
Abstract
Introduction
ZO is an acronym for zonula occludens, the Latin name for TJs, and was given together with number 1 (ZO-1) to the first TJ-specific protein identified in the mid-1980s (158). It was not until the early 1990s that ZO-1 was cloned and sequenced, revealing that the protein has significant homology to disc large (DLG), a tumor suppressor protein of Drosophila septate junctions, and to PSD-95/SAP90, a protein present in postsynaptic densities. Both proteins belong to the membrane-associated guanylate kinase (MAGUK) family (183). ZO-2 (59) and ZO-3 (8) were subsequently identified as proteins that co-immunoprecipitate with ZO-1 (61, 79).
Domain Organization of ZO Proteins
MAGUK proteins are characterized for presenting a core of three domains: PDZ, SH3, and guanylate kinase (GuK) (Fig. 1). The presence of the latter (61, 98) is the origin of the name MAGUK that derives from membrane-associated guanylate kinase homologue. However, in MAGUK proteins the GuK domain is not enzymatically active due to the absence of critical amino acids responsible for GMP and ATP binding (61, 98). The GuK domain instead mediates protein–protein interactions (39, 94, 148, 164), and the intramolecular association with the SH3 domain.
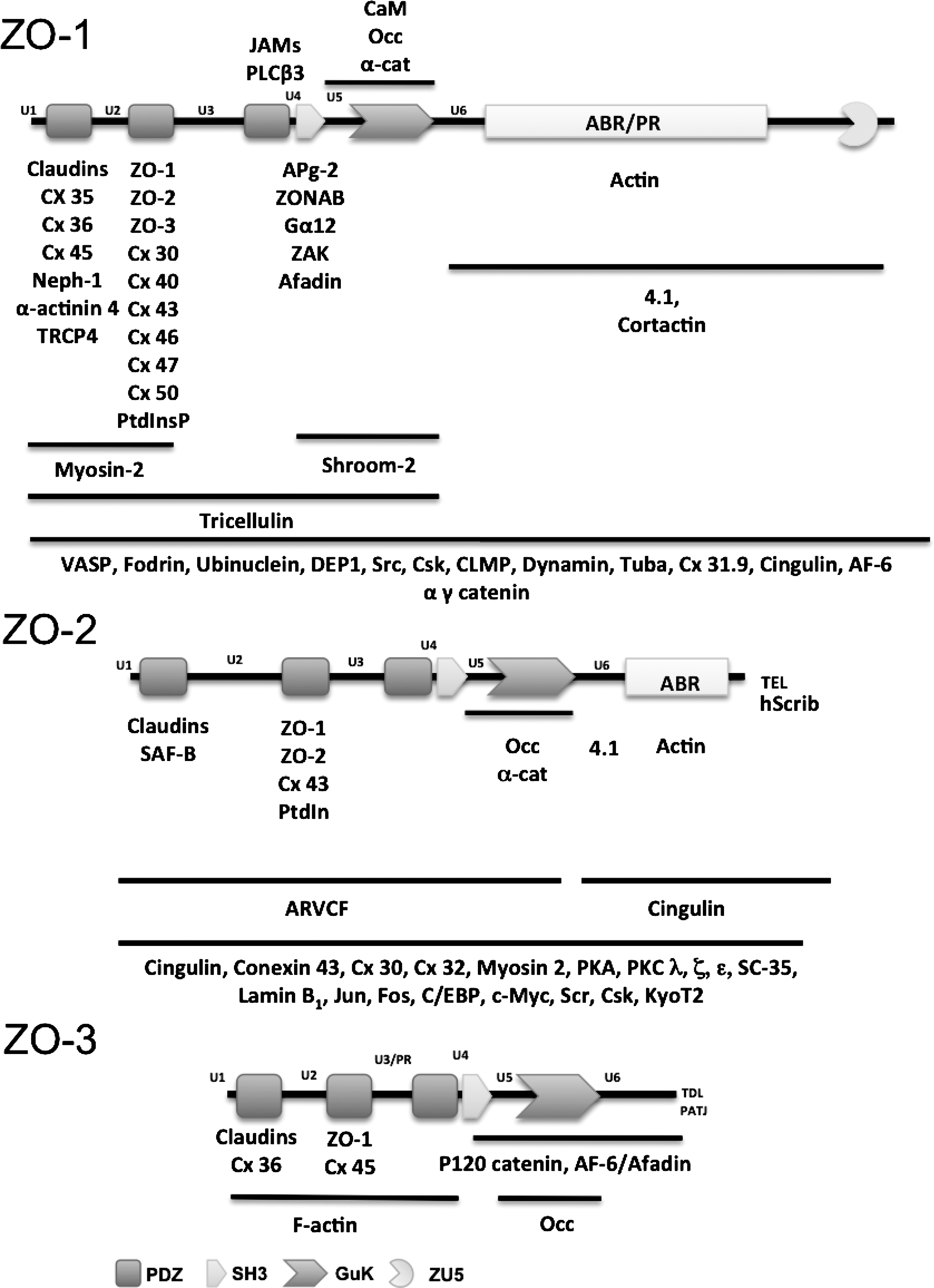
The SH3 module is homologous to a noncatalytic region present in the tyrosine kinase product of the v-Src oncogene. The SH3–GuK interaction was not initially predicted, since the GuK module lacks the PXXP motifs typically required for association with SH3 domains (35). However, crystal structure analysis revealed that the SH3–GuK region, together with its adjacent unique domains 5 and 6 (U5, U6), previously called linkers, are an integrated functional module (112, 117). This explains why mutations that disrupt the interdomain interactions of the SH3–GuK module also disable the association of the GuK domain with its ligands (154, 187). The U5 region that corresponds to the hinge that interconnects the SH3 and GuK modules is required for targeting of ZO-1 to TJs (48) and modulates inter- versus intramolecular interactions of the SH3–GuK unit. Thus, binding of calmodulin or protein 4.1 to the U5 region of the SH3–GuK unit of PSD-95 constrains the flexibility of the hinge region, and promotes the switch from intramolecular interaction to that of SH3 modules with GuK domains present in other molecules, triggering in consequence the formation of heteromultimers (117). In ZO-1, the U5 region functions as an additional binding site for occludin, in addition to the GuK domain. This interaction is based on ionic and helical interactions between basic residues in ZO-1 and acidic residues in occludin, and coiled–coil helix motifs in occludin and the U5 region of ZO-1 (149). Interestingly, α-catenin also binds to the U5 region and the GuK domain of ZO-1, a condition that might explain the successive association of ZO-1 with α-catenin at adherens junctions (AJs) and occludin at the TJs during cell polarity establishment (120). SH3–GuK interaction is not about binding between two independently folded domains, but instead represents the assembly of a unit from subdomains that are separated in sequence. Thus, the U6 region of ZO-1, which is immediately distal to the GuK domain, binds in a divalent metal-dependent manner to a basic surface on the GuK domain that includes helix V and is part of the binding surface for calmodulin. Therefore, the U6 domain is proposed to compete with calmodulin for binding to the GuK domain (120). Since ZO-1 constructs lacking U6 form ectopic TJ strands and the U6 domain inhibits in vitro binding of occludin to the SH3–GuK module (50), it has been proposed that U6 also inhibits occludin binding to the GuK domain of ZO-1 in vivo, preventing in consequence the formation of ectopic TJ strands. This proposal requires the presence of signals capable of displacing U6 in order to permit the binding of occludin to ZO-1 within the TJ region.
Each MAGUK protein also presents other protein–protein binding motifs and domains, as well as various numbers of PDZ repeats. For example, the carboxyl terminal tail of ZO-2 and ZO-3, respectively, exhibit the PDZ binding motifs TEL and TDL, and ZO-1, ZO-2, and ZO-3 have three PDZ domains whereas other MAGUK proteins like MAGI (MAGUK inverted), PATJ (Pals 1 associated tight junction protein) and MUPPI (multi-PDZ domain protein) exhibit 6, 10, and 13 PDZ domains, respectively. ZO proteins additionally posses a proline-rich domain located in ZO-1 and ZO-2 at the carboxyl segment of the protein, whereas in ZO-3 it is found between PDZ-2 and PDZ-3. It is noteworthy that ZO-1 mutants that lack the carboxyl segment with the U6 and proline-rich regions, but maintain the three PDZs, the SH3 and GuK domains (144) or only the three PDZ domains (137), no longer localize at the plasma membrane and induce epithelial to mesenchymal transformation. Similarly, the exogenous expression of the three PDZ containing amino terminal half of ZO-3, perturbs TJ and adherens junction assembly (197). These results hence suggest that the non MAGUK portion of ZO proteins is essential for their pro-differentiation and antioncogenic function.
The carboxyl terminus of ZO-1 also contains a particular domain not yet described in other MAGUKs, named ZU5. This domain, whose function remains unknown, is a region of homology between ZO-1 and the UNC5C/netrin receptor (47). The latter binds the diffusible repellents named netrins that direct migration of various cells and axons in the developing nervous system (103). ZU5 domain is also present in the cytoskeletal protein ankyrin (72).
In summary, the multidomain structure of MAGUK proteins, and of ZOs in particular, is indicative of their scaffolding activity that allows the formation of multiprotein complexes at cell–cell adhesion sites. In addition, the unique combination of domains and motifs present in each ZO protein defines the particular role that each member of this family plays.
ZO Proteins and their Relation to Multicellularity
Although MAGUK proteins play a crucial role in the formation of cell adhesions and synapses, the genes of the MAGUKs MAGI, DLG, and MPP (palmytoylated membrane protein) are present in unicellular protists such as the amoeba Capsaspora owczarzaki (143) and the choanoflagellate Monosiga brevicollis (38).
In contrast, ZO first appears in the Placozoa Tricoplax adhaerens, a flat multicellular animal (Fig. 2). The ZO gene is also present in Cnidaria and Bilateria. In the Craniata an expansion of the gene gave rise to three ZOs: ZO-1, ZO-2, and ZO-3. Interestingly, the ZO gene present in Placozoa, Cnidaria, and Bilateria contains a ZU5 domain, similar to that present in Craniata ZO-1, hence suggesting that ZO-2 and ZO-3 arose as an expansion of ZO-1 that lost the ZU5 domain.

TJs are expected to be present only in animals with true tissue layers. However, the least evolved animals that do have tissue layers are the Cnidaria, and in organisms of this phylum no TJ-like structures have been reported. Hence, it is suspected that ZO-1 in Cnidaria is associated to other cell–cell adhesion complexes in a similar fashion to that observed for ZOO-1 (Zonula occludens ortholog) in C. elegans, where it associates with the cadherin/catenin complex to regulate junctional anchorage to the actin cytoskeleton (109).
In Drosophila, ZO-1 ortholog mutations result in the presence of extra mechanosensory bristles in the notum and head of adult fly (174), and hence the gene was initially given the name Tamou (Tam), as tam in Japanese means “hairy”. Later observations however showed that Tam is the previously described Polychaetoid (Pyd; chaite in greek means hair) gene (180). In Drosophila, Pyd is required for dorsal closure of the embryo (163), sensory organ patterning, and cell fate specification of the developing eye (150) and trachea (80). Pyd protein localizes at the AJs (192) which in insects are located at the uppermost portion of the lateral membrane above the septate junctions that distribute along the lateral membrane (99).
Molecular Interactions of ZO Proteins
In general terms and as can be observed in Figure 1, the C-terminal domains of ZO proteins interact with the cortical actin cytoskeleton (49, 76, 184) and actin binding proteins 4.1 (116) and cortactin (83), while the N-terminal segment that contains the PDZs, SH3, and GuK domains binds to other ZO proteins (172, 184) and to transmembrane proteins, for example, to the TJ proteins occludin (76, 120), claudins (75), tricellulin (138), and JAM (42) [For detailed reviews on claudins, marvel proteins such as occludin and tricellulin, and JAMs, see Overgaard et al. (127a), Blasig et al. (16a), and Bazzoni et al. (12a), respectively, all in this Forum]; to the gap junction conexins(17, 53, 70, 88, 105, 106, 123, 124, 155); to nephrin (68), the main component of the slit diaphragm in the podocytes of the glomerulus; and to the membrane channel TRCP4 (156). Other molecules that associate to ZO proteins include: 1) cingulin (32), afadin (115, 126, 185), and the zonula adherens proteins α-actinin (24) and the p120, α, β, and γ-catenins (120, 133, 185); 2) several signaling molecules such as the kinases ZAK (7), PKC (6), PKA (6), Src and Csk (147), the phosphatase DEP-1 (146), the phospholipase Cβ3 (174), the G protein Gα12 (145), and dynamin (107); the CDC42 specific GEF Tuba (127) and the calcium binding protein calmodulin (128); 3) the transcription factors ZONAB (9), c-Myc (69), Jun, Fos, C/EBP (16), and KyoT2 (67); 4) the tumor suppressor protein Hscrib (119); 5) cytoskeletal proteins such as myosin-2 (190), shroom-2 (46), fodrin (77), and VASP (26); 6) nuclear proteins such as lamin B1 (78), a chromatin component named SAF-B (168) that participates in the assembly of transcriptosome, and the essential pre-mRNA splicing protein Sc-35 (73); 7) phosphoinositides (118); and 8) other proteins whose function is still poorly understood such as ARVCF (87), APg-2 (169), ubinuclein (3) PATJ (141), and CLMP (160).
The multiple molecular interactions above described support the view of ZO proteins as scaffolds that bring together, at specific sites within the cells, groups of proteins involved in a particular cellular function. Thus, the ZO proteins scaffold at the TJ permits the polymerization of claudins at the uppermost portion of the lateral membrane and works as a bridge between transmembrane proteins of the TJ and the actin/myosin cytoskeleton. In addition, this platform of ZO proteins attaches molecules such as kinases and phosphatases that regulate the stability of TJ proteins. At the nucleus, ZO-2, and possibly other ZO proteins as well, forms a scaffold that associates nuclear factors involved in the regulation of mRNA processing and gene transcription with the nuclear matrix.
ZO Proteins Localize at the TJ and the Nucleus
ZO proteins have a dual localization: the nucleus and the TJ (12, 110). ZO-1 and ZO-2 are present in the nucleus of sparse cultures and restricted to the TJ region in confluent monolayers (58, 73). During mitosis when the nuclear membrane is broken, ZO-2 disperses in the cytoplasm and it is not until the late G1 phase of the cell cycle that ZO-2 enters to the nucleus (166). This explains why in cells that are quiescent no ZO-2 is detectable at the nucleus.
ZO-2 inhibits the transcription of promoters regulated by AP-1 sites (16) and the transcription of human cyclin D1 promoter by interaction through cMyc transcription factor with an E box (69). In addition ZO-2 overexpression increases CD1 degradation in the proteosome, reducing in consequence the cellular level of CD1 (166). Thus, in cells transfected with ZO-2, cell cycle progression from G1 to S is blocked and cell proliferation is inhibited.
Redundant and Nonredundant Roles of ZO Proteins
The sequence of human ZO-2 and ZO-3 reveals that they are respectively 56 and 42% identical and 70 and 58% homologous to human ZO-1. Therefore, it is valid to question the redundancy of their function. The three ZO proteins exhibit differential expression in tissues, as ZO-1 and ZO-2 are expressed in both epithelia and endothelia, whereas ZO-3 is exclusively found in epithelia (2). In addition, the RNA transcripts for the three ZO proteins exhibit differential tissue and developmental expression in zebrafish (93).
During mouse blastocyst formation, ZO-1 isoform α− appears at the 8-cell stage, while membrane assembly of ZO-1α+ first occurs during the 32-cell stage, prior to the early blastocyst stage (152). Instead, ZO-2 is present at the cell borders since the 16-cell stage (153). This differential expression of ZO proteins, however, cannot be taken as evidence for nonredundant function, as this could be related to the regulation of the expression either in different tissues or during development, of proteins with redundant function. However, the observation that, in addition to common interaction partners, individual ZO proteins selectively associate with particular proteins (Fig. 1), could suggest nonredundant functions linked to these specific interactions.
Other evidence of nonredundancy among ZO proteins includes the following observations: 1)ZO-1 and ZO-2 play an essential role in mice embryonic development, as ZO-1 and ZO-2 knockout mice respectively die at embryonic days 10.5–11.5 (85) and E7.5 (204). In contrast, mice lacking ZO-3 show no apparent abnormality, and instead are viable and fertile (2, 189). However, it is worth noting that in zebrafish embryos, ZO-3 is critical for epidermal barrier function (92). 2) ZO-1 knockdown generates in mice a more severe inhibition of blastocoel formation than that triggered by ZO-2 silencing (153); 3) In ZO-2 mice chimera generated by injecting ZO-2(-/-) embryonic stem (ES) cells into wild-type blastocysts, the adult males show reduced fertility and pathological changes in the testis. Since the expression levels of other TJ proteins is not affected, it is concluded that ZO-2 is critical for the blood testis barrier (188); and 4) ZO-2 strongly inhibits the expression of cyclin D1 promoter whereas ZO-1 does not (69).
In other aspects such as claudin polymerization at the TJ region, ZO-1 and ZO-2 have redundant roles. Thus, in mammary epithelial cells Eph4 that lack ZO-3 and in which the expression of ZO-1/ZO-2 was suppressed by homologous recombination and RNA interference (1(ko)/2(kd), respectively, a complete lack of TJs is observed. However, when either ZO-1 or ZO-2 is exogenously expressed, TJ filaments are formed. These results hence indicate that ZO-1 and ZO-2 have independently the capacity to initiate claudin polymerization (171).
Oxidative Stress and Hypoxia
Oxidative stress is the result of an imbalance between endogenous oxidants and antioxidants that, by removing free radical intermediates, inhibit oxidation. Reactive oxygen species (ROS) include partially reduced forms of molecular oxygen, such as hydroxyl radical (OH), superoxide anion (O2 −), hydrogen peroxide (H2O2), lipid peroxides, and hypochlorous acid (HClO). The accumulation of ROS is often accompanied by the production of reactive nitrogen species (RNS) such the peroxynitrite anion, a strong oxidant formed by the reaction of O2 − and nitric oxide (NO). However, NO also has antioxidative effects, the best of which is the impairment of lipid oxidation (125).
Hypoxia is a pathological condition in which the body or a region of the body is deprived of adequate oxygen supply. The hypoxic condition occurs with high altitude exposure and more frequently as the result of ischemia, a condition in which there is insufficient blood flow to a certain region of the body, in order to meet the metabolic demand. When the disturbance in the blood supply takes places in the brain due to a blocked or burst blood vessel, it is called a stroke or cerebrovascular accident. Hypoxia induces the breakdown of endothelial and epithelial TJs and in consequence opens the blood brain (BBB) [Lehner et al., (102a)] and blood retinal (BRB) barriers [Frey and Antonetti, (53a)], and induces leakiness in various epithelia. Due to the breakdown of endothelial TJs, intravascular proteins and fluid penetrate into the tissue extracellular space developing edema.
Experimentally hypoxia is produced by ischemia generated by arteries occlusion with clips or by ligation with sutures (e.g., carotid, umbilical cord), or by incubation of animals and cultures in a hypoxic chamber. These experiments are usually followed by reoxygenation in a regular normoxic incubator in order to study ischemia-reperfusion injury. In this regard it should be mentioned that in a wide variety of clinical conditions including circulatory shock, myocardial ischemia, stroke, and organ transplantation, the hypoxic injury is exacerbated after reoxygenation, through mechanisms involving ROS or RNS (104).
Factors and Pathways Activated by Hypoxia that Lead to a Decreased Expression of ZO Proteins and TJ Disruption
HIF-1 is a hypoxia-induced, helix-loop-helix transcription factor that facilitates adaptation to oxygen deprivation by regulating the expression of many hypoxia inducible genes. HIF-1 binds to DNA as a heterodimer composed of an oxygen-sensitive α subunit and a constitutively expressed β subunit, also known as ARNT. Under normal oxygenation conditions, HIF-1α is barely detectable because it is targeted for degradation in the proteosome by the von Hippel-Lindau (VHL) tumor suppressor protein (pVHL) (Fig. 3). The latter is the substrate recognition component of an E3 ubiquitin ligase complex that interacts with HIF-1α in an oxygen-dependent manner, as hydroxylation of a proline residue in HIF-1α mediates pVHL binding and degradation. Instead, in hypoxia, HIF-1α subunits are stabilized and translocated to the nucleus to heterodimerize with ARNT, and to bind to hypoxia response elements within regulatory regions of target genes. The HIF heterodimer activates gene expression at these sites upon recruitment of the transcriptional activators p300 and CBP [for review, see (134)].

It is now well established that hypoxia, through the action of the HIF-1 factor, regulates gene expression of VEGF. VEGF-A, also referred as vascular permeability factor (VPF), is relatively specific for endothelial cells and exhibits two major biological activities: 1) the capacity to stimulate cell proliferation, and 2) the ability to increase vascular permeability (162). These actions are possible through the interaction of VEGF-A with tyrosine kinase receptors VEFGR-1 and VEFGR-2. VEGF-A is known to increase vascular permeability by inducing endothelial fenestration in some vascular beds (140) and by generating a disorganization of endothelial cell–cell adhesion proteins (91).
Oxidative stress action proceeds by several pathways including (Fig. 3): 1) Activation of PLC, followed by increased production of DAG and IP3 that results in intracellular calcium release (50, 51); 2) Activation of classical PKCβII, whereas inhibition of novel PKCδ exacerbates BBB hyperpermeability and TJ disruption (96). These observations are in line with studies that suggest that, in genera,l cPKCs participate in junctional disassembly while novel isoforms regulate junction formation (4, 57); 3) The activity of specific Src family kinases (182); 4) The PI3K/Akt pathway that leads to activation of eNOS (54) and protein kinase G (PKG) (50); 5) The activation of Rho proteins Rac and Cdc42 that, through the mitogen activated protein kinase cascade, activate ERK1/2, c-Jun NH2 terminal kinase (JNK), and p38 (44; 74); 6) Activation of matrix metalloproteinases (MMPs) (11, 111) [For details on MMPs under oxidative stress see Lehner et al. (102a)]; 7) Calcium influx through transient receptor potential channels (TRPC) followed by MLC kinase (MLCK) activation, myosin light chain (MLC) phosphorylation, and actin-myosin contraction (64).
Oxidative Stress-Related Pathologies that Affect ZO Proteins
BBB breakdown
In vertebrates, the barrier properties of the BBB depend on the low rate of fluid-phase endocytosis (136), the absence of fenestrations, and the presence of “tight” TJs in endothelial cells that make up brain capillaries (33). The signals that induce brain endothelial cells to express nonleaky TJs result from the specific interactions between capillary endothelial cells and the surrounding perivascular astrocytes (52). In fact, an astrocyte factor named SSeCKS, whose expression is decreased by hypoxia and strongly upregulated by reoxygenation, can tilt the proliferation/differentiation balance of brain endothelia towards differentiation by inducing the cessation of angiogenesis and the increased expression of TJ proteins ZO-1, ZO-2, claudin-1, and occludin (102) [For details on BBB under oxidative stress, see Lehner et al. (102a)].
Hypoxia-induced HIF-1α activation and VEFG expression decreases ZO-1 protein expression and augments the permeability of brain endothelial cells in both in vitro and animal models (192). In the brain of the MDX mouse, an animal with a genetic defect in the region homologous with the human Duchene muscular dystrophy gene, characterized by a reduction in cerebral oxygenation, BBB opening and cerebral edema, an increased activation of HIF-1α, and overexpression of VEGF, correlates with tyrosine phosphorylation of ZO-1 and reduction in ZO-1 protein content in endothelial cells (122) [For details of BBB under oxidative stress see Lehner et al. (102a) in this Forum].
Blood-retinal barrier disruption
The blood-retinal barrier (BRB) that confers protection or “immune privilege” to the ocular microenvironment is integrated by two separate anatomical sites: 1) the inner BRB (iBRB) formed by the TJs present in the capillaries of the retina, and 2) the outer barrier (oBRB) integrated by TJs present between the retinal pigment epithelial cells as described in detail in the article by Frey and Antonetti in this Forum (53a).
The breakdown of the BRB following retinal ischemia relies on the iBRB as the oBRB remains intact under this condition (86). Disruption of the iBRB results in edema and damage to the retina with adverse effects on vision. In a fashion similar to that observed in the BBB, the capillary endothelial cells of the iBRB rest on a basal lamina covered by pericytes and foot processes of astrocytes and Müller glial cells, which contribute to the proper functioning of the iBRB (Fig. 4). Thus astrocyte-conditioned medium increases the barrier properties of retinal capillary endothelial cells by enhancing the expression of ZO-1 in the vascular epithelium (55). Under normoxia, pericytes conditioned medium (PCM) improves the integrity of the retinal microvascular endothelial cells by inducing occludin and ZO-1 expression (179) and Müller cells secrete pigment epithelium derived growth factor (PEDF) (43) that antagonizes the action of VEGF and hence reduces vascular permeability. Instead under hypoxia, TGF-β expressed by Müller cells become activated, augmenting the endothelial production of MMPs (14) that leads to the proteolytic degradation of TJ proteins (56) and retinal endothelial cell permeability.

Several agents have been employed in experimental or clinical tests, to treat retinal edema associated with hypoxia and acute intraocular inflammatory condition (Fig. 4). Some that affect ZO proteins are: 1) The inhibitor of heat shock protein 90, 17-allylamino-17-demethoxy-geldanamycin (17-AAG), that suppresses the activity of NF-κB, HIF-1α, P38, and PI-3K; and hence diminishes the cellular levels of VEGF, TNF-α, and IL-1β and decreases tyrosine phosphorylation of ZO-1 (131). 2) Clusterin, an extracellular chaperone that stabilizes stress proteins in a folding competent state and restores ZO-1 protein expression in retinal endothelial cells (95).
Epithelial to mesenchymal transition
In epithelial tumors (82) and in kidney (159) and hepatocyte (32) fibrosis, hypoxia is an important stimulus of epithelial to mesenchymal transition (EMT). The EMT induced by hypoxia is characterized by a decreased expression of cell adhesion proteins like E-cadherin and ZOs, and an increased expression of the mesenchymal markers α-smooth muscle actin and vimentin, of metalloproteinases (MMPs), and of transcription factors (TFs) that promote EMT. In hypoxic kidney tubular cells, HIF-1α induces the expression of Twist TF (169); in hepatocytes hypoxia through HIF-1α and TGF-β promotes the expression of snail TF (31), and in a cell line derived from gastric carcinoma, hypoxia induces the expression of snail, slug, Twist, ZEB-1, and ZEB-2. The latter are all TFs involved in EMT.
Anoxia induced in epithelial cells by treatment with metabolic inhibitors that deplete cellular ATP, leads to loss of the permeability barrier and the formation of large and insoluble macromolecular complexes of peripheral TJ proteins that include ZO-1 and ZO-2 (170). The disappearance of ZO-1 from the borders of epithelial cells under hypoxia is mediated by MLCK (178) and can be inhibited in corneal cells by treatment with hepatocyte growth factor (HGF) (97), or keratinocyte growth factor (KGF) in a manner dependent on ERK activation (167) (Fig. 5).

In other several pathological conditions as Diabetes mellitus, lipopolysaccharide-induced damage, obstructive jaundice, inflammatory bowel disease, age-related diseases, vitamin deficiencies, and viral infections, the redox state of the cell is perturbed affecting in consequence the expression of ZO proteins and TJ sealing (Fig. 6).

Diabetes mellitus
Diabetes mellitus is a metabolic disorder characterized by hyperglycemia. Type I diabetes is caused by autoimmune destruction of pancreatic β cells that produce insulin, and Type II diabetes is the result of insulin resistance and/or a β cell secretory defect. The trigger that drives diabetes is the hyperglycemia-induced overproduction of superoxide by the mitochondria. This generation of ROS is central for the onset, progression and pathological consequences of the disease [For review on diabetes mellitus, its metabolic pathways, and resulting oxidative stress, see Frey and Antonetti (53a) in this Forum].
The impact of diabetes on TJs has been explored either in animals where diabetes is induced with streptozotocin (STZ), or in epithelial and endothelial cells cultured with high glucose concentration. Under these conditions, an altered localization and a decreased content and phosphorylation of ZO-1 is observed in kidney glomerular epithelial cells (139). In rat cerebral microvessels the decreased expression of occludin and ZO-1 induced with STZ is concurrent with an increase in BBB permeability and plasma MMP activity (63). Administration of the antioxidant sesamol, a natural organic compound which is a component of sesame oil, improves BBB function and eliminates the decreased ZO-1 and claudin-5 protein expression in STZ-induced diabetic rats (175).
Lipopolysaccharide-induced damage
Lipopolysaccharide (LPS), the main component of the cell wall of Gram-negative bacteria, is an endotoxin involved in inflammatory and immunological reactions. Normally the intestinal epithelium functions as a barrier that avoids LPS translocation, but under conditions of high fat feeding, intestinal permeability increases, producing an increase in plasma concentration of LPS. This condition known as metabolic endotoxemia is involved in the onset of metabolic diseases such as type 2 diabetes. Obese subjects have a higher level of oxidative stress biomarkers than their leaner counterparts and in particular visceral adiposity is significantly correlated with systemic levels of oxidative stress biomarkers (157). Changes of gut microbiota, by antibiotic (20) or prebiotic carbohydrate oligofructose (21) treatment, in high fat fed or ob/ob mice reduces the content of LPS, glucose intolerance, oxidative stress, and macrophage infiltration, and restores the expression of TJ proteins ZO-1 and occludin.
LPS treatment increases the permeability of brain endothelial monolayers and diminishes TER and the expression of ZO-1 and claudin-5 while the production of ROS and NO is increased. The deleterious effects of LPS are significantly reduced upon treatment with pentosan polysulfate, a semisynthetic polysaccharide, structurally related to glycosaminoglycans (176).
Obstructive jaundice
Bile salts are concentrated more than 1000 fold in the biliary canalculi in comparison to portal blood. Therefore, intact TJs at the blood biliary barrier are required for bile secretion without leakage into the blood circulation. Obstructive jaundice causes the collapse of this barrier and leads to significant morbidity and mortality due to liver disfunction and other complications such as peritonitis and sepsis. In rats subjected to common bile duct ligation, an experimental model of obstructive jaundice, gut mucosal permeability and bacterial translocation significantly increase while ZO-1 and occludin expression diminishes (191). In patients with malignant obstructive jaundice, the intestinal mucosa shows a reduced expression of occludin, ZO-1, and claudin-1, whereas claudin-4 is significantly increased (177). The excessive presence of endotoxins in systemic circulation after bile duct ligation stimulates the release of oxygen free radicals and factors like TNF-α and INF-γ (15, 151) that further downregulate the expression of TJ proteins (22). The orally administered probiotic Lactobacillus plantarum (LP) reduces oxidative stress and restores occludin, claudin-1, claudin-4, and ZO-1 levels in hepatocytes of rats subjected to bile duct ligation (193).
Inflammatory bowel disease
Abnormal mucosal permeability is observed in patients with inflammatory bowel disease (IBD) [John et al. (79a) in this Forum]. IFN-γ and TNF-α are elevated in the mucosa of IBD patients and contribute to the proinflammatory cascade that triggers barrier disruption (19). Genetic, immunologic, environmental, and psychological factors contribute to the pathophysiology of inflammatory bowel disease (IBD), however a factor unanimously present in models of IBD is the abnormal levels of antioxidants or oxidized molecules. In fact, the knockout mice lacking the antioxidant enzyme glutathione peroxidase, develops a crypt destructive colitis similar to ulcerative colitis at 11 days of age (45). Likewise, in microscopic colitis, an inflammatory disorder that occurs as a response to a luminal antigen, nitric oxide synthase is induced and IFNγ is significantly upregulated while ZO-1 expression is reduced (161).
Age-related diseases
Age-related macular degeneration is a pathology characterized by the formation of extracellular deposits called drusen that atrophy the retinal pigment epithelium and produce photoreceptor death. The oligomeric form of amyloid-β present in drusen, reduces in retinal pigment epithelial cells, the mitochondrial redox potential, increases the production of ROS, alters transepithelial permeability, and decreases the expression of occludin and ZO-1. Changes in transepithelial permeability and mitochondrial redox potential are reversed with the antioxidant resveratrol (18), present in the skin of red grapes. A further description of how amyloid-β induces capillary angiopathy through oxidative stress is found in Carrano et al. (22a) in this Forum.
Vitamin deficiencies
Oxidative stress plays a major role in the selective neuronal death triggered by thiamine deficiency. An increased production of ROS is reported in brains of rats treated with pyrithiamine (100), an inhibitor of thiamine metabolism. Likewise an increased peroxidase activity is related to Wernicke's encephalopathy (WKS) (34), a cerebral disorder that produces region-selective neuronal loss and BBB breakdown caused by vitamin B1 deficiency. WKS is a common complication of chronic alcoholism and of patients with virus acquired immunodeficiency syndrome (HIV-AIDS). In thiamine deficient mouse brain endothelial cells, permeability is increased, MMP-9 is upregulated, whereas ZO-1 and ZO-2 expression is decreased. Deletion of e-NOS gene restores these BBB alterations, indicating that NO is a major player leading to BBB disruption (13).
Viral infections
HIV Tat protein affects the integrity of the BBB. In mice brain, Tat injection into the hippocampus reduces ZO-1 expression on epithelial cells on the edge of the cerebral peduncle and on endothelial cells of the microvessels of the hippocampus and induces accumulation of inflammatory cells in the brain through a process mediated by ERK1/2 activation. Administration of the antioxidant N-acetylcysteine, a precursor of glutathione, attenuates these effects (132).
Agents that through Oxidative Stress Alter the Expression or Localization of ZO Proteins
Nicotine
Cigarette smoke is a complex mixture of more than 3900 chemical compounds, including free radicals and oxidants (65). Although toxicity exerted by cigarette smoke may be due to a combined action of these compounds, nicotine is probably a mayor player due its presence in tobacco in higher concentrations. Nicotine ingested by humans by smoking or tobacco chewing is an alkaloid that stimulates synaptic transmission acting through nicotine acetylcholine receptors (nAchR) present on neurons and neuromuscular junctions (108) and in a variety of non-neuronal cells, including endothelial cells and lung tissue (130).
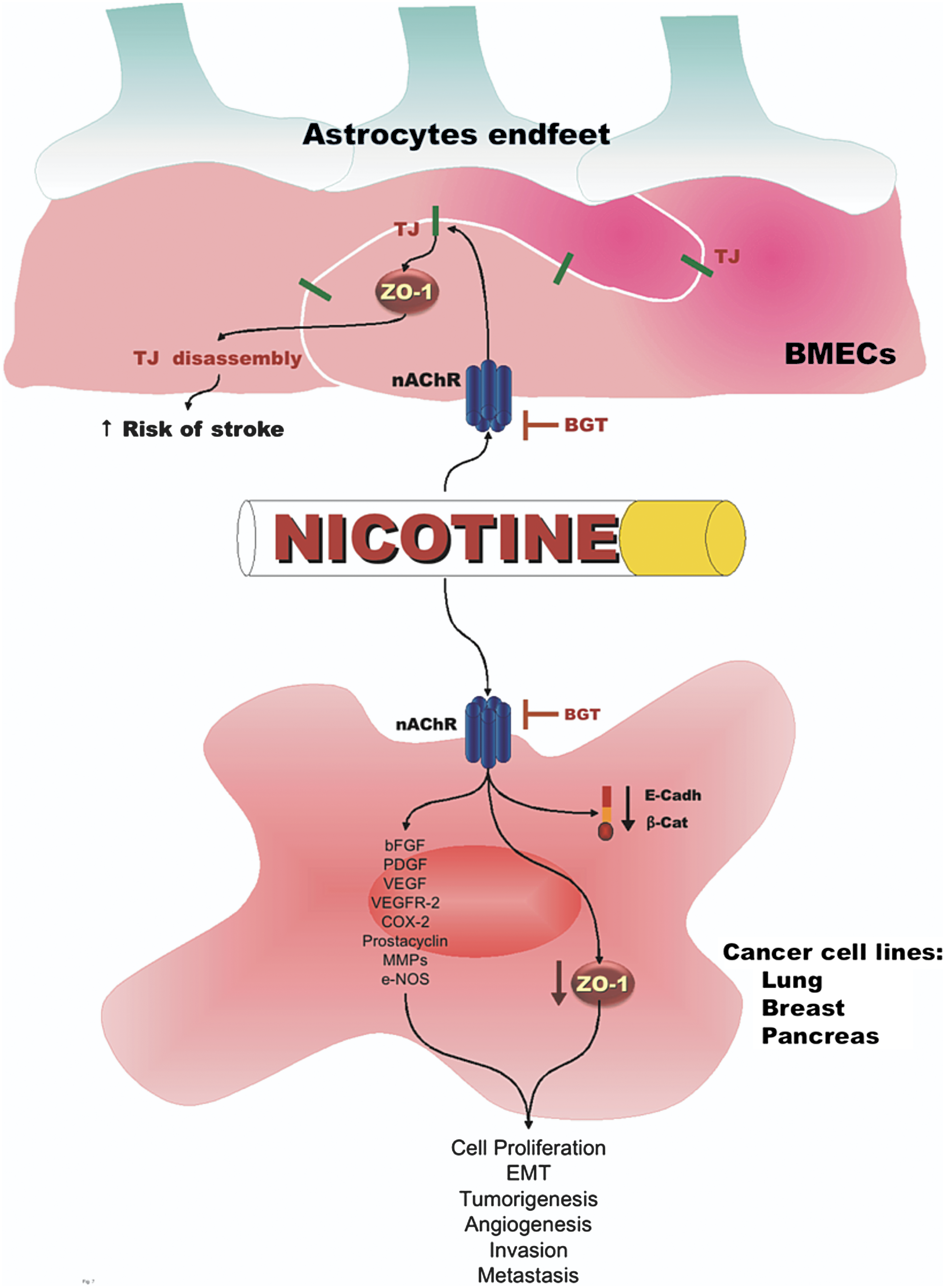
Nicotine induces ROS in a dose-dependent manner and in consequence activates inducible nuclear transcription factor kappa B (NF-κβ) (10). This factor controls the transcription of genes involved in immune response, inflammation, apoptosis, and tumorigenesis. Nicotine induces angiogenesis by upregulating COX-2, prostacyclin, VEGFR-2, MMPs, and e-NOS activity and the cellular levels of VEGF, bFGF, and PDGF (29). Nicotine promotes cell proliferation, invasion, and EMT, characterized by a reduction in the expression of epithelial markers like ZO-1, in a variety of human cancer cell lines, including those from non-small cell lung cancer, breast, and pancreas (36). In mouse models of lung cancer, nicotine promotes the growth and metastasis of tumors pre-initiated by tobacco carcinogens (37). Tobacco not only promotes EMT by acting upon epithelial cells, but also by creating a procarcinogenic stromal environment. Thus fibroblasts exposed to smokeless tobacco extracts, secrete factors that induce partially transformed keratinocytes to lose the expression of cell adhesion proteins such as E-cadherin and ZO-1 (30).
Nicotine and its major metabolite cotinine, through nAchR expressed on brain endothelial cells, alter the distribution and expression of ZO-1 and increase the paracellular permeability of cerebral microvessel endothelial cells and of a hypoxia/aglycemia in vitro model of stroke (1, 62). The increased vascular permeability induced by nicotine, might provide a molecular explanation for the relationship between smoking and stroke severity, incidence, and recovery (Fig 7).
Ethanol
Upon consumption, ethanol is quickly absorbed in the gastrointestinal tract and metabolized in the liver, thus only minor amounts of unmetabolized ethanol are excreted in urine, breath, and sweat (66). The most important pathway for ethanol metabolism involves the cytoplasmic and NAD+-dependent enzyme alcohol dehydrogenase that catalyzes the oxidation of ethanol to acetaldehyde. Ethanol is also metabolized through another pathway involving the peroxisomal heme-containing enzyme catalase, either alone or in combination with NADPH oxidase or xanthine. A third metabolic pathway for ethanol oxidation occurs in the endoplasmic reticulum of hepatocytes and is named microsomal ethanol oxidizing system (MEOS) (28).
In liver mitochondria, ethanol induces the peroxidation of lipids (173) and decreases the content of the most highly unsaturated fatty acids (e.g., arachidonic and docosahexaenoic acids) which are the major peroxidizable substrates (27). In addition, ethanol promotes the formation of F2-isoprostanes that originate from the peroxidation of phospholipids bound to arachidonic acid (121).
The pathological mechanism induced by ethanol is caused by oxidative stress (41), as ethanol and its metabolites react with antioxidant molecules and decrease the antioxidant potential of the tissue (28). This explains why antioxidant pretreatment affords a marked protection against liver damage.
In pancreatic duct (142) and intestinal cells, ethanol and acetaldehyde increase paracellular permeability and disrupt the expression and distribution pattern of ZO-1 and occludin. In intestinal Caco-2 cells, these effects are mediated by: 1) an increased activity of MLCK (113); 2) the inhibition of protein tyrosine phosphatase (PTP) 1B activity that increases tyrosine phosphorylation of ZO-1, E-cadherin, and β-catenin (5); and 3) the overexpression of microRNA 212 that specifically inhibits ZO-1 translation (165).
Chronic alcohol abuse is an important factor for the development of acute lung injury (ALI). Accordingly, in rat alveolar epithelial cells, treatment with ethanol causes a decreased expression and membrane localization of occludin, ZO-1, and E-cadherin that impairs the alveolar barrier function (194). These effects are exacerbated by treatment with ethanol plus LPS and can be markedly attenuated with glutamine supplementation.
H2O2
The impact of oxidative stress induced by ROS has frequently been studied by testing the effect of H2O2. In brain endothelial cells, this is particularly relevant as reoxygenation injury can be prevented by catalase, suggesting that H2O2 is the main mediator of the reoxygenation effect that damages the BBB (51). In brain microvessel endothelial cells (101) and in human umbilical vein endothelial cells (HUVEC), H2O2 causes an altered distribution of TJ proteins, including the dissociation of occludin from ZO-1, through a process that involves activation of cell surface GPCRs, followed by PLC activation and increased intracellular Ca2+ release (51) and MAPK activation (89, 90). In patients with long-term peritoneal dialysis, a hyperpermeable state of their peritoneal mesothelial cells has been observed. Suspecting that this effect is related to oxidative stress, mesothelial monolayers have been supplemented with H2O2, revealing that this treatment induces increased permeability and delocalization of occludin and ZO-1 (81). In several epithelia, such as the human intestinal cell line Caco-2 (89, 144), the bronchial cell line 16HBE (23), and the renal cells MDCK (25), oxidative stress triggered by H2O2 or a mixture of xanthine oxidase and xanthine induces barrier disruption. In Caco-2 and in MDCK cells, this effect is accompanied by induction of tyrosine phosphorylation of numerous cell adhesion proteins including ZO-1. Oxidative stress deleterious effect are prevented in Caco-2 cells by treatment with NO (84) or the tyrosine kinase inhibitor genistein (135), and in 16HBE cells by incubation with KGF (87). In isolated rat hepatocytes couplets the pro-oxidant tert-butylhydroperoxide (tBOOH) that induces the production of ROS, provokes TJ impairment and redistribution of ZO-1 by a PKC-mediated, Ca2+-dependent mechanism that is counteracted by PKA (129).
Chemotherapeutics
Widely used chemotherapeutic agents such as methotrexate (MTX) and cisplatin cause oxidative stress and enhance kidney (40) and intestinal (60, 114) epithelial permeability. Cisplatin chemotherapy in cancer patients induces a fall in plasma antioxidants due to the consumption of antioxidants as well as to the renal loss of small molecular weight antioxidants such as uric acid (181).
In renal epithelial LLC-PK1 cells, cisplatin increases intracellular levels of nitric oxide superoxide anion and peroxynitrite productions, produces TER decrease, and intracellular diffusion of ZO-1. These effects are inhibited by a nitric oxide synthase inhibitor, peroxynitrite scavengers, and by the sodium-dependent glucose transporter (SGLT1) (71). MTX in rat intestine does not change the expression level of ZO-1 and instead produces tyrosine dephosphorylation of the protein and a reduced immunostaining along the cell borders (63).
Conclusion and Outlook
Most studies today correlate oxidative stress to increased epithelial and endothelial permeability and to decreased expression levels of ZO proteins. In this article, we have related how oxidative stress generated in conditions like hypoxia, bacterial and viral infections, vitamin deficiencies, age-related diseases, diabetes and inflammation, alcohol and tobacco consumption, negatively affect the expression of ZO proteins at the TJ, and describe the signaling pathways involved. However, considering that other TJ proteins such as occludin and claudins are often also affected in oxidative stress, it is not clear if changes in ZO proteins levels per se lead to the observed permeability defects, or if they are the consequence of the degradation of the barrier function that affects all proteins that constitute the TJ. Dissecting the pathways triggered by oxidative stress that lead to TJ dysfunction may allow the discovery of molecules that can be targets for therapeutic intervention in a wide array of diseases.
Footnotes
Acknowledgments
This work was supported by Grant 98448 from the Mexican Council for Science and Technology (Consejo Nacional de Ciencia y Tecnología [CONACYT]) and by a Multidisciplinary-10 grant from Cinvestav. Monica Diaz and Miguel Quiros are recipients of doctoral fellowships from CONACYT (267383 and 209822).