
Abstract
Mitochondrial dynamics and mitophagy are recognized as two critical processes underlying mitochondrial homeostasis. Morphological and bioenergetic characterization of the life cycle of an individual mitochondrion reveals several points where fusion, fission, and mitophagy interact. Mitochondrial fission can produce an impaired daughter unit that will be targeted by the autophagic machinery. Mitochondrial fusion, on the other hand, may serve to dilute impaired respiratory components and thereby prevent their removal. The inverse dependency of fusion and mitophagy on membrane potential allows them to act as complementary rather than competitive fates of the daughter mitochondrion after a fission event. We discuss the interplay between mitochondrial dynamics and mitophagy in different tissues and in different disease models under both stress-induced and steady-state conditions. Antioxid. Redox Signal. 14, 1939–1951.
Introduction
Mitophagy refers to the selective removal of mitochondria by the autophagic machinery. Mitophagy appears to be a universal route for the degradation of dysfunctional mitochondria. Under certain physiological settings, mitophagy can also eliminate functional mitochondria as seen during erythroid differentiation (106), oocyte fertilization (98), or during starvation (27). Although there is evidence that other organelles and cellular compartments, such as endoplasmic reticulum (ER) (104), perixosomes (24, 92), and ribosomes (6), also undergo selective autophagy, mitophagy seems to be of special importance for two reasons: (i) mitochondria are one of the main sources of reactive oxygen species (ROS) generation (therefore, they are also the immediate targets of ROS damage). (ii) Dysfunctional mitochondria that are not degraded can produce higher amounts of ROS, be more susceptible to the release of cytochrome c and apoptosis-inducing factor, and thereby, amplify ROS damage (15, 34).
Mitochondrial dynamics refers to repetitive cycles of fusion and fission between mitochondria (56, 97, 119). These opposing processes determine the architecture of the entire mitochondrial population of the cell and influence nearly every aspect of mitochondrial functions, including respiration, calcium buffering, and apoptosis (1, 2, 28, 51, 100). In addition, fusion and fission events per se (and not only the resultant effect on architecture) are suggested to impact mitochondrial homeostasis. Fission events were demonstrated to play a role in segregating dysfunctional mitochondria from the entire mitochondrial web (5, 30, 112) and to sort out mutant mtDNA copies (61, 103). Fusion events were suggested as a complementary route by which mitochondria quickly equilibrate matrix metabolites (43, 44, 47, 86, 111), intact mtDNA copies (3, 73, 80, 95), and mitochondrial membrane components (10, 122).
The simplistic view of a slowly metabolically deteriorating mitochondrion is complicated, given the high rate of mitochondrial content exchange permitted by fusion and fission events. When a small fraction of mitochondria within a cell are tagged by matrix-targeted photoactivatable (PA) GFP, the latter can reach equilibration in some cells within ∼1 h (47, 48, 70, 110). Given that the turnover of mitochondrial proteins is in the range of hours to days (69), it is predicted that the mitochondrial population within a cell will be homogenous in protein content and, consequently, in function. This contradiction was addressed by the understanding that fusion is a selective process and that mitochondria that are destined to mitophagy exist in a preautophagic pool. The preautophagic pool is characterized by mitochondria that are relatively depolarized and are fusion deficient.
During the last 4 years, numerous studies reported on the interaction between mitochondrial dynamics and mitophagy in neurons, skeletal and cardiac myocytes, and pancreatic β-cells. We focus this review on aspects that concern mitochondrial dynamics.
Depolarized Mitochondria Are the Substrate for Mitophagy
Mitochondrial membrane potential (Δψm) is the driving force for mitochondrial ATP synthesis. Depolarization below a certain Δψm may indicate impaired mitochondrial function and is a prerequisite for mitophagy (27, 90, 110). Mitochondrial depolarization appears to precede the translocation of the proteins that tag mitochondria for mitophagy such as Parkin and PTEN-induced putative kinase 1 (Pink1) (75, 76). Yet, although an essential condition depolarization alone is insufficient to trigger mitophagy.
Mitochondrial depolarization may be the result of a gradual or spontaneous deterioration or, alternatively, may occur as a result of a regulated event. To understand the events leading to appearance of depolarized mitochondria and the subsequent mechanism(s) that target them to mitophagy or metabolic rescue, one must characterize the bioenergetic and biochemical properties of the life cycle of mitochondria.
Spontaneous Generation of Depolarized Mitochondria Is Not a Frequent Event (Fission-Independent Pathway)
In both plants and mammalian cells, mitochondria go through continuous cycles of fusion and fission events in a kiss-and-run pattern, that is, a brief fusion event (∼tens of seconds) that is followed by fission (3, 57, 110, 113). As a result, mitochondria spend most of their time in their postfission state as solitary units before entering a subsequent fusion phase. It is therefore suggested that the life cycle of mitochondria can be divided into two periods, the prefusion period (solitary period) and the postfusion period when the mitochondrion is connected to another (networked period).
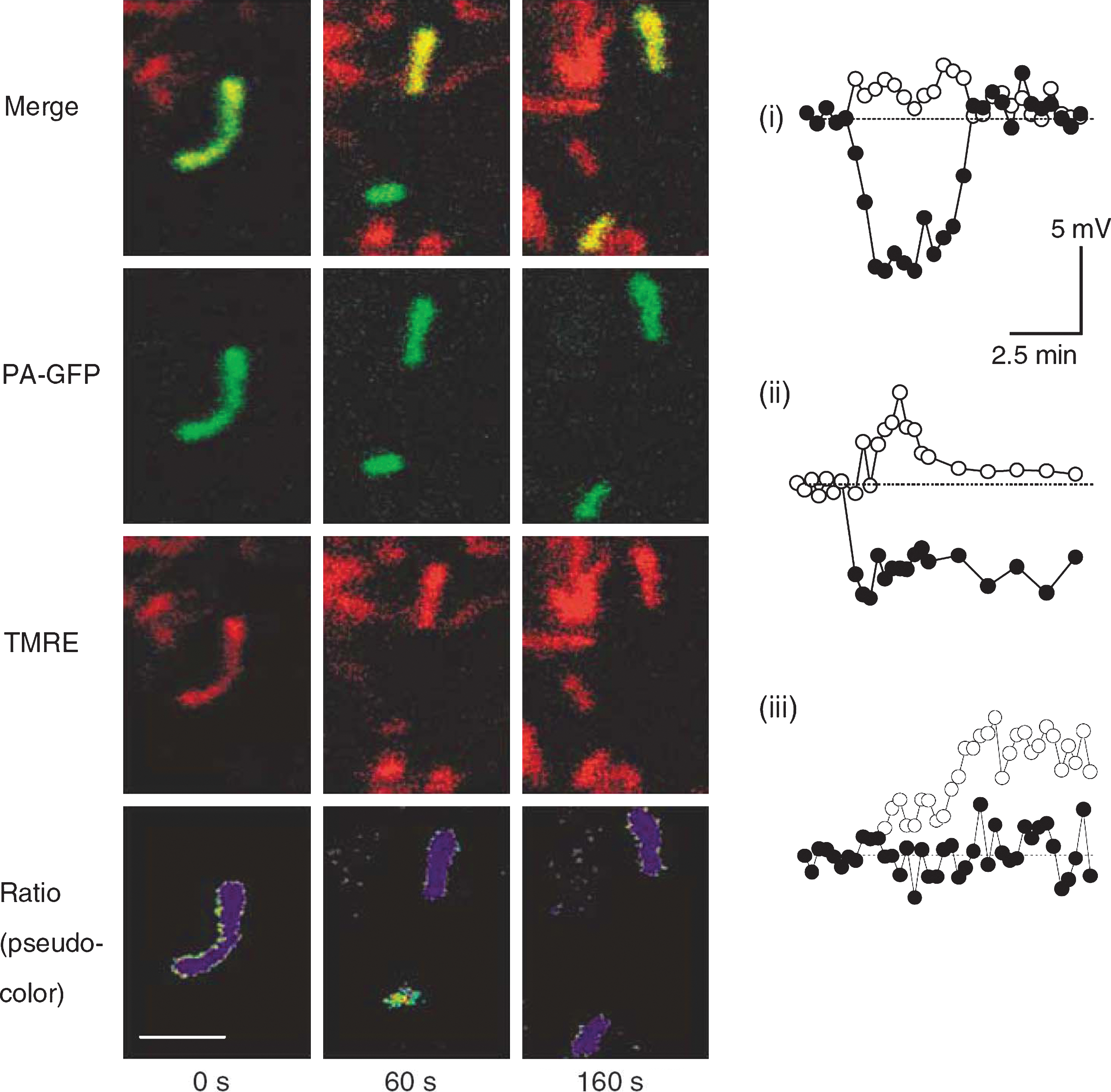
Depolarized mitochondria may therefore be the result of a spontaneous depolarization during the solitary or networked period or during the transition between them. A number of studies have reported on the monitoring of individual mitochondria over time and the observation of a specific depolarization event. Loew et al. provided one of the earliest quantitative measurements of individual mitochondria (using the Δψm-dependent dye TMRE) that were tracked in the z-stack with high temporal resolution. They reported stable Δψm for a period of 40–80 s that could be followed by a drop of >15 mV (58). This pioneering study was, however, limited by the lack of technology to assure that the detected mitochondrion did not go through fusion and/or fission events during the recording time. As fission can occur without movement of the two daughter mitochondria or involve only the inner (but not the outer) mitochondrial membrane (62, 111), it cannot be reliably identified by observation of separation of a mitochondrion into two organelles.
In COS7, INS1, and primary β-cells, prolonged tracking (up to 2 h) revealed that mitochondria retain a stable Δψm during the solitary period (110, 121). During most of the time (95% of the recording period), Δψm of the mitochondrion was within ±2.7 mV of its average baseline. This observation indicates that continuous deterioration in Δψm during the solitary phase is an unlikely (or infrequent) route for the generation of depolarized mitochondria under normal conditions.
Fission-Induced Mitochondrial Depolarization
In contrast to the remarkable stability of Δψm under control conditions during the solitary period, fission events are associated with large changes in Δψm. Interestingly, electron microscope tomography shows that fission events can yield asymmetric daughter mitochondria (5, 64) and unequal nucleoid distribution (3). Direct measurements of Δψm in INS1 cells and COS7 cells show that in the majority of the fission events one of the two daughter mitochondria will leave the fission event with some level of depolarization (Δψm difference >5 mV) (110) (Fig. 1). The depolarization phase is often transient (Fig. 1i, iii), and in only ∼5% of events it is of sustained nature (Fig. 1ii). Given the high frequency of mitochondrial fission in these and other cell lines (47, 113) (a fission event occuring every 22 min or less), mitochondrial fission constitutes a principal route for generating (depolarized) mitochondria that are later targeted by the mitophagy machinery.
Altered Expression of Fusion and Fission Proteins Modify Mitophagy
Table 1 summarizes studies that linked mitochondrial dynamics and mitophagy. The view of mitochondrial fission as a prerequisite for mitophagy is supported by genetic manipulation of the pro-fission proteins Fis1 and Drp1 (Table 1). Knockdown of FIS1 (by siRNA) or overexpression of a dominant negative isoform of DRP1 (DRP1K38A) reduces mitochondrial autophagy exclusively, as confirmed by the lack of an increase in ER mass inside the autophagosomes (APs) (4, 110). Overexpression of hFis1 reduces the mitochondrial (but not ER) mass in HeLa (28, 30) and INS1 cells (83), consistent with the notion that stimulation of fission facilitates mitochondrial autophagy under some circumstances. Arnoult et al. showed that overexpression of Drp1 facilitates mitochondrial elimination under various proapoptotic stimuli (4). Endophilin B1 (also known as Bif-1), a Drp1-dependent mediator of mitochondrial fission (48), interacts with Beclin 1 and colocalizes with AP markers LC3, Atg5, and Atg9 in response to nutrient starvation (105). Its loss is associated with tubulation of mitochondria (48) and suppression of autophagy (105).
Note that in some works mitophagy was assessed directly by colocalization between mitochondrial and AP markers, indirectly as changes in mitochondrial mass, or not distinguished from autophagy.
AP, autophagosome; ER, endoplasmic reticulum; KO, knockout.
Recently, Hailey et al. suggested that under starvation conditions the mitochondrial outer membrane contributes components of the AP (36). In this process, mitofusin 2 (Mfn2) (18) was found to be essential (36). These findings indicate that the proteins that are generally classified as “profusion proteins” have additional roles that affect autophagy. Future studies need to test whether the findings of Hailey et al. can be generalized to other conditions where damaged mitochondria (with potentially damaged components in their membrane) need to be removed by autophagy.
Mitochondrial Fission, Organelle Length, and Mitophagy—Size Does Not Matter
Several studies have linked mitochondrial size to the rate of mitophagy. Studies that visualize mitochondria within APs reported that the organelles' size was small (<1 μm in most cases) in a variety of cell types (27, 31, 37, 75, 77, 112). Yet, reduction of organelle size independently of its energetic status does not trigger mitophagy. Mitophagy rate in INS1 cells is reduced by ∼65%–75% by Opa1 overexpression or by the inhibition of fission (110). The latter is associated with highly ramified architecture, whereas high levels of Opa1 overexpression in these mitochondria are associated with fragmented architecture with an intact fusion capacity and Δψm cellular heterogeneity (70, 110).
Mitochondrial Fragmentation as a Universal Stress Response
The above observations indicate that fission is an event that can alter the energetic state of the mitochondrion and thereby the organelle fate. Although under some conditions, mitochondria hyperfuse as a first line of defense to nonmitochondrial metabolic insults (109), other insults that directly affect mitochondrial metabolism are associated with significant unbalanced fission mainly because of a decreased fusion rate (also referred to as mitochondrial fragmentation). Depletion of ATP either by inhibiting ATP synthase (22, 87), collapsing Δψm (41, 50, 52, 62), or inhibiting the Na+/K+ ATPase (87) triggers general fragmentation of the mitochondrial web due to cleavage of Opa1 (see below). Oxidative stress induced by hyperglycemia (25–50 mM) fragments the mitochondria in cardiac cells (124, 125) and pancreatic β-cells (9). Direct application of hydrogen peroxide is associated with a similar architecture phenotype (60). In an animal model of Alzheimer disease (116, 117) and a culture model of Parkinson disease (17), mitochondrial architecture was fragmented and the relative mitochondrial mass in APs was shown to be selectively increased (71).
Common to the above observations is a stress-induced mitochondrial damage, parallel to fragmentation, and a selective increase of mitochondrial localization in APs. This raises the possibility that mitochondrial fragmentation, which can be induced by various insults, is a common stress response that is principally required to segregate and eliminate dysfunctional mitochondria from the web.
The Dark Side of Excessive and Unbalanced Fission
The above view of fission as a critical process to mitochondrial homeostasis is in accordance with clinical and experimental data testing the role of fission under control conditions. Drp1, a profission protein, is essential for dendritic spine formation during the embryonic period (42). Its mutated form causes fatal encephalopathy in the newborn (118). In neural cultures, Drp1 loss-of-function is associated with reduction in spine mass and altered neural activity (8, 53, 55). Altered respiration was reported in HeLa cells transfected with Drp1 shRNA (7). Altered glucose-stimulated insulin secretion, which is dependent on increased respiration, was reported in pancreatic β cells treated with Drp1 shRNA (110).
On the other hand, excessive and unbalanced fission under a variety of metabolic insults has deleterious effects in a variety of tissues:
Cardiac and skeletal myocytes
Knockdown of Drp1 reduces significantly hyperglycemia-induced apoptosis in H2c9 (124) and HL-1 cardiac cells (125) and prevents ROS formation under increased glucose levels. Knockdown of Drp1 (79) prevents ischemia/reperfusion-induced mitochondrial fragmentation and is associated with reduced cell death in HL-1 cells following ischemia/reperfusion insult. In HL-1 cells, enhancement of autophagy by overexpressing ATG5 protects against Bnip3-mediated cell death, whereas inhibition of autophagy by ATG5K130R enhances cell death (37). These findings suggest a case wherein forcing mitochondria to the fusion state or eliminating mitochondria by autophagy, both have protective effects.
Treatment of adult murine cardiomyocytes with mitochondrial division inhibitor-1 (mdivi1), a pharmacological inhibitor of Drp1, reduces cell death and inhibits mitochondrial permeability transition pore opening after simulated ischemia/reperfusion injury. In vivo treatment with mdivi1 reduces myocardial infarct size in mice subject to coronary artery occlusion and reperfusion (79).
Inhibition of mitochondrial fission during myoblast differentiation is critical for the development of the ramified mitochondrial reticulum in adult myocytes (20). Romanello et al recently suggested that fragmentation of the mitochondrial network triggers the activation of the proteasome-ubiquitin system and the autophagy-lysosome axis, two key players in muscle atrophy (91). In starved mice, knockdown of Fis1 inhibits the activation of MuRF-1 and Atrogin-1 [key genes related to muscle atrophy (94)], results in retained muscle and mitochondrial mass, prevents mitochondrial fragmentation, and decreases autophagy (91). Overexpression of the dominant negative form of Drp1 (Drp1K38A) causes a similar phenotype in muscle with reduced levels of Fis1, even when it is cotransfected with FoxO3, a transcription factor that stimulates mitophagy and remodels the mitochondrial network in atrophying fibers. These results suggest that prevention of fragmentation plays a key role in starvation-induced muscle atrophy. It would be of interest to determine whether overexpression of Opa1 is equivalent to the protective effect of fission inhibition, given its inhibitory effect on autophagy (110), increased fusion rate (57), and its ambiguous effect on mitochondrial architecture as described earlier. In skeletal myocytes, both fusion and fission proteins are induced following exercise. This induction is blunted in type 2 diabetic patients, which makes this experiment physiologically relevant (11, 23, 38).
Pancreatic β-cells
In pancreatic β-cells, exposure to high glucose and fatty acids (also termed glucolipotoxicity) leads to fragmentation of mitochondrial architecture (70). In a type 2 diabetes animal model (ZDF rats), this sequence of events preceded the onset of diabetes (9). We recently showed that knockdown of Fis1 in β-cells prevents the glucolipotoxicity-induced recruitment of Drp1 to mitochondria and restores the ramified mitochondrial architecture as well as the exchange of mitochondrial contents through fusion events. Fis1 knockdown also reduced the level of cell death that is associated with glucolipotoxicity (70). In INS1 and primary β-cells, overexpression of hFis1 or dominant-negative Drp1 reduces both mitochondrial mass and cellular ATP levels by ∼25% and, as a consequence, impairs glucose-stimulated insulin secretion (83). Although ATP is a critical signal for insulin secretion in β-cells, the excessive mitophagy seems to be protective, despite the decrease in mitochondrial mass and overall potential for ATP production. AP formation is increased in β-cells under high-fat diet and prediabetic conditions (26). Mice lacking Atg7 in β-cells show decreased insulin secretion, morphological and functional abnormalities, and faster onset of diabetes under high-fat diet (26, 46).
Neurons
Excessive fragmentation is associated with several chronic and acute neuropathological conditions. In Alzheimer disease, immunoblot analysis of brain tissue reveals that levels of Opa1, Mfn1, and Mfn2 are significantly reduced, whereas levels of Fis1 are significantly increased. Oligomeric amyloid-beta–derived diffusible ligands induce mitochondrial fragmentation as well as mitophagy-mediated reduction in mitochondrial density in the neuronal processes, subsequently leading to loss of dendritic spines (12, 116). In a culture model of Huntington disease, cytotoxicity induced by expanded polyglutamine tracts is mediated, at least in part, by excessive mitochondrial fission (115). Mutations in PINK1 cause autosomal recessive Parkinson disease. These pathogenic mutations also cause a defect in mitochondrial dynamics that can be reversed by the mitochondrial fission inhibitor, mdivi-1 (16), or by overexpressing the dominant negative form of Drp1 (17). In a different model of neuronal cell death, mitochondrial fission was shown to be an early event in ischemic stroke in vivo and in nitric oxide-induced oxidative stress, two conditions that are associated with neural death. In these models, inhibition of fission is associated with neural protection (5, 74). In a glaucoma model, exposure of differentiated retinal ganglion cells (RGC5) to elevated hydrostatic pressure results in mitochondrial fragmentation and decreased ATP (45).
Stem cells
Todd et al. reported recently a novel connection between mitochondrial morphology and homeostasis of mouse embryonic stem cells (108). Knockdown of growth factor erv1-like (Gfer) in embryonic stem cells leads to decreased embryoid body formation, elevated Drp1 levels, and increased mitochondrial fragmentation and mitophagy.
Mitochondrial Fusion—A Selective Rescue Mechanism
As stated earlier, mitochondrial fusion allows diffusion of matrix and membrane components between the two fusion mates (matrix components at a faster rate than the membrane ones). This may serve as a complementary “rescue” mechanism or a first line of defense from autophagy for damaged mitochondria. Thus, fusion may recruit dysfunctional mitochondria into the active pool, whereas autophagy targets depolarized mitochondria for digestion and elimination. This theoretically places mitophagy and fusion as competing fates of the depolarized mitochondria. The principal question is, therefore, what is the selective barrier that prevents any exchange between dysfunctional mitochondria and the intact ones in a manner that would otherwise diminish the efficient segregation of dysfunctional material by mitophagy.
Mitochondria fail to fuse when the Δψm is dissipated by a mitochondrial membrane uncoupler (41, 52, 62, 66 –68). By labeling a group of mitochondria through laser photoconversion of matrix-targeted PAGFP and observing them over time, one can identify nonfusing mitochondria as those that do not share their photocoverted PAGFP. These mitochondria do not dilute the fluorescent signal and can be identified as the brightest mitochondria within the population. Nonfusing mitochondria are found even when numerous mitochondria with intact potential are in their close vicinity (110). Costaining with TMRE or with an anti-Opa1 antibody reveals that nonfusing mitochondria are relatively depolarized compared to average Δψm and their OPA1 content is reduced by ∼50% (110).
Opa1 is a protein with multiple isoforms that is localized in the inner membrane and in the intermembrane space of mitochondria (33, 78). However, it is also involved in outer mitochondrial membrane fusion, as Mfn1 is required for Opa1-mediated fusion (13, 35, 96). The biochemical mechanisms that link Opa1 processing to bioenergetic parameters have been described in different studies. In mammalian cells, the long isoforms (high molecular weight) of Opa1 undergo cleavage (or degradation) when depolarization is induced or ATP is depleted (25, 32, 40, 57, 101). As both long and short isoforms of Opa1 are required for mitochondrial fusion (101), a decrease in the driving force for ATP synthesis (i.e., Δψm depolarization) triggers mitochondrial fragmentation by processing and complete degradation of Opa1 isoforms. Taken together, these findings suggest that mitochondrial fusion is selective for polarized, active mitochondria and that this selectivity, in parallel with asymmetric fission events, determines the fate of the single mitochondrion (survival or degradation).
Selectivity of Mitochondrial Fusion Indirectly Supplies Mitochondria to the Mitophagic Pool
The selectivity of the fusion machinery makes fusion and mitophagy complementary processes, rather than competing ones. There are currently no experimental means to modify the degree of fusion selectivity or to accurately and stably change the rate of dynamic events over time. Recently, the contribution of selectivity and frequency of mitochondrial fusion to mitochondrial function was dissected by an analytical simulation (72) based on the kinetics of mitochondrial dynamics in INS1 β-cells (39, 110, 113). The computer-based simulation predicted mitochondrial activity under changing rates of mitochondrial dynamics and selectivity of the fusion machinery. The program simulated repetitive cycles of fusion and fission events in which intact and damaged mitochondrial contents were redistributed between fusion mates (see Fig. 2 legend for a more detailed description of the model). Redistribution had an impact upon mitochondrial function, thereby influencing the fate of each mitochondrion, that is, to be either destined for subsequent fusion or elimination by autophagy.
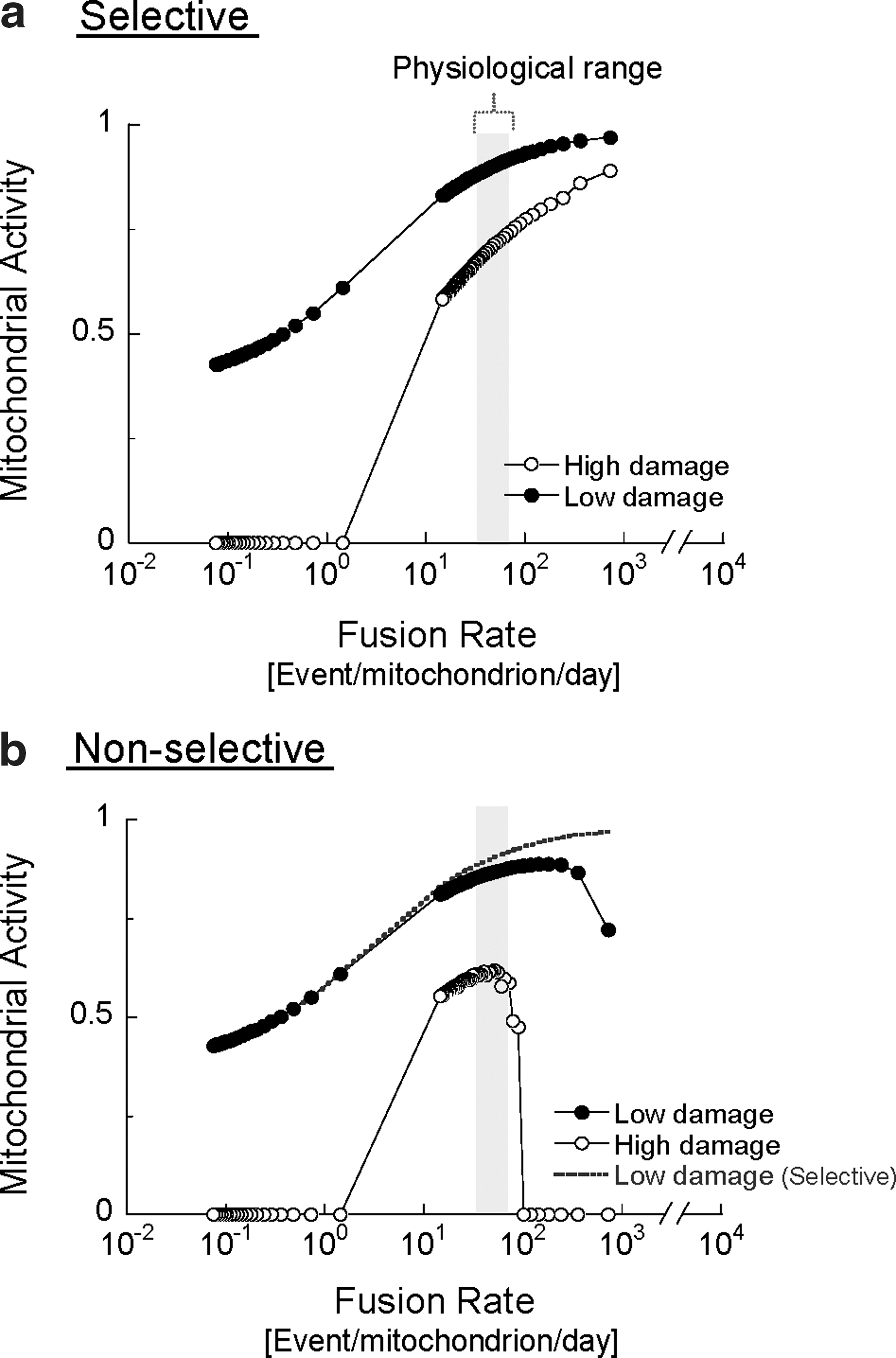
Figure 2 shows the effect of changing the rates of fusion–fission frequency on mitochondrial activity under low- or high-damage rates. The illustration depicts the case of selective fusion (i.e., a mitochondrion can fuse only if its activity is above a certain threshold) and the case of nonselective fusion (fusion occurs independently of mitochondrial activity). Increasing the frequency of either selective or nonselective fusion results in improved mitochondrial activity. This positive relationship, however, is limited to a gap of fusion frequencies for the nonselective case and the gap narrows as the rate of damage increases.
In the case of selective fusion, segregation of the damaged mitochondrion from the fusing population prevents the migration of damaged components into more active mitochondria, but also, and equally important, leaves them available for autophagy. The simulation suggests that the beneficial value of this behavior becomes more apparent under high fusion rates. If fusion is nonselective at high rate of fusion, the duration spent in the solitary state is shortened and, thereby, the probability of autophagy of depolarized mitochondria is reduced (see figure legends) (14, 77, 102 –107). Thus, autophagy is indirectly inhibited when most units are in the fused state, which results in the accumulation of mitochondria containing damaged content (hence reducing the overall activity score of the population). In the case wherein fusion is selective, high fusion frequencies do not prevent the removal of damaged mitochondria, because these mitochondria avoid fusion and are, therefore, constantly accessible to the autophagic machinery.
These findings emphasize the importance of selectivity of mitochondrial fusion not only as an intramitochondrial complementary route, but also as an isolation step preceding autophagy. This property could be viewed as a mechanism that allows the cell to compensate for high rates of mitochondrial damage by increasing fusion frequency without compromising the mitophagy of dysfunctional mitochondria.
Fusion, Fission, and Autophagy as a Bioenergetic Quality Control Mechanism
The observation that mitochondrial fusion is a Δψm-dependent process ensures that potentially dysfunctional organelles will avoid fusion, whereas intact mitochondria will benefit from sharing metabolites. Segregation of dysfunctional mitochondria from the fusing population results in the generation of small and depolarized mitochondria. The finding that autophagy targets depolarized mitochondria places autophagy at the end of the axis of quality control as a receiver of the segregation output. Figure 3 summarizes schematically how the combination of fusion, fission, and mitophagy act as a quality control mechanism. Importantly, this view suggests that the absolute rate of fusion and fission events per se (and not merely the balance between fusion and fission) determines the efficiency of the proposed quality control axis (in accordance with the data presented in Fig. 2). As fission frequency increases, the cumulative probability for the generation of dysfunctional units with sustained depolarization also increases. Under these circumstances, it is expected that the mass of damaged mitochondria that have been segregated from the fusing population and eliminated by mitophagy will be increased.

Mitochondrial Ubiquitin Ligase (PINK1/Parkin, MARCH5, and MULAN) as Molecular Regulators of Selective Fusion
Recent studies of the substrates of mitochondrial ubiquitin ligases, including MULAN, MARCH5, and Parkin, identify them as potential critical mediators that govern selective fusion and mitophagy. Parkin was shown to translocate selectively to energetically impaired mitochondria that were destined to autophagy independent of ROS production and morphology (75, 114). The translocation of Parkin is induced by PINK1 (29, 82). The latter undergoes voltage-dependent proteolysis in polarized mitochondria, but accumulates on depolarized mitochondria overtime (65, 76). This cascade of events accounts for the direct relationship between the level of Parkin on mitochondria and their membrane potential (75).
Studies in Drosophila report that upon recruitment to depolarized mitochondria, Parkin ubiquitinates Mfn (126). In mammalian cells, additional E3 ligases as MULAN and MARCH5 modify mitochondrial dynamics, and in the case of the latter, ubiquitinates Mfn1 (54, 84). Knockout of Parkin or MARCH5 leads to accumulation of Mfn or Mfn1 and to mitochondrial elongation (49, 84, 89, 123, 126). These results suggest that in the absence of PINK1/Parkin or MARCH5, damaged mitochondria are “rescued” by fusion instead of being removed by mitophagy. This compromises mitochondrial quality maintenance. In support of this view, knockdown of Mfn in Drosophila or Mfn1 in HeLa cells was protective from the knockdown of Parkin or MARCH5, respectively (84, 88, 89).
It should be mentioned that the effects of manipulations on PINK1/Parkin system had ambiguous effects on mitochondrial morphology. In Drosophila and COS7 cells, mutants for PINK1 and Parkin had elongated mitochondria (19, 81, 88, 123), whereas in other mammalian systems this manipulation resulted in fragmented phenotypes (17, 59, 93). In these studies, the compromised energetic state induced by PINK1/Parkin mutants could be rescued by preventing mitochondrial fragmentation, for example, by inhibition of fission. (16, 59, 81, 88, 93). The reasons for these conflicting reports remain to be fully elucidated, but, thus far, both technical factors (59) and tissue-specific differences may contribute.
We hypothesize that the reduction in the rate of ubiquitination is finally associated with decreased mitophagy and an increase in unselective fusion (fusion between damaged and metabolically intact mitochondria). Figure 4 illustrates schematically this hypothesis under PINK1/Parkin KO. Under these conditions, mitophagy rate is decreased and damaged mitochondria can reengage (at least to some extent) in fusion events. In a very recent study, Tanaka et al. showed that upon membrane depolarization Parkin mediates selectively the ubiquitination and proteasomal degradation of Mfn1 and Mfn2 by the AAA+ ATPase, p97 (106a). This may provide a mechanism for selective fusion and the segregation of depolarized mitochondria prior to autophagy. Following fission, Parkin prevents or delays refusion of depolarized mitochondria by the elimination of mitofusins and in parallel triggers the accumulation of proteins that facilitate mitophagy. This study also finds that in the absence of fission, Parkin accumulation and mitofusin degradation alone are insufficient to induce mitophagy.

Footnotes
Acknowledgments
The authors are grateful to Drs. Marc Liesa, Linsey Stiles, and Dani Dagan for helpful comments on earlier versions of this manuscript. The authors thank Mrs. Erga Rivis for excellent technical support in producing the schematic illustrations.