
Abstract
This work rests on our previous report (J Cereb Blood Flow Metab 30: 1275–1287, 2010) recognizing that glutamate (Glu) oxaloacetate transaminase (GOT) is induced when brain tissue hypoxia is corrected during acute ischemic stroke (AIS). GOT can metabolize Glu into tricarboxylic acid cycle intermediates and may therefore be useful to harness excess neurotoxic extracellular Glu during AIS as a metabolic substrate. We report that in cultured neural cells challenged with hypoglycemia, extracellular Glu can support cell survival as long as there is sufficient oxygenation. This effect is abrogated by GOT knockdown. In a rodent model of AIS, supplemental oxygen (100% O2 inhaled) during ischemia significantly increased GOT expression and activity in the stroke-affected brain tissue and prevented loss of ATP. Biochemical analyses and in vivo magnetic resonance spectroscopy during stroke demonstrated that such elevated GOT decreased Glu levels at the stroke-affected site. In vivo lentiviral gene delivery of GOT minimized lesion volume, whereas GOT knockdown worsened stroke outcomes. Thus, brain tissue GOT emerges as a novel target in managing stroke outcomes. This work demonstrates that correction of hypoxia during AIS can help clear extracellular neurotoxic Glu by enabling utilization of this amino acid as a metabolic fuel to support survival of the hypoglycemic brain tissue. Strategies to mitigate extracellular Glu-mediated neurodegeneration via blocking receptor-mediated excitotoxicity have failed in clinical trials. We introduce the concept that under hypoglycemic conditions extracellular Glu can be transformed from a neurotoxin to a survival factor by GOT, provided there is sufficient oxygen to sustain cellular respiration. Antioxid. Redox Signal. 14, 1777–1785.
Introduction
Based on a prior transcriptome screening study reported by our laboratory, findings of the current work validate Glu oxaloacetate transaminase (GOT) as an O2-sensitive pathway in neural cells (35). Expression of the Glu metabolizing enzyme GOT increased several fold in stroke-affected tissue that received supplemental oxygen (SO) to correct brain hypoxia (35). GOT transfers the amino group from Glu to oxaloacetate to produce aspartate and α-ketoglutarate, a 5-carbon tricarboxylic acid (TCA) cycle intermediate (18). The current work presents first evidence recognizing GOT as an oxygen-dependent pathway that may switch Glu from a potent neurotoxin to survival factor in the hypoglycemic setting of AIS.
Materials and Methods
Materials
The following materials were obtained from the source indicated: L-glutamic acid monosodium salt, dimethyl sulfoxide, and propidium iodide (Sigma); calcein AM (Invitrogen). For cell culture, Dulbecco's modified Eagle's medium, minimum essential medium, Neurobasal®-A Medium, no D-glucose, L-aspartic acid, L-glutamic acid, L-glutamine, or sodium pyruvate, B-27® Supplement Minus AO (50×), D-glucose, L-glutamine, sodium pyruvate, fetal calf serum, and antibiotic–antimycotic (100×) were purchased from Invitrogen (Carlsbad, CA). Culture dishes were obtained from Nunc (Denmark).
Rodent model of AIS and reperfusion
All experiments were approved by the Institutional Animal Care and Use Committee of the Ohio State University. AIS was induced by the intraluminal suture method of middle cerebral artery occlusion (MCAO) as previously described (25, 35). A drop of > 70% in MCA territory blood flow as measured by laser Doppler flowmetry confirmed successful MCAO.
Correction of AIS-induced brain hypoxia
Rats (Wistar, Harlan) and mice (C57BL6/J; Jackson Laboratories) were randomized into treatment groups before experimental AIS. After confirmation of successful MCAO by laser Doppler flowmetry, rats were subjected to treatment groups (n=5): room air (RA, 21% O2 during AIS) and 90-min hyperbaric oxygen during MCAO (100% oxygen at 2ATA during AIS, SO). Regardless of the treatment mode, all rats were housed in the same experimental hyperbaric chamber under the aforementioned conditions (35). The temperature inside the chamber was maintained at 25°C using a heat lamp.
11.7 T magnetic resonance imaging and infarct volume determination
T2-weighted imaging was performed on stroke-affected mice for infarct volume calcuation using an 11.7T (500 MHz) MR system comprised of a vertical bore magnet (Bruker Biospin) as described previously (33). In brief, mice were anesthetized by 1.5% inhaled isoflurane and a 30 mm birdcage coil was used for image acquisition. A spin echo technique with rapid acquisition with relaxation enhancement (RARE) sequence providing eight echo train length (ETL) was used with the following parameters: field of view (FOV)=30×30 mm, acquisition matrix 256×256, repetition time (TR)=3000 ms, echo time (TE)=30 ms, flip angle (FA)=180 degrees, images in acquisition=15, resolution=8.533 pixels/mm, and number of averages=4. Shim currents were initialized by manual adjustments on all linear and higher order field inhomogeneities. After several localizer scans were completed, a T2-weighted spin echo RARE sequence was applied to generate 15 images corresponding to 15 short axis slices. Postimage processing was done using ImageJ software (NIH) as described previously (33, 35).
9.4T magnetic resonance spectroscopy
Magnetic resonance spectroscopy (MRS) was performed using a 9.4T Bruker Biospin 94/30 magnet (Bruker Biospin). A 3.0-cm-diameter receive-only rat brain coil was placed over the head, and the rat with surface coil was placed inside a 70-mm-diameter linear volume coil. T1-weighted (T1w) and T2-weighted (T2w) images were performed before spectroscopy. Localized spectroscopic data were collected from a 1 mm3 voxel placed over the S1 cortex of the stroke-affected and contralateral hemisphere. Rat underwent T1w imaging with the following parameters: rapid acquisition with relaxation enhancement (RARE) sequences with respiratory gating (RG), repetition time (TR)/echo time (TE) 1200/7.5 ms, Rare Factor (RF) 4, flip angle (FA) 180°, matrix 256×256 pixels, field of view (FOV) 30×30 mm2, and slice thickness 1 mm with no interspaces, one average. T2w imaging were performed with the following parameters: TurboRARE sequences with RG, TR/TE 3500/36, RF 8, FA 180°, matrix 256×256 pixels, FOV 30×30 mm2, and slice thickness 1 mm with no interspaces, two averages. Spectroscopic PRESS (TR/TE=2718/9.9 ms, FOV=13.82 mm3, 512 averages, spectral width 10 ppm with 4096 points) sequence with VAPOR as a water suppression method (bandwidth=350 Hz) and Outer volume suppression was applied. Spectra were analyzed using Bruker TopSpin 1.5 Software.
In vivo GOT knockdown and overexpression
To knock down GOT in mouse brain, iLenti-GFP scramble siRNA and iLenti-GFP GOT-1 siRNA, and for overexpersssion of GOT-1 lentivirus expressing GOT and control lentiviral particles were used. Delivery of lentiviral particles (control or GOT) to the brain were performed using stereotaxic injection as described previously (33). In brief, mice were anesthetized with isoflurane (1%–1.5%) in medical air and secured using a Benchmark Digital Stereotaxic System (Leica) and injection site located using the skull landmark of bregma. A small burr hole (<1.0 mm) was drilled through the mouse skull to deliver control or GOT lentiviral particles (Applied Biological Materials, Inc.). Lentiviral particles were delivered to somatosensory cortex (S1) of the anticipated stroke hemisphere using the following coordinates:−0.5 mm posterior,+3.5 mm lateral, and−1.0 mm ventral to bregma. A 10 μl Hamilton syringe connected to a motorized nano-injector (KD Scientific, Inc.) was used to deliver 5 μl of control or GOT-1 Lentiviral particles (≥1.0×108 IU/ml) at a rate of 0.2 μl/min. After 1 week of delivery, MCAO was performed in mice under RA and SO conditions.
RNA isolation and real-time PCR from brain samples
For GOT-1 mRNA expression, brain tissue was cut into 2-mm coronal sections in a matrix after which it was embedded and frozen in OCT compound (Sakura). Samples were collected for mRNA expression using Laser Microdissection Pressure Catapulting as described previously (35, 36). In brief, to collect neurons from the control and stroke S1 cortex, neurons were quickly immunostained with anti-NeuN antibody (Millipore) and selectively captured as previously described (35, 36). For detection of GOT-1 gene expression, 800,000-μm2 regions of control and stroke-affected S1 cortex were collected from the same brain section. RNA was isolated with PicoPure RNA Isolation kit (Applied Biosystems) and RNA quality was checked as described previously (35, 36). The expression levels of GOT-1 were determined using real-time PCR as described previously (35). Briefly, total RNA (250 ng) was reverse-transcribed into cDNA using an oligo-dT primer and Superscript III (Invitrogen). Reverse transcription-generated DNA was quantified by real-time PCR assay using the double-stranded DNA-binding dye SYBR Green-I. Relative gene expression was standardized to 18s rRNA.
The following primer set was used:
F: 5′-ACCACGAGTACTTGCCCATC-3′
R: 5′-GGCAGAAAACACGCCATTAT-3′
Immunohistochemical detection of GOT-1
After 48 h magnetic resonance imaging (MRI), rat brain tissue was coronal sliced using a brain matrix (Ted Pella, Inc.), formalin-fixed, and embedded in paraffin. Coronal sections (+0.5 mm from bregma, 6 μm thick) were deparaffinized and stained in contralateral control or ipsilateral stroke-hemispheres with anti-GOT1 antibody (LifeSpan Biosciences, Inc.). GOT-1 antibody staining was performed as described previously (35).
Cell culture and treatments
Murine hippocampal HT4 neural cells, originally provided by D.E. Koshland Jr., University of California, Berkeley, were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, antibiotics (100 μg/ml streptomycin, 100 units/ml penicillin, and 0.25 μg/ml amphotericin) at 37°C in a humidified atmosphere containing 95% air and 5% CO2 (22 –25, 37).
Primary cortical neurons
Cells were isolated from the cerebral cortex of rat feti (Sprague-Dawley, day 17 of gestation) as described (24, 31). After isolation from the brain, cells were counted and seeded on culture plates at a density of 1.5–2×106 cells/well of a 12 well plate. Cells were grown in minimal essential medium supplemented with 10% heat-inactivated fetal bovine serum, 40 μM cystine, and antibiotics (100 μg/ml streptomycin, 100 units/ml penicillin, and 0.25 μg/ml amphotericin). Cultures were maintained at 37°C in 5% CO2 in humidified culture incubators where desired O2 (20% oxygen, normoxia, or 0.5% oxygen, hypoxia) ambience was maintained using an electronic OxyCycler (BioSpherix). All experiments were carried out 24 h after plating.
Glu treatment
After 24 h of seeding, culture medium was replaced with Neurobasal®-A Medium media with (25 mM) or without glucose. Glu was added to the medium as an aqueous solution (26, 37). No change in the medium pH was observed in response to the addition of Glu.
Cell viability assay
Viability of primary neural cells was assessed by measuring LDH release from cells into medium after Glu challenge using the in vitro toxicology assay kit (Sigma Chemical Co.) as previously described (22 –25). Survival of primary and HT4 neural cells was also measured by using double staining for simultaneous fluorescence staining of viable (Calcein-AM) and dead cells (propidium iodide). After 18–24 h incubation, medium was removed from each well and cells were washed with warm Dulbecco's phosphate-buffered saline (DPBS) and 3 μM calcein AM and 2.5 μM propidium iodide in sterile DPBS was added. After incubation at 37°C for 15 min cells were washed with DPBS and fluorescent images were collected using a Zeiss Axiovert 200M microscope.
siRNA knockdown of GOT gene expression
HT4 neural cells (0.1×106 cells/well in 12-well plate) were cultured in antibiotic-free medium for 24 h before transfection. DharmaFECT™ 1 transfection reagent (Dharmacon RNA technologies) was used to transfect the cells with 100 nM siRNA pool (Dharmacon RNA Technologies) for 72 h according to the manufacturer's protocol. For control cells, siControl nontargeting siRNA pool (mixture of four siRNAs designed to have ≥4 mismatches with the corresponding mouse gene) was used. A transfection efficiency of >90% was achieved. Cells were then harvested and seeded and 12 h after culturing, the medium was changed followed by treatment with Glu. For determination of mRNA and GOT activity, samples were collected 72 h after siRNA transfection. Total RNA was isolated from cells using the Absolutely RNA® Miniprep kit (Stratagene). The abundance of mRNA for GOT-1 was determined using the real-time PCR. The double-stranded DNA binding dye SYBR green-I was used.
The following primer sets were used:
GAPDH F: 5′- ATGACCACAGTCCATGCCATCACT−3′
GAPDH R: 5′- TGTTGAAGTCGCAGGAGACAACCT−3′
GOT-1 F: 5′- AGAGAAAGATGCGTGGGCTA−3′
GOT-1 R: 5′- TGGACCAGGTGATTCGTACA−3′
GOT activity assay
Cells were collected and pelleted after 72 h of transfection with GOT-1 siRNA to measure GOT activity. Brain samples were snap-frozen in liquid nitrogen. Cell pellets or brain tissue were homogenized and sonicated in 0.05 M phosphate buffer, pH7.4, containing a cocktail of protease inhibitors (Sigma Aldrich), EDTA, 10 mM, and Triton×100 at a final concentration of 1% (10). After sonication homogenates were centrifuged at 21,000 g for 10 min at+4°C to remove debris. Protein concentration was determined with the bicinchoninic acid protein assay from supernatant. Five micrograms protein per samples was used to measure GOT activity using MaxDiscovery™ AST Enzymatic Assay Kit (Bioo Scientific) according to manufacturer's instructions.
Determination of ADP/ATP levels
Changes in the ADP/ATP ratio were measured using bioluminescent assay (EnzyLightTM ADP/ATP Ratio Assay Kit; BioAssay Systems) according to manufacturer's instruction. In brief, brain tissue was dissected to separate stroke affected and contralateral cortex for measurement of ATP concentration. The tissue was immediately homogenized in a manual Dounce-type glass/glass homogenizer on ice in 0.5 ml of a solution containing 0.3% tricloroacetic acid, 1 mM EDTA, and distilled water to precipitate the proteins. This was followed by centrifugation at 10,000 g for 3 min at 4°C. Supernatant was neutralized to pH 7 by the addition of 2 M K2C03 solution at 0°C. The amount of ATP contained in the solution was determined using a bioluminescence assay kit. The luminescence produced in the reaction of ATP and luciferin was detected in a luminometer (Berthold Technologies U.S.A. LLC). The ATP content in each sample was corrected for the protein concentration that was determined with the bicinchoninic acid protein assay (Pierce).
Determination of Glu levels in brain tissue
Frozen brain tissues were homogenized in ultra-pure water on ice using Teflon homogenizer. Homogenates were sonicated three times for 5 s each. After sonication, tissue lysates were centrifuged at 21,000 g for 10 min at+4°C to remove debris. Supernatant were collected and Glu concentration was measured using EnzyChrome™ Glutamate Assay Kit (BioAssay Systems) according to manufacturer's instructions. This assay is a standard spectrophotometric assay based on Glu dehydrogenase catalyzed oxidation of Glu, in which the formed NADH is coupled to the formazan (MTT)/phenazine methosulfate (PMS) reagent. The intensity of the product color measured at 565 nm is proportionate to the Glu concentration in the sample.
Statistical analysis
Statistical analysis of data was performed using SPSS Statistics software (v17.0). All data are reported as mean±S.D. Comparison between groups was tested using Student's t-test.
Results
Our previous effort to characterize the oxygen-sensitive transcriptome in AIS (35) identified the Glu metabolizing enzyme GOT as being oxygen-inducible during cerebral ischemia when the hypoxia component of insult was corrected by SO (Fig. 1A). Microarray outcomes were validated in rats subjected to 90 min of MCAO and kept for the duration of ischemia under RA or SO conditions. Correction of hypoxia by SO during MCAO significantly increased GOT mRNA expression more than threefold in stroke-affected S1 cortex than in contralateral control and RA stroke-affected tissue (Fig. 1B). GOT activity and protein expression were consistently higher (Fig. 1C, D) under SO than under RA conditions.
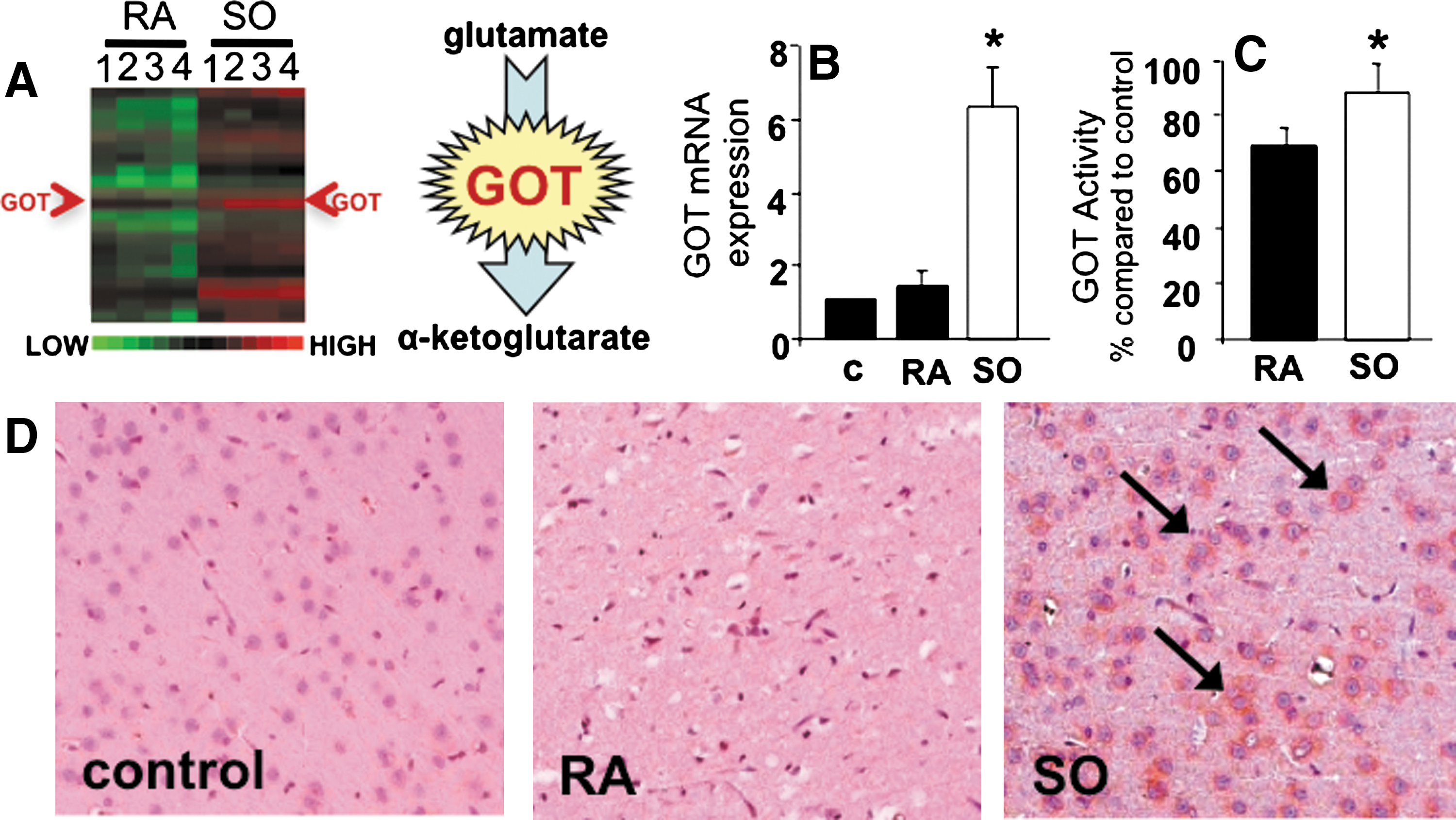
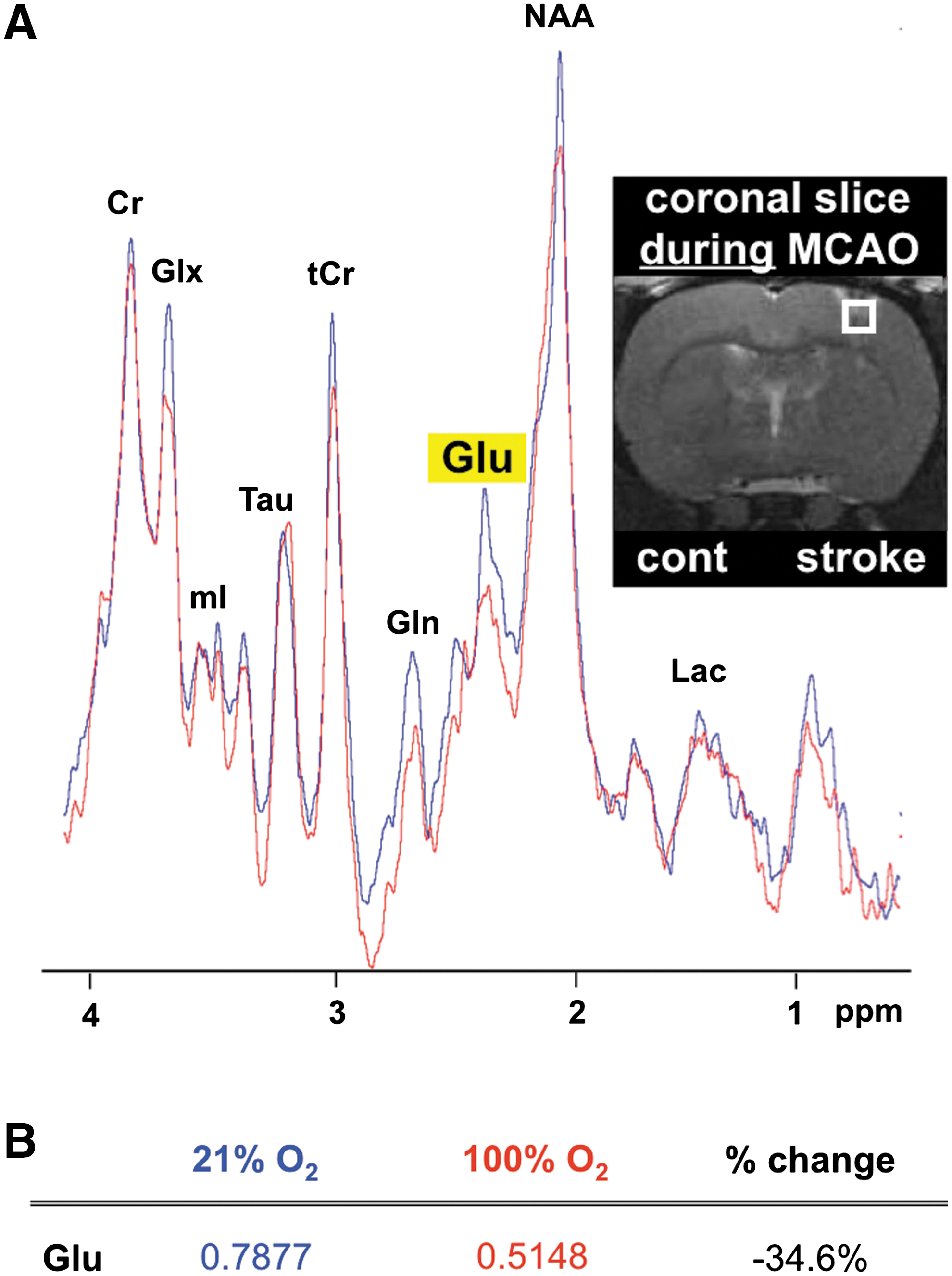
GOT metabolizes Glu to form α-ketoglutarate, a 5-carbon TCA cycle intermediate (18). Because correction of hypoxia during AIS markedly induced GOT expression as well as activity, we sought to test the effects of SO on brain Glu concentration in stroke-affected and contralateral control tissue. No significant difference in S1 cortex Glu concentration was observed between contralateral and stroke-affected hemispheres of RA rats collected immediately after 90 min of MCAO without reperfusion (Fig. 2A). Compared to rats maintained under RA conditions, Glu concentration at the stroke-affected hemisphere was significantly lower in SO-treated rats, suggesting metabolism of Glu by O2-induced GOT. Concurrent with cerebral ischemia interrupting both oxygen and glucose delivery to brain tissue we noted depletion of high-energy phosphate ATP in the stroke-affected hemisphere of RA animals as compared to the contralateral control (Fig. 2B, C). When the hypoxia component of cerebral ischemia was corrected under SO conditions, AIS-dependent depletion of brain tissue ATP was relieved. Outcomes of biochemical Glu quantification were validated during cerebral ischemia using MRS where a 1 mm3 voxel was placed over the S1 cortex of the stroke-affected hemisphere. MRS spectra were acquired after 1 h of MCAO while ischemia still persisted. While the creatine peaks at the stroke-affected cortex of both RA and SO rats were comparable, area under the curve of the corresponding Glu peak was 35% lower in the SO group, supporting improved clearance of Glu at the stroke site by O2-induced GOT (Fig. 3).

To determine the functional significance of GOT on stroke-induced brain lesion volume, we employed a lentiviral approach to overexpress or knock down GOT-1, a cytosolic isoform, in mice before transient MCAO. GFP-tagged lentiviral transduction particles containing GOT-1 overexpression, knockdown, or scrambled (control) siRNA were delivered via stereotaxic injection into the S1 cortex of the right (stroke) hemisphere. One week after delivery, mice were subjected to 90 min MCAO under SO conditions. No GFP was detected in the contralateral control S1 cortex (Fig. 4A) of mice subjected to stroke. Successful transduction was evident in neurons of the stroke-affected S1 cortex by colocalizing GFP with neuron-specific marker NeuN (Fig. 4B). GOT-1 mRNA expression of stroke and contralateral cortex was quantitated by real-time PCR to validate the effects of lentiviral vector delivery (Fig. 4C–E). Control scrambled siRNA had no effect on GOT-1 mRNA expression (Fig. 4C). GOT-1 overexpression significantly increased GOT-1 mRNA expression and attenuated stroke-induced lesion volume as compared to scrambled controls (Fig. 4D, F). GOT-1 knockdown significantly decreased GOT-1 mRNA expression in the stroke-affected hemisphere (Fig. 4E). Such GOT-1 knockdown mice were vulnerable to stroke injury. Only one of six mice receiving GOT-1 knockdown transduction survived to 48 h MRI assessment of lesion volume (Fig. 4G). Other animals died because of massive infarct as evident from MRI of the only surviving mouse (Fig. 4E).

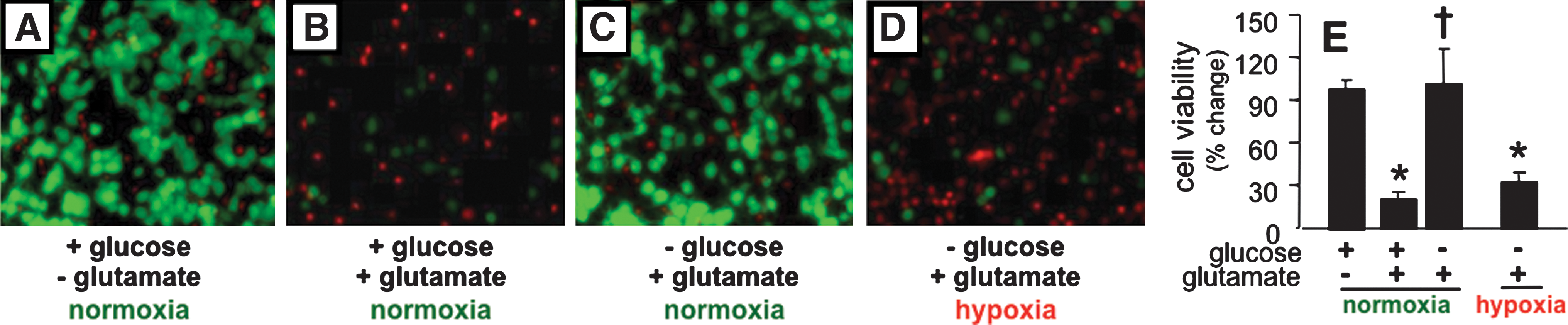
During cerebral ischemia, energetic failure causes sustained neuronal membrane depolarization that contributes to excitotoxic accumulation of extracellular Glu. To model Glu-mediated cell death in vitro, neuronal cultures were incubated in the presence of glucose (25 mM) and excessive extracellular Glu (10 mM) as commonly performed (24, 31). As expected, primary neurons isolated from rat cortex thrived under normoglycemic and normoxic (20% O2) conditions (Fig. 5A). When incubated in the presence of 10 mM extracellular Glu, >75% of neurons died within 24 h (Fig. 5B, E). Strikingly, the same concentration of extracellular Glu did not induce neuronal cell death when glucose was removed from culture media (Fig. 5C, E). In a hypoxic (0.5% O2) and hypoglycemic setting as in ischemic stroke, extracellular Glu failed to sustain cell survival (Fig. 5D, E; Supplementary Fig. S1; Supplementary Data are available online at
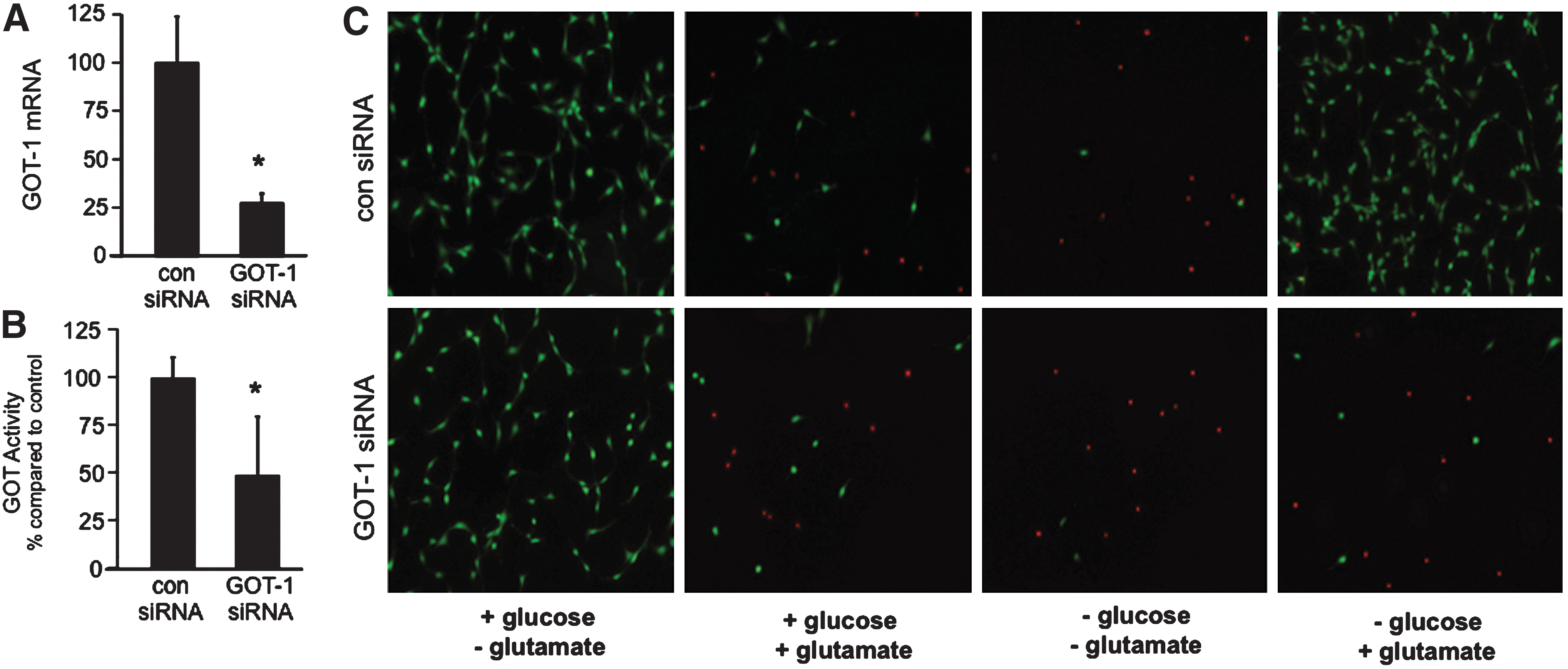
To determine the significance of GOT in the oxygen and glucose-dependent switch of extracellular Glu from neurotoxin to survival factor, HT4 neural cells were subjected to siRNA knockdown of GOT-1. After 72 h of siRNA transfection, GOT-1 siRNA significantly decreased GOT-1 mRNA expression and enzyme activity as compared to control cells (Fig. 6A, B). GOT-1 knockdown resulted in 75% lowering of gene expression (Fig. 6A) and >50% attenuation of catalytic activity (Fig. 6B). At 72 h post-transfection, cells were cultured under normoxic conditions in the presence or absence of glucose (25 mM) and Glu (10 mM). In the presence of glucose and absence of Glu, GOT-1 knockdown had no effect on cell survival. Likewise, siRNA knockdown of GOT-1 had no effect on Glu-mediated cell death in the presence of glucose. In the absence of both Glu and glucose, widespread cell death was observed regardless of GOT-1 knockdown. When glucose was removed from culture media, Glu helped support HT4 neural cell survival (Fig. 6C) consistent with outcomes noted with primary neuronal cell culture (Fig. 5). In GOT-1 knockdown HT4 cells, however, Glu was unable to maintain cell survival in the absence of glucose, demonstrating that GOT-dependent metabolism of Glu is required to maintain cell survival under hypoglycemic conditions.
Discussion
Before the 1959 discovery that Glu plays a central role as an excitatory neurotransmitter (9), and the subsequent developments on synaptic activity and excitotoxicity (8, 20, 28, 32, 39, 40, 42, 44), Glu was viewed as a major metabolic fuel for the brain (30, 41, 45). Resting on that foundation this work proposes a paradigm shift claiming that correction of hypoxia in an ischemic brain injury setting can help clear extracellular neurotoxic Glu by enabling utilization of this amino acid as a metabolic fuel to support survival of the stroke-affected brain tissue. Brain ATP levels decrease by one-third within the first 15 min of cerebral ischemia (21). During ischemic brain injury hypoxia and hypoglycemia represent two major components of insult resulting in accumulation of excitotoxic levels of extracellular Glu (11, 12, 17, 29). To date, strategies to mitigate extracellular Glu-mediated neurodegeneration have primarily focused on blocking receptor-mediated excitotoxicity (i.e., NMDA and calcium antagonists) (43). This approach has failed in clinical trials (1, 14, 15). In this work we introduce the concept that under hypoglycemic conditions extracellular Glu can be transformed from a neurotoxin to a survival factor, provided there is sufficient oxygen to sustain cellular respiration.
From the work of Sir Hans Krebs in 1963 we know that GOT metabolism of Glu generates TCA cycle intermediates under hypoglycemic conditions in brain tissue (18). GOT catalyzes transfer of the amino group from Glu to the 4-carbon TCA cycle intermediate oxaloacetate to generate aspartate and the 5-carbon TCA cycle intermediate α-ketoglutarate (18, 41). Under conditions of stroke, the brain tissue cannot rescue itself from the insult of hypoglycemia by turning to Glu as an alternate source of energy because of inadequate tissue oxygenation. This work demonstrates that the oxidative pathway of Glu metabolism by GOT, as proposed by Krebs, can be utilized as a survival mechanism during stroke as long as tissue hypoxia is corrected. Transamination of Glu by GOT will, on the one hand, clear toxic extracellular Glu from the ischemic site while also feeding cellular respiration to generate energy in glucose-starved energy-deprived cells. Interestingly, strategies to scavenge neurotoxic brain Glu to blood by increasing GOT activity have been shown to protect against stroke in rat (5). Although blood Glu scavenging addresses the problem of neurotoxic Glu handling in brain tissue, it does not resolve the problem of energy deficiency. The current work addresses Glu handling during stroke by repurposing neurotoxic Glu as energy substrate to glucose-starved stroke-affected brain tissue. Taken together, outcomes of the current work suggest a pivotal role for brain GOT in enabling the functional switch of Glu from neurotoxin to survival factor. Findings of this work also recognize brain GOT activity as a therapeutic target in managing stroke outcomes.
Footnotes
Acknowledgments
This work was supported in part by NIH NS42617 and UL1RR025755 from the National Center for Research Resources.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.