
Abstract
Reactive oxygen and nitrogen species (ROS/RNS) are involved in numerous aspects of cellular signaling. Classically ROS/RNS have been associated with cellular dysfunction and disease, but it is now clear that they are also of integral importance under normal conditions. In this review, we discuss ROS/RNS effects in skeletal muscle, with special focus on changes in contractile function. The review deals with the tentative roles of ROS/RNS for acute changes that can occur during strenuous exercise resulting in muscle fatigue, for the recovery from fatigue, and for the effects of training/overtraining. We also discuss two groups of inherited diseases; muscle dystrophies, where recent data suggest that ROS/RNS may be of unexpectedly large importance, and mitochondrial myopathies, where the role of ROS seems more limited than originally thought. Antioxid. Redox Signal. 15, 2487–2499.
Introduction
Superoxide anion (O2 -•) is the primary oxygen free radical. In skeletal muscle, O2 -• is considered to be mainly produced by mitochondria and by NADPH oxidases (76). In mitochondria O2 -• is produced as a by-product of oxidative phosphorylation and hence the rate of production would be expected to increase during physical exercise and in other situations with increased mitochondrial respiration. In mitochondria, O2 -• is mainly produced by complexes I and III, which release O2 -• to the matrix, and complex III, which releases O2 -• to the intermembrane space (68, 76). Early data indicated that the rate of mitochondrial O2 -• production amounts to 1%–2% of the O2 consumption (24). However, this cannot be the situation in exercising skeletal muscle, because prolonged physical exercise and endurance training would then result in dangerously high ROS levels, severe oxidative stress, and muscle damage. Accordingly, more recent measurements of mitochondrial O2 -• production show that the O2 -• production is only ∼0.15% of the O2 consumption (93). It should also be noted that there is no fixed relation between the rate of mitochondrial O2 consumption and ROS production; for instance, uncoupling of oxidative phosphorylation with 2,4-dinitrophenol results in increased O2 consumption accompanied by decreased ROS levels (71) and mitochondrial ROS production appears to be higher during state 4 (basal) as compared to state 3 (maximal ADP-stimulated) respiration, and the latter dominates during aerobic exercise (76).
It has been difficult to detect ROS increases in the mitochondrial matrix with fluorescent indicators during fatiguing stimulation, which increases mitochondrial respiration towards its maximum. For instance, fatigue induced by repeated tetanic stimulation of isolated fast-twitch mouse flexor digitorum brevis (FDB) fibers did not result in increased ROS production in the mitochondrial matrix as measured either by the dihydroethidum-derived fluorescent indicator MitoSOX Red (10, 45) or a novel reversible redox sensitive green fluorescent protein construct targeted to mitochondria (mito-roGFP) (65). On the other hand, increased mitochondrial ROS production during fatigue of FDB fibers is indicated by the fact that recovery after fatiguing stimulation depends on the expression level of superoxide dismutase 2 (SOD2), which is located in the mitochondria and catalyzes the conversion of O2 -• to hydrogen peroxide (H2O2) (22). Taken together, it appears that mitochondrial ROS production increases during intense exercise and, although this increase is likely to be modest and hence difficult to detect, it can induce important changes in contractile function and cellular Ca2+ handling.
Several studies using fluorescent indicators to measure cytosolic ROS levels show moderate increases in muscles during repeated contractions. For instance, contraction-induced increases in ROS have been observed with the general cytosolic ROS indicator dichlorofluorescein in rat diaphragm (81), rat FDB fibers (22), and mouse FDB fibers (65), the latter in which an ROS increase in the mitochondrial matrix was not detected (see above). These results indicate that the increase in ROS production in muscle fibers during repeated contractions mainly involves sources outside of the mitochondrial matrix. One such source is O2 -• generated in mitochondrial complex III and released to intermembrane space (68). Another source is NADPH oxidases; an increased ROS production that appeared, at least partly, to be mediated via NADPH oxidases was observed in experiments with cultured rat myotubes activated by electrical pulses or K+-induced depolarization (34). Thus, an increase in ROS during physical exercise may originate mainly from sources outside of the mitochondrial matrix, such as NADPH oxidases, rather than as a by-product of mitochondrial respiration. Though it must be noted that precise spatial and temporal analyses of ROS production are hampered by method limitations; future experiments with improved techniques are required to achieve a detailed understanding of contraction-mediated ROS production.
Nitric oxide (NO•) is the primary nitrogen free radical. NO• is synthesized from the amino acid L-arginine by nitric oxide synthases (NOS) (50). In addition, it was recently shown that NO• can be formed from the inorganic anions nitrate (NO3 -) and nitrite (NO2 -) (57). Adult skeletal muscle constitutively coexpresses two NOS isoforms: the neuronal (nNOS or type I) and the endothelial (eNOS or type III) isoform (95). A third isoform, inducible NOS (iNOS or type II), is expressed in skeletal muscle in some inflammatory conditions (95). The release of NO• from isolated rat fast-twitch extensor digitorum longes (EDL) muscles and mouse diaphragm and slow-twitch soleus muscles was increased by electrically stimulated contractions (14, 47). The NO• release both under resting conditions and during repeated contractions was similar in eNOS-deficient and wild-type mouse muscles, which indicates that nNOS is the dominating constitutively expressed isoform (47). It should be noted that, with the usage of intact whole muscles, it cannot be excluded that an increased NO• production occurred in other cell types than the skeletal muscle fibers. However, a more recent study showed an increased rate of intracellular NO• production with electrically induced contractions in isolated adult mouse FDB fibers (77). Thus, although less studied than ROS, there is experimental support also for increased RNS production in muscle fibers during repeated contractions.
The contraction of skeletal muscle fibers is initiated by a series of events starting with an action potential propagating along the surface membrane and into the transverse tubules (t-tubules), where it activates specialized voltage sensors, the dihydropyridine receptors (DHPRs). The DHPRs activates the sarcoplasmic reticulum (SR) Ca2+ release channels (ryanodine receptors 1, RyR1) and Ca2+ is released into the cytoplasm where the free [Ca2+] ([Ca2+]i) rapidly increases. Ca2+ subsequently binds to the actin filament regulatory protein troponin C, which leads to cross-bridge cycling and contraction. Ca2+ is actively pumped back into the SR by the SR Ca2+-ATPase (SERCA). When action potentials are no longer generated and SR Ca2+ release ends, SERCA rapidly lowers [Ca2+]i whereby cross-bridge cycling stops and the fiber relaxes. Any of these events can be affected by ROS/RNS, leading to either increased or decreased contractile performance.
In the present review, we will discuss some conditions where ROS/RNS can affect contractile function, with a focus on isometric contractions and studies where cellular mechanisms have been investigated in adult muscle (Fig. 1). We will first discuss acute changes that can occur during strenuous exercise resulting in muscle fatigue. The next section will discuss tentative long-lasting effects of ROS/RNS, which can be observed during the recovery from fatigue and which are likely to be important in relation to endurance training/overtraining. Finally, we will discuss two groups of inherited diseases; muscle dystrophies, where recent data suggest that prolonged effects of increased ROS/RNS may be of unanticipated importance, and mitochondrial myopathies, where the role of ROS seems more limited than originally thought.

Effects of ROS/RNS During the Induction of Skeletal Muscle Fatigue
When repeatedly activated, the contractile performance of skeletal muscle fibers declines, and this is referred to as skeletal muscle fatigue (3). In skeletal muscle fibers, fatigue can be caused by impaired function of any of the events in the excitation–contraction pathway described above. A multitude of factors can contribute to fatigue and the importance of each of these depends on, for instance, the intensity and duration of the activity. Moreover, the susceptibility to develop fatigue, as well as underlying mechanisms, differs between fast-twitch easily fatigued (type IIb), intermediate fatiguable (type IIx), and fatigue resistant (type IIa) fibers (55), and slow-twitch fatigue resistant (type I) fibers (3).
Numerous studies have shown slower fatigue development in the presence of ROS scavengers, but there are also many studies showing no effect of ROS on fatigue development (76). The effects of RNS on fatigue are uncertain with data indicating that NO• can both accelerate and delay fatigue development (76). Adult skeletal muscle constitutively expresses nNOS and eNOS (95). If NO• generated by NOS affects fatigue, this would occur by increased nNOS activity because fatigue development was not affected in mouse muscles deficient of eNOS (47). Interestingly, recent human studies show that an increased dietary intake of nitrate results in decreased O2 consumption during exercise (13, 53, 54) and increased time to exhaustion (12, 13). Dietary nitrate can be metabolized to NO• (57) and the positive effects of increased nitrate intake on exercise performance is considered to be mediated via increased NO•, but the underlying mechanisms remain to be determined.
ROS/RNS effects on fatigue tend to be more marked with submaximal than with near-maximal contractions (76). At the muscle cell level, this suggests effects either on SR Ca2+ handling or myofibrillar Ca2+ sensitivity, because even minor changes in these have large effects on force production during submaximal contractions, which occur on the steep part of the force-[Ca2+]i relationship (see Fig. 6 in Ref. (3)). We will therefore in greater detail discuss how acute fatigue-induced changes in ROS/RNS can affect events related to SR Ca2+ handling and myofibrillar function.
ROS/RNS and Ca2+ release in fatigue
Ca2+ release through RyR1 triggers contraction and there is good evidence that Ca2+ release decreases during fatigue of isolated muscle fibers (4, 58, 96, 109). Given that there are many mechanisms by which this Ca2+ decline can occur (2) and that ROS/RNS scavengers have been shown to prolong the time to fatigue under various experimental conditions (80), it seems possible that post-translational oxidative/nitrosative modification of RyR1 is one mechanism in fatigue. Of course many proteins contribute to the amount of Ca2+ available for release and other tentative targets include the channels contributing to the action potential, the plasma membrane Na+-K+-pumps, and SERCA.
The action potential
The action potential activates DHPR in the t-tubules that interact with RyR1 in the SR. Reductions in the amplitude of the action potential may reduce SR Ca2+ release (27). Extracellular K+ rises in muscle during fatigue and this may reduce the amplitude of the action potential; hence, this is one possible cause of failing SR Ca2+ release during fatigue (89). Numerous studies on many different cell types have shown that the function of Na+-K+-pumps can be adversely affected by ROS (42, 63). Studies from McKenna's laboratory have established that the ROS scavenger N-acetyl cysteine (NAC) can prolong the period of submaximal cycling in trained-humans (64). In a subsequent study, muscle biopsies were taken and the activity of the Na+-K+-pumps was found to be reduced after the exercise period and this reduction was partly ameliorated by NAC (63). In addition, NAC reduced the rise in extracellular K+, presumably through its action on the Na+-K+-pumps. Thus, these studies suggest that increased ROS during fatigue may inhibit the Na+-K+-pumps and exacerbate a K+-induced reduction of the action potential amplitude and thereby contribute to fatigue. However, it appears that decreased membrane excitability is not a limiting factor during most types of intense exercise, which can be explained by a large number of factors counteracting the increase in extracellular K+ and/or its depolarizing effects (for review, see Ref. (3)).
A study on skinned rat EDL fibers electrically stimulated to produce twitches at 1 min intervals showed a markedly accelerated decrease in twitch force at 37°C (∼80% in 7 min) as compared to at 25°C (103). This decrease in twitch force at high temperature was less marked in the presence of the antioxidant Tempol and was ascribed to a ROS-induced decrease in membrane excitablity. However, intact single muscle fibers showed no evidence of action potential failure during tetanic contractions when exposed to ROS (5, 7) or elevated temperature (37°C for FDB and 43°C for soleus fibers) (73). In fact, force was well maintained and there were no signs of action potential failure, even when intact soleus fibers were fatigued at 43°C in the presence of 10 μM H2O2 or tert-butyl hydroperoxide (73). Thus, studies on intact muscle fibers indicate that ROS do not exert any acute inhibitory effects on membrane excitablity.
The SR Ca2+ release channels, RyR1
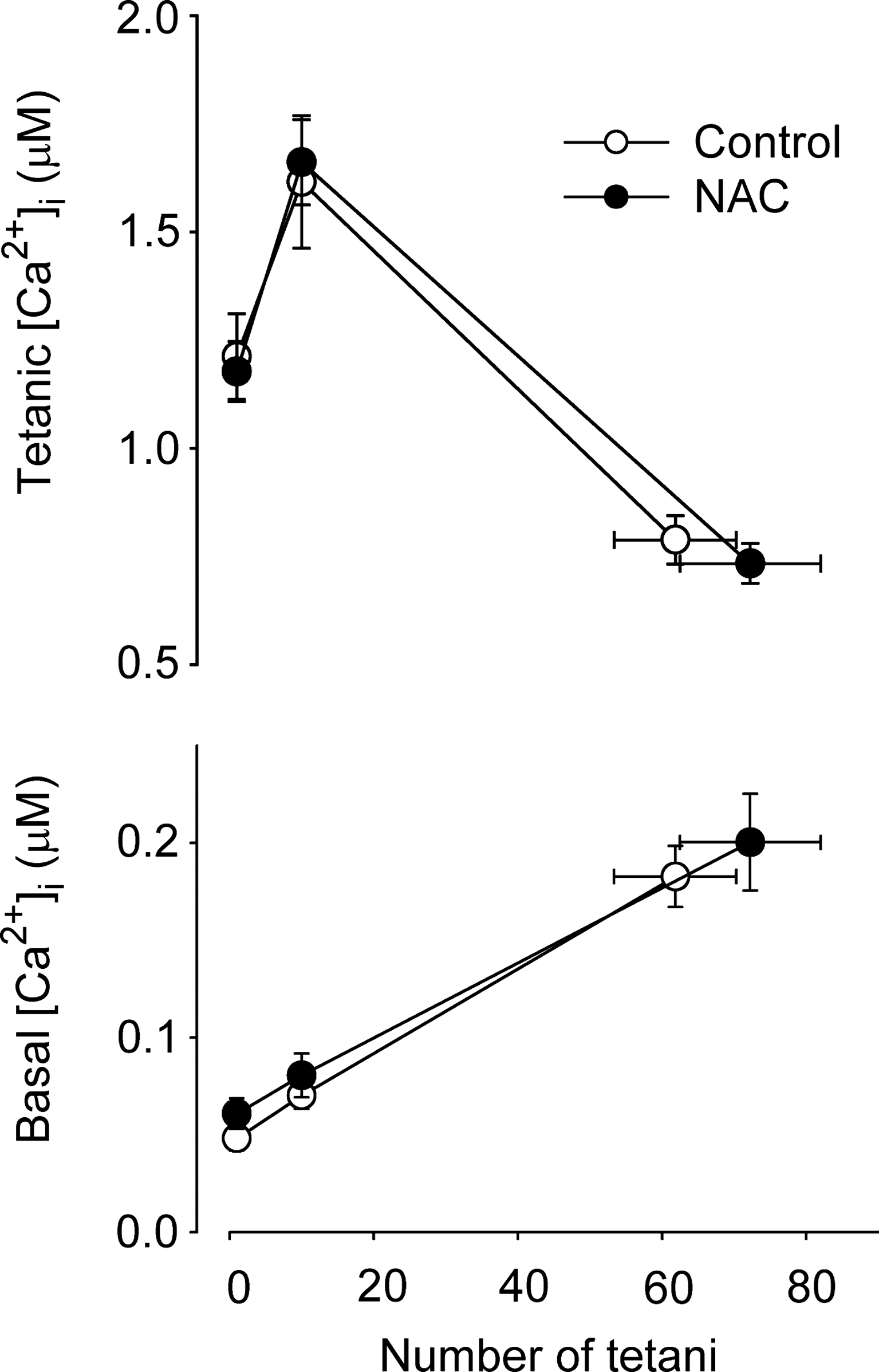
Each RyR1 subunit has 100 cysteines, of which 12 appear capable of modification by S-glutathionylation, S-nitrosylation, or disulfide bridge formation (8). Of these, 9 show signs of endogenous modification while 3 sites appear to respond only to exogenous modification. Various oxidants increase the activity of isolated RyR1 while antioxidants (e.g., glutathione (GSH)) reduce RyR1 activity (8). These data seem to suggest that altered SR Ca2+ release due to ROS-induced changes in RyR1 function might be widespread, but intriguingly this appears not to be the case in the intact system. For instance, while H2O2 depressed depolarization-induced SR Ca2+ release in skinned fibers (20), this effect was not apparent when the release is triggered by action potentials in the t-tubules in either skinned fibers (74) or in intact single fibers (5, 7). Moreover, changes in tetanic [Ca2+]i observed in intact fibers during fatigue induced by repeated tetanic stimulations were similar in control and in conditions with markedly increased ROS levels (66, 73). Furthermore, the number of fatiguing tetani required to decrease force to 40% of the original in single intact FDB fibers was not affected by the presence of the ROS scavanger NAC (22) and the fatigue-induced changes in [Ca2+]i were also similar (Fig. 2).
Recent data suggest that NO• is capable of nitrosylating RyR1 (17), but it is uncertain whether NO• has any acute role in SR Ca2+ release during fatigue. A slight increase in [Ca2+]i during submaximal tetani was observed when intact mouse FDB fibers were exposed to NO• donors (6). On the other hand, NOS inhibitors shifted the voltage dependence of SR Ca2+ release towards negative values in experiments on voltage-clamped mouse FDB fibers (75). Eu et al. suggested that NO• exerts a stimulatory effect on RyR1 Ca2+ fluxes at low O2 pressure and that this effect is missed in many experiments on isolated whole muscles and muscle fibers because these were performed at unphysiologically high O2 pressures (36, 37). Increased NO• during fatigue might then counteract other fatigue-induced effects, resulting in decreased SR Ca2+ release. However, the conclusions of Eu et al. are controversial and other investigators have not observed a critical role of O2 pressure on NO•-induced effects on RyR1 (26). Furthermore, the physiological relevance of fatigue studies performed on isolated whole muscle preparations are likely to be limited by insufficient O2 diffusion to deeper parts of the muscle and hence the development of an artificial hypoxia rather than hyperoxia (15, 117). For instance, force was only reduced by 20% after 120 fatiguing tetani in rat soleus muscles stimulated in situ (85), which is similar to the force decrease observed in single soleus fibers (59, 117) but markedly smaller than the 60% decrease after ∼80 tetani observed in isolated whole rat soleus muscles stimulated with a similar protocol in vitro (58). Thus, although there are no clear signs of altered function induced by hyperoxia in experiments on isolated intact muscle fibers, further studies are required to rule out the possibility that NO• effects on SR Ca2+ release are masked by high O2 pressure in these experiments.
To sum up, both ROS and RNS have the potential to affect RyR1 function but their importance for the decrease in SR Ca2+ release during acute fatigue remains uncertain. A likely reason for this is that metabolic factors (e.g., increased concentration of inorganic phosphate ions (Pi) due to breakdown of creatine phosphate) exert larger depressive effects on SR Ca2+ release during the induction of acute fatigue (3). On the other hand, ROS/RNS-induced modifications of RyR1 may affect the recovery from fatigue (when metabolic changes have been reversed) as well as how the muscle responds to prolonged stress, as discussed below.
ROS/RNS and myofibrillar function in fatigue
Experiments in which intact mouse FDB fibers were exposed to the oxidant H2O2 showed large changes in myobrillar Ca2+ sensitivity, whereas maximum myofibrillar force production was little affected (5, 7). Unexpectedly, the initial response to H2O2 exposure was an increased Ca2+ sensitivity, which was followed by decreased sensitivity with prolonged exposure. Experiments on skinned fibers provide an explanation for this bi-phasic response. Skinned fibers are rather insensitive to application of H2O2 by itself, but a marked decrease in myofibrillar Ca2+ sensitivity was observed when H2O2 was applied together with myoglobin (69); this effect was probably caused by the hydroxyl radical, OH•, produced in the Fenton reaction when Fe2+ in myoglobin reacted with H2O2 (76). Conversely, an increased sensitivity was observed when H2O2 and myoglobin was added in the presence of glutathione (69). Since both myoglobin and glutathione are normal constituents in intact muscle fibers, H2O2 can increase or decrease Ca2+ sensitivity depending on the cellular redox state, for example, reflected by the proportion of reduced to oxidized glutathione (GSH/GSSG).
A marked reduction of the myofibrillar Ca2+ sensitivity is a general feature in fatigued fast-twitch muscle fibers (3, 109). Fatiguing stimulation of fast-twitch muscle is also accompanied by major changes in energy metabolites and an increased concentration of Pi derived from breakdown of creatine phosphate appears to be the major factor underlying the decrease in myofibrillar Ca2+ sensitivity (3). Recently, a very fast force decrease was observed when isolated mouse FDB fibers were stimulated by repeated tetani at increased temperature (37°C). This rapid force decline was due to decreased myofibrillar Ca2+ sensitivity and could be prevented by ROS scavangers (66). Subsequently it was shown that the premature fatigue development was caused by Fe2+ leakage from a stainless steel heat exchanger, and fatigue development was not accelerated at increased temperature when an aluminum heat exchanger was used (79). Moreover, muscles of mice injected with iron showed impaired contractile function and signs of increased ROS production (78). Thus, the combination of increased temperature and Fe2+ had a major force depressing effect via inducing a decrease in myofibrillar Ca2+ sensitivity, most likely mediated by OH• produced in the Fenton reaction (Fig. 3). On the other hand, increased temperature itself does not accelerate fatigue development in isolated muscle fibers (73, 79). To conclude, increased ROS has the potential to decrease myofibrillar Ca2+ sensitivity during induction of acute muscle fatigue but factors related to increased energy metabolism (e.g., increased Pi) are more important under normal conditions.

The rate of NO• production has been shown to increase with electrically stimulated contractions in mouse FDB fibers (77). Intact mouse FDB fibers exposed to NO• donors showed decreased myofibrillar Ca2+ sensitivity (6). Decrease myofibrillar Ca2+ sensitivity was also observed in fast-twitch skinned fibers exposed to NO• donors (92). Thus, a fatigue-induced increase in NO• might contribute to the decrease in myofibrillar Ca2+ sensitivity generally observed during acute fatigue in fast-twitch muscle fibers (109), but, as discussed above, changes in energy metabolites are probably more important (3). Interestingly, NO• donors did not affect the myofibrillar Ca2+ sensitivity in skinned slow-twitch fibers (92), which might contribute to slow-twitch fibers generally being more fatigue resistance than fast-twitch fibers, although other factors such as higher aerobic capacity and lower rate of ATP consumption are likely to be more important (3).
Changes in myofibrillar Ca2+ sensitivity can be caused by altered Ca2+ interaction with the actin filament regulatory troponin–tropomyosin protein complex and/or altered cross-bridge function changing the interaction between cross-bridge attachment and actin filament activation (44). Acute exposure of single mouse FDB fibers to increased ROS or RNS had no significant effect on maximum force production (5 –7), which reflects the ability of cross-bridges to generate force. Moreover, increased NO• had no acute effect on maximum shortening velocity or the rate of force redevelopment after a shortening step in FDB fibers (6). These findings speak against major effects on cross-bridge cycling as the mechanism underlying acute ROS/RNS-induced changes in myofibrillar Ca2+ sensitivity. However, the actual mechanisms by which ROS/RNS alter the Ca2+ activation of the actin filament remain to be established.
Effects of ROS/RNS on the Recovery from Fatigue
The recovery of muscle force production after fatiguing exercise can be very slow, sometimes requiring days for full recovery (33). The force deficit is most marked at low stimulation frequencies and it was therefore initially named “low frequency fatigue” (LFF) (33). The term LFF is now unfortunately being used to describe many different fatigue-related properties, and we recently proposed an alternative term “prolonged low-frequency force depression” (PLFFD) to specifically describe the delayed force recovery (3). In humans, PLFFD has been reported after many different types of muscle activity, such as isometric contractions (33), concentric/eccentric contractions (32), and global exercise such as running (28, 33, 61). PLFFD has important consequences on physical performance and perception of effort because most voluntary contractions are performed with relatively low motoneuron discharge frequencies where the relationship between force production and frequency is steep (19).
At the muscle fiber level, a more marked force decrease preferentially at low stimulation frequencies can be explained by either decreased SR Ca2+ release or reduced myofibrillar Ca2+ sensitivity (see Fig. 6 of Ref. (3)). Although the exact cellular mechanisms underlying PLFFD remain uncertain, there are data supporting a role of ROS-induced changes underlying the slow force recovery after fatigue; for instance, application of the disulfide reducing agent dithiothreitol resulted in increased force production in isolated rat diaphragm strips recovering from fatigue induced by repeated tetani (30). Recent data also indicate a central role for ROS-induced modifications. In a study by Bruton and colleagues (22), the recovery of force after fatigue induced by repeated tetani was followed in FDB fibers of wild-type and transgenic mice overexpressing SOD2. During fatigue, SOD2 overexpressing muscle would be expected to show reduced O2 -• and increased H2O2 levels as compared to wild-types (91). Interestingly, FDB fibers of both groups of mice displayed PLFFD but the defect differed; decreased myofibrillar Ca2+ sensitivity in SOD2 overexpressing fibers and impaired SR Ca2+ release in wild-type fibers (Fig. 4). Furthermore, PLFFD was due to decreased myofibrillar Ca2+ sensitivity in rat FDB fibers; compared to mouse FDB fibers these show higher SOD activity and an increase in cytosolic ROS during fatiguing stimulation (22). These results indicate that increased H2O2, which would dominate in SOD2 overexpressing and in rat FDB fibers, preferentially induces decreased myofibrillar Ca2+ sensitivity, consistent with experiments with exogenous application of H2O2 (5, 7). As discussed above, the ROS that actually mediate the depressant effect on myofibrillar Ca2+ sensitivity is likely to be OH•, which is generated from H2O2 via the Fenton reaction.
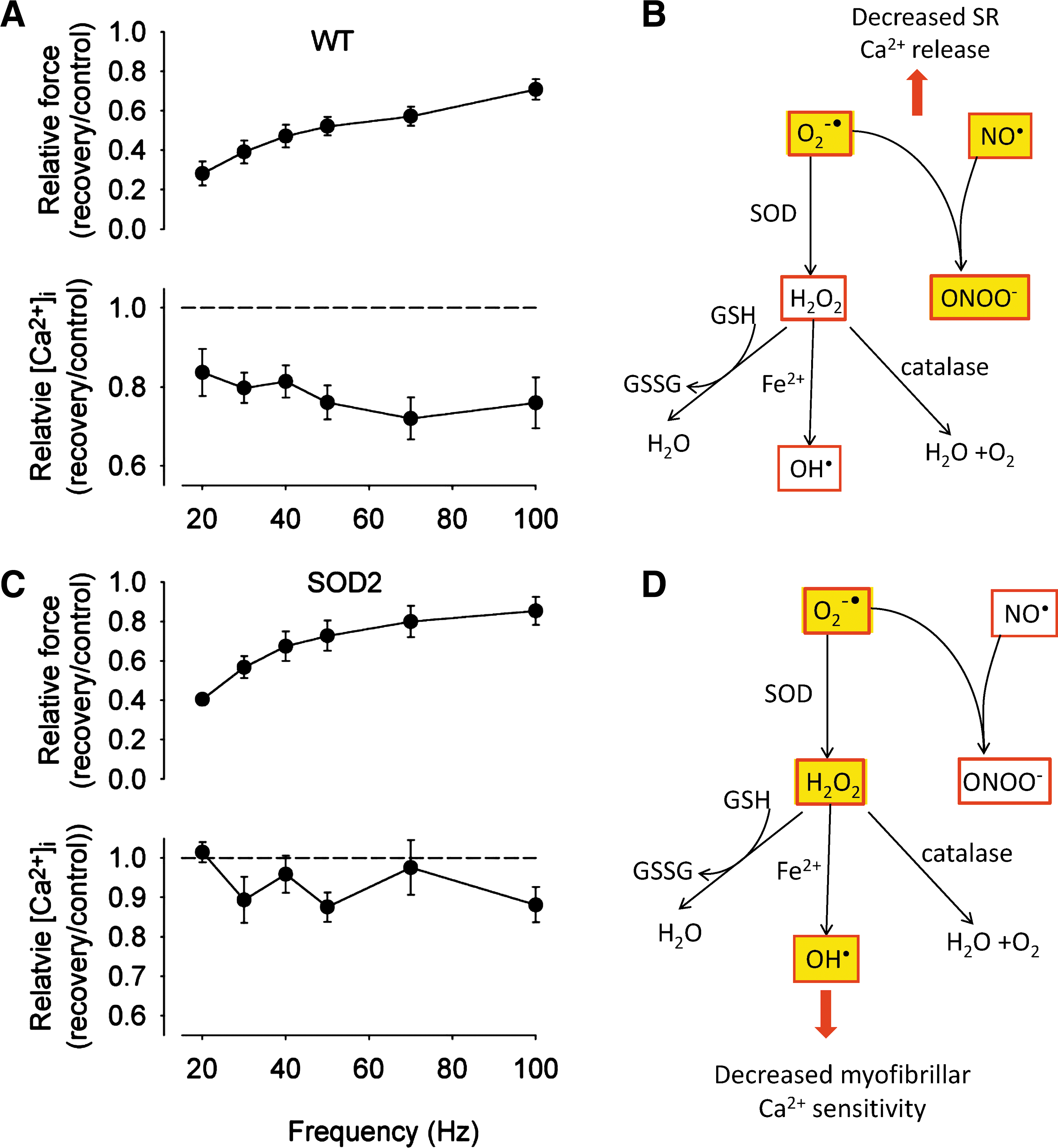
Comparatively higher O2 -• levels, as would be expected during fatigue in wild-type mouse FDB fibers (although not detected with fluorescent indicators; see above), were accompanied by a prolonged decrease in SR Ca2+ release. The fact that different mechanisms underlying PLFFD depend on SOD2, which is located in the mitochondrial matrix, suggests that the O2 -• was of mitochondrial origin. For membrane impermeable O2 -• produced in the mitochondrial matrix to affect SR Ca2+ release, it has to pass through anion channels (101). Alternatively it might react with NO• to form ONOO-, which then directly or indirectly mediates the effect. Thus, while it seems clear that development of PLFFD can be ROS dependent, the exact mechanisms and species involved remain to be established.
Effects of Prolonged ROS/RNS Challenges and Their Relation to Training and Overtraining
Prolonged increases of ROS/RNS are likely to induce post-translational changes in various proteins, which may directly affect contractile function and fatigue resistance. Alternatively, a prolonged elevation of ROS/RNS levels may function indirectly by mediating changes in gene transcription and hence the cellular protein composition. Recently, Bellinger et al. subjected mice to an intense exercise regimen: two 90 min bouts of swimming daily for 3 weeks (18). This, which can be seen as a model for overtraining, resulted in marked changes in RyR1 with serine-2844 phosphorylation, increased nitrosylation of free cysteines, and depletion of the associated proteins 12 kDa FK506 binding protein (FKBP12, also called calstabin-1) and phosphodiesterase 4D3 (PDE4D3). Similar changes in the RyR1 protein complex were seen after a 3-day period of intense exercise in trained cyclists (3 h of 70% V02max). The period of intense exercise increased endothelial NOS in mouse EDL muscles (18), which then provided a potential source of NO• for the RyR1 nitrosylation. At the end of the training period, mice showed reduced exercise capacity and force production was decreased in isolated EDL muscles. Mice with muscle-specific KO of calstabin-1 or PDE4D3 showed reduced exercise capacity similar to that of the exercised mice. A causal relation between the RyR1 changes and the reduced exercise capacity after training was suggested by the use of a drug, S107, which stabilizes the binding of calstabin-1 and PDE4D to RyR1. This drug had no direct effect on muscle performance in control mice but prevented the exercise-induced depletion of calstabin-1 and exercise deficency. Thus, the study by Bellinger et al. provides a model of 'overtraining' in which intense exercise precipitates a decline in performance due to NO•-associated modifications of the RyR1 protein complex. This model shows similarities to PLFFD observed in wild-type mouse fibers and, taken together, these findings indicate that exercise-induced ROS/RNS-mediated changes in RyR1 can cause long-lasting decreases in SR Ca2+ release.
Another example of tentatively ROS/RNS-dependent changes in the RyR1 protein complex was recently described in experiments on muscles of cold-acclimated mice (11). Mammals initially shiver to maintain body temperature when placed in a cold environment (46). With prolonged cold exposure, shivering is normally replaced by the recruitment of uncoupling protein (UCP) 1-dependent heat production in brown adipose tissue (23), whereas UCP1 KO mice continue to depend on heat generated by shivering (43). Soleus muscle fibers of cold-acclimated UCP1 KO mice displayed marked RyR1 phosphorylation and calstabin-1 depletion, accompanied by decreased SR Ca2+ release and impaired contractility (11). Conversely, FDB fibers of cold-acclimated UCP1 KO as well as wild-type mice showed only minor RyR1 phosphorylation and calstabin-1 depletion and these changes were associated with increased SR Ca2+ release (Fig. 5). An important difference between the two muscles is that soleus, but not FDB, muscles participate in the shivering response. Thus, soleus muscles of cold-acclimated UCP1 KO mice were exposed to both prolonged shivering and the stress induced by cold exposure, which includes chronically increased sympathetic activity in skeletal muscle (31). These stressors resulted in a state of muscle dysfunction, which might be equivalent to severe overtraining and similar to that described by Bellinger et al. (18). On the other hand, FDB muscles were only exposed to the general stress induced by a cold environment and this resulted in increased resting and tetanic [Ca2+]i, which were accompanied by an increased mitochondrial biogenesis and increased fatigue resistance (21), (i.e., beneficial changes similar to those obtained with endurance training). As discussed above for intense exercise, the induction of changes in the RyR1 channel complex caused by cold exposure is likely to involve ROS/RNS and provides yet another example of a fine balance between beneficial and deleterious effects of these species.

Heart failure
Humans with heart failure have greatly increased fatiguability of their skeletal muscles (111). Much of this presumably derives from impaired blood flow responses but there is some evidence that intrinsic changes in skeletal muscle also contribute. An early study by Perreault et al. showed that isolated muscle bundles from rats with heart failure exerted less force, fatigued more rapidly, and had reduced [Ca2+]i transients (72). Ward et al. confirmed the reduction in [Ca2+]i transients in muscles of rats with heart failure and they also showed hyperphosphorylation of RyR1 combined with dissociation of calstabin-1 (106). A later study from the same laboratory showed that treating heart failure mice with JTV519, a benzodiazepine that inhibits the dissociation of calstabin-1 from RyR1, improved fatigue performance of isolated soleus muscles (108). Thus, this result provides a link between RyR1 hyperphosphorylation and calstabin-1-depletion to the increased fatiguability of skeletal muscle observed in heart failure. The hyperphosphorylation of RyR1 in these experiments was attributed to increased sympathetic activity associated with heart failure (106, 108). Nevertheless, these changes in the RyR1 protein complex show great similarities with those observed after intense exercise (discussed above) and may then also be related to the increased ROS/RNS frequently observed in heart failure (102, 114). However, it should be noted that Lunde et al. failed to observe changes in the tetanic [Ca2+]i or the fatiguability of fast-twitch skeletal muscle fibers of heart failure rats (58). A later article from this group focused on slow muscles, which were found to be more fatiguable in the presence of heart failure, but the underlying mechanism appeared to be myofibrillar rather than changes in SR Ca2+ handling (59). This myofibrillar impairment appeared to involve a marked reduction in the Ca2+ sensitivity which, as discussed above, can be observed also after intense exercise and which is very sensitive to ROS/RNS. Thus, heart failure is associated with impairments in skeletal muscle related to SR Ca2+ handling and/or myofibrillar function and both these may involve ROS/RNS-induced modifications.
ROS/RNS in Muscle Disease
Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is a disease in which mutations in the dystrophin gene lead to a loss of dystrophin protein. While the chronic effects are clear and include muscle wasting and weakness coupled with inflammation and fibrosis, the early stages of the pathology are still uncertain. It is generally accepted that increased Ca2+ entry into muscle cells, either through damaged membrane or activation of channels that allow Ca2+ entry, contributes to the pathology, notably by activating calpains (41, 70). Increased production of ROS in dystrophic muscle has been recognised for many years (99) and a number of recent studies suggest that ROS-induced modification of RyR1 may play a role in the disease process, possibly by increasing the propensity to SR Ca2+ sparks and leakage.
Ca2+ sparks are temporally and spatially restricted increases of [Ca2+]i induced by the simultanous activation of clusters of RyRs (25, 49). Wang et al. showed that Ca2+ sparks, which are normally hard to detect in mammalian skeletal muscle, can be elicited transiently by osmotic shock (105). These Ca2+ sparks did not require extracellular Ca2+ and are blocked by ryanodine, suggesting that RyR1 is the source. Furthermore, when studied in a mouse model of DMD, the mdx mouse, these sparks were enhanced, became irreversible and occured spontaneously in fibers obtained from mice which were exercised before isolation (105). Wang et al. suggested that the uncontrolled Ca2+ sparks in mdx fibers may result in SR Ca2+ depletion and activation of store-operated Ca2+ entry, which is one of the Ca2+ entry pathways that has frequently been implicated in muscular dystrophy (104). Martins et al. showed that Ca2+ sparks induced by osmotic shock could be largely eliminated by pretreatment with a variety of ROS scavengers or by the NADPH oxidase inhibitors DPI and apocynin (62). A subsequent study from the same group confirmed these findings in mdx fibers and also showed upregulation of gp91phox, the membrane-bound catalytic subunit of NADPH oxidase (90). They also showed increased mitochondrial uptake of Ca2+ and increased ROS generation in mitochondria of mdx fibers indicating a cycle of damaging events.
Another pathway in DMD was recently proposed by Bellinger et al, who showed RyR1 modifications with increased nitrosylation and calstabin-1 dissociation in skeletal muscle of mdx mice (17). Neuronal NOS (nNOS) is normally bound to the dystrophin-associated protein complex, which is lost in dystrophic muscles, and therefore levels of nNOS are reduced (95). In contrast, inducible NOS (iNOS) was greatly increased in mdx muscle fibers and co-immunoprecipitated with RyR1, an association not found in wild-type muscles (17). Bellinger et al. confirmed, as previously shown (8), that exogenous NO donors also cause RyR1 nitrosylation and calstabin-1 depletion. They also showed that the RyR1-stabilizing drug S107 reversed the calstabin-1 depletion and decreased the SR Ca2+ leak without affecting RyR1 nitrosylation. Administration of S107 to mdx mice from 1 month age led to an improvement in muscle strength, reduced serum creatine kinase (a marker of membrane damage), and reduced calpain activity. Thus, Bellinger et al. proposed that Ca2+ leak from the SR elevates [Ca2+]i causing calpain activation and the downstream consequences of muscular dystrophy. However, simply redistribution of Ca2+ from the SR to the myoplasm cannot be the whole explanation because it is widely accepted that total muscle Ca2+ is elevated in muscular dystrophies (41).
Several groups have found evidence that stretch-activated channels are more active and have changed properties in mdx muscle (38, 97). Furthermore, blocking these channels minimizes some of the features of muscle damage, suggesting that this Ca2+ entry pathway is important in early stages of the disease (116). Although stretch-activated channels were discovered by mechanical stretch imposed in patch-clamp experiments, this is not necessarily the mechanism of activation under physiological or pathological circumstances. In fact, several stretch-activated channels seem to be activated by pathways that include ROS (107) and in mdx muscle there is evidence that ROS can activate the same entry pathway (40). Recent work shows that both expression and activity of NADPH oxidase are increased in mdx muscle and confirms that blockers of NADPH oxidase inhibit the Ca2+ entry through stretch-activated channels (110).
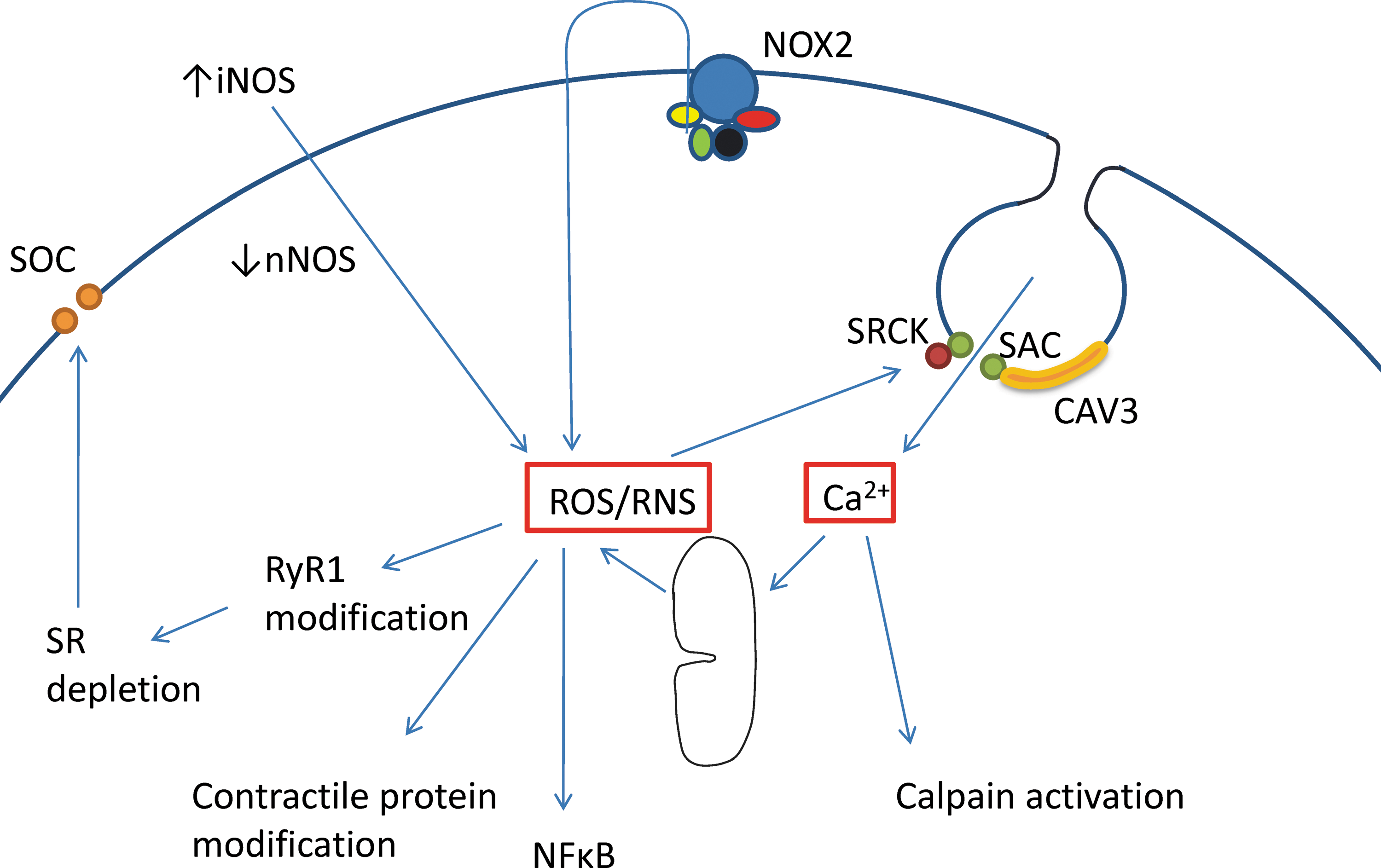
To sum up, there is substantial evidence for an increased ROS production by NADPH oxidase and probably also by mitochondria in dystrophic muscle. Increased NO production by iNOS and decreased production by membrane-bound nNOS are also well established (17, 39, 95). The interaction between ROS/RNS and its potential targets in dystrophic muscle is illustrated in Figure 6. The targets include SR, contractile proteins, and activation of nuclear factor κB (NFκB), which contributes to the inflammation, fibrosis, and atrophy (52). Note that in Figure 6 NADPH oxidase (NOX2) is shown as the membrane-bound, catalytic subunit gp91phox together with its activating subunits (p67phox, p22phox, p47phox, rac 1). Its production of superoxide is initially to the extracellular space (16). Superoxide itself is charged and membrane-impermeant and it is thought to enter the intracellular space either through anion-permeable channels (60) or as the uncharged hydroperoxy radical, HO2 • (86) or as H2O2 after dismutation by extracellular SOD3 (16, 76).
Mitochondrial myopathies
Inherited disorders of mitochondrial oxidative phosphorylation are the most common group of inborn errors of metabolism (48). These mitochondrial diseases are highly variable with respect to age of disease onset and affected organs or tissues (88). Nevertheless, symptoms are most often observed from tissues with high-energy demand, such as skeletal muscle (56, 84), and muscle weakness and exercise intolerance are frequent symptoms in patients with mitochondrial diseases (98). Defective mitochondrial function has been suggested to increase ROS production and cause oxidative stress and these have been suggested to have pivotal roles in the disease process (35, 94, 98). Muscle function has been studied in mice with specific disruption of the nuclear gene for mitochondrial transcription factor A (Tfam KO mice) in fast-twitch muscle fibers. Fast-twitch muscles of these mice display important hallmarks of mitochondrial myopathy, including muscle weakness (112). However, mitochondrial O2 -• (measured with the fluorescent indicator MitoSOX Red) did not increase during fatiguing stimulation of Tfam KO FDB fibers (10). Moreover, increased ROS production has been associated with decomposition of polyunsaturated fatty acids, resulting in formation of reactive carbonyl species that can bind to protein (1), but Tfam KO muscles showed no increase in carbonyl protein adducts (10). These results are in accordance with other recent studies, which also failed to show an increased ROS production or ROS-induced cellular damage in mouse mitochondrial disease models (51, 100). Thus, data from recently developed mouse models do not support an essential role of increased ROS production and oxidative stress in the pathology of mitochondrial diseases.
Concluding Remarks
In this review we have described acute and prolonged effects of ROS/RNS on muscle contractile function. It is clear that the understanding of normal and pathological roles of ROS/RNS in muscle has accelerated dramatically in the past decade. However, there are still areas where knowledge is relatively limited and where rapid progress can be expected in the near future; for example, effects of ROS/RNS on cellular signaling in adult muscle that occur via modulating the activity of kinases and phosphatases (113). There are also major impediments to further progress and these include inadequacy of the tools for detecting, quantifying, and localizing ROS/RNS in cells. In addition, tools for detecting the protein and lipid modifications that are secondary to ROS/RNS are at an early stage of development. Technical developments of measurement tools leading to improved quantification would provide the impetus for a fuller understanding of the regulatory roles of ROS/RNS in normal muscle function and disease.
Footnotes
Acknowledgments
DGA acknowledges support from the Australian National Health and Medical Research Council and the Australian Research Council. HW is supported by the Swedish Research Council, the Swedish National Center for Sports Research, and Association Française contre les Myopathies (AFM).