
Abstract
Diabetic retinopathy (DR) is a leading cause of blindness in Western society. Since the prevalence of diabetes continues to increase dramatically, the impact of DR will only worsen unless new therapeutic options are developed. Recent data demonstrate that oxidative stress contributes to the pathology of DR and inhibition of oxidative stress reduces retinal vascular permeability. However, direct mechanisms by which oxidative stress alters the blood–retinal barrier (BRB) and increases vascular permeability remain to be elucidated. A large body of evidence demonstrates a clear role for altered expression of cytokines and growth factors in DR, resulting in increased vascular permeability, and the molecular mechanisms for these processes are beginning to emerge. The pathology of DR is likely a result of metabolic dysregulation contributing to both oxidative stress and cytokine production. This review will examine the evidence for oxidative stress, growth factors, and other cytokines in tight junction regulation and vascular permeability in DR. Antioxid Redox Signal. 15, 1271–1284.
Introduction
The Retina

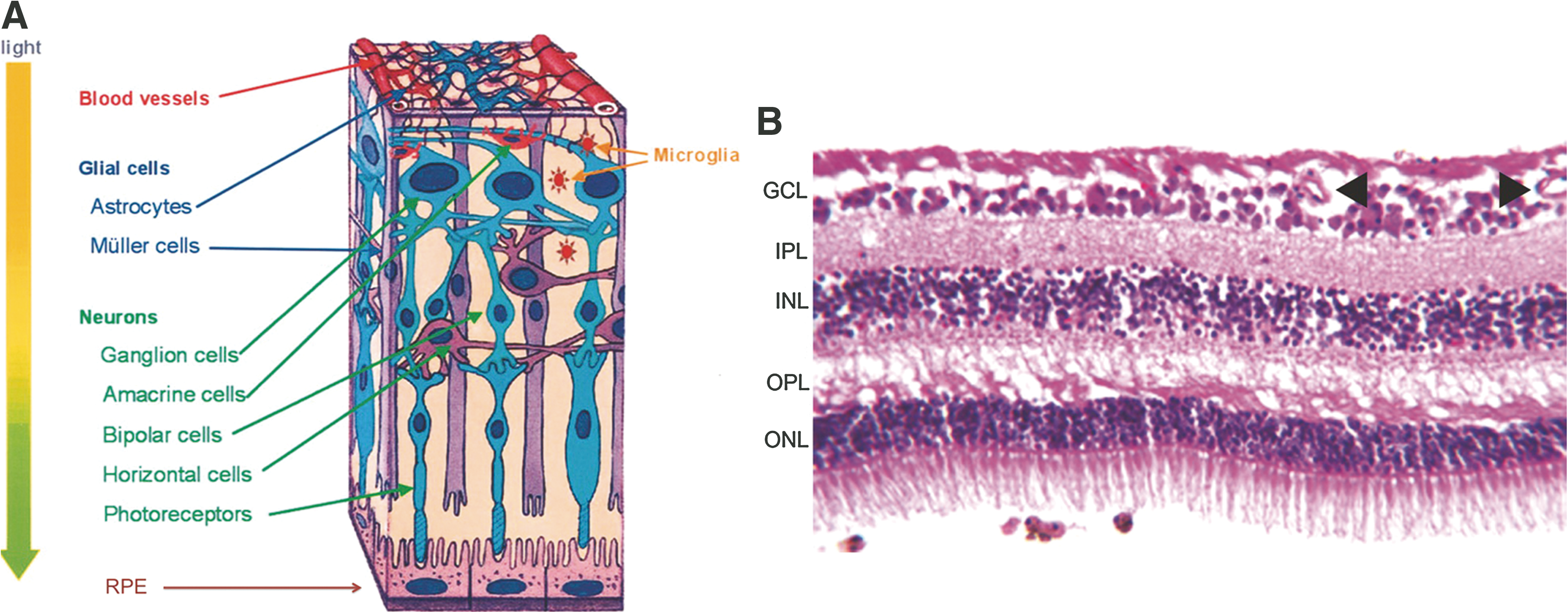
A variety of cell types in the retina coordinate their activity to achieve the conversion of light to neural signals. Neurons including photoreceptors, ganglion, bipolar, horizontal, and amacrine cells; macroglia including Müller cells and astrocytes; microglia or resident macrophages; retinal pigment epithelium (RPE), and microvascular cells including both pericytes and endothelial cells (Fig. 2A) all interact to create the light-stimulated neural impulse interpreted by the brain as vision. The neurons are organized in layers creating the outer nuclear layer (ONL) made of rods and cones, the inner nuclear layer (INL) consisting of bipolar, horizontal, and amacrine cells, and the ganglion cell layer (GCL). These neurons make synaptic connections in the outer plexiform layer (OPL) and the inner plexiform layer (IPL) (Fig. 2B). The rods also interact with the RPE as part of the visual cycle to recover rhodopsin after light signal transduction. Müller cells and astrocytes provide nutritional and regulatory support to neurons and integrate vascular and neuronal signals. Microglia are resident macrophages that monitor the local environment and provide immunomodulatory functions. Finally, the inner retinal circulation stems from the central retinal artery, which branches to three capillary plexuses that anastamose across the inner most superficial region of the retina, through the ganglion cell layer, and throughout the inner nuclear layer (113). Diffusion from the choroidal blood vessels across the RPE provides metabolic support for the outer retina.
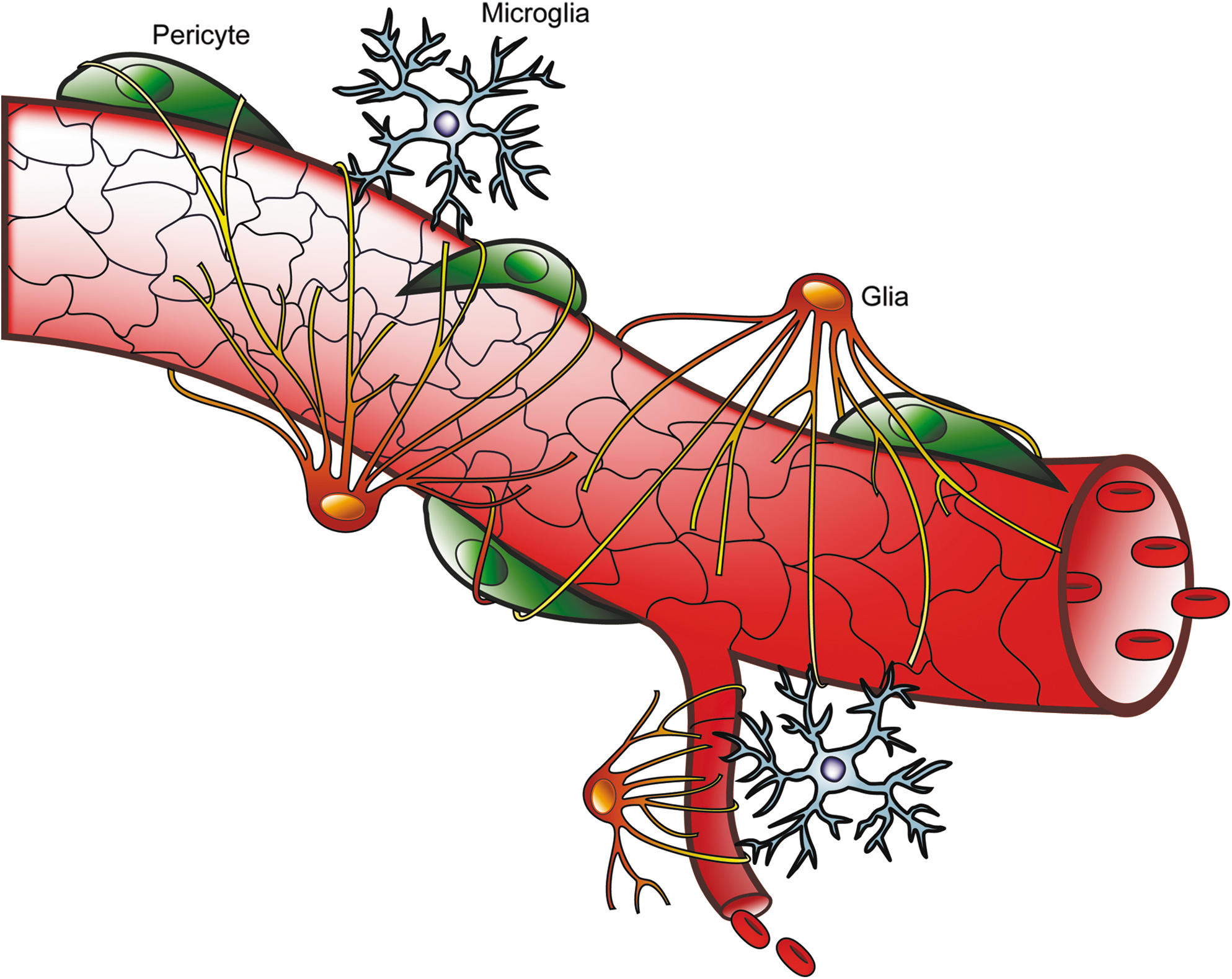
Similar to the central nervous system (CNS), retinal function depends on cellular communication among neurons and metabolic exchange between neurons and support cells. These interactions require a defined environment that is achieved by the formation of the blood–brain barrier (BBB) and blood–cerebrospinal fluid barrier (BCSFB) in the CNS (23a) and the blood–retinal barrier (BRB), thereby separating neural tissue from the circulation. The RPE contributes to the outer BRB and allows oxygen diffusion from the choroidal circulation to the highly metabolic rods and cones. The inner BRB (iBRB) is formed by the blood vessels in the inner retina. The BRB serves as a selective barrier providing immune privilege and regulating osmotic balance, ionic concentration, and the transport of nutrients (sugars, lipids, and amino acids), thereby helping to control the specialized environment of the retina. In addition, pericytes, which are modified smooth muscle cells, share a common basal lamina and directly contact vascular endothelial cells. Formation of the iBRB requires specialized differentiation of the vascular endothelial cells induced by astrocytes, Müller cells, and pericytes (Fig. 3 and reviewed in Ref. 33).
An important component of both the BBB (23a) and BRB is the endothelial tight junction complex. Over 40 proteins have been found to be associated with tight junctions and include transmembrane, scaffolding, and signaling proteins (51). In particular, the transmembrane proteins occludin, tricellulin, the claudin family, and junction adhesion molecules (JAMs), along with the scaffolding zonula occludens proteins (ZO-1, −2, −3) play major roles in the formation and regulation of the tight junction barrier (14a, 16a, 51a, 89a). Alterations to these proteins contribute to the loss of the blood-retinal barrier in diabetic retinopathy.
Diabetic retinopathy
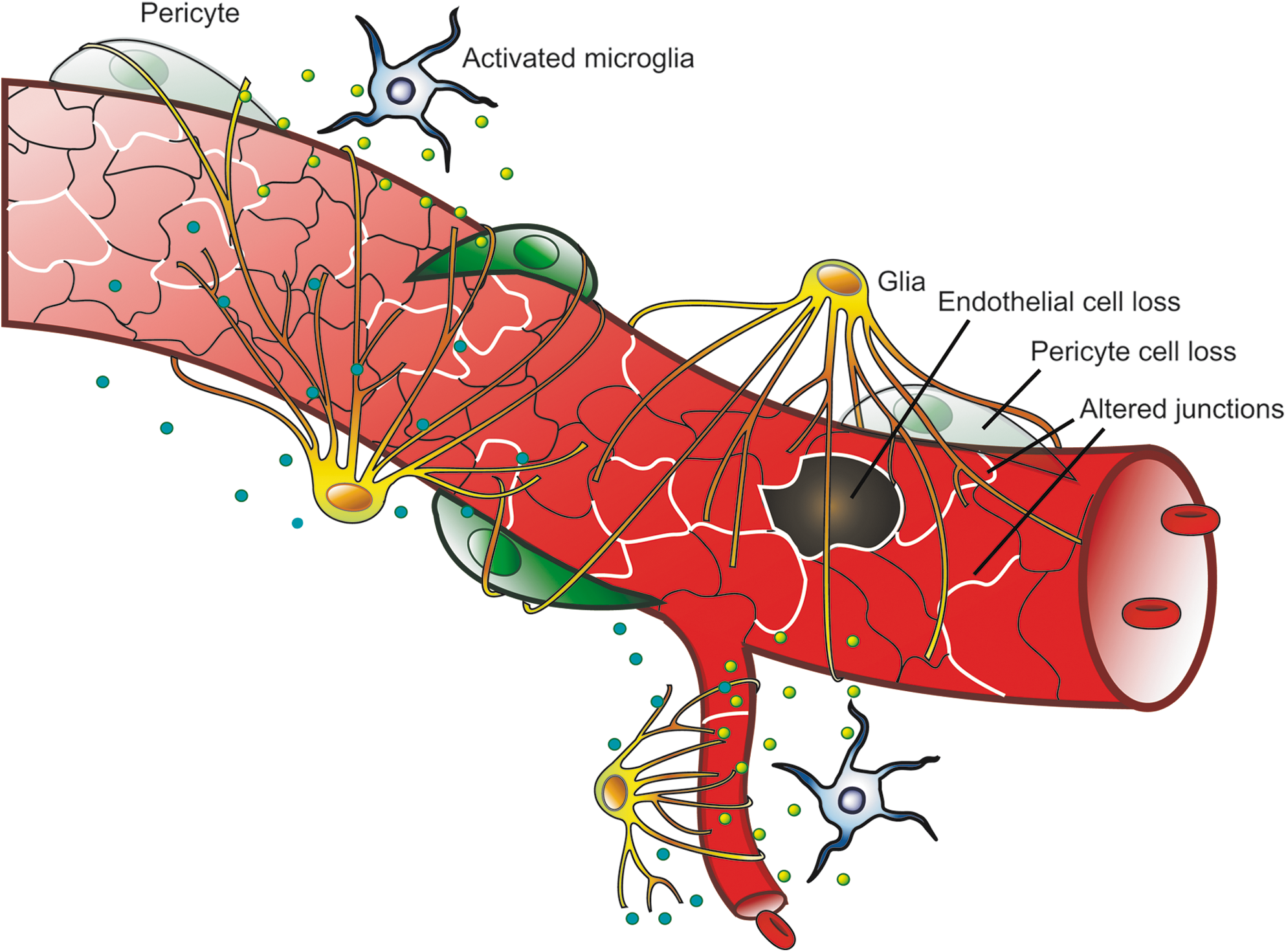
Diabetic retinopathy (DR) is a complication of both type 1 and type 2 diabetes and is the leading cause of acquired blindness in people aged 20–74 years in the United States (91). DR is classified as either nonproliferative (NPDR) or proliferative (PDR). Diabetic complications such as retinopathy are often characterized as microvascular disorders but many retinal cells are affected in diabetes (6). However, vascular changes are clearly linked to loss of visual acuity and observed changes to the retinal vasculature direct clinical care. Early vascular changes include leukostasis, aggregation of platelets, altered blood flow, degeneration of pericytes, and basement membrane thickening (33). Increased retinal vascular permeability is a well-established pathology associated with DR (43) and may result from changes in the tight junction and adherens junction complexes or from increased endothelial cell death (Fig. 4). Macular edema is closely associated with loss of visual acuity in DR (82, 47) and increased permeability of the BRB is believed to contribute to macular edema (93, 100). Therefore, understanding mechanisms of vascular permeability and subsequent macular edema may provide therapeutic options to prevent or reverse loss of retinal function in diabetes.
Hyperglycemia has been proposed to result in the microvascular pathogenesis of DR due to increased alterations in cell signaling pathways, including increased polyol pathway flux, increased advanced glycation end-product (AGE) formation, activation of protein kinase C (PKC) isoforms, and increased hexosamine pathway flux (18). Alternatively, diabetes induces expression of growth factors and inflammatory cytokines in the retina that are the target of multiple phase 3 clinical trials (reviewed in Ref. 46). This review will consider the evidence for the direct effect of hyperglycemia-induced reactive oxygen species (ROS) production and subsequent vascular damage in the diabetic retina and also examine the data suggesting that metabolic dysregulation in diabetes results in cytokine production that alters the BRB and induces vascular permeability. The final section will consider how these two hypotheses may both contribute to the observed disease etiology.
Oxidative Stress and ROS in Diabetic Retinopathy
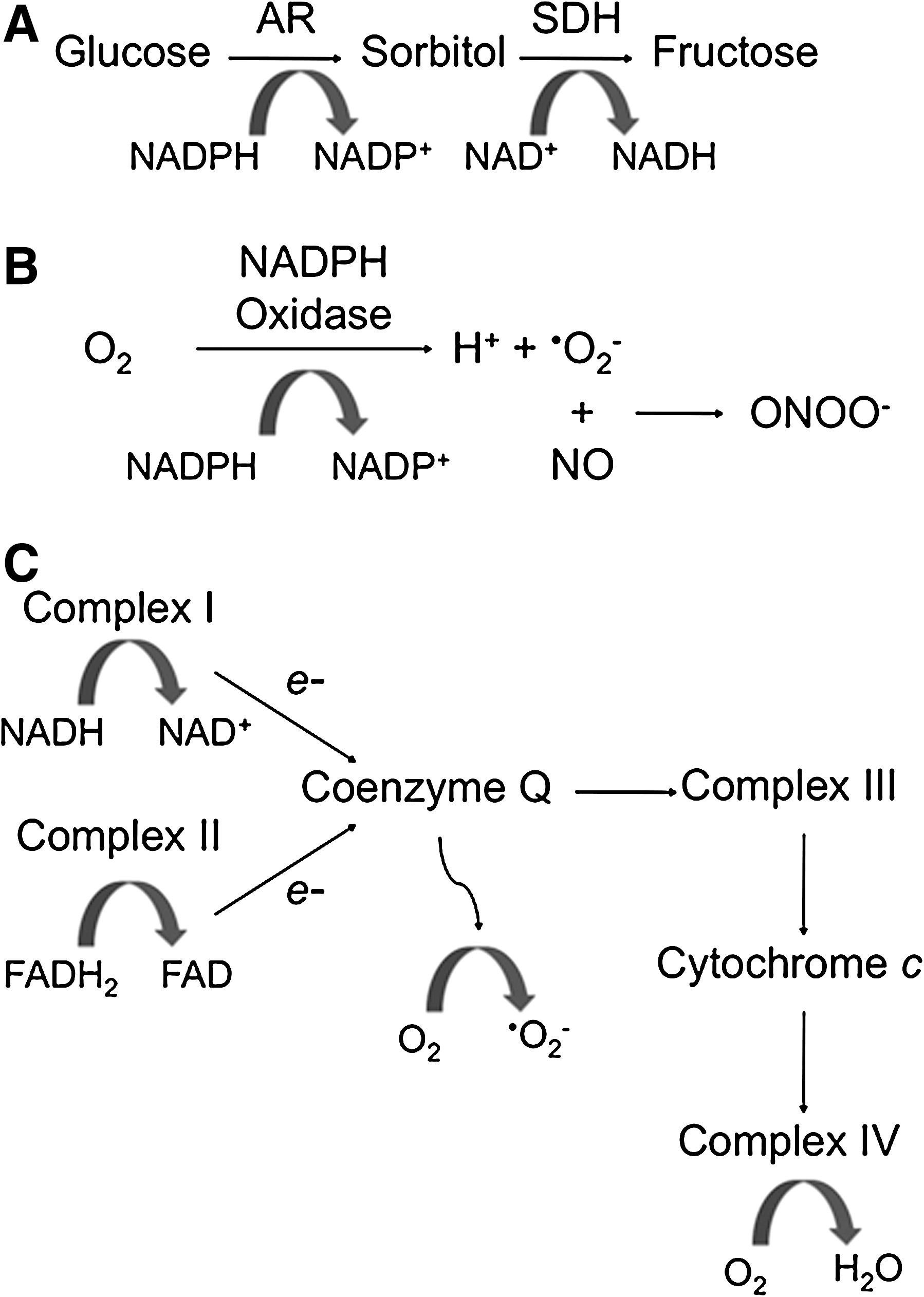
The term oxidative stress has been used to define a number of different processes that are altered in DR. In this review we will refer to oxidative stress as a prolonged or persistent alteration in the NADH (NADPH)/NAD+ (NADP+) ratios, while ROS refers to production of reactive oxygen species. Changes in oxidative stress may impact cell processes by a number of mechanisms. Importantly, loss of NADPH can reduce the activity of glutathione reductase, an enzyme necessary to maintain the intracellular pool of reduced glutathione (GSH). GSH plays a central role in detoxification by reducing hydrogen peroxide, so alterations in NADPH/NADP+ ratio may lessen the ability of cells to respond to ROS (13). There are many potential sources of oxidative stress and ROS production in the diabetic retina, including increased polyol pathway flux, altered mitochondrial metabolism, increased NADPH or xanthine oxidase activity, and the increased formation of reactive nitrogen oxide species (RNOS) such as peroxynitrite (Fig. 5). ROS can oxidize cellular macromolecules, resulting in altered function and contribution to disease pathogenesis. The evidence examining a role for each of these sources of oxidative stress and ROS in models of diabetes, as well as in endothelial cells treated with high glucose, will be examined.
Polyol pathway
Excess intracellular glucose may be metabolized in the polyol pathway (35) with evidence for increasing flux through this pathway in the diabetic rat retina (89). Aldose reductase (AR) first reduces glucose to sorbitol using NADPH as a cofactor, potentially limiting the availability of NADPH for glutathione reductase. The sorbitol is then oxidized to fructose with the corresponding reduction of NAD+ to NADH (Fig. 5). This has been reported to result in an increased NADH/NAD+ ratio that mimics hypoxia (due to impaired oxidation of NADH to NAD+) and has been referred to as hyperglycemic pseudohypoxia (107). The excess NADH may become a substrate for NADH oxidase leading to production of ROS (72). In support of the hyperglycemic pseudohypoxia hypothesis, exposure of retinas from normal rats to high glucose concentrations (20 and 30 mM) for only 2 hours induced a hypoxia-like increase in the lactate-to-pyruvate ratio (indicative of an increased ratio of NADH/NAD+) (85, 103).
A number of preclinical studies support a role for the polyol pathway in the pathology of DR. The accumulation of sorbitol and fructose has been correlated with changes in the activation state of astrocyte and Müller glial cells as measured by immunoreactivity of glial fibrillary acidic protein (GFAP) and an increase in the number of apoptotic neurons. These effects were prevented with the aldose reductase inhibitor (ARI) sorbinil (9). Increased retinal lipid peroxidation and vascular endothelial growth factor (VEGF) protein expression in diabetic rats as well as increased ROS production in high-glucose-treated bovine retinal endothelial cells (BREC) were prevented by the ARI fidarestat (86). ARIs have also been shown to prevent vascular permeability changes (101) and retinal basement membrane thickening (20) in diabetic rats. In addition, sorbinil treatment of diabetic rats prevented both microvascular cell apoptosis and the formation of acellular capillaries, a morphological measure believed to be related to pericyte apoptosis (25). The ARI fidarestat diminished the prevalence of microaneurysms, basement membrane thickening, and pericyte loss in the retinas of diabetic rats in a dose-dependent manner (63). In bovine retinal pericyte cell culture, the ARI SNK-860 prevented high-glucose induced apoptosis and decreased GSH content (78).
Data from AR deficient mice also support a role for the polyol pathway in DR. In mice, AR expression was localized to astrocytes, Müller cells, retinal ganglion cells, and the neurons of the INL. In the blood vessels, AR staining was visualized in the pericytes, but not the endothelial cells. The intensity of staining was increased in the db/db mouse model of type II diabetes, suggesting an increase in expression of AR in the retina of diabetic mice. In AR-deficient mice, a number of diabetes-induced pathologies were normalized as compared to AR-expressing diabetic mice, including pericyte loss, vascular leakage, oxidative-nitrosative stress, VEGF expression, glial cell reactivity, neuronal cell apoptosis, and neovascularization (23).
In contrast to the above results, Winkler et al. found that the content of ATP and lactate in retinas did not differ between fresh tissue and tissue incubated in high glucose (108). Later work revealed no differences in either the NADH/NAD+ or lactate/pyruvate ratios between control and diabetic rat retinas and once again, no differences in lactate or pyruvate levels were observed between fresh retinas and those incubated in media containing high glucose (29). In a study by Ola et al., the lactate/pyruvate ratio was actually found to decrease in rat retinas with increasing duration of diabetes, but this same study found increased flux through the polyol pathway with increasing duration of diabetes, suggesting that depletion of NADPH may make a contribution to oxidative stress in the retina of the diabetic rat (89). Finally, a clinical research trial using sorbinil was not effective in the treatment of retinopathy (1). In summary, there remains uncertainty about the contribution of the polyol pathway to the pathology of DR and in particular loss of the BRB.
Mitochondria
The metabolism of glucose by the tricarboxylic acid (TCA) cycle generates the electron donors NADH and FADH2, which donate electrons to the mitochondrial electron transport chain. The electron transport chain utilizes a series of redox reactions to pump protons across the inner mitochondrial membrane, resulting in a voltage gradient used to drive the synthesis of ATP. Oxygen is the final electron acceptor and thus respiration yields H2O production. While the electron transport chain is essential for energy metabolism, the system has also been implicated in production of ROS due to incomplete and premature reduction of oxygen to form free radicals (Fig. 5).
In 1996, Giardino et al. suggested that hyperglycemia can result in the production of ROS in cultured bovine aortic endothelial cells (BAEC) (50). This work was followed in 2000 by Nishikawa et al. (84), who demonstrated that the hyperglycemia-induced increase in ROS could be prevented by blocking the electron transport chain function with either an inhibitor of electron transport chain complex II, an uncoupler of oxidative phosphorylation, or by overexpression of uncoupling protein-1 (UCP-1). Uncoupling proteins can relieve the proton gradient without ATP generation and thereby regulate ATP production. In addition, overexpression of the mitochondrial ROS scavenger, manganese superoxide dismutase (MnSOD) blocked ROS production. These studies linked hyperglycemia-induced ROS production in BAEC to mitochondrial function (84). This group further demonstrated that normalizing levels of mitochondrial ROS prevented glucose-mediated activation of protein kinase C (PKC), formation of advanced glycation end products (AGEs), and sorbitol accumulation from the polyol pathway, which are three key factors in the pathogenesis of diabetes (84).
Hyperglycemia was subsequently shown to activate a fourth contributor to the pathogenesis of diabetes, the hexosamine biosynthetic pathway, via mitochondrial superoxide production in BAEC (31). Furthermore, through the use of JC-1, a dye that is responsive to increasing mitochondrial membrane potential, and the use of digital imaging, it was demonstrated that hyperglycemia increased the mitochondrial proton electrochemical gradient in BAEC and that overexpression of UCP-1 was able to restore the gradient to normal (30). This accumulation of evidence resulted in the proposal by Brownlee that the single event of increased superoxide production by the mitochondrial electron transport chain results in four important molecular mechanisms implicated in glucose-mediated vascular damage (18).
Mitochondrial generation of ROS may contribute to retinal pathology in DR. Scavenging ROS by administration of the thiol antioxidant, alpha-lipoic acid for the entire duration of diabetes in rats inhibited capillary cell apoptosis, reduced the number of acellular capillaries, and reduced oxidative damage to DNA (8-hydroxy guanosine levels) and proteins (nitrotyrosine) in the retina as compared to nontreated rats after 11 months of diabetes (68). In support of a role for production of ROS in this pathology, MnSOD mRNA expression and activity were reduced in the retinas of diabetic rats, an effect that was prevented by long-term treatment with lipoic acid (65). Overexpression of human MnSOD under the β-actin promoter was achieved in mice with an increase in retinal MnSOD protein and enzyme activity levels of 60% and 70%, respectively. These mice were protected from diabetes-induced oxidative stress as observed by decreased oxidative damage to DNA and proteins (67). Furthermore, mitochondria isolated from diabetic mouse retina had increased superoxide levels and decreased activity of complex III. Overexpression of MnSOD attenuated both of these effects and blocked the increased formation of acellular capillaries in diabetic mice (62). In addition, overexpression of MnSOD prevented apoptosis and oxidative damage to DNA and proteins in high-glucose-treated BREC, indicating that the in vivo observations could at least in part be due to a direct effect on microvascular endothelial cells (65). Cui et al. also observed high glucose induced increase in mitochondrial membrane potential and ROS production in BREC (24). In addition, the hyperglycemia-induced increase in the mitochondrial membrane potential was blocked by the angiotensin-converting enzyme inhibitor perindopril through upregulation of peroxisome proliferator-activated receptor γ (PPARγ) and subsequent UCP-2 expression (114). However, caution must be used when interpreting the cell culture hyperglycemia data in relation to the diabetic retina. Cell culture experiments are carried out with isolated cells and over a time course of hours to days while diabetic animals have a complex metabolic derangement including hyperglycemia for weeks to months.
The above studies suggest that increased ROS production in diabetes is the result of an increase in the mitochondrial membrane potential. However, in a study utilizing human retinal endothelial cells (HREC), oxidative damage of mitochondrial DNA (mtDNA) was observed only 3 hours after high glucose treatment closely followed by decreased expression of mtDNA encoded respiratory chain subunits and decreased mitochondrial membrane potential. Increased mitochondrial ROS production began 12 hours after the start of high glucose treatment, followed by apoptosis at 24 hours. This series of events suggests that oxidative damage of mtDNA is the initiating event in hyperglycemia-induced mitochondrial ROS production and, in contrast to the studies outlined above, is the result of a decrease in mitochondrial membrane potential due to decreased expression of respiration chain subunits (109). Further, high glucose treatment of ex vivo retinas from control and diabetic rats did not result in an increase of TCA cycle flux, suggesting that mitochondrial hyperpolarization is not the source of excess ROS in DR (89). Collectively, while much of the data regarding mitochondrial contribution to ROS production in response to hyperglycemia is compelling, this hypothesis still requires a mechanistic explanation to describe why elevated ATP and NADH fail to prevent excess TCA cycle flux and normal, negative feedback regulation. In addition, it is not yet clear how mitochondrial ROS production regulates such disparate functions as the hexosamine biosynthetic pathway or the polyol pathway in the cytoplasm.
NAD(P)H Oxidase
NAD(P)H oxidases are membrane-associated enzymes that catalyze the 1-electron reduction of oxygen using NADH or NADPH as the electron donor (Fig. 5). NADPH oxidase in phagocytic cells is a multiprotein complex consisting of membrane-bound NOX2 and p22phox, cytoplasmic p47phox and p67phox, and the GTPase Rac. In addition to these subunits, vascular endothelial cells also express the NOX2 homologues NOX1, NOX4, and NOX5 (44). NADPH oxidase can be activated by G protein-coupled receptor agonists, cytokines, growth factors, hypoxia-reoxygenation, and mechanical stimulation (74). Phosphorylation of p47phox is a key post-translational modification involved in the activation of NADPH oxidase and PKC isoforms are believed to be the major kinases responsible for this event (44). This is of particular relevance since the activation of PKC isoforms has been implicated as a key factor in the pathogenesis of diabetes (18).
Increased activity of NAD(P)H oxidase has been observed in diabetic patients and animals. In a model of non-insulin-dependent diabetes (BBZ/Wor rat), higher activity of NADH oxidase-dependent H2O2 production was visualized using electron microscopy of the retinal blood vessels of diabetic rats. In addition, this increase in H2O2 was positively correlated with increased vascular endothelial growth factor (VEGF) expression (40, 41). The activity of NAD(P)H oxidase and expression of enzymatic subunits was increased in the aorta of diabetic rats (64) and in both the saphenous vein and internal mammary artery of diabetic patients (53). Apocynin, an inhibitor of NADPH oxidase, blocked the increased retinal leukostasis observed in the streptozotocin (STZ)-induced diabetic rat model (22). Deletion of NOX2 or treatment with apocynin in a mouse model prevented the diabetes-induced increase in ROS formation, intercellular adhesion molecule (ICAM)-1 expression, leukostasis, and vascular permeability (5). AGE-induced permeability in rat retinas was inhibited by apocynin, indicating a role for NADPH oxidase downstream of RAGE activation (105). Aortic endothelial cells in culture exposed to high glucose increased free radical production as measured by electron spin resonance (ESR), an effect that was abolished when inhibitors of NAD(P)H oxidase and PKC were used (57). These cell culture studies were further supported by in vivo experiments using STZ-induced diabetic rats. Using ESR, an increase in oxidative stress was observed in the abdominal area of diabetic rats in vivo and this effect was normalized by treatment with either a PKC-specific inhibitor or an NAD(P)H oxidase inhibitor (97). Taken together, these studies suggest a key role for PKC-dependent activation of NAD(P)H oxidase in the production of ROS in diabetes.
Statins have also been shown to modulate NADPH oxidase in DR. Statins are a class of drugs widely prescribed to treat hyperlipidemia, and in addition to their ability to reduce the cardiovascular complications of diabetes, statins have also been shown to improve signs of DR (110). Simvastatin was shown to decrease the expression of both NOX2 and p47 phox, signal transducer and activator of transcription 3 (STAT3) activation, the formation of ROS, VEGF expression, and vascular leakage in the retinas of STZ-induced diabetic rats (4). A recent study by Li et al. showed that both NOX4 and VEGF expression were increased in the retinas of db/db mice, an effect that was abrogated by lovastatin. Moreover, siRNA-mediated depletion of NOX4 in db/db mice significantly decreased NADPH oxidase activity, VEGF expression, and retinal vascular permeability. These results were confirmed in hypoxia and high glucose-treated retinal capillary endothelial cells and are consistent with the hypothesis that lovastatin inhibits NOX4, resulting in decreased VEGF expression and reduced vascular permeability in diabetic animals (73). These studies provide evidence that statins, either directly or indirectly, have the ability to affect the function of NADPH oxidase and lower the formation of ROS in the diabetic retina. Statins inhibit 3-hydroxy-3-methyl-glutaryl coenzyme A reductase, one of the rate-limiting steps in cholesterol biosynthesis. In addition to blocking the production of cholesterol in cells, statins also inhibit isoprenoid biosynthesis, which is thought to account for many of the anti-inflammatory effects of these drugs (116). Therefore, more mechanistic studies are required to determine the direct role of statins in BRB regulation.
Xanthine oxidase
The two terminal reactions of purine degradation are catalyzed by the enzyme xanthine oxidoreductase (XOR) that, in the form of xanthine oxidase (XO), produces ROS. XO catalyzes the conversion of hypoxanthine to xanthine and xanthine to uric acid reducing oxygen and generating ROS in each step (55) and the expression of XO has been detected in capillary endothelium (58). Inhibition of xanthine oxidase with allopurinol improved nonretinal vascular and neural function in an STZ rat model of diabetes (56). Moreover, cytokines and hypoxia regulate XOR gene expression in a variety of cell and organ systems (16), indicating a potential role for this enzyme in DR pathogenesis. However, current evidence for a direct role of XO in DR is lacking.
Nrf2/ARE
In addition to production of ROS, the synthesis of ROS scavengers and antioxidant enzymes contributes to the redox state of the cell. A number of transcriptional factors are responsive to cellular redox state and in particular, NF-E2-related factor-2 (Nrf2) acts through the antioxidant response element (ARE) inducing the expression of antioxidant genes in vascular cells (45). These genes include glutathione reductase, NAD(P)H:quinone reductase, and glutathione S-transferase among others (99). Evidence from Nrf2 knockout mice indicate that this transcription factor contributes to the cellular control of ROS production since deletion of Nrf2 enhanced ROS production and amplified the pathology of diabetic nephropathy compared to wild-type mice (59, 111). However, evidence for a direct role of Nrf2 in DR is lacking and is an area for future research.
Growth Factors and Cytokines in Diabetic Retinopathy
The diabetic condition promotes a retinal inflammatory response as observed by nuclear factor-κB (NF-κB) activation (19) and induction of pro-inflammatory molecules such as ICAM-1. Increased ICAM-1 and other cell adhesion molecules promote leukostasis, resulting in increased vascular permeability and further exacerbating the inflammatory milieu of the retina (14, 79). However, no evidence of frank leukocyte infiltration has been observed in the retina. Leukostasis is also thought to contribute to breakdown of the BBB under pathological conditions (23a); however, the exact cell type that adheres to the retinal vasculature in diabetes remains to be elucidated. Thus, the pathology in DR may result from hyperglycemia-induced oxidative stress in endothelial cells, as discussed above or may occur indirectly, in which the diabetic condition induces production of cytokines from a number of potential cell types that act on the vascular endothelium and alter the BRB. A study by Busik et al. (19) in 2008 examined the role of high glucose and cytokines in the generation of ROS and the activation of inflammatory and apoptotic pathways in HREC. Significantly, this study found that treating HREC with high glucose did not increase glucose consumption nor increase the production of ROS, activate NF-κB or mitogen-activated protein kinase (MAPK) pathways, induce tyrosine phosphorylation, nor increase interleukin (IL)-1β, or tumor necrosis factor (TNF)-α production. In contrast, high glucose treatment resulted in increased glucose consumption and IL-1β production in human RPE and Müller cells. Moreover, cytokine treatment of HREC resulted in increased glucose consumption, ROS production, MAPK phosphorylation, NF-κB activation, tyrosine phosphorylation, and caspase activation (19). This study points to an important role for the production of cytokines by nonendothelial retinal cells in the vascular pathogenesis of DR.
Also in support of an indirect effect of hyperglycemia on the endothelial cell tight junction complex, high glucose failed to increase endothelial permeability to 70 kDa rhodamine-B-isothiocyanate (RITC) dextran, 467 Da tetramethylrhodamine, or the transport of water, and failed to alter ZO-1 immunolocalization or claudin-5 expression in BREC (74a). However, high glucose treatment induced Müller cell translational regulation of VEGF both in cell culture and in the retina without affecting VEGF mRNA content and gene deletion of eukaryotic initiating factor 4E binding proteins 1 and 2 blocked the high glucose induction of VEGF protein translation (95).
VEGF
VEGF promotes both vascular permeability and angiogenesis and is elevated in ocular tissues from patients with PDR (2, 3, 75, 76). Recent clinical trials demonstrating the effectiveness of anti-VEGF antibody therapy in promoting visual acuity in conjunction with laser treatment attests to the importance of this cytokine in DR (42). Direct measures of water and solute transport across BREC monolayers demonstrate that VEGF induces permeability to both fluids and solutes (21, 27, 71).
Mechanisms of VEGF-induced vascular permeability are beginning to be elucidated. In particular, occludin regulation by VEGF signal transduction is emerging as an essential component of tight junction regulation in the retina. Mice that do not express occludin form tight junctions with normal morphological appearance and barrier function in intestinal epithelia. However, these mice have abnormalities in several tissues, including calcification in the brain that suggests a complex role for occludin in tight junction regulation (92). STZ-induced diabetes in rats has been shown to increase vascular permeability, reduce occludin content in the retina (8), and alter its distribution from continuous cell border localization to intracellular punctae (11). Subsequently, this redistribution of occludin in diabetes was linked to increased vascular permeability (12). Concanavalin A (ConA) is a plant lectin with a specific binding affinity for α-D-glucosyl and α-D-mannosyl glycoproteins, and will therefore bind to endothelial basement membranes. ConA binding in STZ-diabetic rat retinas was associated with redistribution of occludin staining from the plasma membrane to the cell interior in retinal vascular endothelial cells (Fig. 6), an effect that was also observed after intraocular injection of VEGF. VEGF was also shown to increase occludin phosphorylation in rat retinal vasculature and endothelial cell culture (7). PKC activation by VEGF was later found to be necessary and sufficient for occludin phosphorylation (54). Specifically, the classic β isoform of PKC mediates VEGF-induced phosphorylation of occludin since a PKCβ-specific inhibitor as well as expression of a dominant negative PKCβ mutant abolished VEGF induced occludin phosphorylation and inhibited endothelial permeability (54). Recently, mass spectrometry was used to identify five phosphorylation sites on occludin following VEGF treatment (98). Phosphorylation at serine 490 was subsequently shown to be essential for VEGF-induced ubiquitination, internalization of occludin, and increased endothelial permeability in response to VEGF (Fig. 7) (83). This work provides a mechanistic description of how VEGF, a growth factor known to play a role in DR pathogenesis, stimulates signal transduction in endothelial cells resulting in tight junction alterations and increased permeability.


Inflammatory cytokines
In addition to growth factors, inflammatory cytokines such as IL-1β and TNF-α are increased in DR and may contribute to vascular defects. IL-1β is elevated in the retina of diabetic animals (48, 69). Caspase-1 protease cleaves the precursor form of IL-1β, forming the mature peptide (49) and is activated in the retinas of diabetic mice, humans, and high glucose-treated retinal Müller cells (80, 81, 104). Minocycline, a tetracycline derivative that inhibits caspase-1 as well as a multitude of additional targets, blocked caspase-1 activity and IL-1β production in the retina of mice at 2 months of diabetes and prevented retinal capillary degradation at 6 months of diabetes. Diabetic mice deficient in the IL-1 receptor demonstrate decreased caspase activation and reduced formation of acellular capillaries as compared to diabetic wild-type mice, indicating a role for IL-1β in DR pathogenesis in this animal model (104).
Expression of both TNF-α and IL-1β were elevated in the vitreous and serum of patients with PDR (28) and TNF-α serves as an independent serum marker for PDR in type I diabetic patients (52). TNF-α expression was elevated in NPDR as well (94). Further support for a role of inflammation in DR comes from mice with gene deletions in either ICAM-1 or its binding partner CD18. Studies examining retinal pathology in diabetic mice with either of these gene deletions indicate that these proteins are required for the increased leukocyte adhesion to the retinal vasculature and increased permeability, acellular capillaries, and pericyte ghosts observed in the diabetic retina (60). Increased leukocyte adhesion, vascular leakage, and ICAM-1 expression, but not VEGF expression, were reduced by a soluble TNF-α receptor/Fc construct (etanercept) in diabetic rat retinas indicating a key role for TNF-α in vascular leakage independent of VEGF (61). In addition, increased TNF-α levels were observed and treatment with pegsunercept, a second-generation TNF-α inhibitor, blocked the increased endothelial and pericyte apoptosis, and pericyte ghost and acellular capillary formation in rat models of both type 1 and 2 diabetes (15). An examination of TNF-α induced permeability reveals a mechanism with several unique features compared to VEGF induced permeability. TNF-α does not induce occludin Ser490 phosphorylation and increases occludin mRNA and protein expression. Conversely, TNF-α reduces claudin 5 and ZO-1 mRNA and protein expression in an NF-kB dependent manner and induces a profound disorganization of the tight junction complex (10). Similar to retinal endothelial cells, TNF-α also causes barrier loss in cultured intestinal epithelial monolayers and the TNF-neutralizing antibody infliximab restores the intestinal barrier in Crohn's disease patients (59a, 96). Collectively, these data demonstrate a contribution of inflammatory cytokines to the pathogenesis of DR including loss of BRB function.
The Interaction of Reactive Oxygen Species and Cytokines in DR
ROS appear to contribute to vascular permeability in DR, but a clear mechanism has not yet emerged. Since ROS are known to modulate signal transduction, it is possible that ROS production results in tight junction alterations through the induction of growth factors and cytokines. For example, studies have demonstrated that ROS derived from NADPH oxidase activate kinases such as Akt, Src, and MAPKs as well as the transcription factors NF-κB, activator protein-1, p53, E-twenty six, and hypoxia-inducible factor-1 (44). A number of studies support a role for ROS in the activation of signal transduction pathways that result in increased production of growth factors and cytokines.
The formation of RNOS has been reported to stimulate growth factor expression in DR. Nitric oxide (NO) rapidly reacts with superoxide to form peroxynitrite, which can lead to lipid peroxidation, oxidation of sulfhydryl groups, DNA damage, and disturbed electron transport. The reaction of peroxynitrite with proteins may result in the formation of nitrotyrosine residues that can be used as a marker for peroxynitrite production (77). Inhibition of NOS has been shown to prevent DR pathology in rats (32, 38, 66). Inhibition of NOS or scavenging of peroxynitrite prevented the increased permeability, formation of lipid peroxides, nitrotyrosine, and increased expression of VEGF observed in the retinas of diabetic rats (38). In vitro, high glucose treatment of retinal endothelial cells resulted in increased production of endothelial NOS (eNOS) protein, NOS activity, NO, superoxide, and nitrotyrosine formation. Increased superoxide and peroxynitrite production was shown to be the result of high glucose-induced uncoupling of eNOS leading to the production of superoxide instead of NO and L-citrulline. This uncoupling may occur when the NOS substrate, L-arginine, or its cofactor, tetrahydrobiopterin (BH4), is limited (36). In microvascular endothelial cells, peroxynitrite induced increased expression of VEGF mRNA and protein, an effect that was mediated by the transcription factor STAT3 (90). However, this area remains controversial since other studies have failed to detect hyperglycemia-induced changes to endothelial cell function (19, 74a).
In addition to VEGF expression, peroxynitrite also mediates VEGF signal transduction. High glucose treatment of retinal endothelial cells has been shown to result in apoptosis through the tyrosine nitration of PI 3-kinase by peroxynitrite, resulting in reduced activity of Akt-1 kinase and resultant pro-survival signaling (39). In addition, the peroxynitrite decomposition catalyst 5,10,15,20-tetrakis(4-sulfonatophenyl)prophyrinato iron (III) (FeTPPS) inhibited the sustained phosphorylation of vascular endothelial growth factor receptor (VEGFR)2 induced by VEGF, and subsequent endothelial cell migration and tube formation. These data suggest a role for VEGF-induced peroxynitrite formation in the VEGF signal transduction pathway (37).
There is also evidence for an interaction of ROS and IL-1β. Intraocular injection of IL-1β in normal rats increased oxidative stress as measured by DNA oxidation and NO production. Conversely, the increased levels of IL-1β in the diabetic rat retina were normalized in a prevention trial with antioxidant treatment over a 2-month period (69). In addition, BREC treated with high glucose had increased expression of IL-1β and BREC treated with IL-1β had increased NO expression, caspase-3 activity, and apoptosis (70), suggesting a role for IL-1β in endothelial cell death.
Finally, pigment epithelium-derived factor (PEDF) is a potent inhibitor of angiogenesis and anti-permeability factor originally identified in retinal pigment epithelial (RPE) cells (102,26). Decreased levels of PEDF have been observed in the vitreous and aqueous humor from patients with PDR (17, 34, 87, 88). The decreased levels of PEDF and increased levels VEGF, NADPH oxidase activity, oxidative damage, and vascular leakage observed in the retinas of diabetic rats were prevented by systemic treatment with PEDF (112). In BREC, PEDF blocks the increased expression of VEGF mRNA and protein induced by high glucose through increased expression of UCP-2 and the subsequent decrease in mitochondrial ROS production and Janus kinase 2/STAT3 activation (112). These studies indicate that PEDF acts to prevent generation of ROS and inflammatory mediators in the diabetic retina thereby reducing vascular leakage.
Conclusion
In conclusion, the vascular pathology of DR is likely the result of a complex interaction of growth factors, cytokines, and ROS induced by the action of diabetes-associated metabolic abnormalities. Understanding which cells are primarily affected by these metabolic abnormalities and providing a mechanistic understanding of the resultant pathology will yield novel insight for therapeutic development. One potential model that is emerging suggests that metabolic abnormalities, including hyperglycemia, contribute to oxidative stress and subsequent ROS production in support cells such as glia, microglia, and pericytes. These cells respond with increased cytokine (TNF-α, IL-1β), and growth factor (VEGF) production, potentially as a compensation for the metabolic distress but eventually leading to mal-adaptation and subsequent alterations to the tight junctions increasing the BRB permeability and preventing normal neuronal function. Production of ICAM-1 and leukostasis leads to vascular occlusion and creates regions of hypoxia, further exacerbating the retinal pathology. Ultimately, continued ROS production and inflammation leads to loss of pericytes and vascular endothelial cells, and this vascular degeneration may contribute to loss of visual function (Fig. 8). The relative contribution of each of the diabetes-induced alterations to disease pathology and loss of vision remains to be determined. However, the potential effectiveness of anti-VEGF treatment and anti-inflammatory steroids provide hope that effective, long-term medical intervention to preserve vision is possible.
