig. 8A, B). In control mice, the icv injection of nNOS inhibitors (7 nmol L-NNA; 20 nmol L-NAME), PKC inhibitors (1 nmol Gö7874; 15 nM Chelerythrine), CaMKII inhibitor (10 nmol KN93), or NMDAR antagonists (1 nmol MK801; 3 nmol D-AP5) a few minutes before the initial dose of 10 nmol morphine produces only small changes in the analgesia observed (40). However, these treatments did restore the analgesic potency of morphine in RGSZ2-deficient mice (Fig. 8C).
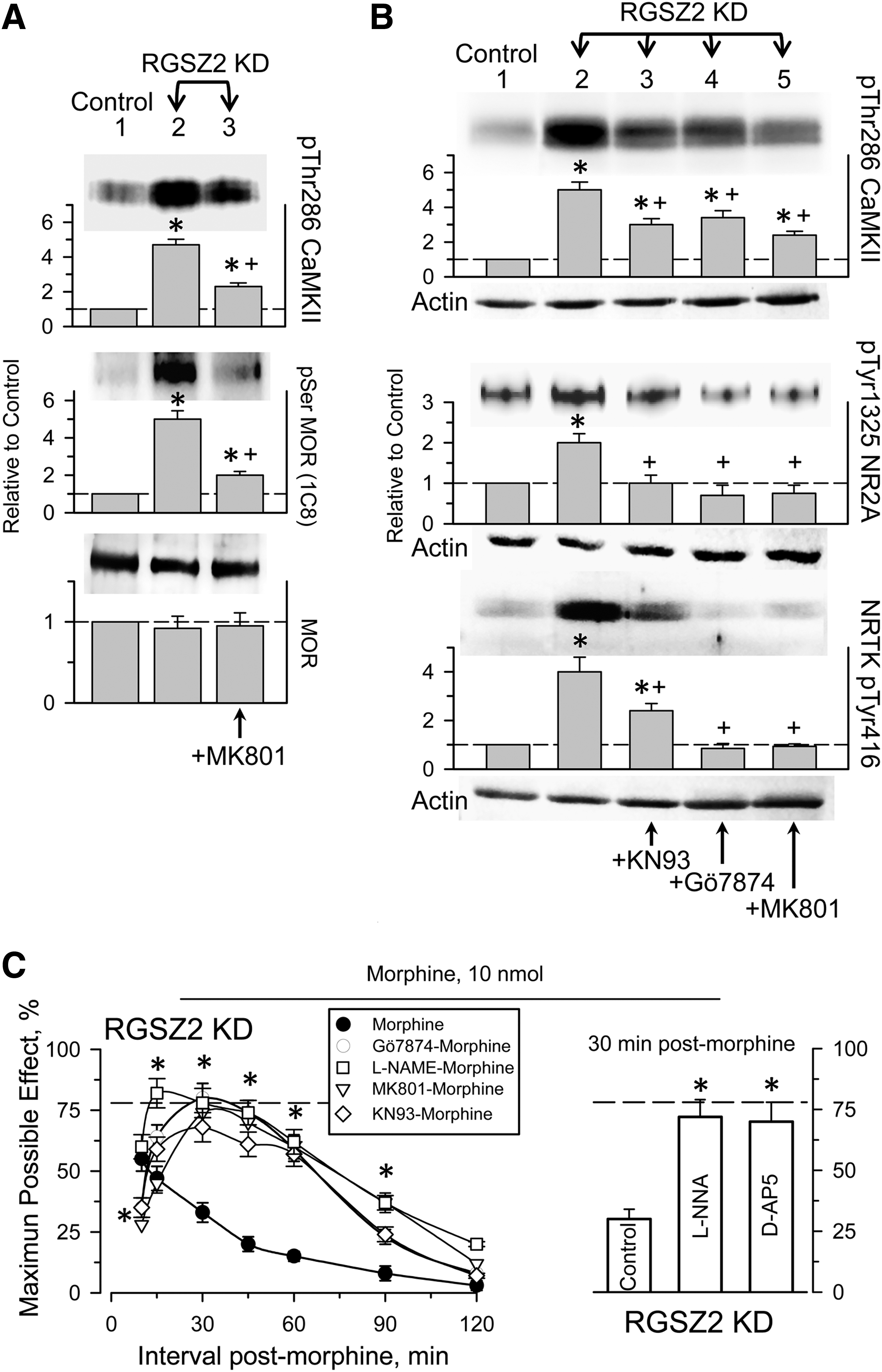
Discussion
The nNOS/NO pathway supports the cross-regulation between the MOR and NMDAR in the control of nociception (32). Morphine regulates the enzymatic activity of nNOS by a push–pull mechanism, whereby it promotes nNOS activation via the MOR/PI3K/Akt pathway and it diminishes NO production by protecting nNOS from the effects of Akt (39). Our results indicate that morphine-induced production of NO is efficiently counterbalanced by nNOS binding to the MOR-coupled RGSZ2 protein. The MOR bears the HINT1-RGSZ2 complex in its CT, and while HINT1 elimination abolishes nNOS recruitment to the MOR, it increases morphine effects but it does not prevent morphine from producing analgesic tolerance (18, 36). Therefore, morphine induces nNOS binding to RGSZ2 proteins, and in absence of HINT1 this complex is formed but separated from the MOR. The nNOS-RGSZ2 interaction controls NO signaling and accordingly, disruption of this association results in the overactivation of nNOS/NO-regulated signaling pathways (e.g., the NMDAR/CaMKII cascade), provoking fast and lasting tolerance to morphine. In these circumstances, strong serine phosphorylation of the MOR probably prevents it from productively regulating of G proteins, thereby contributing to the long-lasting, weak analgesic effects of morphine.
The specificity of NO signaling is regulated by the targeting of nNOS to the precise neuronal environment. This is achieved by means of adapter proteins that interact with its PDZ domain. The cytoskeletal protein PSD95/93 links nNOS to the NR2 subunits of NMDAR, whereas the carboxy-terminal PDZ ligand of nNOS (CAPON) links nNOS to synapsin and Dexras1. Other proteins like PIN, NOSIP, Hsp90, and caveolin-3 bind outside the PDZ domain and they apparently either regulate NO production or modulate the localization of nNOS (49). The canonical nNOS PDZ domain contains ∼100 amino-acid residues, forming a compact globular structure of a six-stranded antiparallel β-barrel flanked by two α-helices able to bind either the carboxyl-terminal sequences of proteins or internal peptide sequences. The interaction between a PDZ domain and its target may be constitutive; however, agonist-dependent activation of cell surface receptors is sometimes required to promote interaction with PDZ proteins. The nNOS PDZ-containing N terminus facilitates the regulatory binding of this enzyme to the MOR-coupled RGSZ2 protein. This canonical nNOS PDZ domain binds to an internal peptide sequence in RGSZ2 upstream of the RGS domain that fulfils the D-X-V requirement (45). This type of PDZ interaction is promoted by GPCR activation, and thus, after acting on the MOR, morphine promotes the activation of the PI3K/AKT/nNOS pathway and the binding of nNOS to RGSZ2 at the MOR CT. Since active nNOS forms a homodimer (10), the binding of RGSZ2 could alter the productive formation of the nNOS complex.
In PAG neurons, morphine-activated MOR increases NMDAR-induced calcium fluxes by the concatenated activation of PI3K/Akt/nNOS pathway (39). NO promote the release of zinc ions from endogenous stores, for example, metallothioneins, which is required for the binding of inactive PKCγ to the HINT1-RGSZ module at the MOR CT (36). This action reduces PKCγ sensitivity to local concentrations of DAG, enabling it to be regulated by morphine-released Gβγ dimers that act on PLCβ to produce the DAG and calcium ions needed to activate this kinase (30). Subsequently, PKCγ recruits Src to phosphorylate NMDAR subunits (6, 7, 38), contributing to the sustained potentiation of NMDAR-mediated glutamate responses (8, 24, 31). This provokes the activation of CaMKII, which directly or indirectly uncouples MOR from its regulated transduction and favors the development of morphine tolerance (40). This regulatory loop is blocked by nNOS inhibition (39), PKC inhibition, NMDAR antagonism, and CaMKII inhibition [reviewed in Garzón et al. (16)]. These pharmacological interventions restore the coupling of MOR to G proteins, but they uncouple MOR from the nNOS/PKC/NMDAR pathway, whereby morphine maintains its analgesic potency.
In the absence of morphine, the phosphorylation of signaling proteins implicated in the MOR-NMDAR connection increases in PAG synaptosomes from RGSZ2-deficient mice. This is probably a consequence of endogenous substances, opioids included, acting on GPCRs functionally coupled to NMDARs via PKC/Src (16, 26). Such abnormal increases in protein phosphorylation were efficiently reduced by restoring RGSZ2 levels and through pharmacological approaches, such as the inhibition of nNOS, PKC, NMDAR, or CaMKII. These treatments disrupt the hyperactivity of the NMDAR/CaMKII pathway and restored the potency of morphine. Therefore, RGSZ2 dysfunction affects nNOS/NO, PKC, NMDAR, and CaMKII, which act in the negative feedback loop that controls morphine-activated MOR signaling.
In the light of these data, NO is clearly implicated in the hyperactivation of the NMDAR/CaMKII cascade in RGSZ2-deficient mice. The loss of RGSZ2 unleashes nNOS activity, increasing NO levels and probably the presence of free zinc ions. Moreover, disruption of the HINT1-RGSZ2 complex prevents zinc-mediated recruitment of inactive PKCγ to the MOR environment (36). When liberated of the negative influence of the MOR, PKCγ is more rapidly activated, and since high zinc stabilizes the binding of DAG to PKC regulatory domain, this activation persists for longer (36). These changes contribute to the abnormal enhancement of NMDAR/CaMKII function mediated by PKC/Src. On the other hand, nNOS also contributes to overregulation of NMDAR signaling in those RGSZ2-deficient mice by participating in MOR-extracellular signal-regulated kinase (ERK)1/2 pathway (1, 4). Indeed, NO can directly activate H-Ras (19), and the nNOS-CAPON complex activates Dexras1 (49). As a result, the Ras/Raf-1/MEK/ERK1/2 cassette becomes active (48). ERK1/2 activity increases the surface expression of GluR1 and GluR2L subunits of the glutamate-driven α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor, which contributes to the enhancement of NMDAR activity and to long-term potentiation of the postsynapse (33, 50). Consequently, the increased activity of nNOS in absence of RGSZ2 promotes NMDAR-mediated fast MOR desensitization and long-term analgesic tolerance to morphine develops.
The MOR-NMDAR relationship explains the clinical efficacy of NMDAR antagonists in reducing opioid tolerance and their successful use in diminishing the consumption of analgesics (21, 27). Pharmacological, electrophysiological, and behavioral studies have demonstrated a relevant role of the NMDAR in the genesis and/or maintenance of chronic/persistent pain states (3, 14). The attenuation of opioid efficacy observed in states of persistent pain appears to be caused by abnormally strong NMDAR activity. Therefore, the genesis and/or maintenance of persistent pain could arise from alterations to MOR/NO/PKC/NMDAR coupling. Silencing of the MOR-associated RGSZ2 helps increase nNOS/NMDAR activity and it promotes profound and lasting morphine-induced MOR desensitization. Thus, RGSZ2 emerges as a potential target to treatment of neural diseases in which an enhanced NMDAR activity depresses that of the associated GPCR.
Materials and Methods
Preparation of recombinant proteins
Constructs of the murine full-length RGSZ2 (AF191555) and the PDZ domain of murine nNOS (NM_008712.2) were generated by polymerase chain reaction (PCR) using mouse PAG cDNA as a template (High Fidelity PCR enzyme mix; Fermentas # K0191). The primers included SgfI and PmeI restriction sites and corresponded to the 5′ and 3′ ends of the coding region: RGSZ2-specific primers: 5′-GACCGCGATCGCCAGAAAACGGCAGCAGTCACA-3′ (forward) and 5′-GATGGTTTAAACTTAGGATTCAGAAGTACAGCTGGTG-3′ (reverse); nNOS primers: 5′-AGGAGCGATCGCCATGCAGATCCAACCCAACGTCATT-3′ (forward) and 5′-GATGGTTTAAACTTAAACATCCCCTGTGAAGGTG-3′ (reverse). Glutathione S-transferase (GST) fusion proteins were generated by inserting the PCR product into the pFN2A (GST) Flexi vector (Promega, #C8461) at the SgfI and PmeI sites. The vector was introduced into Escherichia coli BL21 (KRX; Promega, #L3002) and the GST fusion proteins were purified on glutathione-sepharose 4B columns (GE Healthcare Bio-Sciences, #27-4570). The proteins retained were eluted with a 0–20 mM glutathione gradient as GST fusion proteins, or they were cleaved in the column with ProTEV protease (Promega, #V605A).
Site-directed mutagenesis of the RGSZ2 protein (Glu 62 to Ala and Ser 63 to Gly) was performed by PCR mutagenesis using the following primers: 5′-CCAAAATGG
BiFC analysis
The plasmid pPD49.83 was used to generate two cloning vectors for BiFC analysis. Constructs containing hsp-16.41 heat shock promoter, an Myc or hemagglutinin tag for detection of BiFC fusion proteins, a multiple cloning site, a linker sequence, and the N-terminal fragment of Venus truncated at residue 173 (VN173) or the C-terminal fragment of Venus starting at residue 155 (VC155), were generously provided by Dr. Chang-Deng Hu (Purdue University). Full-length murine RGSZ2 and the PDZ domain of nNOS were subcloned in-frame into pCE-BiFC-VC155 or pCE-BiFC-VN173 plasmids using standard cloning strategies. Fragments were PCR amplified using the following primers: RGSZ2, AGC
RGSZ2-nNOS interaction
RGSZ2 (200 nM) and nNOS (15–98) (200 nM) were incubated alone (negative control) or together in 450 μL of buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% P20) mixed by rotation for 30 min at room temperature. Glutathione sepharose 4B (GE Healthcare Bio-Sciences AB #17-0756-01) beads were added, and after mild centrifugation, the pellets were washed three times and solubilized in 2× Laemmli buffer. The presence of RGSZ2 or nNOS (15–98) was analyzed by Western blots.
Surface plasmon resonance analysis of RGSZ2 and nNOS interactions was carried out on a BIACORE X (GE Healthcare Bio-Sciences). The PDZ domain of nNOS (50 μg/ml) was amine-coupled to channel 2 of a CM5 sensor chip (GE, BR-1000-14) and channel 1 was the blank. The sensor surface was equilibrated with HBS-EP buffer (GE, BR-1001-88) and sensorgrams were collected at 25°C at a flow rate of 10 μL/min following the passage of RGSZ2 (75 μL). The CM5 sensor chip was regenerated after each cycle with two 15 μL pulses of 10 mM glycine provided at 30 s intervals (pH 2.5, GE, BR-1003-56). Increasing analyte concentrations were studied and the results were plotted using BIAevaluation software (v 4.1).
Effect of RGSZ2 on nNOS activity
The activity of recombinant nNOS (Alexis Biochem., ALX-201-028) was monitored by the conversion of arginine to citrulline. The nNOS (50 nM) was incubated for 10 min at 25°C with increasing concentrations of RGSZ2 (10–300 nM), and the NOS was assayed (Calbiochem, 482700) with 14C-arginine (Amersham CFB63, 319 mCi/mmol) in a final volume of 50 μL containing: 50 mM Tris-HCl pH 7.4, 3 μM tetrahydrobiopterin, 1 μM FAD and 1 μM FMN, 1 mM NADPH, 0.6 mM CaCl2, 14C-arginine (50 μCi/ml), and 0.1 μM calmodulin. The NOS inhibitor, L-NAME, was added as a negative control. The reaction was stopped by adding stop buffer (400 μL) and then the equilibrated resin (100 μL) was added into each reaction mixture before the samples were transferred to spin cups. After centrifugation the eluates were counted in a liquid scintillation counter.
Animals, intracerebroventricular injections, and evaluation of antinociception
Male albino CD-1 mice weighing 22–25 g were housed and used strictly in accordance with the guidelines of the European Community for the Care and Use of Laboratory Animals (Council Directive 86/609/EEC). The experiments performed were previously approved by the Bioethics Committee of the “Consejo Superior de Investigaciones Científicas (CSIC).” The response of the animals to nociceptive stimuli and the influence of morphine sulfate (Merck) were determined using the warm water (52°C) tail-flick test using a cut-off time of 10 s to minimize the risk of tissue damage. Baseline latencies ranged from 1.5 to 2.0 s and they were not significantly affected by the inhibitors used: L-NG-Nitroarginine (LNNA, Tocris 0664; Biogen), Gö7874 (Calbiochem, #365252; VWR), Chelerythrine (Calbiochem #220285), MK801 (Tocris, #0924), D-AP5 (Tocris, #0106), KN93 (Calbiochem #422711), or their solvents. Antinociception is expressed as a percentage of the maximum possible effect (=100×[test latency-baseline latency]/[cut-off time-baseline latency]). Groups of 10 mice were lightly anesthetized with ether and received 10 nmol morphine sulfate (Merck) injected in a volume of 4 μL into the lateral ventricle. Thereafter, antinociception was assessed at different time intervals.
Previously characterized synthetic end-capped phosphorothioate (indicated as *) antisense ODN (synthesized by Sigma-Aldrich) were used to reduce the expression of the target RGSZ2 protein (NM_019958), generating RGSZ2-deficient mice (15) (Supplementary Fig. S3). The mouse KO with a targeted disruption of HINT1 (on a 129SvJ background) was generously supplied by I.B. Weinstein/J.B. Wang. Breeding pairs of homozygous WT and KO mice were obtained from hybrid mutant mice (originally generated on a 129SvJ-C57BL/6 background) by backcrossing 129SvJ mice for several generations. The genotype was confirmed by PCR analysis of DNA isolated from tail biopsies.
Acute tolerance and interval required to recover the analgesic response
The animals were icv-injected with 10 nmol morphine, a dose that produces 70%–80% of the maximum effect detected in this test, comparing the controls with the RGSZ2-deficient mice. Morphine antinociception was evaluated after 30 min when the peak effect of the compound was achieved. To study the recovery from acute analgesic tolerance and to avoid repeated injections of the mice, the animals were then divided into groups of eight mice each. Acute tolerance was determined in one of these groups by icv-injecting of a test dose of 10 nmol morphine 24 h after the priming dose. Afterward, additional doses of 10 nmol morphine were administered every 24 h but to a different group of mice. In RGSZ2-deficient mice, the recovery from acute tolerance was studied with and without concomitant ODN treatment. In all cases the mice were evaluated 30 min after morphine injection. Data are expressed as the mean±standard error of the mean of groups of eight mice.
Immunocytochemistry
Deeply anaesthetized animals (Equithesin, Janssen Laboratories, 2.5 ml/kg intraperitoneally) were ventilated and perfused through the left ventricle with 0.9% saline, followed by 250 ml of a fixative solution containing 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were then removed, cut into small blocks, and postfixed for 4 h at room temperature in the same fixative. The blocks were then cryoprotected at 4°C in a 30% sucrose solution in phosphate buffer, and serial 40-μm-thick transverse frozen sections were obtained on a 2800 Frigocut (Reichert-Jung) microtome that were then processed for immunocytochemistry. The antibodies used in this study have already been characterized and they fulfill the recommended criteria for use in immunohistochemistry (41). All these antibodies were diluted in phosphate-buffered saline containing 0.2% Triton X-100. The expression of the nNOS protein was evaluated with a goat polyclonal antibody suitable for immunohistochemistry (Abcam ab1376). Endogenous RGSZ2 protein was identified with a rabbit polyclonal antibody that has been adequately characterized (15, 37). Immunofluorescence staining for confocal microscopy was carried out by incubating tissue sections overnight at 4°C with the primary antibodies against nNOS and RGSZ2 diluted 1:2500 and 1:500, respectively. After washing thoroughly in phosphate-buffered saline, antibody staining was observed with Alexa Fluor 488 donkey anti-goat IgG and Alexa Fluor 568 donkey anti-rabbit IgG (Molecular Probes), incubated for 1 h at room temperature. Confocal images were acquired on a Leica TCS SP5 scanner (Leica Microsystems GmbH). Controls for immunohistochemistry were performed following standard protocols (43).
Coimmunoprecipitation of signaling proteins
The PAGs were obtained from groups of eight mice at various intervals after icv injection of morphine, and synaptosomal membranes were obtained as described previously (12, 35). Briefly, the PAGs were collected and homogenized in a buffer containing 25 mM Tris-HCl (pH 7.4), 1 mM EGTA, and 0.32 M sucrose supplemented with a phosphatase and protease inhibitors. The homogenate was centrifuged at 1000 g for 10 min to remove the nuclear fraction, pellet 1 (P1), and the supernatant (S1) was centrifuged at 20,000 g for 20 min to obtain the crude synaptosomal pellet (P2) and the supernatant S2. The pellet P2 was diluted in Tris buffer supplemented with a mixture of protease inhibitors and added to the top layer of a discontinuous step Percoll gradient (3%, 10%, 15%, and 23%). The tubes were centrifuged at 30,000 g at 4°C for 5 min and to diminish the presence of Percoll, the fraction containing the synaptosomes was diluted fourfold with ice-cold sucrose/EDTA buffer and centrifuged at 20,000 g for 30 min at 4°C. To obtain the synaptosomal membranes the pellet was placed in a hypotonic buffer and centrifuged at 20,000 g for 30 min at 4°C. The resulting pellet was then used in the subsequent experiments of MOR immunoprecipitation and analysis of coprecipitated proteins. To determine the possible internalization of the MOR as a consequence of morphine treatment, the supernatant (S2) was centrifuged at 105,000 g for 1 h to obtain the crude microsomal pellet (P3). The S3 supernatant was concentrated in Amicon Ultra-4 centrifugal filter devices (nominal molecular weight limit NMWL of 10,000 #UFC8 01024; Millipore Iberica S.A.), the proteins recovered were resolved by SDS-PAGE and the MORs analyzed in Western blots.
Antibodies against the second extracellular loop of the MOR and against the CT of RGSZ2 proteins were labeled with biotin (Pierce #21217 & 21339; Fisher Scientific). The target proteins were then immunoprecipitated from solubilized membranes and analyzed as described previously (34). Since unidentified proteins might coprecipitate with the MORs and interfere with the phosphoserine analysis (clone 1C8; Calbiochem, 525281), the existing protein interactions were disrupted under denaturing conditions before immunoprecipitation.
Detection of signaling proteins
Western blots were probed with affinity-purified IgG antibodies directed against peptide sequences in: the murine MOR, anti-MOR second extracellular loop, and anti-MOR CT (diluted 1:1000) (35); anti-RGS17(Z2) CT (aa: 192–215; GenScript), anti-RGS17(Z2) IQ (aa:46–60; GenScript) (15, 34), and anti-RGS17(Z2) W15 (sc-48286) internal region; anti-phospho-MOR (Ser375) (1:1000; Cell Signaling 3451; Izasa); anti-nNOS (1:1000; Santa Cruz sc1025; Quimigen); anti-phospho-nNOS (Ser847) (1 μg/ml; Abcam ab16650); anti-phospho-nNOS (Ser1417) (2 μg/ml; ab5583); anti-NMDAR1 (1:1000; ab1880); anti-phospho NMDAR1 (Ser890) (1:1000; Cell Signaling 3381); anti-NMDAR2A (1:1000, ab14596); anti-NMDAR2A phospho-Y1325 (1:300, ab16646); anti-Tyr416 phospho-nonreceptor tyrosine kinase (1:1000; Cell Signaling 2101); mouse monoclonal IgG1 anti-CaMKII (1:3000; BD Transduction labs, 611292; BD); anti-phospho-CaMKII (Thr286) (1:2000; Cell Signaling, 3361); anti-S-nitrosocysteine (1:1000; Abcam ab50185); anti-Actin (1:3000; Stressgen, CSA-400; Bionova). Other antibodies included a mouse monoclonal antibody (IgM) to detect clone 1C8 [goat anti-mouse IgM (Calbiochem, #401295) (H+L) conjugated to horseradish peroxidase. The antibodies were diluted in TBS+0.05% Tween 20 (TTBS) and incubated with polyvinylidene fluoride membranes for 24 h at 6°C. The primary antibodies were detected using the corresponding secondary antibodies conjugated to horseradish peroxidase (diluted 1:10,000 in TTBS). Antibody binding was observed using ECL Plus Western Blotting Detection System (GE #RPN2132), and chemiluminescence was recorded with a ChemiImager IS-5500 (Alpha Innotech). The assays were typically performed twice or three times using samples obtained from independent groups of mice, and the results were consistent.
Statistical significance
Analysis of variance, followed by the Student-Newman-Keuls test (SigmaStat; SPSS Science Software), was performed and significance was defined as p<0.05.
Footnotes
Acknowledgments
This research was supported by FIS PI080417 (P.S.B.), PS09/00332 (J.G.), and “Instituto de Salud Carlos III, Centro de Investigación Biomédica en Red de Salud Mental, CIBERSAM.” M.R.M. is currently supported by a CIBERSAM contract, and A.V.S. is a predoctoral fellow from the Spanish Ministry of Science and Innovation. We would like to thank Beatriz Fraile and Gabriela de Alba for their excellent technical assistance.
Author Disclosure Statement
The authors declare that, excluding income received from our primary employer “Ministerio de Ciencia y Tecnología,” no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional services and that there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.