
Abstract
Introduction
All modern organisms possess a variety of mechanisms to react upon an imbalance in the redox state. These mechanisms split into stress response processes, which grant the survival of a cell during an acute stress situation, and balancing processes, which continuously maintain the redox state and compensate naturally occurring levels of oxidants and reductants (8, 21).
Upon an oxidative burst, the eukaryotic cell slows down cell division and growth, and induces several enzymatic as well as nonenzymatic antioxidative defense systems. These processes help to restore the natural redox balance (21, 24), and repair damage that has already occurred (24, 40). Although many biochemical details of the defense systems have been elucidated, the sensor systems that monitor the redox state and are responsible for induction and control of the redox-state balancers are only marginally understood. The best studied mechanism is the transcriptional induction of stress response genes via transcription factors that are regulated by redox-sensing cysteines. The oxidation of the thiol function in cysteine, whether directly or indirectly by the transfer of redox equivalents from a regulatory interaction partner (such as the glutathione peroxidase 3 [Gpx3]), induces the expression of antioxidant enzymes (3, 15, 41). However, such a system is not capable of responding to stress caused by reducing agents and this indicates that additional sensors exist.
Innovation
The pentose phosphate pathway (PPP) is a known donor of NAPDH during oxidative stress conditions. Here, we provide evidence for a second role of the pathway during the antistress response. The deletion of yeast PPP enzymes caused growth phenotypes on oxidant-containing media that were not attributable to aberrant NADPH formation, and had defects in the regulation of gene expression upon an oxidant-treatment. Since augmented PPP flux provoked gene expression profiles that resembled oxidative stress conditions, it can be concluded that the PPP is a metabolic redox sensor and involved in adapting the transcriptome to counteract oxidative stress.
Beside transcriptome and proteome, also the metabolome has been identified as an integral component of the cellular stress defense machinery (12). A major target under oxidative conditions is the pentose phosphate pathway (PPP), a pathway in primary carbon metabolism that is closely interconnected with glycolysis. The PPP divides into an oxidative and a nonoxidative branch, two independently operating metabolic modules (45) (Fig. 1a, and Table 1 for overview of reactions/enzymes). The oxidative branch is composed of irreversible reactions and reduces NADP+ to NADPH. In contrast, reactions of the nonoxidative branch are reversible, and neither NAD(H) nor NADP(H) dependent.
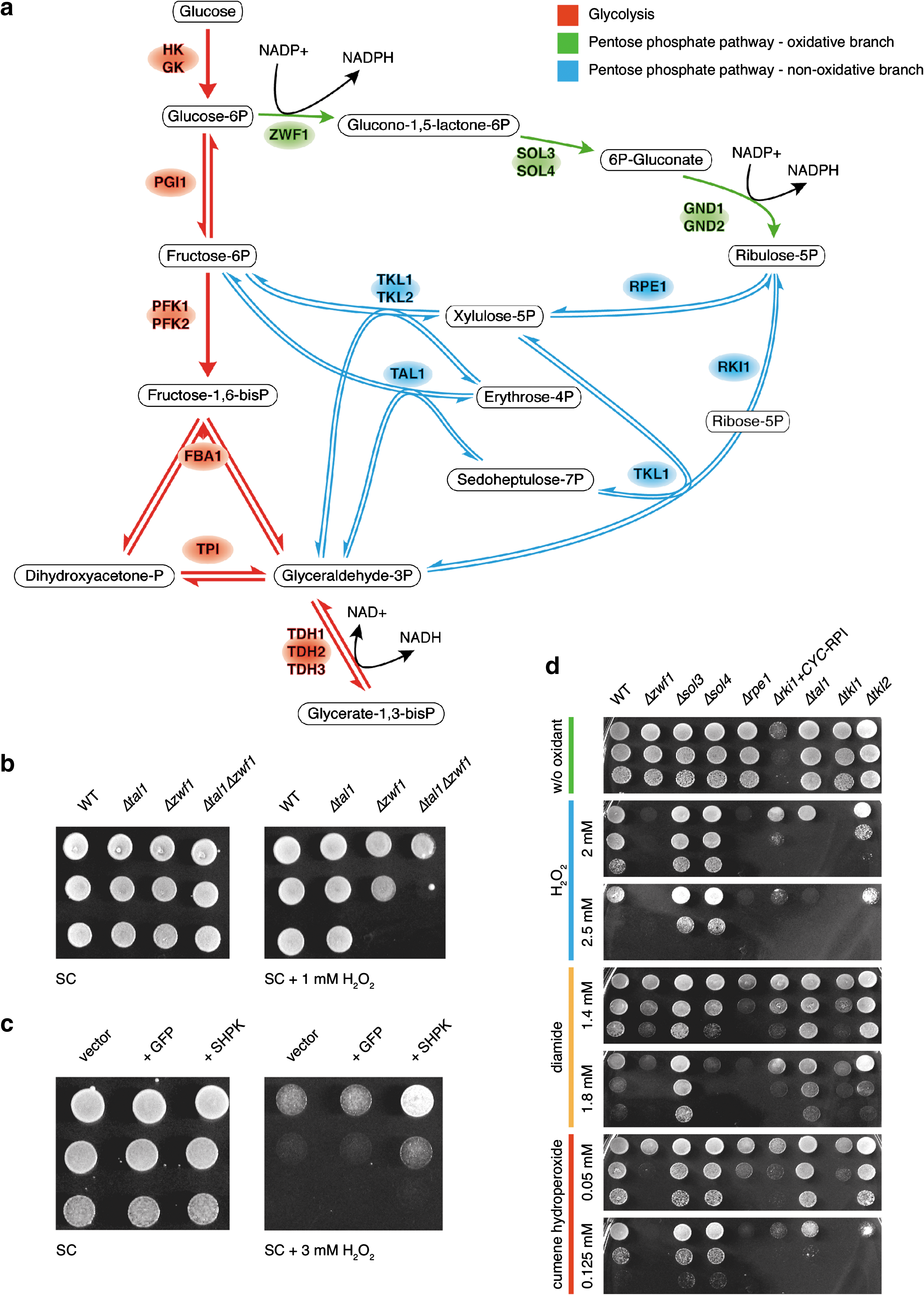
Treatment with an oxidant blocks glycolysis, but glucose uptake continues, leading to an increased metabolic load of the redox-resistant PPP during the initial phase of the stress response (16, 33, 37). Yeast cells exposed to hydrogen peroxide (H2O2) show a strong increase in the concentration of PPP intermediates. In addition, oxidative treatments induce PPP enzymes on transcript and protein levels (7, 10, 37). The benefits of this metabolic transition are presently explained by the production of NADPH in the oxidative PPP branch, the primary redox-cofactor for glutathione synthesis and for enzymes of the antioxidative machinery (11, 17, 29). Cells with an increased influx to the PPP have a higher NADPH/NADP+ ratio and are more resistant to oxidants (33). Vice versa, deletion of the first and rate limiting enzyme of the oxidative PPP, the glucose 6-phosphate dehydrogenase (G6PDH, Zwf1 [ZWischenFerment]), causes hypersensitivity to multiple oxidants in various organisms (14, 17). The redox-protective effects of the PPP are also diminished when the NADPH oxidizing enzyme GDP1 (an NADP(H)-dependent form of glyceraldehyde-phosphate dehydrogenase found in the fungus Kluyveromyces lactis) is expressed (33, 42). Castegna and colleagues determined NADPH/NADP+ ratios in wild-type and Δzwf1 cells, without and upon the addition of H2O2, by liquid chromatography tandem mass spectrometry (LC-MS/MS). In the wild-type strain, NADPH/NADP+ ratio fall from 1.5 to ∼1.0 after adding H2O2 for 60 min. The Δzwf1 mutant had a ratio of 1.0, which collapsed to <0.2 when exposed to H2O2 (4). Hence, NADPH is not sufficiently recovered in cells deficient for the oxidative PPP when exposed to H2O2.
Here, we demonstrate that the PPP has a second function in the oxidative stress response. We describe redox phenotypes of PPP enzyme mutants that are not a result of altered NADPH metabolism, and found evidence that the PPP is an integral component of the gene expression regulome during stress conditions. Our data, showing disturbed regulation of gene expression during the stress response in PPP deficient yeast, support the hypothesis of PPP being a metabolic redox sensor. Further, we provide evidence that the glycolysis/PPP transition is essential for functioning timing of antioxidative mRNA and protein expression.
Results
An NADP(H)-independent role of the PPP during the antioxidant response
The requirement of the PPP during the oxidative stress response has been explained with its function as source of redox equivalents in form of NADPH. However, even though NADPH is solely produced in the oxidative branch of the PPP, sensitivity toward oxidants has also been reported for mutants deficient in nonoxidative PPP enzymes (17). Further, in our previous LC-MS/MS studies we observed accumulation of nonoxidative PPP intermediates upon exposing cells to oxidants (33, 34). Together, these observations indicated that the involvement of the PPP in the stress response might go beyond its function as NADPH donor.
To investigate this hypothesis, we first generated double-deletion strains lacking one of the nonoxidative PPP enzymes, TKL1, RPE1, or TAL1, and the limiting entry enzyme of the oxidative PPP (Zwf1). Since the oxidative part of the PPP is not reversible, Δzwf1 strains cannot use the PPP to reduce NADP+. Δtkl1Δzwf1 and Δrpe1Δzwf1 double mutants were not viable (Supplementary Fig. S1; Supplementary data are available online at
Complementary to this experiment, we then increased the flow into the non-oxidative PPP by transgenic expression of sedoheptulokinase (SHPK). SHPK is a mammalian enzyme, not present in yeast, and converts the non-PPP sugar sedoheptulose to the central nonoxidative PPP intermediate sedoheptulose 7-phosphate (18). As illustrated in Figure 1c, yeast cells having this second route to produce nonoxidative PPP intermediates were more resistant to H2O2 treatment than wild-type cells and isogenic controls expressing GFP. Thus, distortion of the nonoxidative PPP decreased the cellular tolerance to H2O2, whereas additional flow into this pathway resulted in an increased redox tolerance.
The investigations were continued by comparing the resistance of various PPP deletion mutants to multiple oxidants. Oxidants vary in respect of their primary targets and redox (Nernst) potential, and therefore, each compound triggers a specific cellular response (39). This raised the question whether the deletion of oxidative and nonoxidative PPP enzymes provokes divergent/specific or similar redox phenotypes. Isogenic mutants of both the oxidative (zwf1, sol3, and sol4) and the nonoxidative (rpe1, rki1, tal1, tkl1, and tkl2) PPP branches were spotted as serial dilution series on agar plates with increasing concentration of the oxidants H2O2, diamide, and cumene-hydroperoxide (CHP). As shown in Figure 1d, yeast strains deleted for zwf1, rpe1, and tkl1 were sensitive to the three tested oxidants; Δtkl2 and Δsol4 selectively to at least one. In contrast, lack of sol3 increased the overall oxidant tolerance. The remaining strains had divergent phenotypes: Δtal1 cells were sensitive H2O2 but resistant to diamide, Δsol4 cells were resistant to H2O2 and CHP, but sensitive to diamide. A strain with lowered ribose 5-phosphate isomerase (RPI) activity [a Δrki1 knockout is unviable, the strain with reduced Rpi activity was engineered by low-level expression of the human Rki1p ortholog RPI1A in a RKI1 null strain (44)] showed slow growth on control plates, but its growth was largely unaffected by the oxidant treatments.
To test if these phenotypes were specific to oxidants, we tested the resistance of Δzwf1, Δsol3, Δsol4, Δrpe1, Δtal1, Δtkl1, and Δtkl2 yeast against other stressors. The strains were spotted on media with different concentrations of dithiothreitol (DTT) to simulate a reductive stress, NaCl for salt stress, sorbitol for osmotic stress, and MnCl2 for a heavy metal stress. Except slight growth advantages on NaCl, no other stress phenotypes of PPP deletion strains were observed (Supplementary Fig. S2). Further, we measured the ratio of [NADPH] to [NADP+ + NADPH] (=NADPHtotal) in cell extracts of nonoxidative PPP knockout strains. These ratios were unchanged compared to wild-type under the chosen growth conditions, indicating that the altered oxidant resistance of nonoxidative PPP knockouts is not the result of lack or excess of NADPH (Supplementary Fig. S3).
In summary, depletion of both oxidative and nonoxidative PPP enzymes strongly affected oxidant resistance of yeast, but did not cause broad growth phenotypes with unrelated stressors. Remarkably, depleting enzymes of both PPP branches produced specific phenotypes. For instance, Δtal1 yeast was sensitive to H2O2, resistant to diamide, and tolerated CHP like the wild-type. A comparable phenotype was not seen among deletion strains of oxidative PPP enzymes.
Taken these findings together, it could be demonstrated that the PPP plays a specific role in the oxidative stress response, and that not only its NADPH-producing oxidative branch but also its nonoxidative part is required for maintaining the natural oxidant tolerance of yeast. It was therefore concluded that the function of the PPP in stress response is not restricted to provide NADPH.
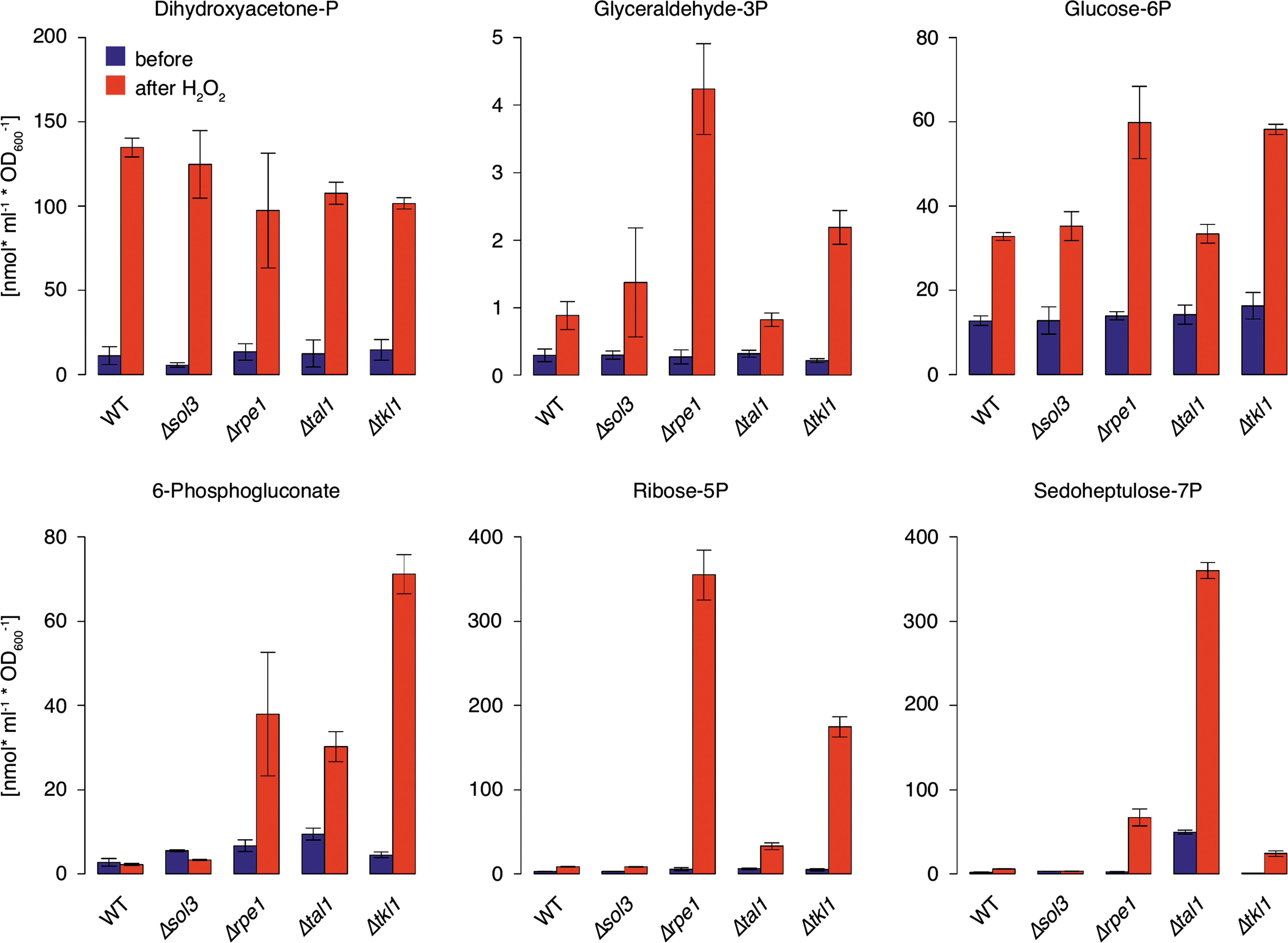
Redox-sensitive mutants accumulate nonoxidative PPP intermediates upon an oxidative burst
To investigate whether redox sensitivity of non-NADP+ reducing PPP deletion mutants correlates with metabolic changes, which occur during the stress response, six PPP intermediates were quantified using LC-MS/MS. The three most redox-sensitive nonoxidative PPP deletion strains (Δrpe1, Δtal1, and Δtkl1) as well as the most resistant (Δsol3) strain and their wild-type parent BY4741 were profiled. Additionally, these metabolites were quantified after exposure to H2O2. As reported earlier (33), H2O2 treatment triggered an increase in the concentrations of PPP intermediates in the wild-type strain. Similar metabolic changes were observed in Δsol3 yeast (Fig. 2). However, this response was substantially different in Δtal1, Δrpe1, and Δtkl1 cells. Here, PPP intermediates 6-phosphogluconate, ribose 5-phosphate, and sedoheptulose 7-phosphate accumulated up to 100-fold compared to the wild-type. Thus, it can be deduced that a block in the nonoxidative PPP, which causes oxidant sensitivity, has a strong impact on the concentration of its intermediates during an acute stress response.
The glycolysis/PPP transition induces transcriptional rearrangements that are part of the antioxidant response
The glycolysis/PPP transition is induced very quickly; a raise in the concentration of PPP intermediates is detected within seconds after the oxidative burst. This fast induction is attributable to oxidative inactivation of glycolytic enzymes (34, 37). Since changes in transcript levels are detected much later (minutes) (7) and metabolic pathways are in intensive crosstalk with the gene expression machinery (12), we speculated that the early metabolic shift could be connected to the regulation of gene expression during the stress response.
To pursue this hypothesis, we profiled the transcriptome of a mutant yeast strain (MR105) that has increased activity of the PPP due to reduced activity of a central glycolytic enzyme, triosephosphate isomerase (Tpi1).
MR105 is deleted for endogeneous TPI1, and expresses a mutant allele (TPIIle170Val) from a centromeric plasmid (31). The TPIIle170Val allele, identified as heterozygous polymorphism in TPI deficiency (1), has 30% of the wild-type TPI activity. Previously, we have shown that PPP intermediates are increased in MR105, and found that this strain is resistant to oxidants and has an increased NADPH/NADP+ ratio (31, 33).
The transcriptome analysis was performed to compare changes in gene expression that follow the activation of the PPP, induced by the TPI mutation or by H2O2 exposure. Global mRNA expression profiles were obtained by quantitative shotgun sequencing on a Genome Analyzer II (Illumina). The wild-type strain with and without H2O2, the MR105 strain, and yeast strains deleted for the PPP enzymes Tal1, Tkl1, and Zwf1 were profiled. cDNA was synthesized from total RNA using an oligo-dT primer and processed to obtain strand-specific sequencing profiles, which were obtained using a 51-bp paired-end sequencing-by-synthesis approach as described earlier (26). Each experiment yielded 10–14 million single reads, which could be aligned to 97% of the annotated yeast open reading frames. Compared to the mutant strains, grown without interruption, the oxidant treatment caused broader rearrangements in the transcriptome as illustrated by Pearson's pairwise correlation of all profiles (Fig. 3a, upper panel) and individual scatter plots (lower panel). By normalizing the transcriptome of the TPI mutant MR105 and the H2O2-treated strain, significant overlaps of the transcriptome profiles were detected. About 40% of the targeted genes were shared between the TPI mutant and the H2O2-treated strain (Fig. 3b). These two unrelated conditions, both increasing the concentration of PPP intermediates, triggered expression changes in an overlapping number of genes.

Whereas H2O2 treatment creates an oxidative environment, a drop in TPI activity shifts the NADPH/NADP+ ratio toward a reducing environment (33). Therefore, it was possible that this overlap was an indirect consequence of redox-balancing processes. However, it was found that only 60 of the 140 transcripts correlated directly with the redox state (were upregulated in the H2O2-treated sample and downregulated in the TPI mutant or vice versa [Fig. 3b]). The larger number of transcripts (80), however, responded equally to the H2O2 treatment and the reduced TPI activation. Thus, a large fraction of the transcriptional changes seen upon H2O2 treatment did not directly correlate with the redox state, but with the shift in primary carbon metabolism.
The expression tendency of these genes in the PPP deletion strains was studied to assess whether their regulation was a consequence of altered PPP activity. The RNASeq expression profiles of Δtal1, Δtkl1, and Δzwf1 yeast were hierarchically clustered with the profiles of the genes regulated upon a H2O2 treatment or in the TPI mutant. Interestingly, a substantial fraction of transcripts targeted by H2O2 treatment were also found targeted in the PPP mutants (Fig. 3c). We noticed that the profile of this set of genes in Δtkl1 cells clustered closer to the H2O2 profile than the TPI strain profile (Fig. 3c, left panel). To exclude the potential effects of a changing redox state, we narrowed the analysis to transcripts regulated similarly in the TPI and H2O2 profiles (Fig. 3c, right panel) and detected strong overlap between the TPI/H2O2 set and the PPP deletion strains: 71% of the transcripts correlating with the activation of the PPP were found altered in all three, and 92% in at least two, of the profiled PPP deletion strains. This demonstrates that transcriptional changes induced by H2O2, which correlate with a shift in primary carbon metabolism, respond to a depletion of PPP enzymes. By performing a gene ontology (GO) comparison of these transcripts, using the targets of reduced TPI activity and H2O2 as background set, GO IDs 6333 (chromatin assembly and disassembly) and 45333 (cellular respiration) were found to be enriched (p<0.01, Fig. 3c, lower panel). The other gene sets implicate the metabolic transition in the regulation of protein biosynthesis. Comparing the list of all redox-state correlating transcripts using the entire transcriptome as background, GO terms relating to translation and protein biosynthesis were enriched (Fig. 3d, upper panel). Reversely, transcripts directly responding to the metabolic transition were enriched for its preceding processes, namely, the assembly and biosynthesis of ribosomes (Fig. 3d, lower panel).
The metabolic transition regulates the expression of respiratory chain and chromatin components during the stress response
With given evidence for a role of the PPP in transcriptional regulation upon an oxidative burst, it was then examined how the targeted transcripts may behave upon its perturbation. The expression of identified targets belonging to the two enriched GO categories (Fig. 3c), cellular respiration (transcripts COX1, COX2, COX3, and CIT1) and chromatin assembly and disassembly (transcripts HHT1+2, HTA1+2, HHF1+2, and HHO1), was analyzed by quantitative real-time-polymerase chain reaction (qRT-PCR) and targeted proteomics. Control transcripts for internal normalization (ACN9, ATG27, and TAF10) were selected from the RNASeq data, fulfilling the criteria that they were minimal affected in their expression levels, neither by the H2O2 treatment nor by the TPI- or PPP mutations. Triplicate time courses for the expression of all transcripts in exponentially growing wild-type, Δzwf1, Δtkl1, and Δtal1 strains were generated by determining their relative expression levels before and 3, 5, 10, 15, and 30 min upon the addition of H2O2. The relative expression levels were normalized to the geometric mean of the EΔCt values of three reference transcripts (28).
The H2O2 treatment provoked downregulation of mitochondrial-encoded cytochrome c oxidase (COX) genes to ∼50% of their initial level in wild-type cells, whereas citrate synthase (CIT1) was virtually unaffected for 15 min (Fig. 4a, upper panel).
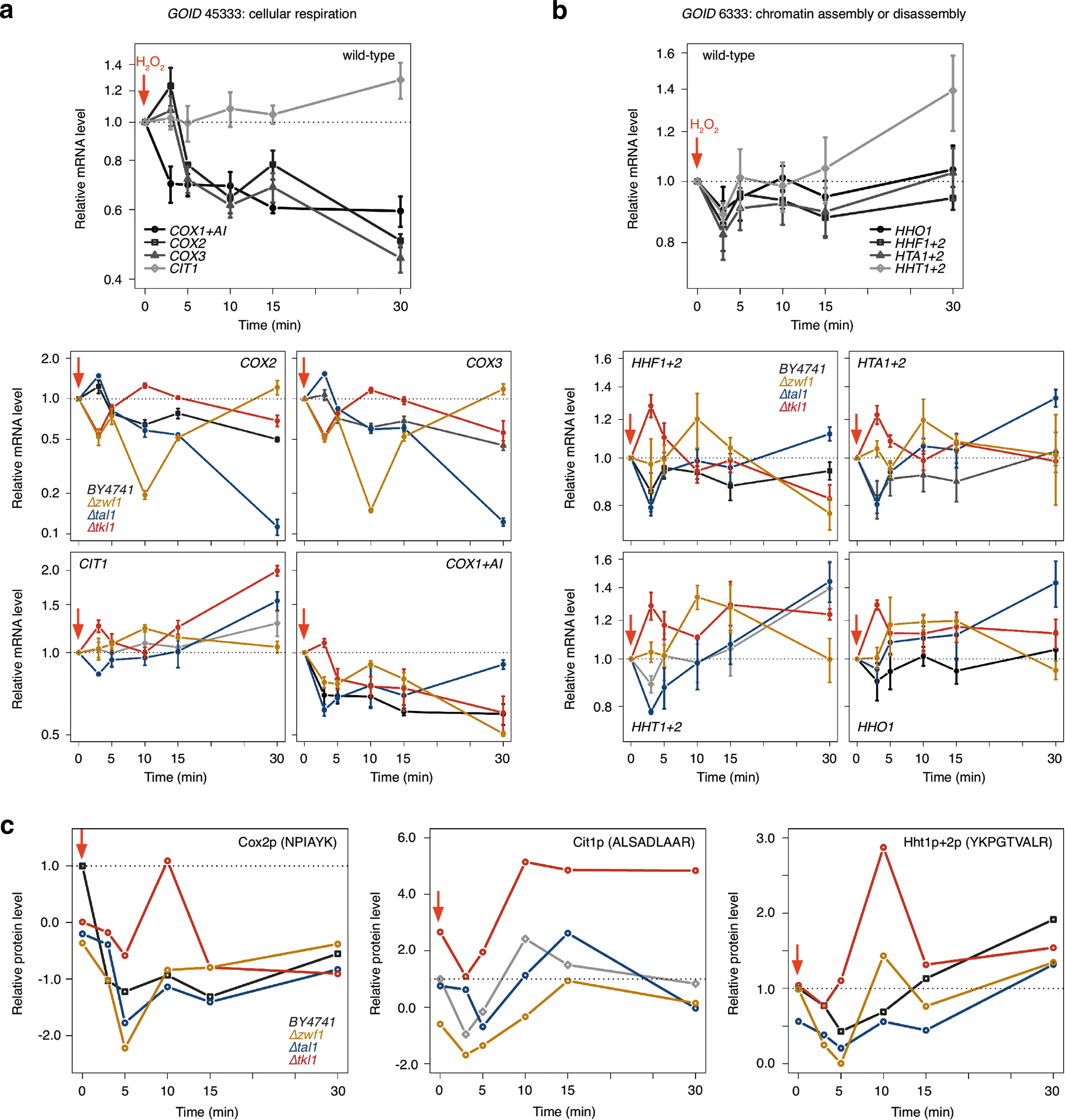
Remarkably, the expression courses of these transcripts were strongly disturbed in the PPP deletion strains (Fig 4a, lower panels). In Δzwf1 yeast, COX2 and COX3 mRNAs lost ∼80% of their initial levels within 10 min. At the same time, these transcripts were upregulated in the Δtkl1 strain. In contrast, in Δtal1 yeast, COX3, COX2, and COX1 transcripts [the latter also encodes for a reverse transcriptase AI (19)], maintained the wild-type trend for 15 min, but then their expression levels dropped drastically. The time courses for CIT1 were primarily affected by the removal of TAL1 and TKL1. In these strains, CIT1 accumulated stronger and faster than in the wild-type situation.
In the next step, a mass Western approach was used to verify whether these transcriptional changes were also represented on protein level. Multiple reaction monitoring (MRM) experiments with internal normalization were performed on a nanoLC system coupled to a triple quadrupole/hybrid ion trap mass spectrometer (QTRAP5500) as described earlier (2). A Cox2p and a Cit1p-specific peptide was quantified in wild-type, Δzwf1, Δtkl1, and Δtal1 strains upon H2O2 treatment for 0, 3, 5, 10, 15, and 30 min.
As shown in Figure 4c, like the corresponding transcripts, the abundance of Cox2p and Cit1p specific peptides were affected by the deletion of PPP enzymes. The protein expression trend was much more stable in the wild-type strain. The concentration varied strongly in the PPP mutants, or was delayed as in the case of CIT1 transcript levels in Δtal1 cells.
To further corroborate the influence of PPP deletions in oxidant-induced gene expression, a similar analysis was performed with members of the second enriched GO term, chromatin assembly and disassembly, containing yeast histone genes. In difference to metazoans, yeast histone transcripts are polyadenylated and are therefore present in oligo-dT-primed cDNA. Figure 4b illustrates the wild-type time-course of histone mRNA expression upon the addition of H2O2 to an exponentially growing yeast culture. In the wild-type strain, changes in histone gene expression were very moderate. Transcripts encoding HTA1+2 (analyzed with a primer pair that amplifies both paralogs), HHF1+2 and HHO1, exhibited a slight decline in their expression directly upon the addition of the oxidant. Their levels then stabilized rapidly and returned to basal levels. The trend in HHT1+2 expression levels was identical during the first minutes, but then increased slightly and continuously until the end of the experiment.
Depletion of PPP enzymes also disturbed their time-course. At time points <10 min (=all histone mRNAs were decreased in the wild-type), they were increased in Δtkl1 cell; after 10 min, all PPP deletion strains showed abnormalities in their expression trends. After 30 min (i) HTA1+2 transcripts were at wild-type levels, (ii) HHF1+2 transcripts were downregulated, or (iii) upregulated (HHT1+2 and HHO1+2 transcripts). Their course in Δzwf1 cells was comparable, but regulation of histone mRNA expression in this strain had a delayed onset. The Δtal1 course, which followed the wild-type trend initially, was above wild-type levels in the end of the experiment.
Thus, alike transcripts of respiratory metabolism, stress-induced regulation of histone mRNA expression was disturbed upon the depletion of PPP enzymes. Although the relative differences in expression level were moderate when compared to those of the respiratory metabolism components, they were pictured on the protein level. In the wild-type strain, the level of an Hht1p+2p-specific peptide decreased during the first 10 min after addition of the oxidant, then stabilized, and reached wild-type levels and more (Fig. 4c, right panel). In contrast, in Δtkl1 and Δzwf1 yeast, the Htt-peptide accumulated and reached a maximal signal 10 min after the oxidative burst, and returned to a wild-type-like concentration at min 15–30.
In summary, the different time-course experiments reveal defects in timing and induction of gene expression during the stress response when different steps of the nonoxidative PPP are disturbed. Thus, the activation of the PPP, which occurs upon an oxidative burst, is required for accurate regulation of gene expression during the stress response.
Discussion
The metabolic network is of modular architecture and closely interconnected with transcriptional machinery (12, 13). This is necessary to balance metabolic perturbations or to react to a changing nutrient status, and thus changes in the metabolic network can stimulate the transcriptome and proteome (5, 12, 13). Recent discoveries unveil the metabolic network as an integral component of the cellular signaling system, and it is becoming clear that metabolic control goes far beyond self-regulation of metabolic pathways. For instance, it has recently been shown that inhibition of the eicosanoid pathway promotes pluripotency in embryonic stem cells, and, vice versa, the substrates of pro-oxidative reactions (e.g., acyl-carnitines) promote neuronal and cardiac differentiation (48)
The PPP, a biochemical module within the central carbon metabolism, is of major importance in the chemical redox balance of the cell. When cells get in contact with an oxidant, this pathway is upregulated at the transcript, protein, enzyme-activity, and metabolite level (10, 16, 33, 37). As the oxidative part of the PPP reduces two NADP+ molecules to NADPH for every metabolized glucose equivalent, this metabolic re-configuration provides the cellular antioxidative machinery (including glutathione and peroxiredoxin systems) with its primary redox cofactor (29, 49). This role of the PPP in the stress response is consistent with several experimental observations, including that the depletion of PPP enzymes can dramatically reduce the oxidant-resistance of yeast and other organisms (17).
The data presented here provide evidence for an additional, and NADP(H)-independent role of the PPP in the stress response. The LC-MS/MS measurements showed that intermediates of the nonoxidative PPP accumulate upon an oxidative treatment (33, 34), and the deletion of several nonoxidative PPP reactions impacts on the resistance against oxidants (17, 39).
We demonstrated that the deletion of a nonoxidative PPP enzyme (Tal1p) decreased oxidant sensitivity in a yeast strain that cannot use the PPP for NADP+ reduction. Vice versa, increased flow into the nonoxidative PPP via the expression of a foreign enzyme (SHPK), which generates the PPP intermediate sedoheptulose-7P from the non-PPP metabolite sedoheptulose, increased yeast's H2O2 tolerance. Additionally, the exposure to different oxidants revealed divergent phenotypes among deletion strains of the oxidative and nonoxidative PPP branch. Testing three oxidants, we identified a phenotype for all assayed PPP enzyme knockouts. These phenotypes were specific, and occurred irrespectively whether the deleted enzyme was part of the oxidative and nonoxidative branch. All together, these results gave reason to postulate the existence of a second, NAPD(H)-independent function of the PPP in the stress response.
A unique feature of PPP activation is the speed at which this process occurs. An increase in the concentration of intermediate metabolites is detectable within seconds upon the cell's exposure to the oxidant (34). This rapid flux is triggered by the oxidative inactivation of at least one glycolytic enzyme, glyceraldehyde 3-phosphate dehydrogenase (GAPDH); GAPDH inactivation is completed within 10–15 s upon cellular contact with the oxidant (34) Both mathematical modeling and experimental results showed that GAPDH inactivation is capable to increase the concentration of PPP intermediates (33).
Transcriptional adaptation to oxidative conditions is much slower than this metabolic response. In yeast, early transcripts are induced ∼2–3 min after exposure to the oxidant. All later transcriptional changes adhere to a strict timeline, resulting in characteristic time-courses for stress-induced mRNAs (7). A strong relation between the binding affinity of a transcription factor and the activation timing of its targets has been reported (6), but most mechanisms that are crucial for the dynamic timing of the stress program remain yet to be elucidated. A cell proceeds with its transcriptional program after the metabolic transition. Therefore, we considered that this process could be connected to the induction of transcriptional changes during the stress response. The comparison of mRNA expression profiles of yeast treated with oxidants and a TPI mutant with a constitutively activated PPP revealed that both conditions had a substantial overlap in their set of targets. Studying the expression of these genes in other PPP deletion strains demonstrated a PPP dependence of their transcriptional regulation.
Currently, the best understood sensing system detects a collapse in the redox state via oxidation of thiol groups in regulatory cysteines, present in oxidant-responsive transcription factors, or their binding proteins (3, 15, 41). The results presented here are consistent with this model, but also demonstrate that this current picture is still incomplete. The oxidation of sensory thiols cannot explain expression changes provoked by the activation of the PPP when no oxidant was added to the cells. For instance, one or more intermediates of the PPP could have a direct regulatory function and bind to stress responsive transcriptional regulators, thereby leading to their activation. The so-called reporter metabolites, metabolic intermediates with regulatory functions that entail transcriptional changes, have been found in several pathways, including glycolysis, but have not been discovered among PPP intermediates yet (12, 27). This assumption could be proven by the identification of one or more regulatory small molecule(s) among PPP intermediates, and by elucidating its/their interaction with the stress responsive regulome. An alternative possibility is classic, post-translational modifications and/or protein–protein interaction-based signaling cascades that might be initiated by a PPP enzyme. Such a mechanism, for instance, has been reported for GAPDH that activates mTOR signaling under conditions of high glycolytic flux. In the presence of high levels of the GAPDH substrate glyceraldehyde 3-phosphate, a complex between this enzyme and the mTOR activator Rheb is destabilized; released Rheb is then capable to activate the mTORC1 complex (23)
The primary targets of the metabolic transition were processes involved or directly dependent on energy metabolism. When PPP mutants were exposed to oxidants, transcripts related to ribosome biogenesis and translation [considered as the most energy-consuming processes of the cell (47)], subunits of the respiratory chain, and chromatin components did not show expression time-courses like in the wild-type strain.
This could have consequences on stress signaling, since dynamic changes in chromatin structure are central to mediating energy deprivation; histone modifications are under the direct control of the central carbon metabolism (46). Chromatin is central for spreading response to metabolic changes and stress (20). Heat stress, for instance, leads to a domain-wide displacement of histones. This stimulates the expression of heat shock genes independent from the activity of Swi/Snf transcriptional regulators (50). Similar processes are likely to play a role during the oxidative stress response and help the cell to establish and maintain a system-wide antioxidative response.
We provided examples of transcripts that were improperly regulated or timed upon the oxidant treatment of PPP-deficient yeast strains. For instance, HHT1 mRNA levels are slightly decreased in the wild-type cells 3 min after exposure to the oxidant but reached the basal level 2 min later. The same process took much longer (15 min) in Δtal1 yeast (Fig. 4b). In most cases, the effects of the metabolic shift were strongest within the first 15 min after addition of the oxidant. This result matches the time-course of the metabolic transition itself, which has its amplitude quite early (∼1 min) after an oxidative burst (34). The observations reveal that the metabolic transition plays its primary role in controlling gene regulation during early events of stress response. Later, this function may be taken over by other mechanisms. Indeed, some of the transition-responsive gene promoters contain binding sites for known oxidant-inactivated transcription factors [e.g., the AP-1-like transcription factor YAP1 (15)]. Thus, gene expression regulation by the metabolic shift (glycolysis/PPP) and redox-responsive transcription factors seem to be complementary and/or overlapping processes that are induced at a different kinetic. In this respect, it is possible that the PPP is involved in the regulation of gene expression upon a strong oxidative burst, but not during the endurance of chronic, low-level oxidative stress.
In summary, we demonstrated that a metabolic transition in central carbon metabolism is part of the cellular redox sensor and a regulatory component of the early antistress response. Not only does the increased metabolic activity of the PPP, a consequence of carbon flux rerouting that occurs upon the addition of a toxic oxidant concentration, increase the reduction rate of NADP+, but at the same time, it is also required for accurate induction of the transcriptional stress response. Consequently, the PPP has a pivotal role upon an oxidative burst; it provides redox cofactors for the antioxidative machinery and is a regulator of gene expression (Fig. 5). This finding extends our knowledge regarding the induction of stress response with a dynamic, time-dependent system showing for the first time that a shift in a metabolic pathway is used to sense and transmit a stress stimulus. The current understanding of the dynamics in the cellular metabolome and the knowledge regarding functional interactions between small molecules of primary metabolism with regulatory macromolecules are still marginal. However, the dynamic nature of metabolic pathways make them ideal candidates to sense and control flexible systems, and, consequently, similar mechanisms could play a common role in the regulation, fine-tuning, and timing of dynamic biological processes.

Materials and Methods
Yeast strain generation and cultivation
Yeast was cultivated in yeast-extract peptone dextrose (YPD; [20 g/l peptone BD Difco], 10 g/l yeast extract [BD Difco], 2% glucose) or synthetic complete (6.7 g/l yeast nitrogen base with ammonium sulfate [Wickerham formula; BD Difco], 0.59 g/l complete supplement mixture [CSM-ADE -HIS–LEU–TRP–URA, MPBiomedicals], 10 mg/ml adenine, 20 mg/l uracil, 20 mg/l histidine, 60 mg/l leucine, 40 mg/l tryptophan) agar (20 g/l), or liquid media at 28°C–30°C. Oxidant-bursts in liquid cultures were induced by adding H2O2 to a final concentration of 2 mM. BY4741 (S288c)-descendant yeast strains deleted for ZWF1, TKL1, TAL1 (35), transgenic yeast expressing TPIIle170Val (33), and low-level RPI (44) were described previously. RPE1 and TKL2 were deleted by a single-gene-replacement approach using the nourseothricin-resistance marker natMX4, and SOL3/SOL4 using kanMX4. The Δtal1Δzwf1 double mutant was generated by replacing ZWF1 in the Δtal1 yeast with kanMX4. Because a similar approach failed to create Δtkl1Δzwf1 and Δrpe1Δzwf1 double-knockouts, Tkl1p and Rpe1p were expressed from an URA3 centromeric plasmid in Δtkl1 or Δrpe1 cells. Δzwf1 was deleted in these transformants by single-gene replacement with kanMX4. Counter selection with 0.15% 5-fluoroorotic acid of TKL1 or RPE1 encoding URA3 plasmids demonstrated a synthetic-lethal phenotype of Δtkl1Δzwf1 and Δrpe1Δzwf1 double-knockouts (Supplementary Fig. S1).
An URA3 2μ plasmid for the expression of GFP (p426GPD-GFP) was described earlier (32). The vector for the expression of SHPK was generated by PCR amplification of the mouse SHPK coding sequence from an E. coli expression plasmid (18) and sub-cloning it into the URA3 2μ vector p426GPD (25). The generated plasmid was verified by sequencing.
NADP(H) and NADPH measurements
For determination of the ratio of NADPH to the total NADP(H) concentration, yeast pellets were obtained from exponentially growing YPD cultures. Nucleotides were extracted by vigorous shaking of the frozen pellet on a Fast-Prep-24 machine (MP Biomedicals) after covering it with water:phenol:chloroform and zirconium beads. After centrifugation, the aqueous phase was transferred to a fresh tube and cleaned with phaselock gel (5Prime) to remove remaining organic solvent. Then, high-molecular components were removed by filtration through a 3-kDa-cutoff spinfilter (Millipore). NADPH and NADP(H) total concentrations in the filtrate were analyzed with a colorimetric NADP+/NADPH assay kit (Abcam ab65349).
Expression profiling by deep sequencing of the transcriptome
For obtaining expression profiles, five yeast cultures (2×BY4741, Δtal1, Δtkl1, and Δzwf1) were grown in parallel in YPD media to mid-log phase. Thirty minutes before collecting the cells, H2O2 was added to one BY4741 culture to a final concentration of 2 mM. The cultures were centrifuged, pellets were washed, and total RNA was isolated using the RiboPure-Yeast kit (Ambion). mRNA was transcribed into cDNA and libraries for strand-specific Illumina sequencing were prepared as described earlier (26). In brief, polyA+ RNA was purified with the Dynabeads mRNA purification kit (Invitrogen) following the manufacturer's instructions and treated with TURBO DNase (Ambion) (0.2 units per 1 μg of RNA) for 30 min at 37°C. The first-strand synthesis reaction was performed with 0.5 μg of polyA+ RNA, d(N)6 primers and an oligo(dT) primer, reverse transcription buffer, dNTPs, MgCl2, DTT, actinomycin D, RNase, OUT'inhibitors, as well as SuperScript III reverse transcriptase (Invitrogen). dNTPs had been removed using a self-made 200 μl G-50 gel filtration spin-column (26) before the second strand was synthesized with DNA polymerase I as described.
Sequencing of the cDNA yielded 5–7-million paired-end reads (length 51 bp) and 10–14-million single reads. These reads were aligned using bowtie (22) on yeast genome SGD1.01 (
Quantitative RT-PCR
Two μg total yeast RNA was transcribed into cDNA using a 12–18 oligo dT primer and Moloney Murine Leukemia virus (M-MuLV) reverse transcriptase (NEB) according to the manufacturer's instructions. qRT-PCR were performed on an Prism 7900HT cycler (Applied Biosystems) with gene-specific primers (Table 2) as described previously (44). The relative expression ratio of the target genes was normalized to the geometric mean of the three endogenous references genes (ACN9, ATG27, and TAF10) by the method of Pfaffl (28). Each assay was performed in independent triplicate runs with templates from independent reverse transcription reactions.
Quantitative mass spectrometry
Sugar-phosphate intermediates were quantified by LC-MS/MS as described by Wamelink et al. (43). In brief, metabolites were extracted in Hanks balanced salt solution containing 2% perchloric acid, and spiked with an isotope-labeled internal standard (13C6-glucose-6P). Proteins were precipitated after neutralization with a phosphate buffer. Then, samples were then separated with a linear water/acetonitrile gradient using octylammonium acetate (500 mg/l, pH 7.5) ion pairing reagent on a C18 column at a flow rate of 1 ml/min. The on-line coupling of the HPLC system to an API-3000 mass spectrometer (AB/Sciex), operating in MRM mode, allowed quantification of sugar-phosphate intermediates.
Peptide quantification was conducted on a hybrid triple quadrupole/ion trap mass spectrometer coupled to a 2D ultra nanoLC (Eksigent) as described previously (2). In brief, whole yeast protein extracts were digested with trypsin and separated with a 90 min water/acetonitrile gradient on a Zorbax 300SB-C18, 0.0075×150 mm column (Agilent) at a flow rate of 300nl/min. Quantification was carried out on a QTRAP5500 (AB/Sciex) instrument equipped with Nanospray III ion source (AB/Sciex) operating in MRM mode. Each peptide was quantified with two MRM transitions and internally normalized to a tryptic peptide of 60S ribosomal protein L36-B (RPL36B). MRM (Q1/Q3) transitions are given in Table 3.
Footnotes
Acknowledgments
The authors would like to thank Beata Lukaszewska-McGreal, Erwin Jansen, and Serkan Ceyhan for help with mass spectrometry and sample preparation, our lab members for critical discussions, and Emile van Schaftingen (Université catholique de Louvain) for providing a mouse SHPK plasmid. Part of this work was funded by the Max Planck society.
Author Disclosure Statement
All authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.