
Abstract
Oxidative stress has been shown to convert endothelial nitric oxide synthase (eNOS) from an NO-producing enzyme to an enzyme that generates superoxide, a process termed NOS uncoupling. This uncoupling of eNOS converts it to function as an NADPH oxidase with superoxide and hydrogen peroxide generation. eNOS uncoupling has been associated with many pathophysiologic conditions, such as heart failure, ischemia/reperfusion injury, hypertension, atherosclerosis, and diabetes. The mechanisms implicated in the uncoupling of eNOS include oxidation of the critical NOS cofactor tetrahydrobiopterin, depletion of
Endothelial NOS (eNOS) is responsible for the enzymatic generation of NO within the endothelium. eNOS is a homodimeric heme-containing enzyme that converts
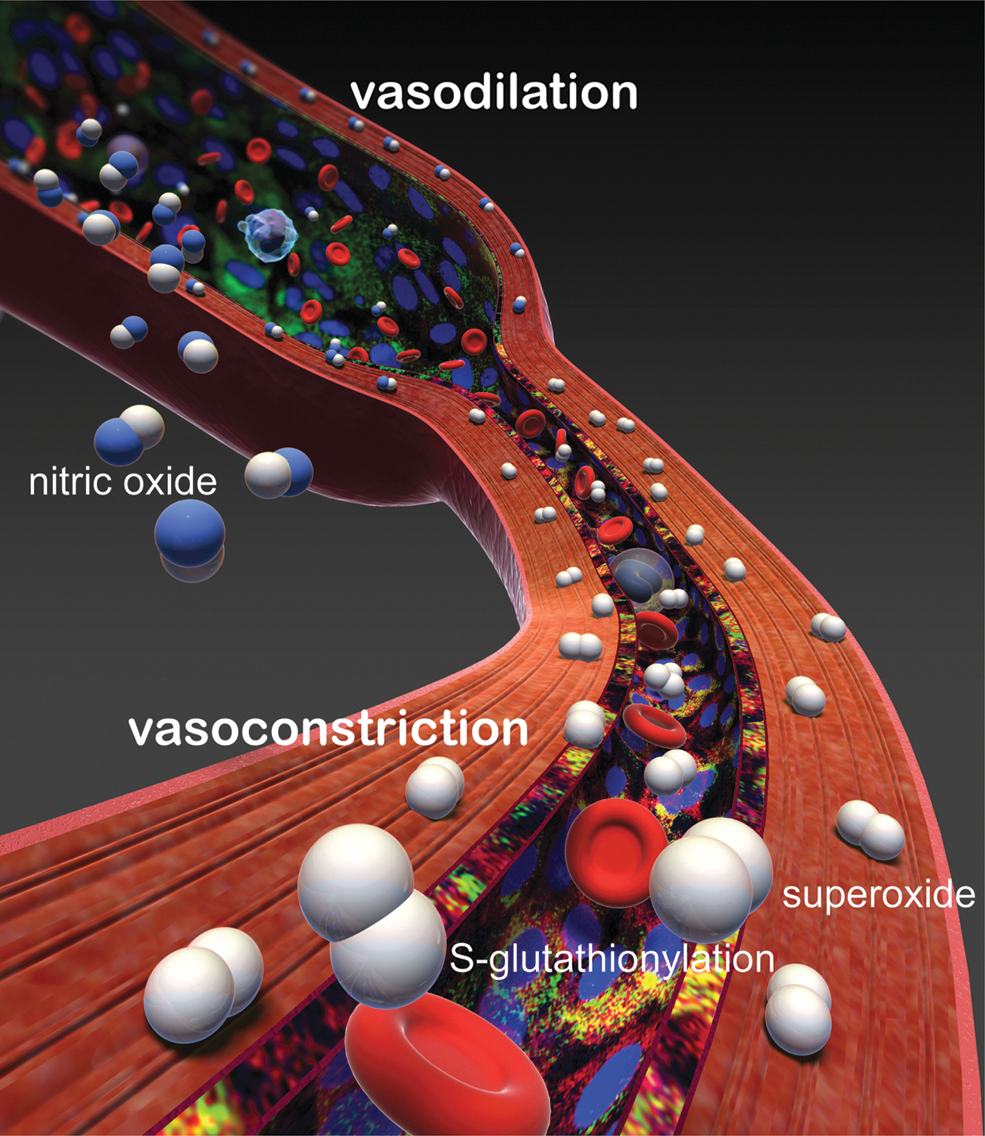
NO is involved in a number of roles within the endothelium, regulating vascular tone, vascular growth, platelet aggregation, and modulation of inflammation (23, 29, 32). As such, endothelial dysfunction produced by a decrease in bioavailable NO is a characteristic feature in many disease states, and one of the mechanisms underlying this dysfunction is the production of reactive oxygen species (ROS) in the vasculature (2, 35, 48). A general mechanism by which ROS elicit endothelial dysfunction is via the impairment of NO signaling due to the ROS-dependent inactivation of eNOS (10, 34, 53). Further, studies have shown that eNOS itself can also become a source of ROS, in a process termed eNOS uncoupling (25, 43, 49). Under normal circumstances, the oxidation of NADPH is tightly coupled to the production of NO by eNOS. However, when the oxidation of NADPH is uncoupled from the production of NO, eNOS generates •O2
We provide a brief review of the mechanisms underlying eNOS uncoupling, with a special focus on the newly identified mechanism involving the S-glutathionylation of eNOS (8).
eNOS Uncoupling via BH4 Oxidation
BH4 is crucial for proper eNOS function and is involved in stabilizing NOS protein structure. It fosters dimer formation and stabilizes the formed dimer. The transfer of electrons to the heme is an interdomain transfer, from the reductase domain of one monomer to the oxygenase domain of the second monomer of the eNOS dimer (30). As such, the dimer stability provided by BH4 binding facilitates eNOS coupling. BH4 binding also shifts the spin state of the heme iron and modifies the heme redox potential, making the transfer of electrons from the reductase domain more efficient. The binding of oxygen is also affected by BH4. Moreover, BH4 is absolutely required for the correct and timely activation of oxygen necessary for catalytic activity. The catalytic cycle of eNOS involves two mono-oxygenation steps, each requiring the formation of a two-electron reduced iron-oxo species at the NOS heme (36). First, an electron is transferred from the reductase domain to the heme, forming the ferrous heme, which then binds oxygen. BH4 delivers one electron to the oxygen-bound ferrous heme iron, producing the iron-oxo species necessary for catalysis. The one-electron oxidized BH4 (the BH3 • radical) is reduced by the reductase domain to regenerate BH4, and the catalytic cycle continues (47). In the absence of BH4, the oxygen-bound ferrous heme dissociates, producing •O2 − and the ferric heme. Two-electron oxidized BH4, dihydrobiopterin (BH2), can bind to NOS but does not support NO formation; rather, when BH2 is bound, eNOS produces •O2 − (44). Thus, when BH4 is oxidized and/or catabolized, eNOS will become uncoupled and produce •O2 − instead of NO.
It has been demonstrated in vivo and in vitro that eNOS is uncoupled when BH4 is limiting. The mechanism leading to BH4 depletion is generally attributed to oxidation of BH4 by ROS and/or ONOO−, the product of the reaction of NO with •O2 −. •O2 − can oxidize the NOS-bound BH4, and supplementation with BH4 has been found to restore NOS activity (15, 41). The in vivo source of the ROS that may lead to BH4 depletion has been attributed to pathways including NADPH oxidase, xanthine oxidase, and the mitochondrial electron transfer chain (27, 35, 57). ONOO− does rapidly oxidize BH4; however, it can also irreversibly inactivate the NOS enzymes, likely by a direct reaction with the NOS heme, producing an inactive enzyme rather than an uncoupled enzyme (10, 37, 38).
The oxidation of BH4 can result in eNOS uncoupling by two mechanisms, by reducing the total biopterin pool or by increasing the BH2:BH4 ratio (37, 38, 43, 44). A two-electron oxidation of BH4 produces the quinoid form of BH2 (qBH2), which can either rearrange to produce BH2 or decompose to form dihydropterin. Dihydropterin is subject to catabolism, and thus, oxidation of BH4 can result in a decrease in the total biopterin pool. BH2 can be recycled back to BH4 by the action of dihydrofolate reductase (DHFR), and this enzyme has been shown to be critical in the regulation of the BH2:BH4 ratio in endothelial cells (13).
eNOS Uncoupling via l -Arginine Depletion or Increase in Methylarginines
It has been well established that accumulation of methylarginines is associated with an increase in ROS production (24, 39, 40). This has also been identified in patient populations demonstrating endothelial dysfunction (5). Moreover, the activities of both the enzymes responsible for formation of methylarginines and the enzymes responsible for clearance of methylarginines (dimethylarginine dimethylaminohydrolase) have been shown to be positively and negatively regulated by changes in the redox environment, respectively (5, 16). Accordingly, oxidative stress can lead to the accumulation of methylarginines. Moreover, it has been shown that the accumulation of methylarginines in endothelial cells to the levels found in pathophysiologic settings can reach the concentrations necessary to significantly inhibit eNOS activity (7).
Methylarginines inhibit NO generation by competitive inhibition, competing with the substrate
eNOS Uncoupling via S-Glutathionylation
In our recent report, we showed for the first time that S-glutathionylation of eNOS reversibly uncouples eNOS (8). It is clear that NOS dysfunction occurs in diseases associated with oxidative stress; however, supplementation with BH4 and/or
This S-glutathionylation–directed uncoupling is unique among the eNOS uncoupling mechanisms in that the resultant •O2
− generation is from the reductase domain. Two cysteine residues (Cys689 and Cys908) that are highly conserved within the NOS family of proteins are S-glutathionylated, both in vitro and in vivo. These two cysteines are critical for normal eNOS function, and when these residues are modified, the enzyme produces •O2
− and this ROS production is not inhibited by N-nitro-
Mechanisms of S-Glutathionylation
Several mechanisms can lead to protein S-glutathionylation. Cysteines can be attacked by ROS to form thiyl radicals that in turn react with glutathione (GSH). Alternatively, protein thiols can be glutathionylated by oxidized glutathione (GSSG) through disulfide exchange (4, 17 –19). Human eNOS (heNOS) contains 29 cysteines, 27 of which are conserved in all known mammals, and prior reports indicate (20 –22) that NOS requires reducing agents, such as dithiothreitol, cysteine, or GSH, to promote NO production and maintain enzyme stability. We observed in vitro in the presence of GSSG that heNOS S-glutathionylation occurs. Thus, it is clear that eNOS possesses redox-sensitive thiols that are readily S-glutathionylated with profound modulation of the function of this critical enzyme. Protein thiols are of vital importance in redox signaling and regulation (4, 17 –19). They maintain correct folding and tertiary protein structure and serve as redox sensors. At normal physiological pH, protein thiols are in the reduced state and are difficult to oxidize because of their protonation. However, the local electrostatic environments of some protein thiols can cause a shift to lower pK a, rendering these sites more susceptible to oxidation (4).
Although the X-ray crystal structure for the reductase domain of eNOS is not available, we predicted the three-dimensional structure of the heNOS reductase domain based on the reductase domain of rat neuronal NOS (1F20) using the Swiss Model First Approach Mode. This modeling showed that Cys689 and Cys908 are located on the domain surface surrounded by several positively charged residues. Cys689 is surrounded by Arg898, Lys904, and Arg1174. Cys908 is located in a pocket that contains His972, Arg1174, and the adenine ring of FAD (Fig. 2). Because of this positively charged environment, both Cys689 and Cys908 would likely be in the deprotonated states because of a lowered thiol pK a via electrostatic interactions, and as such these cysteine residues are predicted to be susceptible to oxidation by GSSG. However, other potential mechanisms, including the formation of protein thiyl radicals or protein nitrosothiols, followed by reaction with GSH can also occur. Indeed, the loss of eNOS activity in endothelial cells exposed to NO has been postulated to be due to thiol S-nitrosylation, which in turn reacts with vicinal or free thiols (including GSH) forming intramolecular or intermolecular disulfide bonds (42).
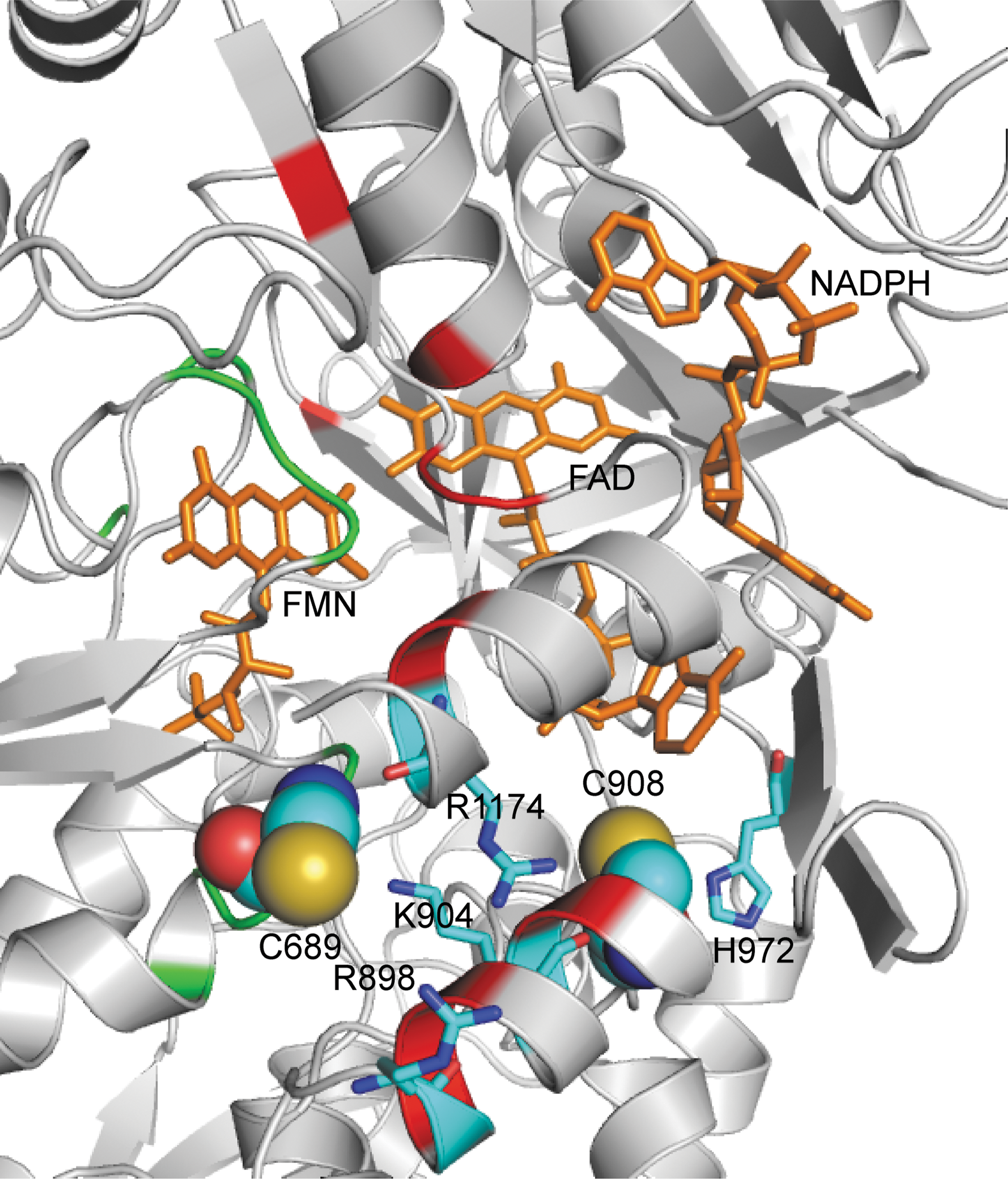
Glutathione is a tripeptide, consisting of glycine, cysteine, and glutamic acid, present in cells at 1–10 mM concentrations (9). S-Glutathionylation results in formation of a mixed disulfide bond between the reactive Cys-thiol and GSH. The additional bulky negatively charged GS group would alter the protein structure and function by both steric and electrostatic interactions. Our molecular modeling predicted that both Cys689 and Cys908 are located along the interface of the FAD- and FMN-binding domains, as such their modification would be predicted to alter FAD-FMN coupling, potentially by increasing their solvent accessibility (Fig. 2) (8). By exposing these flavins to the solvent environment, oxygen would be able to access and accept an electron from the reduced flavin, leading to •O2 − formation, and by default decrease the electron transfer to heme, leading to eNOS uncoupling.
Potential Cellular Function of eNOS S-Glutathionylation
In response to oxidative stress, the formation of mixed disulfide bonds, such as S-glutathionylation, can functionally activate or inactivate enzymatic activity to combat or enhance dysfunction. For example, S-glutathionylation of creatine kinase inactivates the conversion of ADP to ATP, and this process can be reversed by GSH or DTT (33). In contrast, other enzymes, such as matrix metalloproteinase, can be activated by S-glutathionylation (28). It has been well documented that NOS requires thiol-specific reducing agents for proper function, indicating that the enzyme contains cysteine residues that are critical for its function. Although S-glutathionylation induces a loss of NO synthesis with enzyme uncoupling, we observe that this process is reversible. As such, during oxidative stress, eNOS S-glutathionylation could function to prevent irreversible oxidative damage of the thiols critical for eNOS function, and with restoration of cellular reducing state, S-glutathionylation would be reversed, restoring proper eNOS function.
As described above, S-glutathionylation of eNOS uncouples the enzyme, leading to enhanced production of •O2 −, which is a redox signaling molecule known to trigger cell proliferation, apoptosis, and senescence (9, 12). Therefore, S-glutathionylation of eNOS could play a role in switching the enzyme from the generation of one signaling molecule (NO) to the generation of a very different signaling molecule (•O2 −). Thus, this protein modification provides a molecular redox-regulated switch for the control of cellular signaling. Indeed, we observed that with a shift of cellular redox state, induced by inhibition of glutathione reductase, eNOS S-glutathionylation occurs in endothelial cells with marked decrease in NO production and enhanced •O2 − generation (8).
NO produced from NOS can react with •O2 − to generate the potent oxidant, ONOO− (46, 51, 54, 55), and the abnormal cellular production of ONOO− has been linked with many diseases. As such, the decrease in NO production from S-glutathionylated heNOS may provide a regulatory mechanism to prevent irreversible oxidation in cells, caused by overproduction of ONOO−. Therefore, S-glutathionylation of eNOS has a pivotal role in switching the enzyme from NO to •O2 − between generation, and this may control cellular redox signaling under oxidative stress.
Redox Regulation by Thioredoxin and Glutaredoxin
The identification of the S-glutathionylation–directed regulation of eNOS function opens a new avenue of potential enzymatic regulation of NO production. The sulfhydryl group of cysteine not only is important in maintaining protein structure, but also plays an important role as an antioxidant by reversible formation of disulfide or mixed disulfide bonds when it reacts with ROS. In cells, there are two major antioxidant systems, namely Thioredoxin (Trx) and Glutaredoxin (Grx), for maintaining the reduced state of protein thiols and cellular reducing environment after oxidative stress. The Trx system, consisting of Trx, NADPH, and Trx reductase, is a NADPH-dependent disulfide reductase that catalyzes disulfide reduction. The Grx system, consisting of Grx, GSH, glutathione reductase, and NADPH, is a GSH-dependent oxidoreductase that catalyzes the reduction of disulfides or mixed disulfides.
Numerous studies have shown that Trx exerts a protective effect against oxidative stress-induced damage in a number of tissues (45). Infusion of human Trx in an isolated rat heart subjected to ischemia/reperfusion protected the heart against reperfusion-induced arrhythmias (1). Overexpression of Trx in lung endothelial cells has been shown to prevent NO-induced reduction of NOS activity (52). For the Grx system, the results from mouse models with Grx1 knockout, as well as overexpression of Grx1 and Grx2, suggested that Grx exerts a cardioprotective effect through protein deglutathionylation after oxidative stress (11, 26). Therefore, the deglutathionylation of eNOS by Grx could provide a potential mechanism of enzyme-directed redox regulation of eNOS function, which could more specifically modulate cardiovascular function and disease.
Uniqueness of S-Glutathionylation–Induced Superoxide Generation
Classically, eNOS-derived •O2
− generation is inhibited by the NOS inhibitor N-nitro-
We have shown that S-glutathionylation of critical thiols uncouples heNOS, and the resultant •O2
− generation of S-glutathionylated eNOS is not suppressed by L-NAME or EGTA. As such, this eNOS uncoupling mechanism is different from that induced by lack of substrate
Conclusion
In conclusion, the oxidant stress that occurs in many diseases including heart attack, stroke, diabetes, and cancer can trigger a newly discovered molecular switch, S-glutathionylation, which alters the function of the critical cell-signaling enzyme eNOS. S-Glutathionylation uncouples eNOS, switching it from NO to •O2 − generation in a reversible fashion. Two highly conserved cysteinyl residues at the interface between the FMN- and FAD-binding domains are S-glutathionylated and this leads to a novel mechanism of uncoupling resulting from the leak of electrons to molecular oxygen with •O2 − generation at the reductase domain. This posttranslational modification can function as a regulatory mechanism for the prevention of the irreversible oxidation of cellular components by limiting the formation of ONOO−. It serves as a molecular switch to flip eNOS from forming the potent anti-inflammatory vasodilator NO to the pro-inflammatory vasoconstrictor •O2 − and provides a unique redox mechanism by which eNOS can be regulated.
Thus, S-glutathionylation of eNOS plays an important role in modulation of cellular signaling and vascular function. In view of the importance of NO and eNOS-mediated endothelial dysfunction in disease, identification of this novel redox-signaling pathway provides new insights regarding therapeutic approaches. Indeed, these observations indicate the potential for a new class of therapeutics that reset this S-glutathionylation–dependent redox switch, restoring normal NO synthase function, and such compounds could hold great promise in the prevention, improved treatment, or cure of many of the most prevalent human diseases including heart attack, stroke, atherosclerosis, diabetes, and cancer.
Footnotes
Acknowledgments
This work was supported by R01 grants HL63744, HL65608, HL38324 (J.L.Z.), HL081734 (L.J.D.), and HL103846 (C.-A.C.) from the National Institutes of Health.