
Abstract
Introduction
Because protein aggregation requires at least local backbone fluctuations and in many cases partial unfolding (12, 14), the deposition propensity of globular proteins appears to be linked to their thermodynamic and/or kinetic stability (10), in such a way that mutations or environmental conditions that destabilize the native structure or increase unfolding rates are associated to pathological phenotypes in several protein models (6, 24). This could be one of the underlying reasons why proteins that function in harsh environments, such as the extracellular space, have evolved disulfide bonds (48). Natural disulfide bonds can stabilize proteins to a large extent and strongly reduce conformational fluctuations (1). In some cases, the stabilizing effect is so high that the protein readily unfolds when its disulfides are reduced (19). By crosslinking sequentially distant regions of the polypeptide chain, disulfide bonds are assumed to decrease the entropy of the unfolded ensemble, making it less favorable compared with the folded conformation. Thus, the maximum stabilization will be attained when the disulfide bond links the two termini of the molecule; natural proteins like cyclotides exploit this strategy to become exceptionally stable (9). Crosslinking the polypeptide chain affects not only the stability of the protein at equilibrium but also, in many cases, its mechanism and/or kinetics of folding (25, 49). In fact, engineering of new disulfide bonds and analysis of the corresponding changes in folding kinetics is a useful approach to study the proximity of specific protein regions in the transition state ensemble (27).
Innovation
Cyclization significantly stabilizes SH3 domain of p85α subunit of bovine phosphatidyl-inositol-3′-kinase (PI3-SH3) and strongly accelerates its folding rate while having a minor effect on its unfolding rate, promoting the fast attainment of the folded state and a higher proportion of native molecules at equilibrium, contributing thus to decrease the population of aggregation-prone species. In addition, even when amyloidogenic intermediates are populated, cyclization slow downs PI3-SH3 self-assembly and renders the fibrils less cytotoxic. Thus, disulfide bonds likely reduce the chances of pathogenic protein aggregation, especially in the extracellular space, where polypeptides are not subjected to the surveillance of molecular chaperones. Prevention of protein aggregation is an important selective pressure acting on the evolution of protein sequences (11); the protective effect of disulfide bonds against toxic protein deposition in the oxidative extracellular background might explain why the sequences of secreted SH3 domains can support a higher intrinsic aggregation load than those of their less stable intracellular counterparts. Importantly, during the revision of this work, a study by Mossuto et al. showed that reduction of disulfides in human lysozyme increases the toxicity of the resulting fibrils and, more generally, that disulfide-containing proteins in the human proteome display a high aggregation propensity (36). Overall, it appears that the impact that covalent polypeptide crosslinking exerts in the complex processes of protein folding and aggregation might be more relevant from the physiological point of view than previously thought. In this sense, the design of new disulfide bonds might be an effective strategy to reduce the aggregation of target proteins of biotechnological interest.
SRC homology 3 domain (SH3) domains are good model systems for folding and aggregation analysis due to their monomeric state, their small size, and the absence of cofactors and, usually, disulfide bonds. The SH3 domain of p85α subunit of bovine phosphatidyl-inositol-3′-kinase (PI3-SH3) consists of 83 residues that fold into five β-strands and two short helix-like turns. The five β-strands are arranged in two β-sheets that are orthogonal to each other forming a β-sandwich (33) (Fig. 1). PI3-SH3 appears to fold/unfold following a canonical two state mechanism (49), although recent studies suggest the existence of a transient intermediate after the rate-limiting step of folding (47). PI3-SH3 constitutes one of the best-characterized globular proteins able to form ordered amyloid fibrils starting from an acid-unfolded state (28, 46). Therefore, this domain provides a framework to study the effect of backbone cyclization on protein stability, folding, and aggregation.
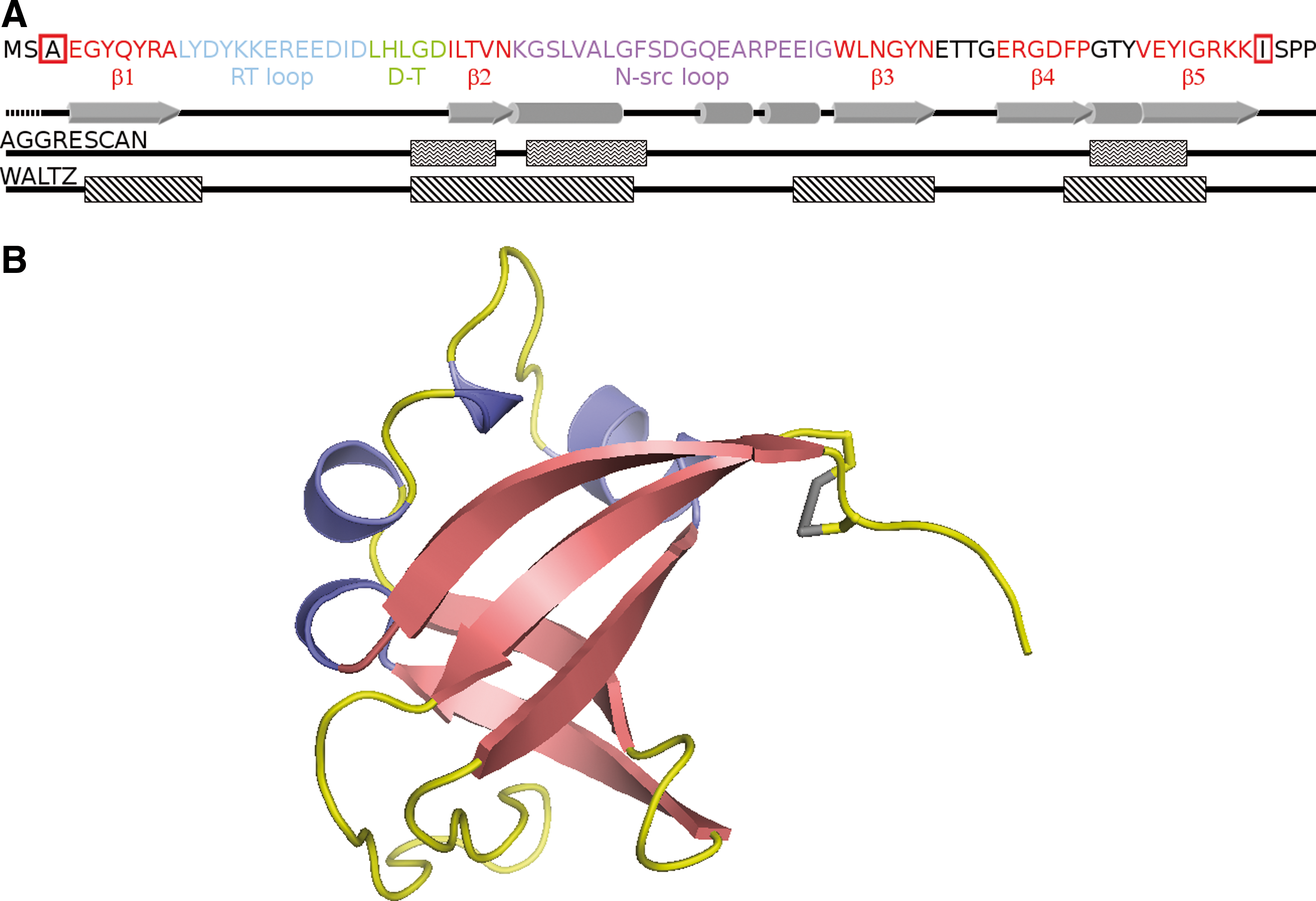
Here, we investigate the properties of a PI3-SH3 mutational variant in which the N- and C- termini have been crosslinked by a designed disulfide bond. Overall, the data collected here indicate that, by altering polypeptide chain connectivity, disulfide bridges have a high impact in protein folding as well as in amyloid formation and suggest that they may play a major role in minimizing misfolding and aggregation phenomena.
Results
Crosslinking the N and C termini in the PI3-SH3 domain
We used FoldX (42) to design a PI3-SH3 domain with the N and C terminal ends linked by a disulfide bridge. We searched for two residues close enough in the native structure to permit the formation of the desired covalent interaction after their mutation to Cys. The best positions were Ala3 and Ile82, due to their proximity (distance between the β-carbons=4.2 Å), face-to-face orientation, and proper dihedral angles. Therefore, we cloned, produced, and purified an SH3-PI3 mutant with both residues replaced by Cys (PI3-SH3-SS). The absence of detectable alkylation by vinylpyridine confirmed the formation of the designed disulfide bond. The introduced covalent linkage closes off a loop of 78 residues (Fig. 1).
Conformational analysis of PI3-SH3 domains under native conditions
The wild-type domain (PI3-SH3-WT) and PI3-SH3-SS exhibit analogous far-UV circular dichroism (CD) spectra under native conditions (pH 7.0 and 298 K) with a characteristic minimum at 222 nm (46) (Fig. 2A). However, the PI3-SH3-WT maximum at 235 nm is absent in PI3-SH3-SS. The far-UV CD spectrum of PI3-SH3-SS under reducing conditions does not change significantly, suggesting that the change in the spectrum is associated to the sequential changes and not to disulfide bond promoted strain. Accordingly, Cys residues are known to contribute negatively to the CD signal of peptides in this region of the spectrum (32). We will refer to PI3-SH3-SS in its reduced form as PI3-SH3-(SH)2.
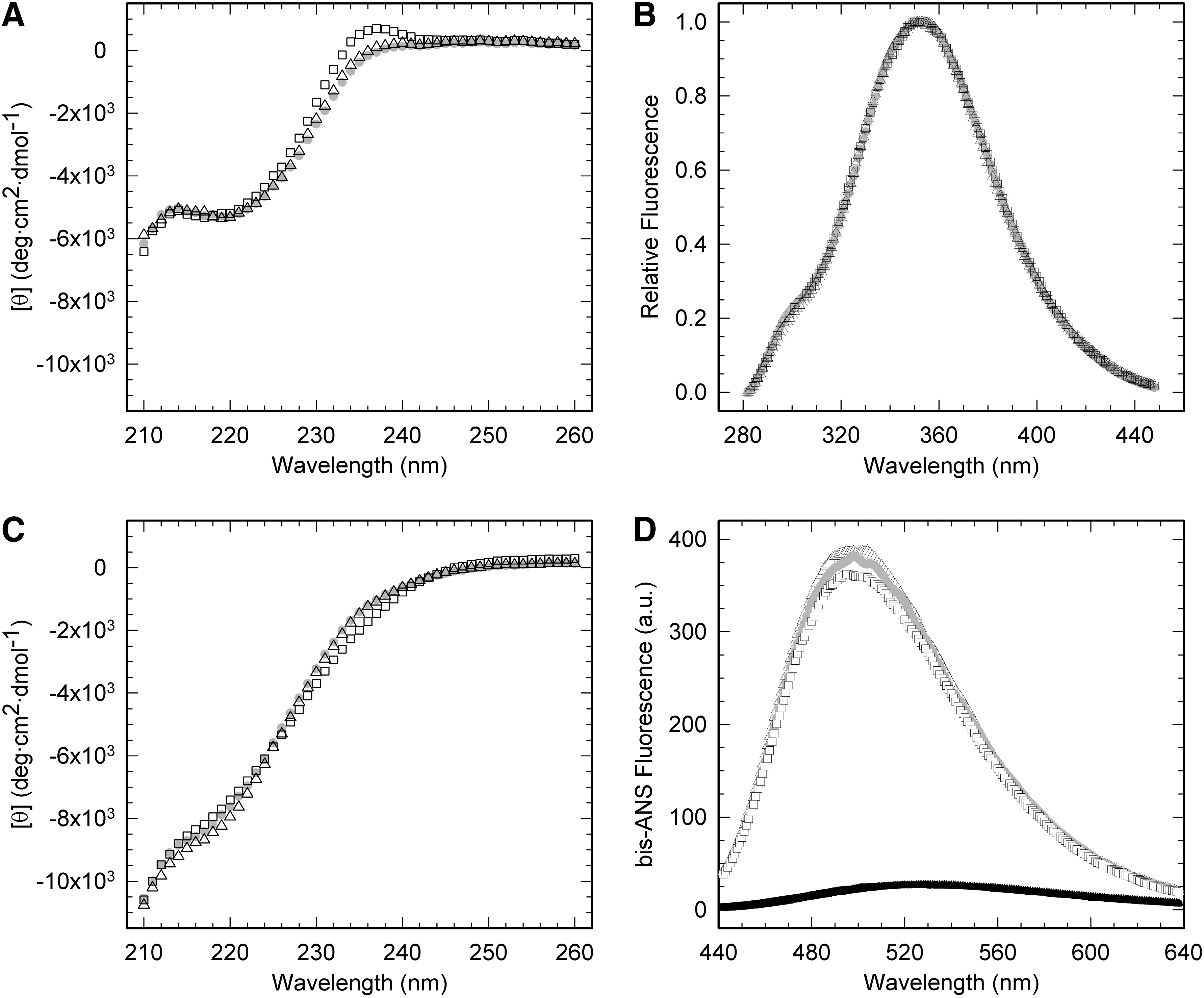
PI3-SH3 contains a single Trp in position 55 and 4 Tyr residues at positions 6, 8, 12, and 59. We excited the different proteins at 268 nm and recorded their intrinsic fluorescence emission spectra under native and reducing conditions. No significant differences were detected between domains (Fig. 2B). The structural features of the PI3-SH3-WT and PI3-SH3-SS were also evaluated by NMR spectroscopy. The one-dimensional NMR (1H-NMR) spectrum of both proteins at pH 7.0 and 298 K display a wide signal dispersion of resonances at both low (amide and aromatic region) and high (methyl region) fields, with good peak sharpness, characteristic of folded molecules (Fig. 3). The two spectra are almost identical, which together with the CD and fluorescence data indicate that the introduced mutations do no affect significantly the PI3-SH3-fold.
Thermal unfolding of PI3-SH3 domains
The thermal stability of PI3-SH3 domains at pH 7.0 was analyzed by intrinsic fluorescence, CD, differential scanning calorimetry (DSC), and 1H-NMR (Fig. 4).
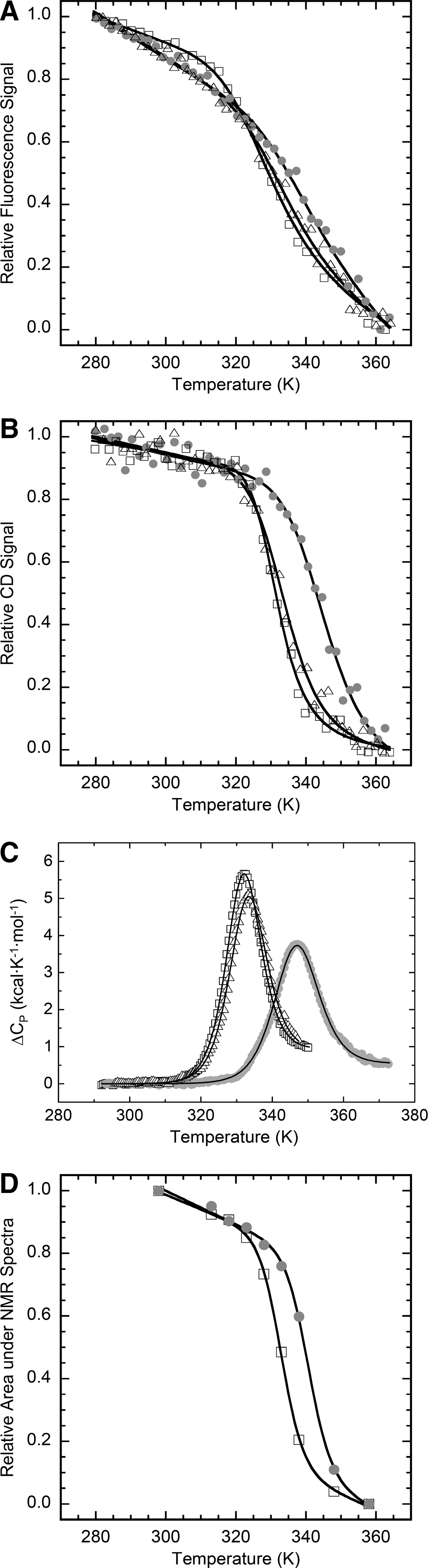
The transition curves from heat-induced fluorescence emission changes in PI3-SH3 domains are shown in Figure 4A. The thermal denaturation curves followed by far-UV CD at 235 nm are show in Figure 4B. In both cases a single cooperative transition was observed and the data could be fitted accurately to a two-state temperature-induced unfolding model (R>0.99). DSC scans of PI3-SH3 exhibited a single cooperative transition corresponding to a two-state unfolding process without a significant population of intermediate partially unfolded states, according to the ratio between the van't Hoff and calorimetric enthalpies (Fig. 4C). As shown in Table 1, the transition temperatures of unfolding obtained by the three independent methods are very similar, coinciding to indicate that PI3-SH3-SS is considerably more stable toward thermal denaturation than the wild-type and reduced forms. PI3-SH3-(SH)2 is somewhat more stable than the wild-type domain.
DSC.
Intrinsic fluorescence.
CD.
1H-NMR.
ΔC p, heat capacity change of unfolding; CD, circular dichroism; DSC, differential scanning calorimetry; PI3-SH3, SH3 domain of p85α subunit of bovine phosphatidyl-inositol-3′-kinase.
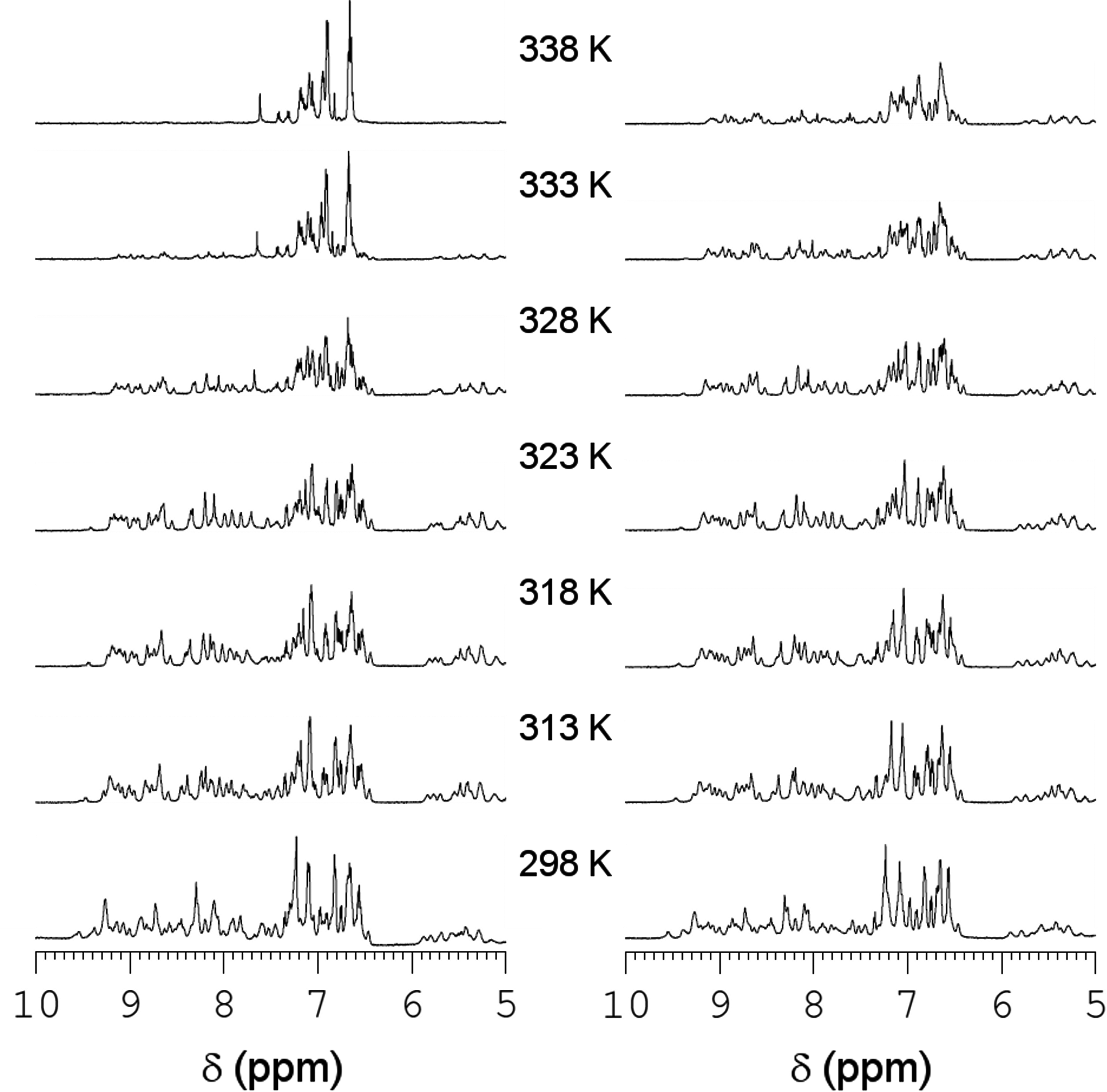
The temperature-induced unfolding of PI3-SH3-WT and PI3-SH3-SS was also analyzed by monitoring the changes in their 1H-NMR spectra (Fig. 3). PI3-SH3-WT displays native signal dispersion and peak sharpness until 318 K; above this temperature the intensity of the signals steadily decays until at 338 K the spectrum collapses and the resonances at low fields are hardly detectable, indicating the absence of a preferential folded conformation. In the case of PI3-SH3-SS, the spectra remain essentially unchanged until 328 K and even at 338 K the spectrum presents good signal dispersion, although with decreased peak intensity. Both proteins appear to be completely unfolded at 358 K (data not shown). The area of the NMR signals in the aliphatic region was plotted against temperature and the resultant curves were adjusted to a two-state model (Fig. 4D). The derived melting temperatures confirm the stabilization of the PI3-SH3-SS domain against thermal unfolding (Table 1).
Equilibrium denaturation of PI3-SH3 domains
The urea denaturation at equilibrium of the different PI3-SH3 domains was analyzed at pH 7.0 and 298 K. Protein unfolding was monitored by following the changes in intrinsic fluorescence at increasing urea concentrations. All three domains display a single detectable transition indicating the cooperativity of the unfolding process (Fig. 5A). The main thermodynamic parameters of the unfolding reaction were calculated from the equilibrium curves assuming a two-state model (R>0.99 in both cases) (Table 2). The stability of PI3-SH3-WT is 4.4 kcal/mol with a [urea]50% of 3.8 M. In agreement with the thermal denaturation data, PI3-SH3-(SH)2 is slightly more stable. Both domains display similar m-values. The value of [urea]50% for PI3-SH3-SS is ∼7.5 M; this value is so high that a final baseline for the fully unfolded state cannot be accurately measured and the m-value could not be determined. Therefore, we used guanidine hydrochloride (Gdn·HCl) for equilibrium denaturation of PI3-SH3-SS and compared it with that of the two other domains using the same denaturant (Fig. 5B). As detailed in Table 2, crosslinking stabilizes PI3-SH3-SS by ∼3.5 kcal/mol relative to PI3-SH3-WT and shifts [Gdn·HCl]50% from 1.7 to 2.9 M. Overall, thermal and chemical denaturation data coincide to indicate that the introduced 3–82 disulfide bond is strongly stabilizing both relative to the wild-type and the dithiol forms and therefore that the mutations are sterically acceptable.
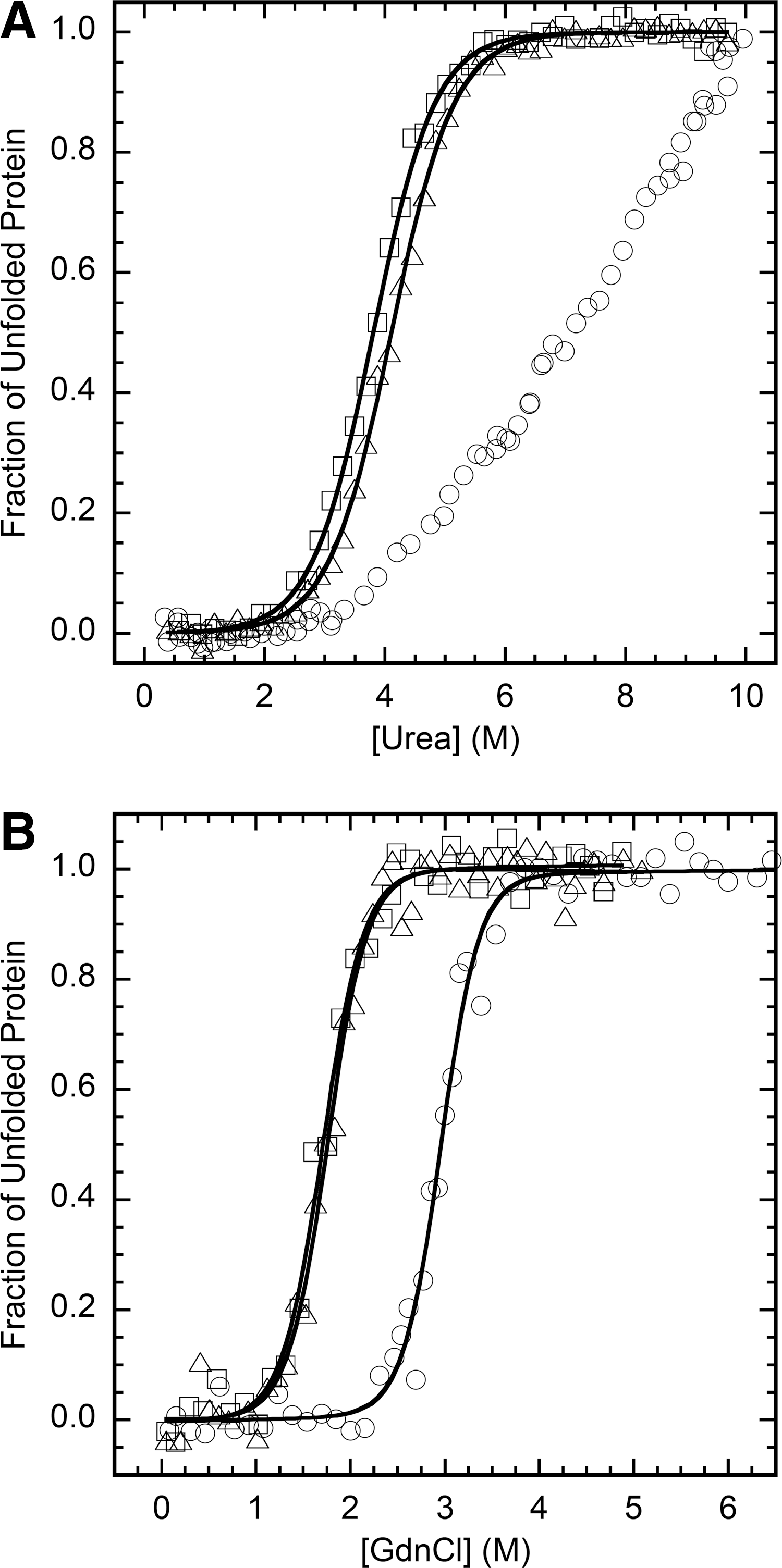
ΔG U-F (free energy of unfolding) and ΔΔG U-F (the difference in ΔGU-F between the particular mutant protein and WT) were calculated from the equilibrium parameters as described in the Materials and Methods section.
Folding and unfolding kinetics of PI3-SH3 domains
The kinetics of folding and unfolding of the different PI3-SH3 domains were determined by stopped-flow at neutral pH and 298 K under a wide range of denaturant conditions. In all cases, the folding and unfolding traces by fluorescence fit well into single exponentials, indicating the lack of detectable intermediates according with a two-state model. The chevron plots for the different domains appear to be linear in the complete range of urea concentrations studied (Fig. 6). The rate constants for folding (k f) and unfolding (k u) and their dependence on the denaturant concentration (m f and m u) are shown in Table 3. The kinetic data agree with the equilibrium stabilities. Interestingly, PI3-SH3-SS folds over 35 times faster than PI3-SH3-WT (Fig. 6). This high acceleration of the folding reaction can be univocally attributed to the presence of the covalent bond, since PI3-SH3-(SH)2 exhibits a k f close to that of the wild-type domain. In contrast, cyclization has only a minor effect on the unfolding rate. The calculated ΦF-U value of 0.85 for the disulfide crosslink suggested that the N and C termini might be ordered in the transition state for folding. We analyzed the folding and unfolding kinetics of Y8A and V74A mutants in the N- and C-terminal β-strands to probe the degree of association between PI3-SH3-WT ends in the transition state (Fig. 6B). The side-chains of Tyr8 and Val47 face each other in the native structure. The two mutations are highly destabilizing (Table 3), indicating that these residues play an important role in maintaining the native conformation. However, the ΦF-U values of 0.16 and 0.20 calculated for Y8A and V74A, respectively, indicate that, in the wild-type domain, the terminal β-strands 1 and 5 are not part of the folding nucleus.

Kinetics of folding and unfolding were followed by changes in intrinsic fluorescence on a stopped flow instrument at 298 K; k f is reported in 0.5 M urea and k u is in 8 M urea to avoid extrapolation; m f and m u are the dependences of the folding and the unfolding rates, respectively, on urea. ΔG U-F (free energy of unfolding) and ΔΔG U-F (the difference in ΔGU-F between the particular mutant protein and WT) were calculated from the kinetic parameters as described in the Materials and Methods section.
Reductive unfolding and oxidative folding
In the presence of 1 mM dithiothreitol (DTT), the disulfide bond in native PI3-SH3-SS is completely reduced in less than 1 min, indicating that it is highly exposed to solvent (Supplementary Fig. S1A; Supplementary Data are available online at
Conformational analysis of PI3-SH3 domains at low pH
PI3-SH3-WT readily forms amyloid-like fibrils in vitro when incubated under acidic conditions. At pH 2.0 the CD spectra of all three domains display transitions to more unfolded conformations (Fig. 2C). These transitions do not reflect global unfolding, as when adding a strong chemical denaturant, but rather partial unfolding, since it is known that at pH 2.0 PI3-SH3-WT forms the so-called A-state, a conformation that is more compact that the denatured state but less than the native one (28, 46). The A-state contains little secondary structure and exposes hydrophobic surfaces to solvent; a property that can be monitored using 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid (bis-ANS). The A-state, from which amyloid assembly starts, seems to be accessible to the three domains, since in all cases we could detect a significant increase in the fluorescence emission of bis-ANS and a blue shift of its λ max in the presence of the proteins at low pH (Fig. 2D). The native domains had a negligible effect on the spectral properties of the dye.
Sequential aggregation propensity of PI3-SH3 domains
We used AGGRESCAN (18) and WALTZ (35) algorithms to estimate the effect of mutations on the intrinsic aggregation propensity of PI3-SH3. AGGRESCAN predicts three identical aggregation-prone regions for the wild-type and mutant proteins, comprising residues G27-V32, G35-F42, and G71-I77 (Fig. 1A). The first and last regions correspond with the second and last β-strands and the second region to the α-helix at the N-src loop. This algorithm calculates aggregation propensities of −19.9 and −20.7 for PI3-SH3-WT and PI3-SH3-(SH)2, respectively. WALTZ identifies four identical amyloid-prone regions in both domains (Fig. 1A), comprising residues G5-Y12 in β-strand 1, G27-G41 in the RT-loop, E52-N60 in the N-src loop, and β-strand 3 and F69-G78 in the last β-strand. Overall, the introduced mutations are not expected to promote by themselves large changes in the aggregation propensity of the domain.
Amyloid fibril formation by PI3-SH3 domains
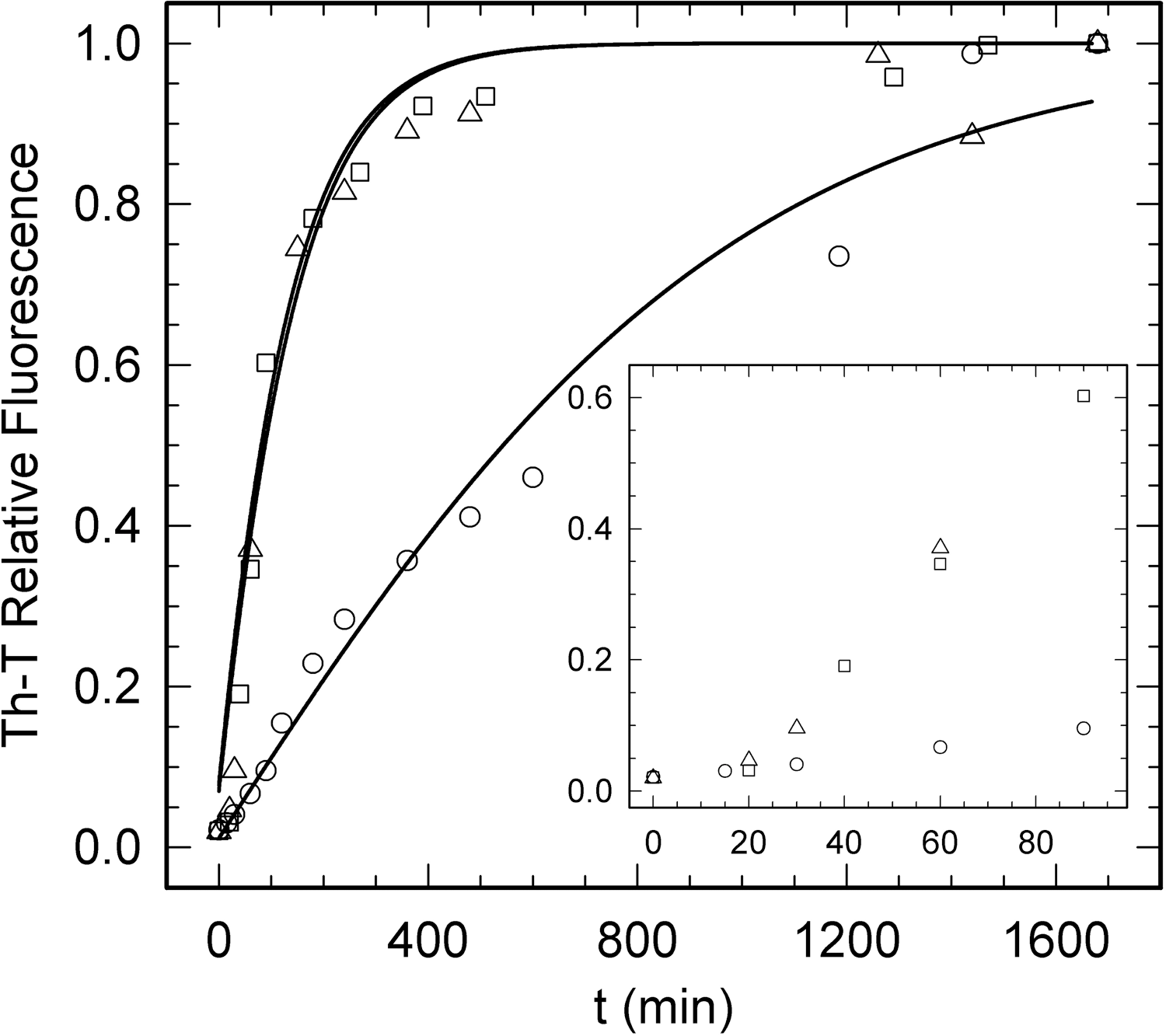
We analyzed the kinetics of PI3-SH3 domains amyloid fibril formation at acidic pH by monitoring the changes over time in the fluorescence of the amyloid staining dye Th-T (Fig. 7). In all cases we obtained characteristic polymerization sigmoidal curves, reflecting a nucleation-polymerization process that can be analyzed as an autocatalytic reaction (40). In agreement with the theoretical predictions, PI3-SH3-WT and PI3-SH3-(SH)2 aggregation curves overlap exhibiting similar nucleation (7.10×103 s−1 and 6.29×103 s−1, respectively) and elongation rates (1.76×10−3 M −1 s−1 and 1.02×10−3 M −1 s−1, respectively). In contrast, the aggregation reaction of PI3-SH3-SS is significantly slower, with a 7-fold slower nucleation rate (1.01×103 M −1 s−1) and 4-fold slower elongation rate (4.09×10−4 M −1 s−1) than the wild-type protein.
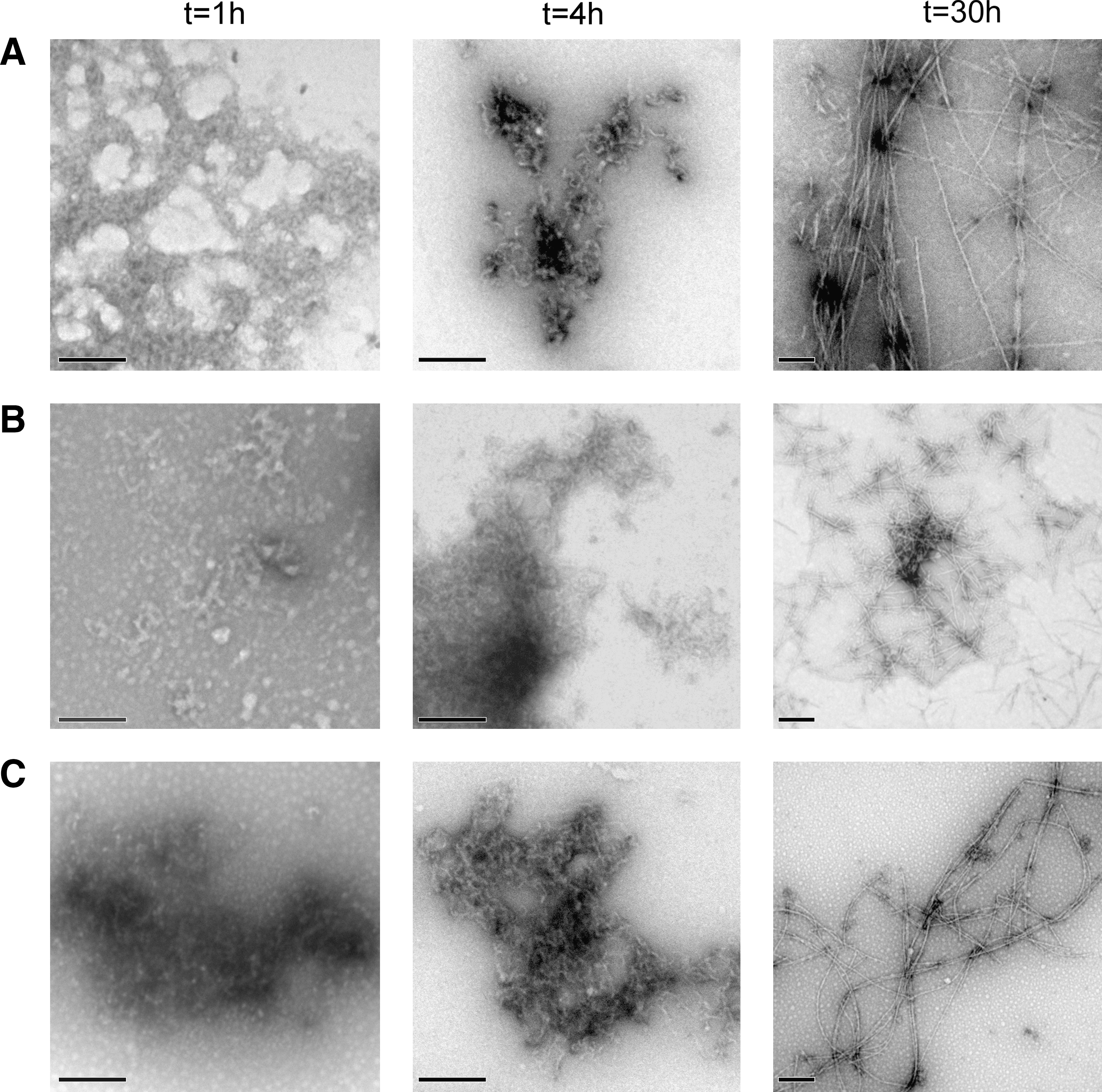
Analysis of the different aggregation reactions by transmission electron microscopy (TEM) (Fig. 8) shows that the formation of protofibrilar aggregates in PI3-SH3-SS samples is delayed relative to that in PI3-SH3-WT and PI3-SH3-(SH)2 solutions. After 42 h incubation, the three domains form typical amyloid fibrils (Fig. 8). However, differences in the morphology of the fibrils were observed. PI3-SH3-WT and PI3-SH3-(SH)2 fibrils were long and eventually tended to twist around each other, whereas PI3-SH3-SS formed shorter, discrete, and straight structures (Fig. 8). No evident depolymerization or shift in morphology could be observed when mature PI3-SH3-SS fibrils were incubated for 5 h under reducing conditions (data not shown).
Conformational and toxicity properties of PI3-SH3 amyloid fibrils
The conformational stability of the fibrils formed by the different domains after 42 h was assessed by equilibrium chemical denaturation with Gdn·HCl at pH 2.0 and 298 K following the changes in Th-T fluorescence (Fig. 9A) and light scattering (Fig. 9B). The fibrils seem to denature in a cooperative manner. The [Gdn·HCl]50% calculated by measuring Th-T fluorescence were 3.2, 2.4, and 2.2 for PI3-SH3-SS, PI3-SH3-(SH)2, and PI3-SH3-WT fibrils, respectively. Light scattering measurements rendered [Gdn·HCl]50% values of 3.9, 3.1, and 2.9 for PI3-SH3-SS, PI3-SH3-(SH)2, and PI3-SH3-WT fibrils, respectively. Therefore, the fibrils of PI3-SH3-SS appear to be more stable than those of the wild-type and reduced forms. In all cases, the stability calculated from Th-T measurements is lower than that obtained from light scattering. The difference in the two transition midpoints suggests the presence of intermediate states in which the ordered β-sheet is disrupted, in such a way that it does not longer bind Th-T, but still remains in an aggregated state, contributing thus to the light scattering signal. The population of these species can be approximated from the normalized denaturation data and fitted to a Gaussian distribution (Fig. 9C). The intermediate species distributions suggest that the β-sheet structure in the wild-type fibrils is less stable than in the fibrils of the disulfide crosslinked domain. To further explore whether the observed differences in fibrils morphology and stability are related to differences in the interactions supporting their architecture, we analyzed the conformational properties of PI3-SH3 mature fibrils by attenuated total reflectance–Fourier transformed infrared spectroscopy (ATR-FTIR) and bis-ANS binding and limited proteolysis. The ATR-FTIR spectra of all three fibrils in the amide I region is dominated by a main band at 1615–1640 cm−1 corresponding to β-sheet conformations (Fig. 9D–F). The secondary structure composition of PI3-SH3-WT (Fig. 9D) and PI3-SH3-(SH)2 (Fig. 9E) fibrils are almost identical and differ from that of PI3-SH3-SS fibrils (Fig. 9F). The proportion of amyloid-like intermolecular β-sheet secondary structure is higher in the fibrils formed by PI3-SH3-SS (72%) than in those formed by PI3-SH3-WT (61%) and PI3-SH3-(SH)2 (60%) (Supplementary Table S1). In agreement with these data, PI3-SH3-WT and PI3-SH3-(SH)2 fibrils exhibit higher binding to bis-ANS than the fibrils formed by the disulfide-containing domain, indicating the existence of a lower proportion of hydrophobic residues exposed to solvent in the fibrils formed by PI3-SH3-SS (Fig. 9G). Accordingly, we observed that, in our conditions, the PI3-SH3-SS fibrils were totally resistant to pepsin digestion, whereas the wild-type fibrils were susceptible to proteolysis (Fig. 9H).
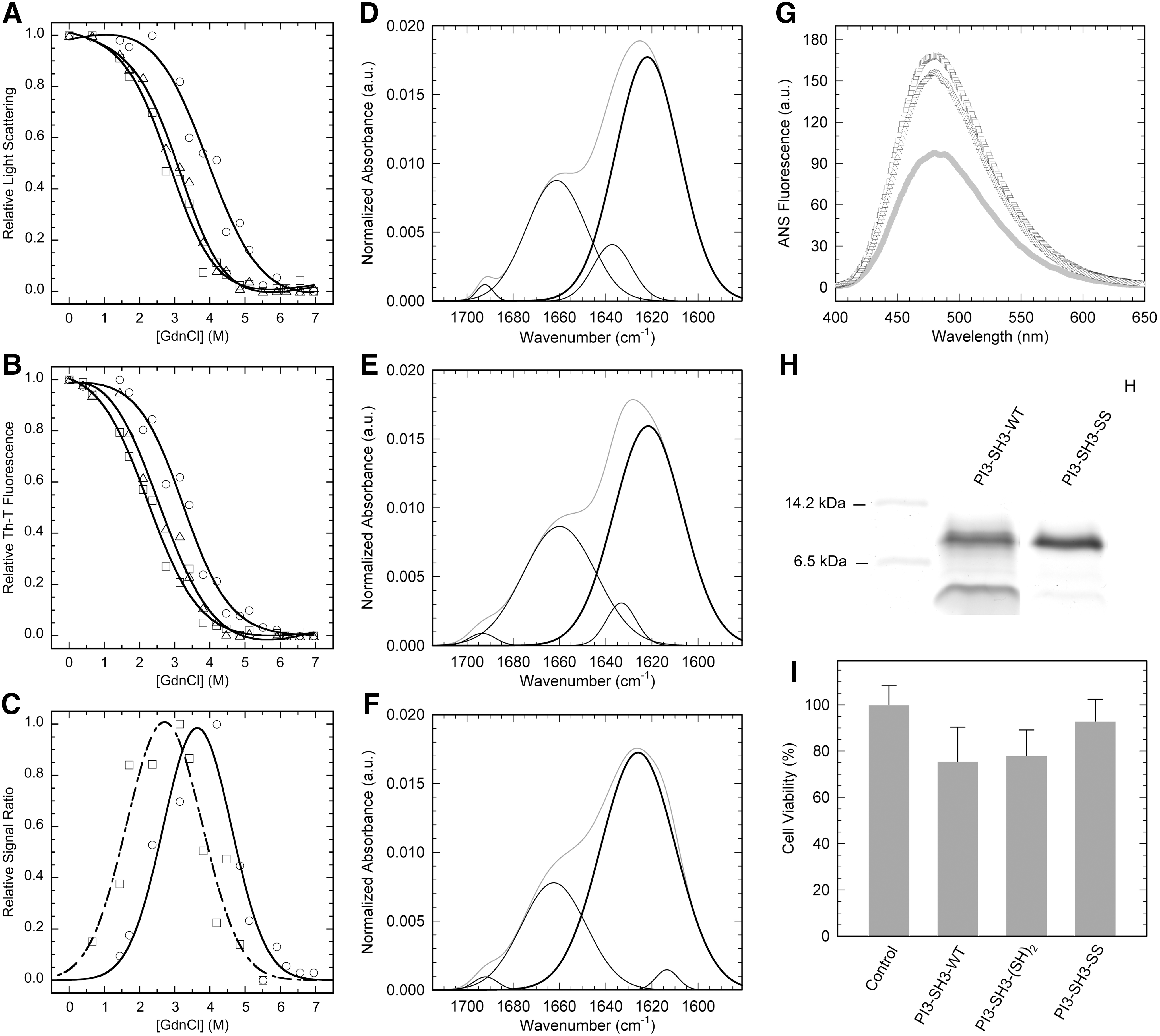
The toxicity of amyloid fibrils is related to their conformational properties. It has been shown for different and unrelated proteins that the binding to ANS-like dyes correlates with the toxicity of amyloid species, suggesting that the exposure of hydrophobic regions is a critical characteristic of these pathogenic assemblies (5). Although the aggregation of PI3-SH3 is not associated to any known disease, its amyloid assemblies are inherently cytotoxic (7). We analyzed the toxicity of PI3-SH3 fibrils on cultured neuroblastoma cells from the SH-SY5Y cell line. In excellent agreement with bis-ANS binding data, the fibrils formed by PI3-SH3-SS were less toxic than those formed by the PI3-SH3-WT and PI3-SH3-(SH)2 domains (Fig. 9I).
Extracellular SH3 domains display disulfide bonds and a high sequential intrinsic aggregation propensity
SH3 domains have been considered traditionally as structural elements of multidomain intracellular signaling proteins and devoid of disulfide bonds. However, it has been shown recently that the extracellular melanoma inhibitory activity protein (MIA) consists of a single domain adopting an SH3-fold covalently linked by two intramolecular disulfide bonds. Moreover, three MIA homologous extracellular proteins with the four conserved Cys have been identified (TANGO, MIA-2, and OTOR) (44). Since our data suggest that the presence of disulfide bonds might modulate the aggregation propensity of SH3 domains, we compared the intrinsic aggregation properties of these extracellular SH3 disulfide-containing domains with those of classical intracellular ones using AGGRESCAN (Fig. 10). The analysis indicates that extracellular SH3 domains display a higher number of aggregation-prone regions in their sequence and support a higher intrinsic aggregation load than intracellular domains.

Discussion
Cyclization increases PI3-SH3 domain stability
Biophysical data indicate that the three PI3-SH3 domains analyzed in the present study exhibit a properly folded conformation in their native states. The dithiol form is slightly more stable than the wild-type domain. DSC data indicate that ΔH is lower, in absolute value, in PI3-SH3-(SH)2 than in PI3-SH3-WT and therefore that stabilization does not arise from the establishment of favorable interactions by the thiol groups but rather from a decrease in ΔS in the mutant form. The ProtSA server (21) indicates that, in the wild-type domain, Ala3 and Ile82 are more exposed to solvent than expected for these hydrophobic residues. Their mutation to Cys displaying polar free thiols in the reduced state would be entropically favorable and might well account for the observed increase in stability. Oxidative folding experiments confirm that the Cys are exposed to solvent and in a flexible environment in PI3-SH3-(SH)2.
Direct comparison of PI3-SH3-SS with its dithiol form allows differentiating the effect of the mutations from the effect of the disulfide bond. Crosslinking of PI3-SH3 stabilizes the domain against both thermal and chemical denaturation, with an increase in T
m of ∼15 K and a ΔΔG
U-F at 298 K of ∼3.5 kcal/mol, relative to the dithiol form. It is generally assumed that the stability provided by a disulfide bond results largely from a reduction in the configurational entropy (ΔS
conf) of the unfolded state. The ΔS
conf is related to the size of the polypeptide loop enclosed by the disulfide bond (N) and can be approximated theoretically according to the equation (37):
where R is the gas constant.
According to Equation (1) the expected stabilization of PI3-SH3-SS, relative to its reduced form, is 4.5 kcal/mol. Therefore, although the experimentally measured increase in stability is among the highest reported for an SH3 domain (27) it is lower than the theoretical estimate, indicating that loop entropy is not the only factor determining the stability of the cycled domain. Together with a decrease in ΔS, as measured by DSC, the disulfide bond causes a decrease in ΔH, indicating enthalpy–entropy compensation. The same effect has been observed for a number of disulfide mutants, including those of a camelid antibody (29), cytochrome c (4), and barnase (31), demonstrating that the precise thermodynamic effects of introducing a covalent link in a polypeptide are hardly predictable from simple theoretical considerations (16). In these protein models, disulfide bonds tend to decrease the heat capacity change of unfolding (ΔC p). The ΔC p between the folded and unfolded states of proteins is thought to arise from the exposure of buried side-chains to solvent, with minor contributions from changes in configurational entropy. As proposed by Doig and Williams (20), the 45% decrease in the ΔC p of PI3-SH3-SS likely results from a more compact unfolded state with restricted hydration of side chains.
Cyclization accelerates PI3-SH3-folding
Cyclization is always expected to increase folding rates due to the larger decrease in configurational entropy of the unfolded state relative to that in the transition state. Unfolding rates will decrease only if the polypeptide termini become apart at the transition state. Connection of the N- and C- termini in PI3-SH3-SS causes a large increase, by over 1 order of magnitude, of the folding rate and has, by contrast, only a small influence on the unfolding rate. This behavior has also been observed for disulfide mutants of barnase (15), subtilisin (45), and acylphosphatase (38). However, cyclization in the structurally homologous SH3 domain of src kinase (src-SH3) domain affected roughly equally its folding and unfolding rates (27), suggesting that its ends are not as structured in the transition state as they are in the native conformation and, likely, that a rate-limiting step in unfolding is the dissociation of the N- and C- termini (27). This view is consistent with the results obtained by Φ analysis for this protein (39) and the spectrin-SH3 domain (34). The two studies indicate that the folding transition state of these domains involves the association of the distal β-hairpin and the diverging turn, whereas the N and C termini are completely disordered. The low Φ values obtained here for mutants in the β-strands 1 and 5 indicate that the protein ends are also unstructured in the transition state of PI3-SH3-WT, confirming thus a robust energy landscape for the folding of SH3 domains. Cyclization makes the topology of the protein symmetric and tends to reduce the polarization of the transition state. The dissimilar Φ values obtained by circularization and point mutation of N- and C- regions in PI3-SH3 indicate that the free energy landscape of this particular domain is severely affected by the disulfide bond promoted topology change, confirming that despite the folding landscape of this protein family is relatively insensitive to sequential variations it is sensitive to topological constrains (26).
Cyclization slows down PI3-SH3 aggregation
The effect of polypeptide chain crosslinking in amyloid fibril formation is still poorly understood. Disulfide crosslinking does not prevent self-association into amyloid structures. Therefore, the conformational restrictions introduced by the covalent bond do not prevent the accommodation of the polypeptide backbone into the highly ordered cross-β-sheet structure characteristic of amyloid fibrils. However, it strongly decreases the nucleation and elongation rates, slowing down the lag phase as well as the overall aggregation reaction. The aggregation of PI3-SH3-WT has been shown to be consistent with the nucleated conformational conversion mechanism (8), in which initially soluble monomers coalesce to form amorphous assemblies that later reorganize into ordered oligomers and finally β-sheet enriched amyloid fibrils (43). Hydrophobic forces are thought to drive the initial unspecific condensation of monomers into amorphous oligomers. According to the conformational properties and hydrophobic residues clustering of PI3-SH3-SS and PI3-SH3-(SH)2 domains, there is not obvious reason to assume differences in their association rates at this stage. Upon the initial collapse, polypeptides within the disordered oligomers realign slowly to establish hydrogen bonds that favor the formation of β-sheets. The dynamic flexibility of the polypeptide chain might play an important role in this second step, since multiple rounds of association and dissociation between adjacent protein regions likely occur during the lag phase, before an initially ordered and stable enough β-sheet conformation is attained (2). The reduced flexibility of PI3-SH3-SS might difficult this rearrangement stage delaying the formation of productive oligomers, resulting in a lower kn.
The elongation step in amyloid formation is thought to involve a “dock and lock” mechanism, in which a significant conformational rearrangement of incoming monomers is necessary before they become incorporated into preformed fibrils (17). The slower elongation rate of PI3-SH3-SS suggests that dynamic protein flexibility might be also important at the polymerization stage.
Cyclization reduces the toxicity of PI3-SH3 amyloid assemblies
High-resolution MAS NMR analysis has allowed Bayro et al. to propose a structural model for PI3-SH3-WT amyloid fibrils (3). In the model, four β-sheets form the core of two distinct protofilaments and lateral contacts between PI3-SH3-WT subunits in adjacent protofilaments allow their association to form the fibrils (Supplementary Fig. S2). The N-terminus adopts a rigid β-strand structure in the core of the fibril. In contrast, the C-terminal residues appear to be dynamically and structurally disordered and located in the two external sides of the fibril (3). Restriction of the C-end entropy by crosslinking might allow its integration in the core β-sheet structure (Supplementary Fig. S2). This would explain the highest stability of the β-sheet structure in PI3-SH3-SS fibrils, the insensitivity of these fibrils to highly reducing conditions, the higher proportion of intermolecular β-sheet and the resistance of fibrils to proteolysis, as well as the reduced exposition of hydrophobic residues to solvent. This cyclization-promoted structural gain has a crucial functional consequence: it significantly reduces the cytotoxicity of the resulting fibrils.
Materials and Methods
Disulfide bond design
A PI3-SH3 domain with the N and C terminal regions linked by a disulfide bridge (PI3-SH3-SS) was designed using FoldX (version 2.65) (
Cloning, mutagenesis, and expression
The PI3-SH3-WT protein consists of 83 residues from the SH3 domain of bovine PI3-kinase plus a Met at the N-terminus. Its cDNA was cloned in pBAT-4 (46). The Ala3 and Ile82 residues were mutated to Cys, the resulting plasmid was transformed in BL21(DE3) Escherichia coli cells and the proteins were expressed and purified as described previously (46). The mutant cDNA was sequenced and the protein identity was checked by mass spectrometry. Gel filtration chromatography indicated that the mutant protein is a monomer. Unless otherwise indicated, experiments were performed in 50 mM sodium phosphate at pH 7.0 (buffer-N).
Disulfide crosslinking and reduction
To confirm the formation of the designed disulfide bond, PI3-SH3-SS was incubated in 0.1 M Tris-HCl buffer, pH 8.5, containing 0.1 M 4-vinylpyridine for 1 h at room temperature, diluted 1:10 in 0.1% aqueous trifluoroacetic acid, and analyzed by MALDI-TOF MS in an Ultraflex spectrometer (Bruker). When required, the disulfide bond was reduced by incubation in the presence of 10 mM DTT or Tris (2-carboxyethyl)phosphine (TCEP) for at least 1 h.
Oxidative folding and reductive unfolding
For oxidative folding experiments PI3-SH3-SS was reduced and unfolded in 0.5 M Tris buffer pH 8.4, containing 10 mM DTT and 7 M guanidinium chloride for 2 h at room temperature. To initiate folding, the sample was loaded onto a desalting column equilibrated with 0.1 M Tris buffer, pH 8.4. The protein was eluted in the same buffer to a final protein concentration of 0.3 mg/ml, in the absence and in the presence of 0.5 mM GSSG. Folding species were trapped in a time-course manner by alkylation with 0.1 M sodium iodoacetate for 2 min at room temperature. The trapped intermediates were subsequently analyzed by SDS-PAGE under nondenaturing conditions. For reductive unfolding experiments PI3-SH3-SS (0.5 mg/ml) was dissolved at room temperature in 0.1 M Tris buffer, pH 8.4, containing 1 mM DTT and the species were trapped and analyzed as described above.
Circular dichroism
CD spectra were measured in a Jasco-710 spectropolarimeter thermostated at 298 K. Spectra were recorded from 260 to 205 nm, at 1 nm intervals, 1 nm bandwidth, and a scan speed of 10 nm/min. Twenty accumulations were averaged for each spectrum. Protein concentrations were 20 μM in buffer-N. Thermal denaturation was monitored at 235 nm each 0.1 K, with 2 min temperature equilibrium between measures. Experimental data were fitted to a two-state transition curve for which the signals of the folded and unfolded states are dependent on the temperature using the nonlinear least squares algorithm provided with Kaleidagraph (Abelbeck Software).
Intrinsic fluorescence
Fluorescence was measured in a Varian Cary Eclipse spectrofluorometer using an excitation wavelength of 268 nm. Slit widths were typically 5 nm for excitation and 10 nm for emission. Spectra were acquired at 1 nm intervals, a 600 nm/min rate, and 0.1 s averaging time.
In PI3-SH3 domains, Trp55 is exposed to solvent and its fluorescence exhibits a linear dependence on the temperature or chaotropic agents concentration. Tyr residues are responsive to conformational changes, but their signal is strongly influenced by the fluorescence of Trp55. The ratio 303/350 nm provides an accurate measurement of structural changes in PI3-SH3 domains. Thermal denaturation was analyzed in buffer-N at 20 μM protein concentration each 0.1 K with 2 min equilibration between measures. For chemical denaturation the native protein was dissolved at 20 μM in buffer-N containing selected concentrations of denaturants (0–9 M urea or Gdn·HCl). The reaction was equilibrated for 20 h at room temperature. Experimental data were fitted as described for CD measurements.
Differential scanning calorimetry
The heat capacity of PI3-SH3 as a function of temperature was measured with a high-sensitivity differential scanning VP-DSC microcalorimeter (MicroCal). Protein samples and reference solutions were properly degassed and carefully loaded into the cells to avoid bubble formation. Thermal denaturation scans were performed with freshly prepared buffer-exchanged protein solutions. The baseline of the instrument was routinely recorded before the experiments. Experiments were performed in buffer-N, at a scanning rate of 1 K/min. Experiments were carried out with 40–50 μM of protein. Thermal denaturations were reversible (as judged by the agreement of the thermogram shapes and sizes between the first and the second scans). Pre- and post-transition baselines were considered linear with temperature.
A model-free analysis indicated that the calorimetric enthalpy is equal to the van't Hoff enthalpy, within experimental error. Therefore, protein stability parameters were obtained by analyzing thermal scans considering a two-state unfolding model using software developed in our laboratory implemented in Origin 7 (OriginLab). The unfolding thermodynamic parameters are not influenced by the ionization properties of the buffer as long as the pH of the experiment is close to the buffer pKa (7.2 for phosphate), and the buffer ionization enthalpy is small (0.86 kcal/mol for phosphate).
NMR spectroscopy
Samples were prepared at 40 μM in buffer-N, using a 9:1 H2O/D2O. One-dimensional NMR spectra were acquired at selected temperatures on a Bruker AVANCE 600-MHz spectrometer using solvent suppression WATERGATE techniques. The collected spectra were processed and analyzed using the TopSpinv2.0 software packages from Bruker Biospin. The area under the NMR signals at the aliphatic region (form 10 to 5 ppm) was calculated and plotted to follow the temperature-induced denaturation of PI3-SH3 domains.
Determination of folding kinetic parameters
The kinetics of the folding and unfolding reactions at 298 K were followed in a Bio-Logic SFM-3 stopped-flow instrument using excitation at 268 and a 300 nm fluorescence cut-off filter. Starting with the protein in buffer-N, the unfolding reaction was promoted by dilution with appropriate volumes of the same buffer containing 9.5 M urea. For the folding reaction, appropriate volumes of urea-free buffer were added to an initial protein solution in buffer-N containing 9.5 M urea. Kinetic traces were fitted to single-exponential functions and the resulting rate constants to a two-state transition equation to determine the kinetic and free energy values:
where k
f and k
u are the rates of folding and unfolding, respectively, at denaturant concentrations experimentally accessible for the domain. The differences in the free energy of unfolding (ΔΔG
F-U) between the wild-type protein and each mutant are calculated as:
The parameter ΦF is defined as:
and is interpreted as the fraction of the mutated residue's interactions that are formed in the transition state.
Amyloid fibril formation
Lyophilized PI3-SH3 domains were dissolved in buffer-N at 7 mg/ml. After filtration through a 0.22 μm filter to remove residual aggregates, protein solutions were diluted to 3.5 mg/ml adding an equal volume of 50 mM glycine, and the pH adjusted immediately with 5 M HCl to pH 2.0. Fibril formation was promoted by incubating the samples at 298 K and 500 rpm agitation. The 10 mM TCEP was used as reducing agent in these experiments.
Aggregation kinetics
After different incubation intervals samples were diluted in glycine buffer containing 60 μM Thioflavin-T (Th-T) and equilibrated 5 min at 298 K. They were excited at 450 nm and the fluorescence emission spectrum recorded between 470 and 600 nm with slit widths of 5 and 10 nm for excitation and emission, respectively. The kinetic parameters for the aggregation process were calculated according to an autocatalytic reaction, as previously described (40).
bis-ANS binding
The fluorescence emission of bis-ANS was recorded at 298 K. Protein samples (40 μM) at pH 7.0 or pH 2.0 were diluted tenfold into the corresponding buffer containing bis-ANS (2.6 μM). Spectra were collected after 5 min incubation. The samples were excited at 365 nm and emission measured between 440 and 640 nm with slit widths of 5 and 10 nm for excitation and emission, respectively.
Electron microscopy
Aggregated protein samples were diluted 20 times in the same buffer, 10 μl placed on a carbon-coated copper grid and allowed to stand for 5 min. The grid was washed with distilled water and the sample stained with 2% uranyl acetate for 1 min. The samples were imaged in a Hitachi H-7000 transmission electron microscope operating at an accelerating voltage of 75 kV.
Amyloid fibrils chemical denaturation
Mature fibril solutions were incubated in 0–7 M Gdn·HCl at pH 2 and for 16 h. Subsequently, they were diluted in glycine buffer containing 60 μM Th-T. The changes in light scattering at 340 nm and Th-T fluorescence at 480 nm were followed with a Varian Cary Eclipse spectrofluorimeter. Experimental data were fitted as described for CD measurements.
Limited proteolysis
Limited proteolysis was performed using pepsin in 50 mM glycine buffer at pH 2.0 and 298 K using an E/S ratio of 1:200 (by weight). The reactions were quenched after 5 min by adding an appropriate volume of 5 M NaOH. The proteolytic mixtures were analyzed by SDS-PAGE.
Attenuated total reflectance–Fourier transformed infrared spectroscopy
ATR-FTIR analyses of SH3 aggregates were performed using a Bruker Tensor 27 FTIR Spectrometer (Bruker Optics) with a Golden Gate MKII ATR accessory as previously described (41).
Cell viability assays
The toxicity of PI3-SH3 aggregates (10 μM) was analyzed on cultured neuroblastoma cells (SH-SY5Y cell line) by measuring formazan formation by mitochondrial dehydrogenases as previously described (41). Four independent assays were performed for each protein sample.
Footnotes
Acknowledgments
This work was supported by grants BFU2010-14901, BFU2010-19451, and BFU2010-16297 from Ministerio de Ciencia e Innovación (Spain), 2009-SGR-760 and 2009-CTP-00004 from AGAUR (Generalitat de Catalunya), and grupo consolidado Protein Targets (B89) (Gobierno de Aragón). S.V. has been granted an ICREA Academia award (ICREA).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.