
Abstract
Claudins are a family of nearly two dozen transmembrane proteins that are a key part of the tight junction barrier that regulates solute movement across polarized epithelia. Claudin family members interact with each other, as well as with other transmembrane tight junction proteins (such as occludin) and cytosolic scaffolding proteins (such as zonula occludens-1 (ZO-1)). Although the interplay between all of these different classes of proteins is critical for tight junction formation and function, claudin family proteins are directly responsible for forming the equivalent of paracellular ion selective channels (or pores) with specific permeability and thus are essential for barrier function. In this review, we summarize current progress in identifying structural elements of claudins that regulate their transport, assembly, and function. The effects of oxidant stress on claudins are also examined, with particular emphasis on lung epithelial barrier function and oxidant stress induced by chronic alcohol abuse. Antioxid. Redox Signal. 15, 1179–1193.
Introduction
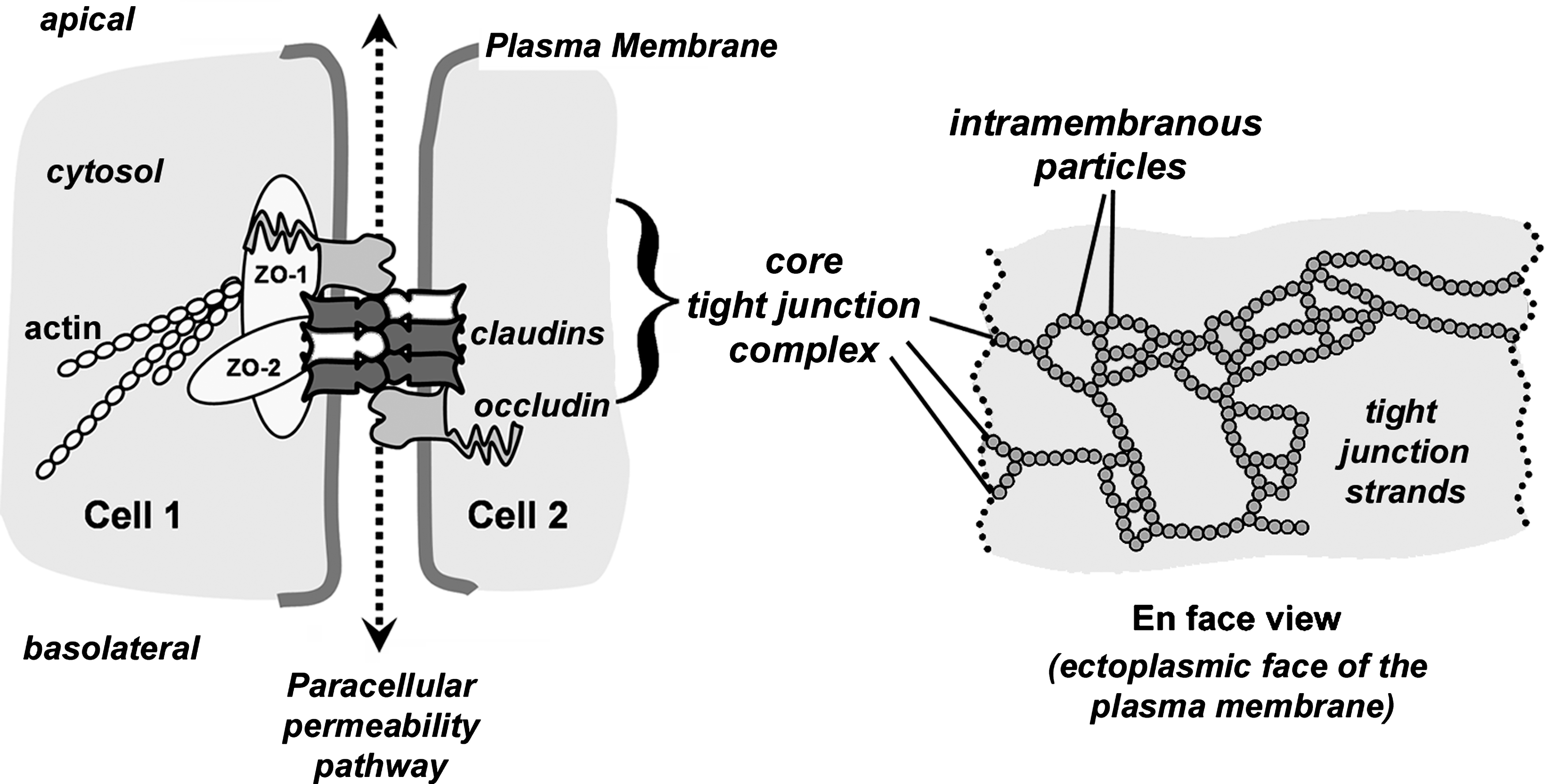
Tight junction protein complex
Tight junctions are composed of several different classes of proteins which interact in a coordinated manner to form epithelial barriers. Of these, the major functional transmembrane proteins that confer specific paracellular barrier properties are tetraspan proteins known as claudins (3, 5, 73). The indispensable role for claudin-family transmembrane proteins in forming tight junctions was demonstrated by showing that strands of intramembranous particles appear when claudin-null fibroblasts are transfected to express different claudins (40).
For claudins to form functional tight junction strands requires additional proteins. Most notably are the cytoplasmic scaffold proteins ZO-1 and ZO-2 which bind directly to claudins and link them to the actin cytoskeletal network (Fig. 1) [for review, see Gonzalez–Mariscal et al. (42a)] (121). Another tetraspan transmembrane protein, occludin, interacts directly with claudins and helps to regulate tight junction formation [reviewed by Blasig et al. (15a)]. While claudins, occludin, ZO-1, and ZO-2 can be thought of as a core tight junction protein complex, specialized tight junctions contain other proteins. For instance, tricellulin, which is structurally similar to occludin, promotes tight junction assembly by interacting with occludin and remains associated localized to sites where three cells are in direct contact (74, 132). Tight junctions are also regulated by other plasma membrane proteins which are not part of the core tight junction complex, such as single pass transmembrane immunoglobulin superfamily proteins such as junction adhesion molecules (77) [reviewed by Bazzoni (11a)]. Despite the complex network of proteins which are needed to properly regulate the paracellular barrier, claudins are clearly essential tight junction proteins.
Claudin family proteins and paracellular permeability
To date, 23 human claudins have been identified, including two splice variants (Fig. 2). Epithelial and endothelial cells express anywhere from two to ten claudin isoforms in a tissue-specific manner; expression of multiple claudins by the same cell provides the ability to form tight junction strands of unique composition. Thus, claudin composition determines whether tight junctions will be relatively permeable, as is the case for kidney epithelium (47), or relatively impermeable, as is the case for microvascular endothelium of the blood–brain barrier (120).

Claudins fall into two functional subcategories: 1) claudins which specifically increase paracellular permeability through the formation of paracellular channels (pore forming claudins), and 2) claudins which more generally reduce paracellular permeability (sealing claudins) (73). Of the pore forming claudins, one of the best characterized is claudin-2 (cldn-2), which was initially discovered to cause Madin Darby Canine Kidney (MDCK) cells to assume a “leaky” phenotype (38) and was subsequently found to form pores specific for monovalent cations (1) and water (104). Other pore forming claudins include cldn-7 and cldn-15 and −16. Cldn-10 is a particularly interesting pore forming claudin in that the two mRNA splice variants, cldn-10a and cldn-10b, form pores with preferential permeability for ions of opposite charge (125).
Claudins that have been associated with a more general barrier tightening function, include cldn-1, −3, −5, −11, and −19. However, the ability of these claudins to enhance barrier function is not universal. For instance, in the vascular endothelium, cldn-5 is a critical component to maintain the blood brain barrier [reviewed by Carrano et al., (17a) and Lehner et al. (77a)] (88). While cldn-5 has also been associated with a tightening of the paracellular barrier in other cell types (2, 131), this is not universal since increased cldn-5 expression by lung airway and alveolar epithelia correlates with decreased barrier function (25, 32, 127). The most likely explanation for this disparity is that cldn-5, and claudins in general, are sensitive to the tight junction microenvironment and their function depends upon interactions with other claudins present. Thus, the context of claudin expression needs to be considered in addition to the absolute level of expression when considering their effects of paracellular permeability.
Claudin Structure
Claudins are proteins that are composed of four transmembrane domains, two extracellular loops, a short cytosolic N-terminus, and longer cytosolic C-terminus. Although detailed high resolution structures for claudins are not yet available, the four transmembrane domains are predicted to form individual alpha helices which span the membrane bilayer (Fig. 3). Although typically drawn as an extended “snake” in the membrane, the alpha helices of claudins are much more likely to be tightly packed, based on structures of other tetra-span membrane proteins, such as Cluster of Differentiation protein 81 (CD81), and Connexin26 (Cx26) (81, 107) and modeling of the claudin second extracellular loop domain (EL2) (99).
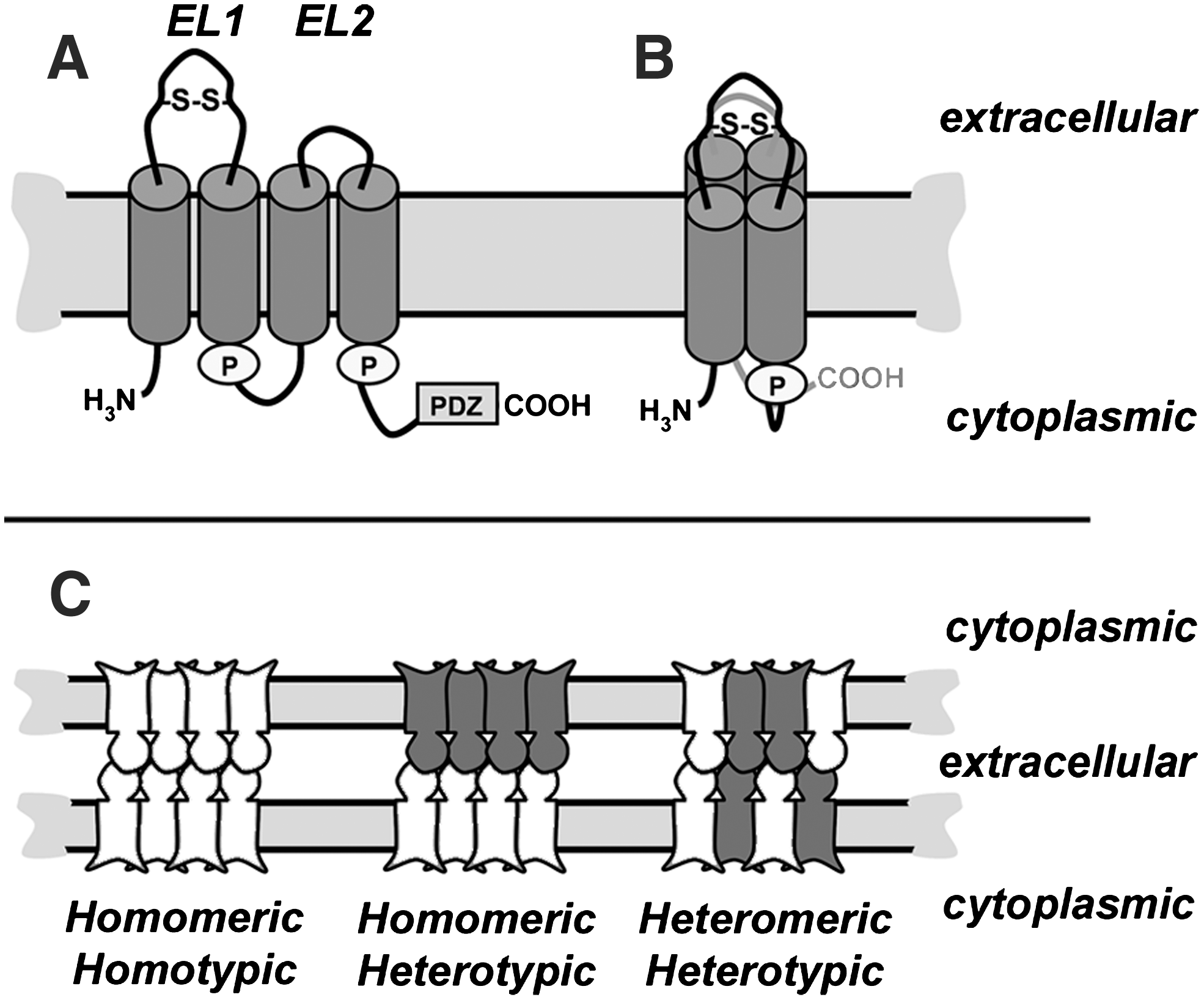
Claudins are oriented in the membrane so that they have both N- and C-terminal cytosolic domains. The N-terminus is short, ∼7 amino acids, and has no known function. By contrast, the C-terminus varies in both length (21 to 63 amino acids) and sequence between the various claudin isoforms. Despite sequence divergence amongst family members, nearly all claudin family members contain a Post-synaptic density 95, Discs large, ZO-1 (PDZ) binding motif in the C-terminal tail (122). The PDZ binding motif interacts most notably with the scaffold proteins ZO-1 and ZO-2, which tethers claudins into tight junction strands by linking them to the actin cytoskeleton (121, 123). However, other scaffold proteins such as multi-PDZ domain protein (MUPP)-1 (50), and Pals1 Associated Tight Junction protein (PATJ) (103) have been found to associate with claudins and regulate their function.
The first extracellular loop is ∼ 50 amino acids and is responsible for the selective permeability of tight junctions (23, 131). Additionally, the first extracellular loop also contains a conserved motif [Gly-Leu-Trp-x-x-Cys-(8–10aa)-Cys] which also is found in the closely-related peripheral myelin protein 22 (PMP22), membrane protein 40 (MP40), and the epithelial membrane proteins (EMPs) (68). It is hypothesized that the cysteines in this motif form a disulfide bond which stabilizes the secondary structure of the first extracellular loop. While the presence of a disulfide bond in the first extracellular loop has not been conclusively demonstrated, the paracellular barrier function of cldn-2 is resistant to thiol-reactive agents, such as methanethiosulfonate (8). In addition, mutating either or both of the cysteines in the first extracellular loop to serine disrupts the ability of cldn-5 to form high resistance tight junctions (131).
A disulfide bond in the extracellular loop of claudins is expected to be necessary for intercellular binding to occur, analogous to the requirement for disulfide bonds in connexin extracellular loop domains (11). Thus, the first extracellular loop represents a site which could be sensitive to changes in redox potential (61). The second extracellular loop is smaller (∼16 amino acids) and has been modeled to form a helix-turn-helix structure (99). The selectivity of claudin paracellular permeability is predominantly dictated by the first extracellular loop domain, however, both extracellular loop domains are critical for the regulation of claudin–claudin interactions. Claudin motifs which regulate the ion selectivity of paracellular diffusion and formation of tight junction pores have been discussed in recent reviews (3, 5, 73); here we will focus on interactions between claudins and pathways for claudin assembly into tight junction strands.
Interactions Between Claudins
Homotypic and heterotypic interactions
Claudin extracellular loop domains bridge the space between adjacent cells at tight junctions. While it is known that claudins on abutting cells interact with each other, the specific characteristics of these interactions are an active area of study.
Head-to-head interactions between claudins on opposing cells are known as either homotypic or heterotypic, depending on whether they are between the same type of claudin or between different claudin gene products. By and large, most claudins seem to interact homotypically, based largely on co-localization at cell–cell contact sites when expressed by fibroblasts or other claudin-null cells. However, this has not been exhaustively examined.
Heterotypic interactions are much less frequent (Table 1). To date, the only heterotypic claudin interactions that have been characterized have involved cldn-3 (25, 33, 41). By contrast, several pairwise combinations of claudins have been found to be heterotypically incompatible, even though they are heteromerically compatible when expressed by the same cell (e.g., cldn-3 and cldn-4 or cldn-16 and cldn-19). In addition to the claudins listed in Table 1, there is also evidence from transfected MDCK cells that cldn-2 and cldn-8 are heterotypically incompatible, although this is difficult to interpret since cells transfected with cldn-8 show decreased expression of endogenous cldn-2 (6).
One of the few claudins shown to be heterotypically incompatible with cldn-3 is cldn-4, despite the fact that these two claudins have nearly identical extracellular loop domains (33). Heterotypic incompatibility was found to require a single amino acid residue, Asn44 which, when converted to Thr, created a mutant form of cldn-3 which was able to interact with cldn-4 (33). Notably, this point mutation did not ablate the ability of cldn-3(N44T) to interact with cldn-1 or cldn-5, underscoring that several elements of the extracellular loop domains help mediate heterotypic interactions. Consistent with these findings, a conserved motif in the second extracellular loop of cldn-5 contains several bulky hydrophobic amino acids which have been shown to stabilize antiparallel homotypic interactions (99) that could also play a role in stabilizing heterotypic interactions as well.
The functional ramifications of heterotypic claudin compatibility are just beginning to be explored, however, it is most likely to enable epithelial cells to fine tune their paracellular permeability, comparable to heteromeric gap junction channels (24). As an example of this, MDCK cells transfected to overexpress cldn-3 showed a decrease in cldn-2 based cation pore activity (84), which could be due to heterotypic interaction between these two compatible claudins (41). Interestingly, MDCK cells overexpressing cldn-3 had comparable paracellular water pore activity as untransfected MDCK cells (104), indicating that pores formed by heterotypic cldn-2/cldn-3 had unique properties unattainable from a homomeric cldn-2 channels. By contrast, claudins with limited or no heterotypic compatibility should be able to form pores which are less influenced by other claudins present in tight junction strands. This could be important to enable an acute change in paracellular permeability, such as that recently demonstrated for cldn-4, which is upregulated in response to acute lung injury (70, 134). A complete heterotypic compatibility profile will help determine whether this is the case.
Heteromeric interactions
In addition to head-to-head interactions, claudins also interact side by side in the plane of the membrane, referred to as homomeric and heteromeric interactions. Homomeric interactions have been most extensively studied using cldn-5 (15, 99). Using fluorescence resonance energy transfer (FRET), which offers the ability to assess claudin homomers in the native context of the cell membrane, C-terminal Cyan Fluorescent Protein (CFP) or Yellow Fluorescent Protein (YFP) tagged cldn-5 proteins were shown to interact at a distance of <6 angstroms at the plasma membrane (15, 99). Importantly, the C-terminal tag blocked the ability of these claudins to interact with ZO-1 or ZO-2, ruling out a contribution of the tight junction scaffold in mediating homotypic interactions.
By contrast with heterotypic interactions, heteromeric interactions are more frequent. For example, cldn-3 and cldn-4 heteromerically interact, as do cldn-16 and cldn-19, despite the fact that these pairs are heterotypically incompatible. In fact, heteromeric interactions are likely to be required for these pairs of claudins to form functional tight junction strands by enabling a homotypic/heteromeric complex to form.
Cldn-16 (110) and cldn-19 (67) have both been implicated in familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC). When co-expressed in LLC-PK1 cells, cldn-16 and cldn-19 acted synergistically to produce sodium-selective pores in the tight junction barrier rather than magnesium-selective pores (53). Thus, FHHNC is associated with a decrease with sodium permeability rather than a direct change in magnesium, which suggests that when either cldn-16 or cldn-19 are disrupted in the kidney, the defect in magnesium resorption is indirect and due to a decrease in transepithelial potential by decreased sodium permeability. Interestingly, incorporation of cldn-16 to tight junctions depends on cldn-19 and vice versa, suggesting that these claudins act as co-chaperones (53). Other claudins expressed by the kidney, such as cldn-10a and cldn-18, retain the ability to form tight junctions, regardless of whether cldn-16 or −19 are present, consistent with a model where subsets of claudins are coordinately regulated.
How the specificity of claudin hetero-oligomerization is regulated by the kidney remains an open question. In particular, a yeast two-hybrid assay was used to demonstrate that cldn-16 is unable to homo-oligomerize and seems to be restricted to hetero-oligomerization with cldn-19, whereas cldn-19 homo-oligomerizes and also hetero-oligomerizes with cldn-10a and cldn-18 (53). Assessing claudin compatibility using yeast two-hybrid assays should be interpreted with caution; for instance, cldn-16 homo-oligomerizes when expressed by L cell fibroblasts (54). Regardless of mechanism, one implication of this model is that tight junction strands are not composed of simple homogeneous mixtures of claudins and instead have fine structure which could have functional ramifications for epithelial barriers.
Claudin-Occludin Interactions
Ever since occludin-deficient mice were found to have near normal barrier function (105), it has become clear that occludin is more prominently involved in regulating tight junctions as opposed to being a strict structural element [for review, see Blasig et al. (15a)]. Differential roles for claudins and occludin in maintaining tight junctions is further underscored by fluorescence recovery after photobleaching (FRAP) and fluorescence loss In photobleaching (FLIP) experiments performed with Green Fluorescent Protein (GFP) fusion proteins, where cldn-1 is a relatively stable component of tight junctions, with a mobile fraction of ∼ 25% of the total signal (109). Interestingly, cldn-1 stability was unaffected by deleting the C terminal PDZ binding domain, suggesting that a direct interaction with ZO-1 or ZO-2 is not needed to stabilize cldn-1 once assembled into tight junction strands; instead, claudins lacking the PDZ binding motif are more likely to be stabilized by heterotypically binding to claudins on adjacent cells with the capacity to interact with ZO-1. By contrast, occludin is significantly more dynamic and has a mobile fraction approaching ∼ 80%. The rate of cldn-1 recovery is about twice as rapid as that of occludin, suggesting that cldn-1 is more freely diffusing through the plasma membrane than occludin.
Despite these differences in mobility, an extracellular loop mimetic peptide corresponding to the EL1 domain of cldn-1 stably binds to cldn-1, cldn-3, and occludin, which correlates with the ability of the peptide to disrupt tight junctions (91). While not definitive, this does suggest that the EL domains of claudins and occludin are close enough to interact which could help strengthen the tight junction barrier. Other evidence in support of direct claudin–occludin interactions comes from studies of the mechanism of action for Clostridium perfringens enterotoxin (CPE), which binds to several claudin EL2 domains with high affinity. There are three major biochemically isolatable complexes that represent the sequence of interaction of CPE with cell tight junction proteins, a small complex containing CPE and claudins which subsequently oligomerizes and then incorporates occludin (111). Since CPE lacks a high affinity binding site for occludin, recruitment of occludin into the CPE complex is most likely to reflect a claudin–occludin interaction, rather than a direct interaction with CPE.
Post-Translational Modification of Claudins
Palmitoylation
Palmitoylation is likely a universal feature of claudin assembly, since signature di-cysteine palmitoylation motifs are conserved throughout the claudin protein family (124) (Fig. 3). Palmitoylated cldn-14 more efficiently partitioned into detergent-resistant membranes as compared to a mutant, nonpalmitoylated cldn-14 construct (124), which suggests that membrane microdomains, or lipid “rafts”, play a role in tight junction assembly (34, 97). Although claudin partitioning into microdomains is established, roles for microdomains in regulating the trafficking and oligomerization of claudins are not well understood at present.
Tetraspanins, transmembrane proteins that enhance membrane assembly and trafficking to the plasma membrane, are also palmitoylated which enhances tetraspanin partitioning into microdomains and oligomerization (52, 78). Intriguingly, tetraspanin palmitoylation occurs in the Golgi apparatus and is linked to the formation of higher order membrane protein complexes (136). Although tetraspanins and claudins both span the bilayer four times and have similar protein orientation in the bilayer, they are structurally distinct and not homologous (107). Despite these differences in structure, claudins were found to weakly interact with the tetraspanin CD9, indicating that these two classes of proteins partition into comparable membrane microdomains (72). However, siRNA depletion of CD9 has little effect on epithelial barrier function, thus the functional role for a putative CD9-claudin interaction remains obscure.
Palmitoylation of the tetraspanin CD81 is inhibited by oxidant stress induced by glutathione depletion or treatment with hydrogen peroxide (21). This, in turn, promotes CD81 binding to a 14-3-3 scaffold protein and alters downstream signaling (21). Whether claudin palmitoylation is regulated by oxidative stress is an open question. However, this possibility is underscored by the demonstration that treatment of MDCK cells with hydrogen peroxide disrupts claudin localization to tight junctions and decreases barrier function (42).
Phosphorylation
Members of the claudin family are also directly phosphorylated on the C-terminal cytosolic domain which has dramatic and divergent effects on claudin activity and integration into tight junctions. For instance, phosphorylation of cldn-5 (57) or cldn-16 (56) by protein kinase A stimulates a redistribution of intracellular pools of these claudins to incorporate into tight junction strands, as assessed by immunofluorescence microscopy. These increases in claudin assembly into tight junctions are also associated with increased barrier function. However, this effect is not universal, since lysophosphatidic acid stimulates rho kinase-mediated phosphorylation of cldn-5 which causes decreased incorporation into tight junctions and decreased barrier function (135). Further, protein kinase A has been shown to decrease assembly of cldn-3 into tight junctions resulting in decreased barrier function (26).
Protein kinase C shows a comparable specificity in regulating the phosphorylation and integration of cldn-1 and cldn-4, but not other claudins, into intestinal epithelial tight junctions (10). Consistent with a role for cldn-1 phosphorylation in promoting assembly into tight junctions and increasing barrier function, protein phosphatase 2A has been shown to dephosphorylate cldn-1 and decrease barrier function (96).
Clearly, the potential for claudin phosphorylation to affect their function is just beginning to be addressed, and tools such as antibodies that recognize claudin-specific phosphorylation events will prove to be valuable reagents (135). Although phosphorylation has been correlated with changes in claudin assembly and function, the mechanisms which underlie this are not well defined. In particular, little is known about whether claudin phosphorylation affects interactions with scaffold proteins or other tight junction components.
Claudin Trafficking and Oligomerization
Based on freeze-fracture electron microscopy analysis, claudins are organized into tight junctions as a network of “beaded” strands, where the individual beads are roughly the diameter of a gap junction channel (117). This led to the suggestion that claudins are organized into a basic hexameric unit, similar to connexins in gap junction hemichannels (69). However, there is little biochemical evidence in support of claudins organizing into stable hexamers. For instance, native gel electrophoresis shows that claudins form ladders of stable oligomers in increasing in molecular mass up to a hexamer (25, 85). These results should be interpreted with caution, since this method of analysis does not distinguish claudin aggregates or heterotypic complexes from bona fide hexamers that would be properly assembled in a membrane bilayer.
There are several examples where claudin mutations disrupt delivery to the plasma membrane and instead accumulate in intracellular compartments, mainly the endoplasmic reticulum (ER) and Golgi apparatus, presumably due to misfolding (64, 99). Inefficient trafficking is not restricted to mutant claudins. For instance, cldn-16 and cldn-19 act as apparent co-chaperones, since trafficking to the plasma membrane is more efficient in cells expressing both of these claudins as opposed to cells expressing only one or the other (53). All of these observations have direct relevance to how claudin misprocessing can disrupt tissue barrier function, since the ability of one class of mutant claudin to perturb other normal claudins may play a significant role in the pathologic weakening of tight junctions in human disease.
Little is known about how claudins interact prior to assembly into tight junction strands. If claudins parallel classical multimeric transmembrane proteins, then proper folding and oligomerization would be a prerequisite to further transport along the secretory pathway (35, 112). However, it is not known whether claudins interact as early as the endoplasmic reticulum.
Connexin family gap junction proteins may provide one clue to whether claudins oligomerize early in the secretory pathway. Connexins represent an interesting exception to the classical pathway, since it is well established that Cx43 is transported from the endoplasmic reticulum as a monomer to the Golgi apparatus where it subsequently oligomerizes into biochemically stable hexameric hemichannels (69, 75). The recent discovery that a chaperone protein, ERp29, stabilizes monomeric Cx43 underscores the existence of a connexin specific quality control pathway which promotes oligomerization in the trans Golgi network (27). The extent of claudin oligomerization which occurs prior to delivery to the plasma membrane remains an open question.
As a tool to study connexin trafficking, we previously developed a series of connexins tagged with an ER retention/retrieval motif, His-Lys-Lys-Ser-Leu (HKKSL) (83) (Fig. 4). Cx43-HKKSL shows steady state localization to the endoplasmic reticulum as monomers stabilized by ERp29 (27 –29, 82, 83). However, Cx43-HKKSL also has the capacity to act as a dominant negative and prevent the transport of untagged connexins to the plasma membrane, provided that they are compatible to hetero-oligomerize and can be retrieved from the Golgi apparatus (Fig. 4B). Specifically, Cx43-HKKSL was previously found to prevent the transport of Cx46 to the plasma membrane due to a direct interaction (Figs. 5A–5C) (30, 31). Thus, HKKSL-tagged connexins provide a method to define heteromeric compatibility amongst connexins in the context of a native membrane microenvironment.

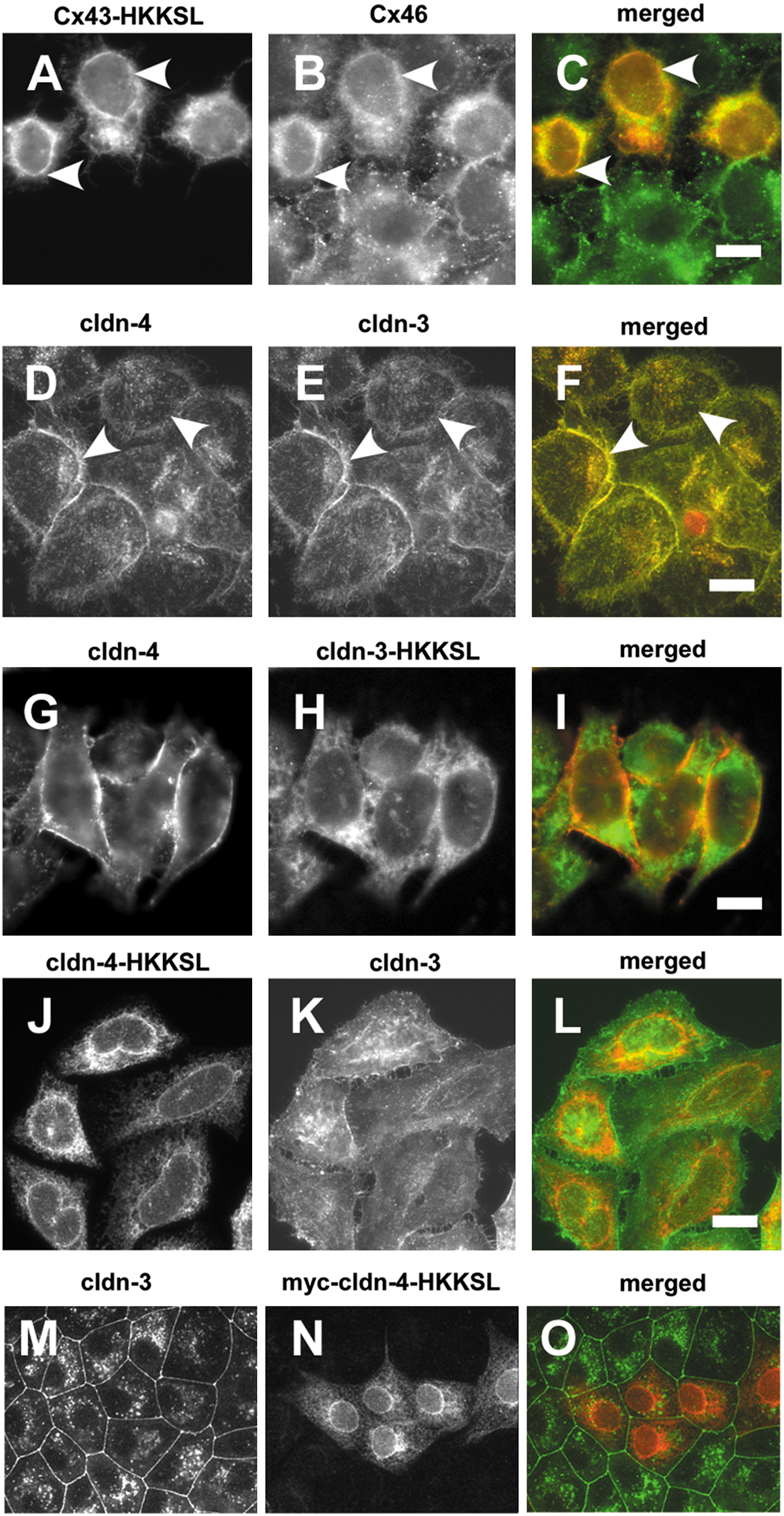
With the original intention of performing a parallel analysis to identify subclasses of heteromerically compatible claudins, we produced a series of HKKSL-tagged claudins. To our surprise, we found that, regardless of context, none of the cldn-HKKSL constructs we developed had the capacity to act as dominant negatives against native claudins. As shown in Figure 5, despite the fact that cldn-3 and cldn-4 are heteromerically compatible claudins (33), neither cldn-3-HKKSL nor cldn-4-HKKSL had a dominant negative effect on cldn-4 or cldn-3, respectively. Note that cldn-4-HKKSL also had no effect on endogenous cldn-3 expressed by MDCK cells (Fig. 5). In fact, expression of cldn-3-HKKSL or cldn-5-HKKSL lacked the ability to inhibit transport of any endogenous claudins expressed by MDCK cells (not shown).
The inability of cldn-HKKSL constructs to act as dominant negatives is in striking contrast to Cx43-HKKSL, which inhibits Cx46 trafficking to the plasma membrane, even though Cx43 and Cx46 oligomerize in the trans Golgi network (71). One potential interpretation of this result is that claudins oligomerize in an extremely late secretory subcompartment, or even at the plasma membrane. For this to occur would require a quality control pathway with the capacity to prevent premature claudin–claudin interactions, analogous to the connexin quality control pathway (69). Alternatively, claudins may not readily form stable oligomers in isolation and instead require interactions with the tight junction scaffold and/or heterotypic interactions with other claudins to maintain a higher order structure. Finally, these observations are subject to the caveat that the HKKSL tag could interfere with normal claudin–claudin interactions, even though this was not the case for connexins. One potential approach to distinguish between these possibilities would be to determine whether claudin overexpression drives premature oligomerization or tight junction fibril formation in an intracellular compartment, since this would imply a saturation of a putative quality control pathway, provided that overexpression does not cause the claudins to aggregate. Alternatively, a detailed analysis of claudin–claudin interactions in a native membrane context using techniques such as FRET could help establish whether claudins are close enough to assemble in the endoplasmic reticulum and/or Golgi apparatus.
Claudin Mutations in the Pathology of Human Disease
The importance of claudins in mammalian physiology is reflected by human diseases due to claudin mutations, as well as the phenotype of claudin-deficient mice (Table 2). Although claudins are frequently expressed in several different tissues, the phenotype resulting from claudin mutations or deficiencies tends to reflect a major effect on a single or limited set of organ systems. For instance, mutations in cldn-1 have been associated with neonatal ichthyosis, a rare skin disorder combined with sclerosing cholangitis, a liver disorder caused by blockage of bile ducts (49). Additionally, cldn-1 null mice have an embryonic lethal phenotype due to increased skin permeability which leads to neonatal dehydration (39). Cldn-14 mutations have been shown to cause nonsyndromic deafness in both humans and mice (129). It is currently hypothesized that cldn-14 is a vital component in the cation restrictive barrier responsible for maintaining proper cation levels in endolymph in the cochlea of the ear (13).
Unless otherwise stated, the human diseases and mouse phenotype refer to claudin mutations or claudin-deficient mouse models.
Failure to absorb magnesium in the thick ascending loop of Henle was shown to be the underlying cause of FHHNC (14, 102). Mutations in cldn-16 lead to recessive renal hypomagnesaemia (67) and cldn-19 mutations show a similar deficiency in regulating magnesium reabsorption (130). Interestingly, cldn-16 interacts with cldn-19 to increase paracellular selectivity to sodium, rather than having a direct effect on paracellular magnesium permeability (54). This leads to a model for indirect control of ion permeability where a gradient of cldn-16/cldn-19 expression in the thick ascending limb of the kidney establishes a NaCl concentration gradient which creates a lumen positive transepithelial diffusion potential that drives paracellular magnesium reabsorption, as opposed to a direct change in magnesium permeability (7).
To date, the only mutant claudins to be associated with human disease are cldn-1, −14, −16, and −19. Based on the phenotypes for claudin-deficient mice that have been discovered (Table 2), it seems likely that mutations in other claudins will also be found to have human pathology, although another strong possibility is that claudin mutations will result in developmental lethality rather than disease.
Effects of Oxidant Stress on Claudins
Alcoholic lung syndrome and glutathione depletion
Oxidant stress is associated with several different classes of barrier failure where claudin misregulation plays a critical role, including inflammatory bowel disease [reviewed by John et al. (59a)] and disruption of the blood-brain barrier [reviewed by Carrano et al. (17a) and Lehner et al. (77a)]. However, the lung is uniquely susceptible to oxidant stress in that it is directly exposed to environmental oxygen.
Of particular relevance to public health, clinical studies have linked excess dietary ethanol to oxidant stress in the lung (89) which contributes to the severity of acute respiratory distress syndrome (ARDS) (62, 90). Since lungs are particularly sensitive to oxidant stress, high levels of the antioxidant glutathione are required in the airspaces as a protective mechanism (59). As a significant factor in alcoholic lung syndrome, ethanol metabolism to acetaldehyde depletes the reduced glutathione pool and is a direct source of oxidant stress in response to chronic alcohol abuse (17, 90). Conversely, a diet enriched in the glutathione precursor procystine prevents the alcoholic lung phenotype from developing in an animal model for chronic alcohol ingestion (16, 44).
The intense oxidant load on alcoholic lungs provides causes the alveolar epithelium to be prone to injury and apoptosis (16). Since inflammation, the concomitant infiltration of neutrophils and activation of alveolar macrophages all further contribute to oxidant stress in response to acute lung injury (20), the alcoholic lung is highly susceptible to a second hit leading to more severe ARDS than a healthy lung which is not already under an oxidant burden.
A primary risk factor in the severity of ARDS is a defect in alveolar barrier function, since this promotes alveolar flooding (128). Consistent with this, prolonged ethanol ingestion creates a baseline condition where an otherwise healthy alcoholic has defective lung barrier function due to a dysregulation of lung claudin expression, based on an analysis of claudin expression by rats fed alcohol for 6 weeks (36, 46). The effect of alcohol was most striking for type I alveolar epithelial cells (Fig. 6), which comprise the majority of the lung air–liquid barrier. Specifically, chronic alcohol ingestion decreased expression of several claudins, including cldn-1, cldn-7, and cldn-18 (36). The effect of alcohol on claudin expression is not restricted to lung epithelium, since dietary ethanol has also been shown to increase gut permeability through a decrease in cldn-1, primarily in the ileum, which was also accompanied by decreases in occludin and ZO-1 (138). Cldn-1 expression by intestinal epithelia showed a linear correlation with expression of the transcription factor hepatocyte nuclear factor 4α; whether this is the case in pulmonary epithelia remains to be determined.
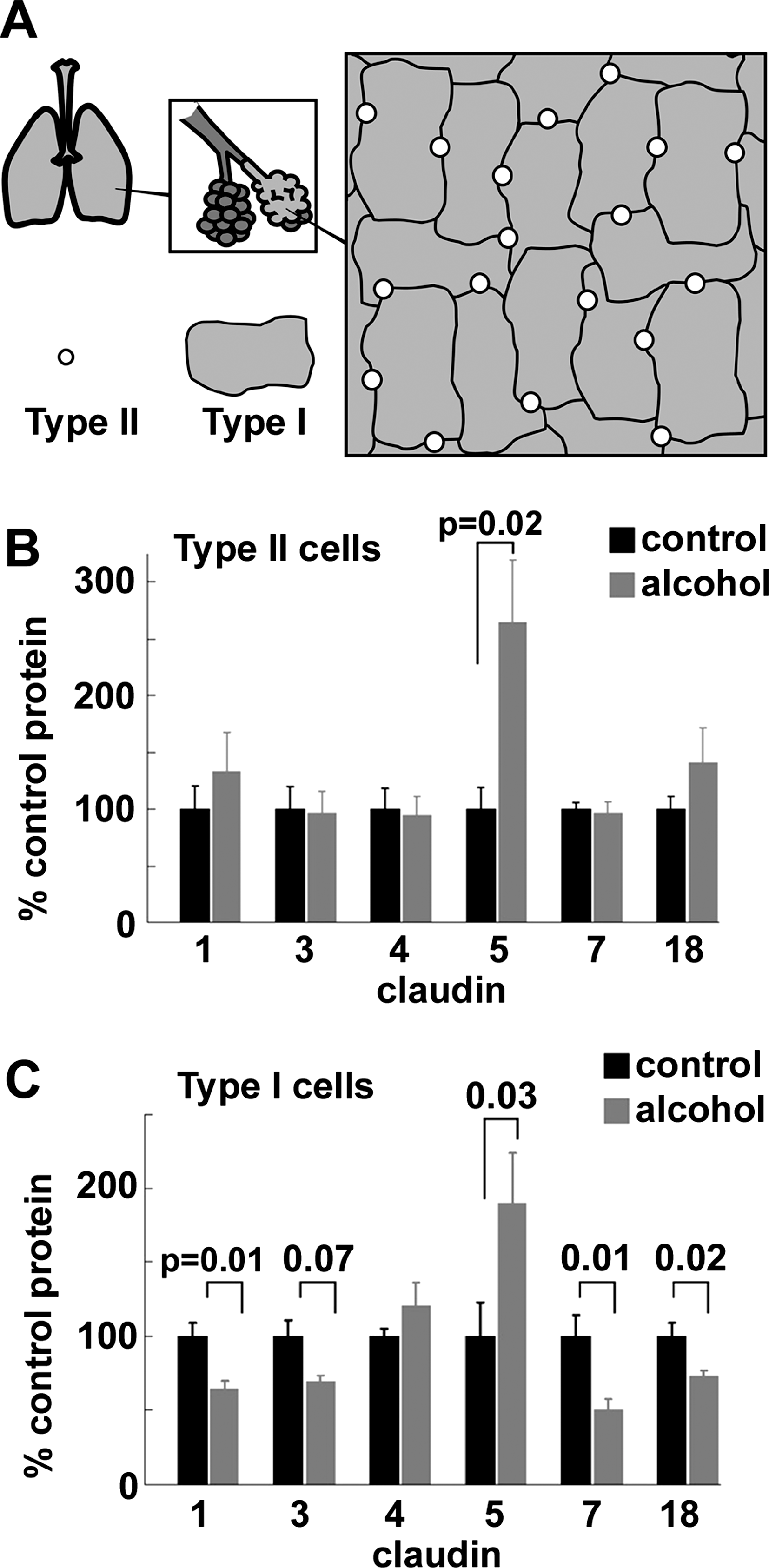
In parallel to the alcohol-induced decreases in claudins expressed by alveolar epithelial cells, alcohol also increased the protein expression of cldn-5 (36). Surprisingly, we and others have found increased cldn-5 to be associated with increasing the permeability of lung epithelial barriers (25, 33, 127). The mechanism for increased barrier permeability associated with increased cldn-5 expression by alveolar epithelia is not known at present and may reflect a tissue specific role for cldn-5, since cldn-5 is crucial for endothelial barrier function (95) and overexpression of cldn-5 by other epithelia has also been found to improve the function tight junction barriers (2). Nonetheless, these results underscore the concept that the effect of alcohol on alveolar barrier function is due to a remodeling of tight junction claudin composition as opposed to a generalized decrease in tight junction assembly.
Several studies from other systems suggest that incorporation of claudins into tight junction strands is sensitive to cell glutathione content, as well as the relative pool sizes for sulfur-containing amino acids. For instance, renal epithelial cells cultured in medium containing reduced levels of cysteine show altered pattern of claudin expression, and a modest increase in barrier function (114). Feeding mice a mixture of probiotic bacteria (Lactobacillus and Bifidobacterium strains), which produce significant levels of glutathione in the gut, significantly increased systemic plasma glutathione, and enabled mice to resist the deleterious effects of acute pancreatitis on intestinal barrier function. Probiotic bacteria helped intestinal epithelium maintain normal levels of cldn-1 and cldn-2 incorporated into tight junctions, which is expected to help maintain the intestinal barrier and reduce the potential for sepsis (80). Ingested probiotic bacteria (Lactobacillus plantarum) were also found to prevent the downregulation of cldn-1 and cldn-4 by hepatocytes in an animal model of obstructive jaundice (137). In this model, Lactobacillus prevented plasma glutathione oxidation, further underscoring the ability of probiotic bacteria to have beneficial systemic effects. Nonetheless, it remains an open question how probiotic signals from bacteria can be transmitted from the gut lumen to the circulatory system.
Angiotensin II
In addition to its metabolic effects on the antioxidant glutathione pool, ethanol also induces cell-signaling pathways that contribute to oxidant stress that are linked to alterations in tight junction permeability. In particular, ethanol stimulates angiotensin II activity (12) which, in turn, upregulates nicotinamide adenine dinucleotide phosphate-oxidase (Nox) (108). Angiotensin II has also been shown to reduce blood-brain barrier function through a protein kinase C-mediated pathway, although there was no obvious effect on cldn-5 expression or localization (37). Thus, by analogy angiotensin II may play a direct role in compromising the barrier function of the pulmonary circulation as well.
Transforming Growth Factor β
As a response to oxidant stress, alveolar epithelial cells increase expression and secretion of transforming growth factor β (TGF-β) (12, 62). TGF-β has previously been shown to exacerbate the acute phase of lung injury (92, 100) and influence alveolar epithelial function by promoting epithelial-to-mesenchyme transition (EMT) (63, 65, 133). Alveolar epithelial cells undergoing EMT will directly affect the lung barrier by simply downregulating expression of tight junction proteins, including claudins.
TGF-β also increases oxidant stress by decreasing γ-glutamylcysteine synthetase expression (9, 58), thus reducing the antioxidant glutathione reserves of the lung. TGF-β also increases oxidant stress by increasing Nox expression (55, 115) and H2O2 production (126). Increased oxidant stress has the added potential to exacerbate alveolar injury by creating a positive-feedback loop, particularly if TGF-β expression and activation are driven by a second insult, such as sepsis or direct trauma (45). In addition to reactive oxygen species, reactive nitrogen species, including peroxynitrite, are generated during acute lung injury (48, 113) which could potentially interfere with tyrosine phosphorylation and function of claudins.
Other redox sensitive pathways affecting claudins
Although it remains to be determined whether claudins are direct targets of oxidants, there are several lines of evidence which show that oxidant stress can impair barrier function. For instance, several oxidized lipids have the capacity to disrupt tight junctions (18), most notably 1-palmitoyl-2-(5-oxo-valeroyl)-sn-glycero-3-phosphocholine, which is relevant to cardiovascular disease (60). Oxidants such as hydrogen peroxide and chloramine have also been found to disrupt tight junctions when tested on cultured cells. This is most frequently manifested as a disruption of claudin localization to tight junctions (93, 98). A similar effect has also been observed in alveolar epithelial cells isolated from alcohol fed rats (36).
In one study, the effect of oxidant stress on claudin localization was attributed to p38 mitogen activated protein (MAP) kinase which correlated with an increase in the ability to extract cldn-4 using Triton X-100, indicating a change in association with the tight junction scaffold (98). Moreover, ZO-1 expression was found to decrease in response to chloramine which also disrupts tight junction morphology (93), consistent with the central role for ZO-1 in stabilizing claudins to tight junctions (121, 123) [for review, see Gonzalez–Mariscal et al. (42a)]. However, the effect of oxidant stress is not necessarily due to a global effect of tight junction proteins. For instance, gastric epithelial cells challenged with hydrogen peroxide show a specific decrease in barrier function and cldn-3 expression, while expression of other claudins such as cldn-4 and cldn-7 are unchanged (51).
Oxidant stress has also been found to be induced by human immunodeficiency virus proteins, such as glycoprotein 120 (HIV gp120), which can have an adverse effect on tissue barrier function (4, 76, 79). In particular, HIV gp120-induced oxidant stress increases expression of matrix metalloproteinases which degrade cldn-5 and thus compromise the blood-brain barrier (79). Antioxidant enzymes had a protective effect (79), suggesting a potential therapeutic approach to the prevention of HIV-associated dementia.
Conclusions
In the past decade, there has been considerable progress from identifying claudins as key components that regulate tight junction permeability to understanding how claudins function at a detailed molecular level. Several areas require further investigation. For example, the compatibility profile for claudin–claudin interactions remains incomplete. Current understanding of claudin assembly into tight junctions and identifying roles for other transmembrane tight junction proteins also requires additional study. Of particular importance is to determine how the complexity of claudin expression and organization into tight junctions can have significant effects on paracellular permeability. There also is an emerging literature correlating changes in tumor cell claudin expression to their metastatic potential (87, 118). Thus, in addition to their central role in regulating tight junctions, it seems likely that future studies will elucidate other aspects of cell behavior regulated by claudins.
Alterations in redox state clearly have the capacity to alter paracellular barrier function. There are both direct and indirect potential pathways where claudins can be affected by redox state, ranging from intracellular signaling via reactive oxygen species to redox sensitive motifs present in claudins such as extracellular loop domains stabilized by disulfide bonds. Identifying control points where redox alters claudin expression and function is anticipated to enable antioxidant strategies to be developed which might prove as suitable approaches to improve barrier function.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health Grants HL-083120, AA-013757 (to MK) and AA-013528 (to CEO and LAM).