
Abstract
Introduction
The mechanistic basis for the anatomically specific destruction induced by complex I inhibitors is not entirely clear. In the case of MPTP, evidence indicates that after astrocytes take up MPTP, convert it to MPP+, and release it to the extracellular space that neuron dopamine transporters preferentially absorb the toxin (105). This should of course ensure that MPP+ concentrations in dopaminergic neurons exceed those of other neuron types, which could lead to a disproportionally greater complex I inhibition and hence toxicity in dopaminergic neurons. Still, after MPTP administration it is found distributed throughout the brain for at least a brief period and the degree of complex I inhibition that results throughout the brain can reach easily detectable levels (43). Another relevant point is that rotenone does not appear to show preferential uptake by dopaminergic neurons (10). Therefore, an argument can be made that for unknown reasons dopaminergic neurons are less able than other cell types to tolerate complex I dysfunction (97).
In 1989 several studies reported evidence of altered complex I function in persons with idiopathic PD (11, 83, 94, 112). From a methodologic perspective these studies used either spectrophotometric Vmax assays or immunochemistry. Although in some respect their essential observations were consistent and could have unified the PD research community, what resulted was a long-running controversy over the complex I defect's anatomic distribution. The reasons for this controversy arose from the fact that one early report claimed in brain the complex I defect was restricted to the substantia nigra (113). Two of the other reports, though, identified deficient complex I activities in both PD subject platelets and muscle (11, 94). This was a debate with important implications. If the PD subject complex I activity reduction was in fact restricted to the substantia nigra, it would reduce the likelihood that the phenomenon was genetically mediated. If the PD complex I defect was systemic, it would conversely suggest a genetic basis. Two decades later, the controversy over the anatomic distribution of the PD complex I defect seems to have been settled (128, 131). It has repeatedly been demonstrated in PD subject platelets and muscle, and has also been demonstrated in fibroblasts and lymphocytes (7, 9, 12, 13, 18, 34, 42, 65, 85, 87, 94, 119, 162). More recently, it was shown that the original controversy likely arose entirely as a consequence of methodological issues (128) (Fig. 1). Because it is difficult to prepare enriched mitochondrial fractions from small brain regions, the study of Schapira et al. necessarily determined complex I Vmax activities in tissue homogenates (113). Unfortunately, determining electron transport chain (ETC) Vmax activities in brain homogenates provides a substantially reduced reaction signal, which makes it a somewhat insensitive way to detect inter-group ETC Vmax differences. This limitation would predictably be exacerbated by the use of autopsy brain tissue, which is subject to postmortem artifacts that further reduce assay sensitivity. Indeed, a 2008 study demonstrated that when complex I activity was determined in frontal cortex homogenates of PD subject autopsy brains, no difference was detected compared with the activity in control brains, but that with increasing enrichment of the assayed mitochondrial fractions a clear difference emerged (96).

During the 1990s investigators, nevertheless, began to test for a potential genetic contribution to the PD complex I defect. Complex I functions as a large multimeric holoenzyme that contains 46 known subunits all of which are encoded by different genes, thus complicating a comprehensive evaluation of this possibility (37) (Fig. 2). Most initial attempts to demonstrate a genetic basis for the PD complex I defect were sequence based and either negative or, if positive, not supported through replication (62, 118, 122, 123, 139). The first durable data to strongly make a case for mitochondrial DNA (mtDNA) as an important idiopathic PD mediator come not from sequence based studies, but rather from studies of cytoplasmic hybrids, or “cybrids”(130).

What Are Cybrids?
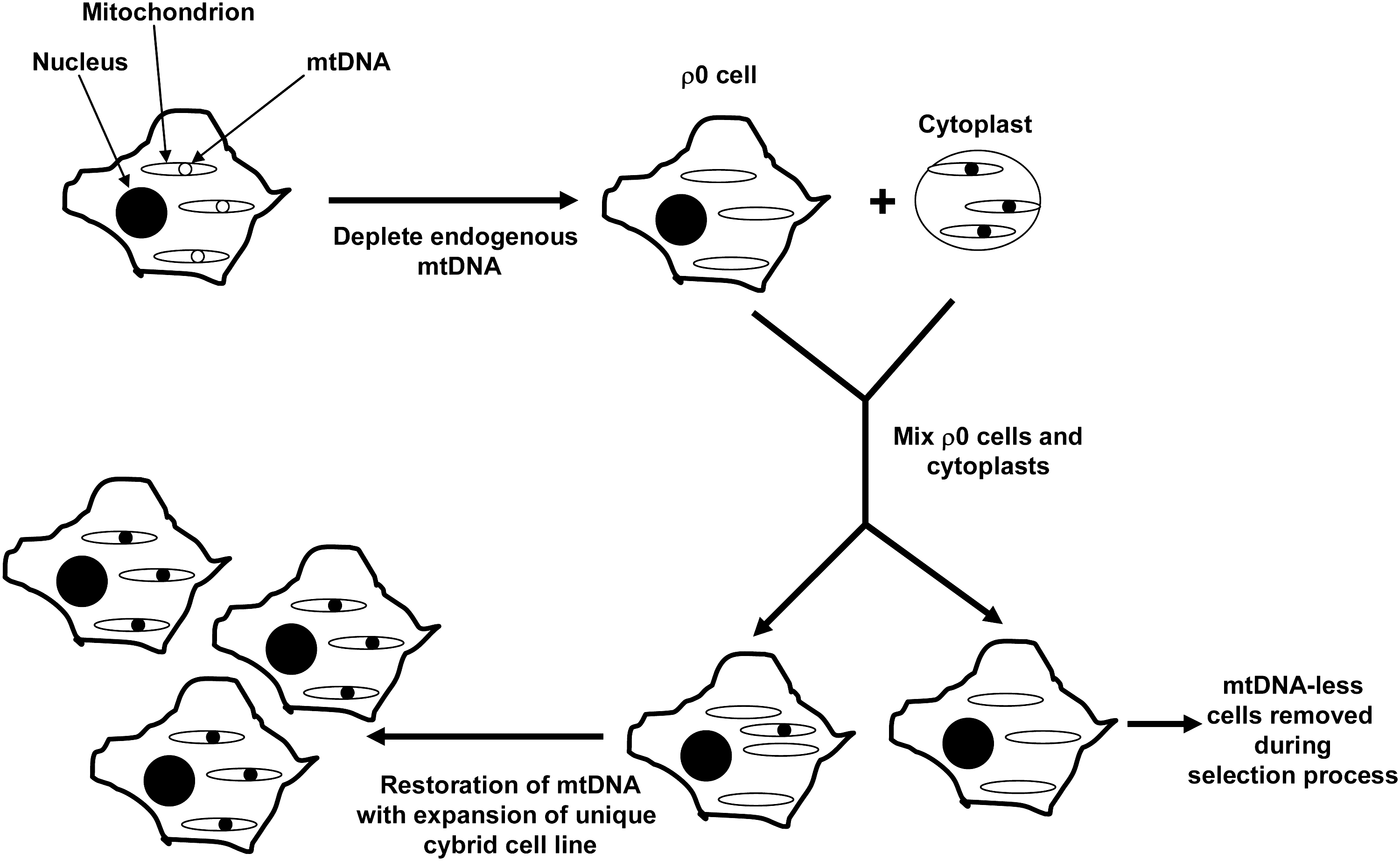
Decades ago investigators succeeded in fusing two nucleated cells together to generate binucleated hybrid cells. Cybrid cells differ from hybrid cells because they are generated by mixing the contents of a non-nucleated cell, or cytoplast, with a nucleated cell (101) (Fig. 3). Cybrid cells, therefore, have just one nucleus but contain the cytosolic contents of two different cells. This approach was developed during the 1970s to explore the functional consequences of unique mtDNA species (16). In one early cybrid study, enucleated cytoplasts from a chloramphenicol-resistant HeLa cell strain were mixed with the intact cells of a chloramphenicol-sensitive HeLa strain. The resulting cybrid cell line showed stable chloramphenicol resistance and illustrated in cultured cells choramphenicol sensitivity versus tolerance is determined by mtDNA (157).
The creation of mtDNA-depleted cell lines facilitated adaptations of this basic approach. During the 1970s and 1980s it was shown that chronic exposure to cationic DNA mutagens, such as ethidium bromide, can selectively block mtDNA replication (84, 160). Selective mtDNA toxicity in this case results from the fact that cationic mutagens concentrate within negatively charged mitochondrial matrices where they can reach concentrations capable of disrupting mtDNA replication while sparing nuclear DNA replication.
Through a series of experiments it was found that with proper metabolic support it was possible to completely eliminate the endogenous mtDNA from some cell lines. Because before its identification as mtDNA cytoplasmic DNA was referred to as ρ DNA (30), cell lines completely depleted of endogenous mtDNA are typically called ρ0 cells. First, it was shown that mtDNA-depleted cells require uridine (84). The accepted reason for this is that one step of the pyrimidine de novo pathway, the dihydorotate dehydrogenase-mediated dehydrogenation of dihydroorotate to orotate, is coupled to the reduction of ubiquinone to ubiquinol (51, 84, 150, 163). Cells that lack a functional ETC theoretically cannot regenerate ubiquinone, which subsequently blocks de novo pyrimidine synthesis. Uridine, a nucleoside created through the N1-glycosidic linkage of uracil, a pyrimidine base, to a ribose sugar increases flux through the pyrimidine salvage pathway and compensates for the loss of de novo pyrimidine synthesis. Pyruvate appears to further promote the expansion of mtDNA-depleted cell lines, and it has been proposed to accomplish this by shifting the cell redox balance to a more oxidized state (58). This would theoretically increase glycolysis pathway flux, which is relevant because without a functional ETC cell ATP demand must be met entirely through glycolysis. It is worth noting that most successfully generated ρ0 cell lines are tumor cell lines (130), which have already shifted to some degree their energy production from mitochondrial respiration to glycolysis, a phenomenon widely known as the Warburg Effect (152, 158).
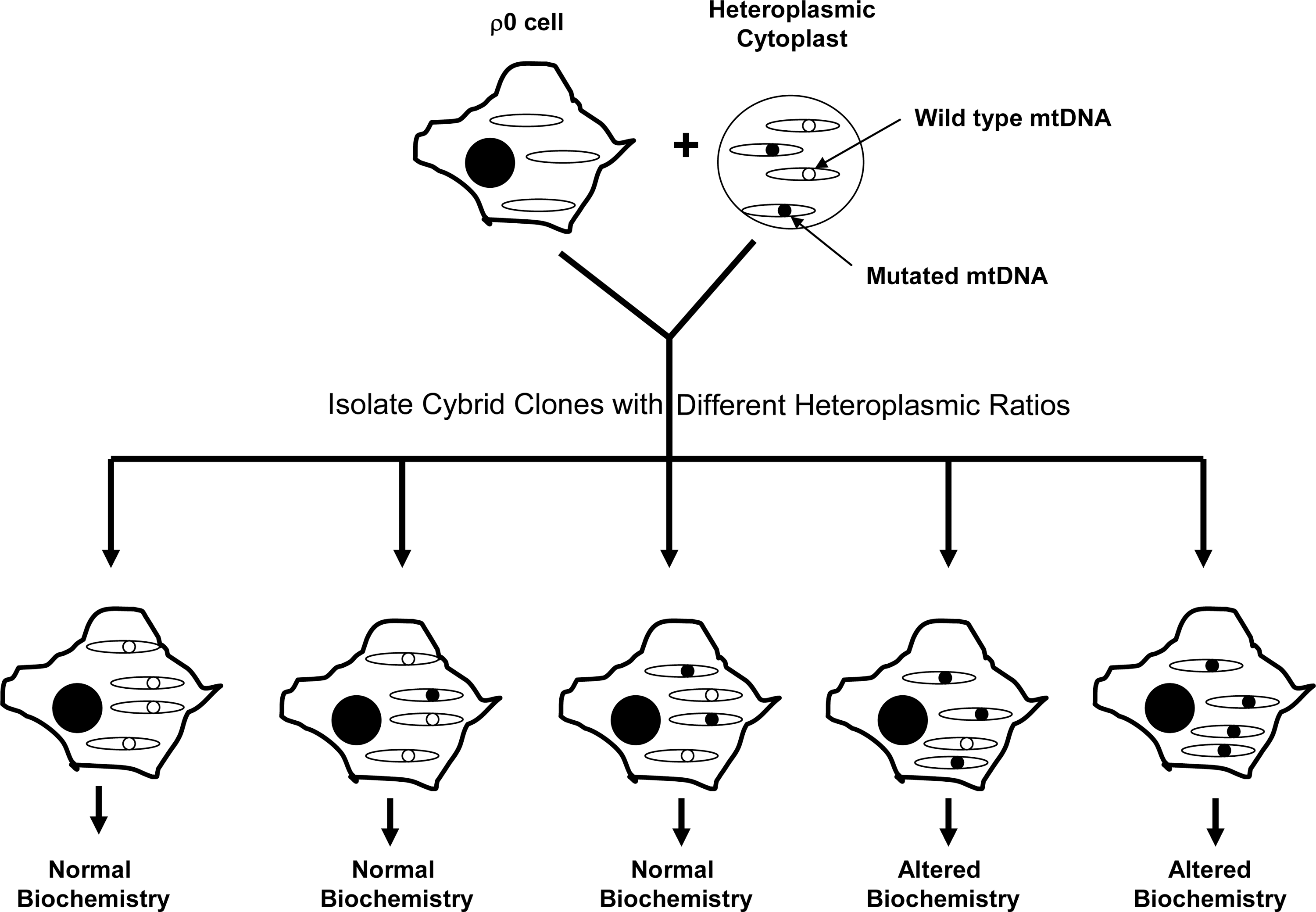
In 1989, King and Attardi reported the creation of an osteosarcoma ρ0 cell line that expanded when its medium was supplemented with uridine and pyruvate, but which was not viable in their absence (58). Uridine and pyruvate auxotrophy was eliminated following either the “fusion” or injection of these osteosarcoma ρ0 cells with enucleated fibroblast cytoplasts. Both approaches restored aerobic competency by repopulating the osteosarcoma cells with cytoplast mitochondria and hence mtDNA. The ability to generate cybrid cell lines from ρ0 cells allowed investigators to better control the mtDNA content of the cybrid lines they were generating, and the technique was soon adapted for studies of mtDNA mutational threshold (Fig. 4). For example, osteosarcoma ρ0 cell lines were fused with fibroblast cytoplasts or platelets obtained from individuals with heteroplasmic point mutations responsible for causing the mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes syndrome and the myoclonic epilepsy and ragged red fiber syndrome (22, 59, 76).
In 1994, a variation of the cybrid approach was described in which platelets were substituted for fibroblast cytoplasts (21). Platelets lack nuclei and are easy to isolate, since they can readily be separated from whole blood samples via centrifugation. Several milliliters of whole blood can generate enough platelets to generate a cybrid cell line. The ability to use platelets as a source of mitochondrial and therefore mtDNA transfer meant that virtually any desired subject at any time could serve as an mtDNA donor when creating a cybrid cell line. In 1996 the first human neuronal ρ0 cell line, the SH-SY5Y neuroblastoma cell line, was reported (81).
Why Were Cybrids Used to Study PD?
By the mid-1990s several groups had reported that PD subject platelet mitochondria have reduced complex I activity, but the explanation for this phenomenon was unknown (9, 42, 65, 94, 162). It was proposed that environmental or endogenously generated toxins could account for this, and several candidate toxins were considered (28, 44, 47, 73, 77, 86, 89, 104). No potential toxic agent, though, seemed able to generate sustained enthusiasm.
Although initial efforts to identify a genetic explanation for the PD complex I defect were unsuccessful, a compelling rationale for a genetic and especially an mtDNA-related genetic basis remained. Part of this rationale was based on the epidemiology of PD, which revealed that although individuals with a family history of PD generally have an increased risk of developing PD, only in rare cases does this increased risk demonstrate recognizable autosomal dominant or autosomal recessive Mendelian inheritance patterns (97, 140). Monozygotic twin studies also reveal clinical discordance, although PD sub-clinical endophenotypes may develop in the otherwise asymptomatic monozygotic twin siblings of PD subjects (46, 67, 103, 141, 159). For these reasons most idiopathic PD is said to show “sporadic” epidemiology. Sporadic epidemiology for a disease, though, does not mean that genes do not contribute to its risk or pathophysiology. It simply suggests that autosomal dominant or autosomal recessive inheritance is unlikely to account for the majority of the cases. Under such circumstances it is worth considering the relevance of other genetic mechanisms that can influence disease risk yet not produce recognizable Mendelian inheritance patterns.
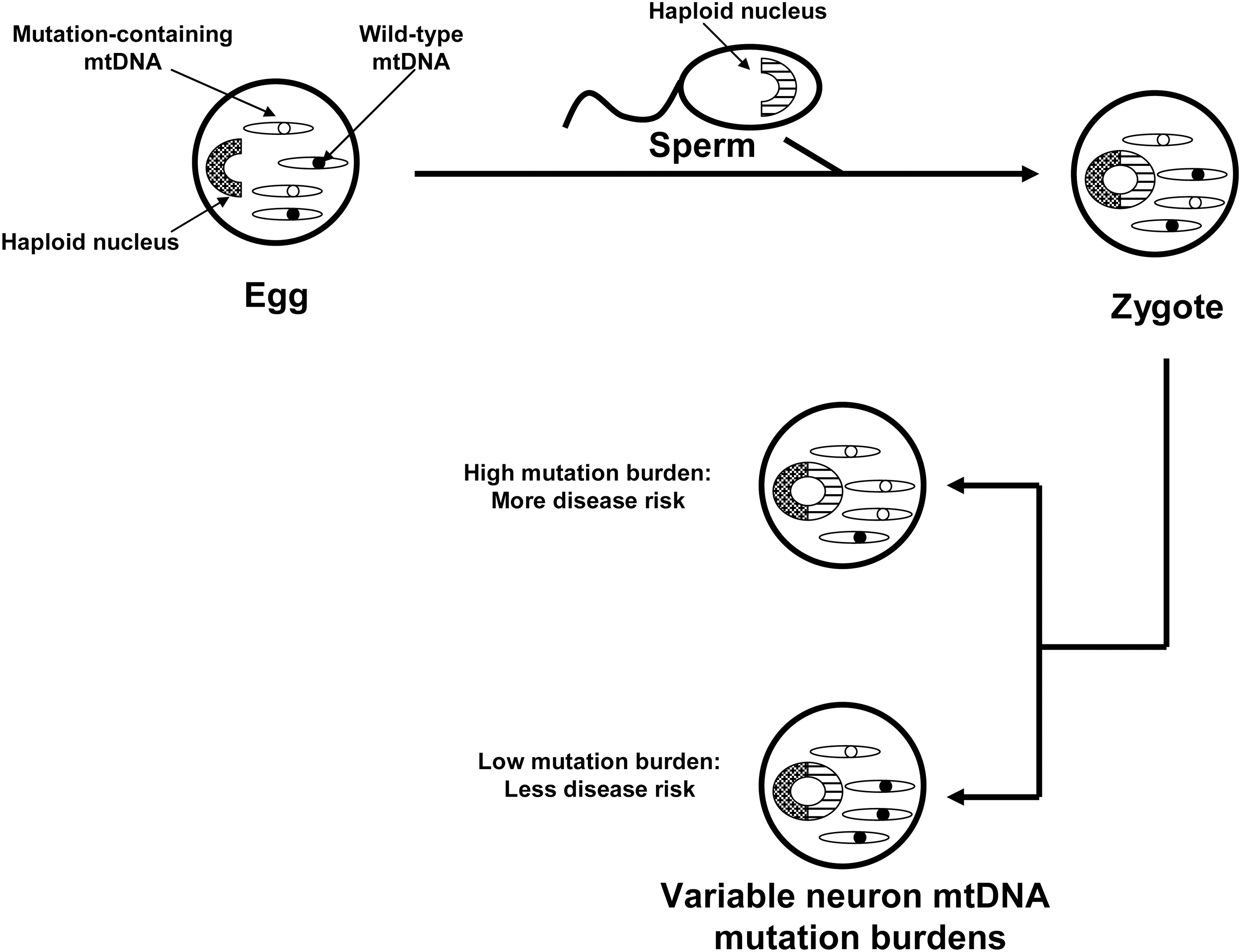
In the 1980s mtDNA was demonstrated to show almost exclusive maternal inheritance (39). During that decade primary mtDNA disorders were expected to exclusively show recognizable maternal inheritance that is distinguishable from Mendelian inheritance. This assumption was questioned by Parker, who hypothesized that because mtDNA shows heteroplasmy, threshold effects and mitotic segregation even maternally inherited mtDNA disorders would present in a sporadic fashion (93) (Fig. 5). According to this hypothesis inherited mtDNA signatures could confer disease risk within an obvious matrilineal context, within a context in which maternal inheritance is reflected only by a subtle maternal inheritance bias, or in a completely sporadic fashion. Based on this, Parker et al. proposed that mtDNA could specifically explain the finding that complex I activity is systemically lower in PD subjects than it is in non-PD subjects (94). Parker et al. further postulated that because most PD is sporadic, low abundance mtDNA heteroplasmic mutations were of particular interest.
The Parker hypothesis was somewhat radical. By the mid-1990s, low abundance mtDNA heteroplasmies had not been shown to exist, but this was mostly due to the fact that low abundance heteroplasmies were not detected by the routine dideoxynucleotide sequencing protocols used at the time (52). Of course, since then many of the key technical limitations responsible for this have been resolved, and it has been clearly demonstrated that low abundance heteroplasmies not only exist but are in fact commonplace (17, 23, 69, 80, 121, 125).
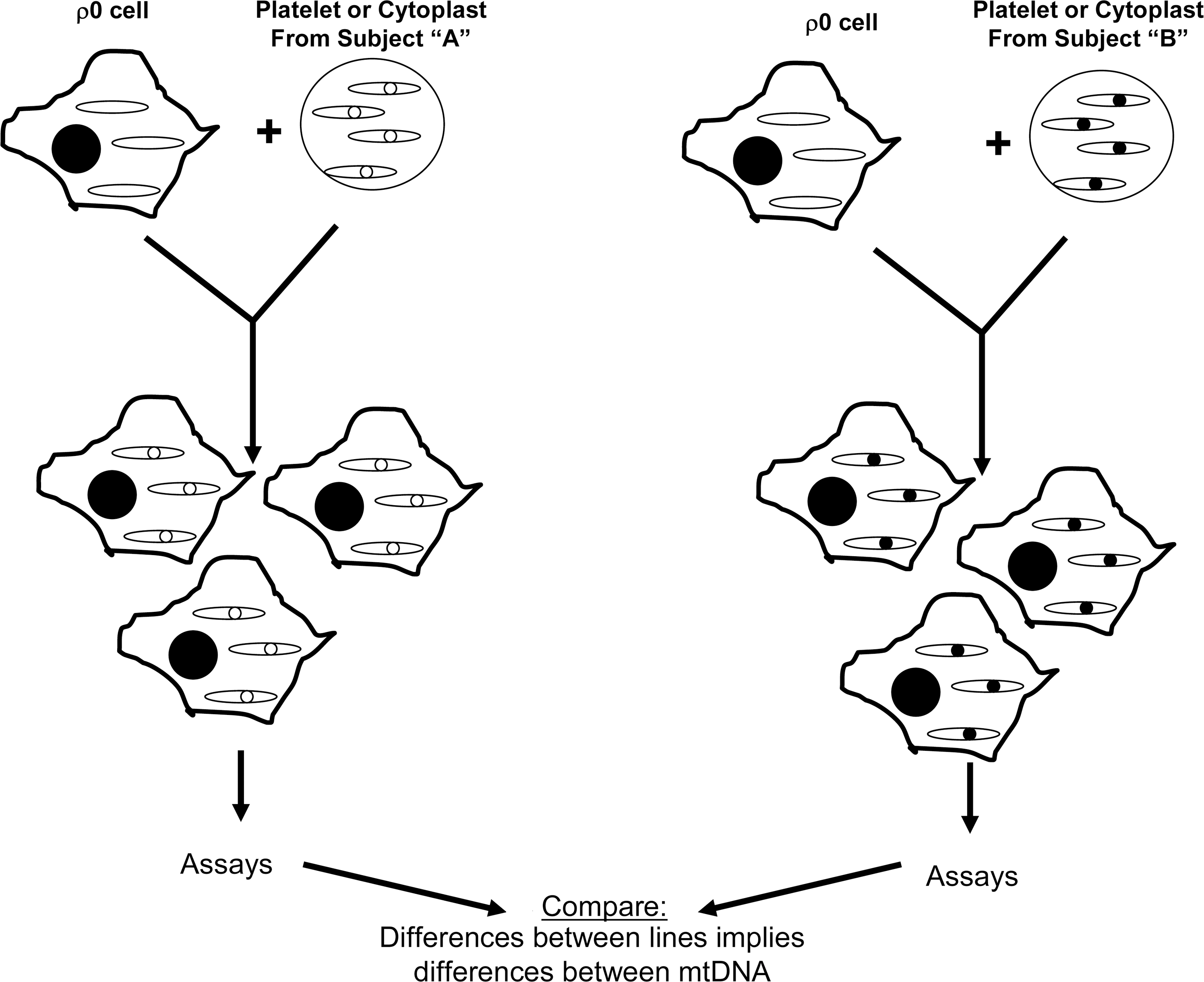
In the mid-1990s, though, the cybrid technique was felt to represent a reasonable approach for testing whether mtDNA might account for the PD complex I defect. The idea of using cybrids to study this question was, nevertheless, greeted with some skepticism as it represented a departure from the way cybrids were currently being used at the time, which was mostly to study threshold and functional effects of already-defined mtDNA mutations (130). Rather, the use of cybrids to study PD mtDNA was more reminiscent of the chloramphenicol resistance experiments of two decades prior, in which cybrid cell lines were generated to simply address the question of whether mtDNA could induce a particular cell biology, physiologic, or biochemical phenotype (16, 157) (Fig. 6). This type of application can infer a distinct mtDNA signature exists, but cannot identify the actual sequence signature. The results of what has now become an extensive body of PD cybrid work strongly argue that sporadic PD subject mtDNA differs in some way or ways from the mtDNA of control individuals. This body of work is summarized in the following sections.
Complex I Activity Is Reduced in PD Cybrids
The first PD cybrid study was reported in 1996 (138). The SH-SY5Y ρ0 cell line was used to generate a total of 52 distinct cybrid cell lines. mtDNA reconstitution for each cell line was accomplished by mixing platelets obtained from a different individual with an aliquot of ρ0 cells. In this study, 24 platelet samples were obtained from sporadic PD subjects, and 28 platelet samples were obtained from age-matched control subjects. The mixing was performed in a polypropylene tube using gentle agitation and polyethylene glycol to enhance the exchange of cytosolic contents between platelets and ρ0 cells. Following the mixing procedure, tube contents were plated in a cell culture flask and placed in medium that was supplemented with neither uridine nor pyruvate. Over the next several weeks nontransformed cells detached and died, whereas transformed cells grew initially as visible colonies that were eventually dispersed by routine passaging of the flask contents. The selection for transformed cells was completed over a 6-week period, during which time the cells underwent multiple cycles of cell replication. Therefore, although the initial cytoplasmic mixing between platelets and ρ0 cells likely involved more than just platelet mitochondria, over the many weeks of cell selection and many cycles of cell division the potential effects of any nonperpetuating, nonreplicating platelet components were theoretically eliminated through degradation and dilution. Theoretically, the only perpetuating, replicating component transferred from platelets was platelet mtDNA. While this mtDNA was incorporated as a component of the whole platelet mitochondria that were transferred from the platelets, by the end of the selection period the only unique mitochondrial feature carried over from the transfer event was ideally the mtDNA. By the end of the selection process, virtually all mitochondrial membrane was newly synthesized, and all nuclear-encoded mitochondrial proteins, whether constituting part of the ETC, an outer or inner mitochondrial membrane protein, or an inter-membrane or matrix space protein was a product of the SH-SY5Y cell line's nuclear DNA. On the other hand, the 13 mtDNA-encoded ETC components were translated according to the sequence specifications of the platelet donor subject's mtDNA. Therefore, the cybrid cell lines that were generated differed from each other only in the origin of their mtDNA, and hence their mtDNA sequence.
At the conclusion of the selection process enriched mitochondrial fractions were prepared from each individual cybrid line and complex I and complex IV Vmax activities were spectrophotometrically determined. The mean complex I activity of the PD cybrid group was only 80% of that of the control cybrid group. The mean complex IV activities between the groups were comparable. This study concluded mtDNA at least partly accounts for the low systemic complex I activity observed in PD subjects.
Since this initial report, other PD and control cybrid series have been generated using SH-SY5Y ρ0 cells and obtained the same outcome. Shults and Miller reported the results of their SH-SY5Y cybrid study in 1998 (120). Also in 1998, Swerdlow et al. prepared SH-SY5Y cybrid cell lines from members of an extended family in which PD appeared to pass through maternal lines of inheritance (137). In that study, cybrid cell lines prepared using platelet mitochondria from PD-affected kindred members and the young, asymptomatic descendents of PD mothers had lower complex I activities than cybrid cell lines prepared from platelet mitochondria obtained from asymptomatic kindred members descended through paternal lines.
In 1998, Gu et al. reported a study of PD cybrid cell lines that were produced on an A549 lung adenocarcinoma epithelial cell background (41). In this cybrid series the mean complex I activity was 75% of the control mean complex I activity. This study also directly measured the platelet mitochondria complex I activities from the subjects it studied, and concretely established that low complex I activities in PD subject platelet mitochondria are carried over into and persist within cybrid cell lines created through mitochondrial and mtDNA transfer from the platelets of those subjects.
In 2008, Esteves et al. reported a study of PD cybrid cell lines that were produced on an NT2 teratocarcinoma cell background (34). The mean complex I activity in the PD cybrid lines was approximately half of what it was in the control cybrid lines. As was the case with the Gu et al. study, complex I activities were also directly measured in platelet mitochondria from subjects whose platelet mitochondria were used to accomplish mtDNA reconstitution of the ρ0 cells. Again it was found that low complex I activities in PD subject platelet mitochondria are carried over into and persist within cybrid cell lines. Indeed, the magnitude of the complex I defect seen in the PD cybrid cell lines was equivalent to what was seen in the direct platelet mitochondria measurements. In 2010, Esteves et al. reported data from a separate series of NT2-based PD cybrids in which the complex I activity was ∼60% that of the control cybrid group (35).
Challenges to the fundamental finding of these studies, which is that platelet mtDNA transfer from PD subjects to ρ0 cells restores complex I activity in the resulting cybrid cells to levels below those seen when mtDNA is transferred from control subjects, have been relatively minor. After the first PD cybrid study was published the primary criticism was that perhaps there was some unique, unrecognized property of the SH-SY5Y ρ0 cell line that produced an inaccurate result. This possibility has been addressed by the fact that the essential finding has now been seen in cybrid cell lines with three different nuclear backgrounds. Another early criticism was that the cell colonies, or clones, that resulted from a particular cybrid fusion were grown in the same flask and not separated from each other during the selection process (114). At the time, clonal isolation was a standard approach that was used to generate cell lines with different heteroplasmic burdens of mtDNA mutation. The studies described above, though, were not designed to study strict questions of heteroplasmy although the results certainly do not rule out heteroplasmy. The Gu et al. study also considered whether clonal cybrid populations might behave differently than mixed cybrid populations, and found no evidence to support that possibility (41).
In 2000, a study of PD cybrids produced on a HeLa cell background failed to find reduced complex I activity (3). The authors concluded that mtDNA must not contribute to the PD complex I defect. In light of the fact that multiple studies by multiple groups using other ρ0 cell lines have obtained different results suggests this is an over-generalization. This possibility is further supported by the fact that in studies of other diseases HeLa-based cybrid cell lines also yield results that diverge from those of other investigators who use different ρ0 cell lines (50, 115, 130, 136).
Concerns have also been raised by the fact that in the positive cybrid studies mtDNA reconstitution of the ρ0 cell lines was accomplished via transfer of intact platelet mitochondria, and other components of the platelet cytosol could have also undergone transfer as well. This is certainly likely and does imply a conceptual concern, although the likelihood that mtDNA does not account for differences in ETC activities between cybrid cell lines that have different mtDNA sequences would seem to be minor. To date, no alternative explanation for the PD cybrid results has been demonstrated, and in fact a study in which PD-affected members of the Contursi kindred provided platelet mitochondria did not reveal a cybrid complex I defect (135). Contursi kindred members have an autosomal dominant version of PD that is caused by a mutation in the α-synuclein gene, and therefore have a distinct and identifiable etiology that does not need to implicate mtDNA (100). The fact that a persistent complex I defect was not seen in the Contursi kindred cybrids indirectly argues against the possibility that a nongenetic, cytosolic factor transferred during the fusion process accounts for the complex I defect that is seen when cybrids are generated from sporadic PD subject platelet mitochondria. Regardless, this criticism does lead to the biggest remaining question about the cybrid studies discussed above, which is if mtDNA does account at least in part for the PD complex I defect, what specifically is different about the mtDNA of sporadic PD subjects?
PD Cybrid Modeling of Other PD-Associated Pathologies
The PD and control cybrids produced by Swerdlow et al., Gu et al., Esteves et al., Esteves et al., and others have been used to study a variety of physiologic phenomena (34, 35, 41, 138) and have shown that a number of pathologies observed in PD subject brains are actually recapitulated by PD cybrids. When considering these pathologies, it is important to keep several basic tenets in mind. First, the only recognized difference between PD and control cell lines is the origin of their mtDNA and their mtDNA sequence. Second, mtDNA only encodes only 13 proteins, 22 tRNAs, and 2 rRNAs. Since all mtDNA-encoded proteins are ETC constituents, mtDNA-mediated cell differences must arise through differences in ETC function. Third, as is the case with PD subjects, PD cybrids have altered complex I function. Therefore, all unique physiologic phenomena observed within PD cybrids must result from their apparent mtDNA-dependent ETC peturbations and especially to perturbation of the complex I holoenzyme (Fig. 7).
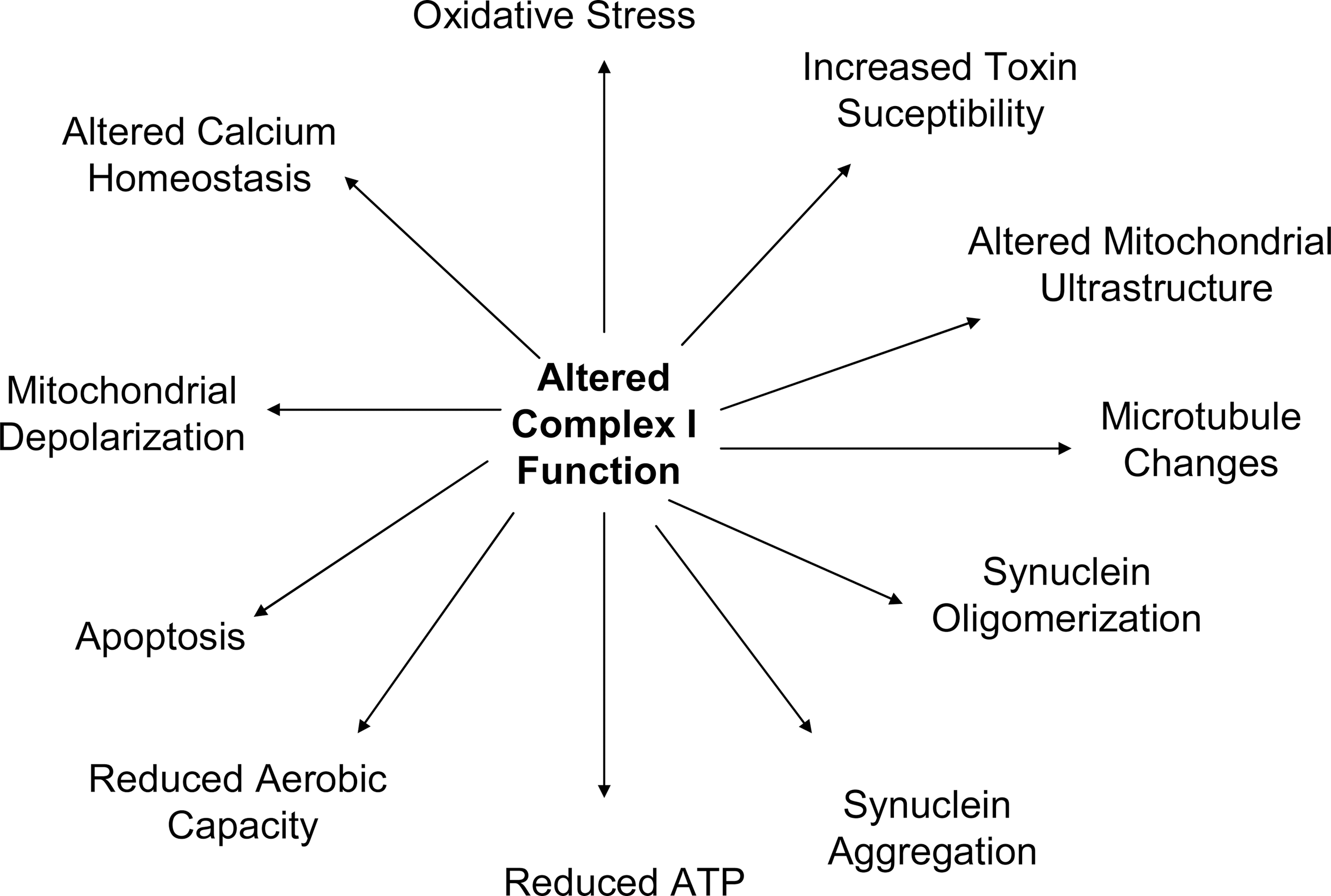
PD cybrid mitochondrial ultrastructure is altered (34, 137, 148). Mitochondria tend to be larger and are more likely to show disruption of cristae than mitochondria in control cybrids. PD cybrid mitochondria show more frequent inclusions.
PD cybrid viability is different from that of control cybrids. Under basal conditions PD cybrid cultures show increased LDH release, as well as an increased activation state of the intrinsic apoptosis pathway, altered levels of apoptosis-relevant proteins such as the bcl proteins, PARP activation, and condensed nuclei (34, 90, 91, 153). PD cybrids are also more likely than control cybrids to show apoptotic changes when exposed to equivalent concentrations of the MPP+toxin (34, 138). Related findings include the fact that PD cybrid mitochondria are relatively depolarized (34). This probably accounts for the reduced ability of their mitochondria to buffer cytosolic calcium (32, 116).
PD cybrid cell line ATP levels are reduced compared to those of control cybrids (31, 33, 34). The status of pathways influenced by cell bioenergetics is also altered. PD cybrids show reduced SIRT1 phosphorylation, reduced peroxisome proliferator-activated receptor-gamma coactivator-1alpha levels, and increased NF-kB activation (20, 35). Some of these redox-dependent changes may also arise in part as a downstream consequence of increased oxidative stress, as PD cybrids show enhanced rates of peroxide formation, decreased glutathione, and increased protein oxidation despite the presence of a presumably compensatory increase in antioxidant enzyme activities (19, 31, 91, 138). Recapitulation of oxidative stress in PD cybrid lines is of particular interest as oxidative stress occurs in the brains and peripheral tissues of PD subjects (25, 27, 53, 75, 110, 111). The PD cybrid model therefore implicates a specific potential source for the oxidative stress that is observed in this disease. Cell mtDNA content is reduced in PD cybrids, which is perhaps consistent with their lower proliferator-activated receptor-gamma coactivator-1alpha levels (14, 55).
PD cybrids appear to have ATP-dependent cytoskeletal changes, which manifest as microtubule depolymerization (31, 33). α-synuclein protein is also altered in PD cybrids. α-synuclein oligomer levels are increased (31 –33). When neuronal PD cybrids are differentiated the resultant neurites show detectable increases in α-synuclein (145). α-synuclein aggregates reminiscent of Lewy bodies are also commonly observed in PD cybrids, while they are far less commonly detected in control cybrids (31, 146). In conjunction with altered mitochondrial function, cytoskeletal changes may account for reductions in mitochondrial movement that are seen in PD cybrids (147).
Recently, improvements in respirometer technology have facilitated oxygen consumption measurements in whole cells. In a study of NT2 PD cybrids, basal oxygen consumption between PD and control cybrid lines was comparable. However, when cell mitochondria were pushed via chemical-induced uncoupling into a state of maximum oxygen consumption, the control cybrid lines showed a greater increase (35). PD cybrids, therefore, have a reduced respiratory reserve capacity. It would be interesting to know if this is also the case in the brains of PD subjects, but of course this parameter would be difficult to test.
Are Specific mtDNA Signatures Typical of PD?
A standard mtDNA sequence called the Cambridge Sequence was agreed upon several decades ago (1, 2). During the 1990s investigators reported homoplasmic Cambridge Sequence deviations are common in PD subjects, although homoplasmic Cambridge Sequence deviations are also common in control populations (49, 61, 155). Association studies have occasionally concluded particular homoplasmic Cambridge Sequence deviations may be more frequent or less frequent in a particular PD cohort as compared to a particular control cohort. In most cases, though, replication of such reported associations is inconsistent or awaits confirmation. For example, an A4336G transition in the mtDNA tRNA (Gln) gene has been found to be over-represented in PD subjects in some but not other studies (78, 118, 122, 139). More recently, one large study found that a common nt10398G change is underrepresented in PD subjects, but a second study did not (123, 151). A G5460A transition was found in one study to associate with PD, but this was not seen in other cohorts (62, 122, 139). The ND1 gene T4216C polymorphism has reportedly associated with an elevated risk of PD (60, 107).
Certain mtDNA variations are typical of particular geographical regions or populations (108, 109). Some variations tend to occur in combination and are used to define mtDNA haplogroups. The possibility that haplogroups might influence risk for particular conditions or diseases has been raised (74, 106). One study reported that among the classic European mtDNA haplogroups, as compared to persons with haplogroup H persons with haplogroups J and K have reduced PD risk (151). Additional haplogroup association studies also conclude that particular mtDNA haplogroups or haplogroup clusters may influence PD risk although risk attributions between the different populations analyzed do vary (5, 38, 102, 151). The question of whether mtDNA haplogroups are functionally relevant has also been raised, and one recent cybrid study did find differences in mitochondrial respiratory capacity in cells containing haplogroup H versus haplogroup Uk (40). Other cybrid studies have similarly reported that common mtDNA polymorphisms may influence mitochondrial function (54, 99).
Although individual case or family reports do suggest that particular homoplasmic or high-abundance mtDNA mutations can cause parkinsonism (124, 143), the association studies thus far mentioned in this section do not persuasively argue that any particular homoplasmic mtDNA sequence deviation is an outright cause of PD in a large percentage of the cases. While common variations may turn out to influence risk, with particular mtDNA and nuclear complex I gene polymorphism combinations having additive or subtractive effects on someone's overall risk (139), common variations do not appear to be PD-deterministic.
If actual mtDNA mutations are deterministic in a large percentage of PD cases, then those mutations would, therefore, have to exist in the form of low-abundance heteroplasmies. Although recent advances are making the detection of low abundance heteroplasmies more practical, to this point the sensitive and accurate identification of low abundance mtDNA heteroplasmies has been technically challenging. Regardless, several relevant studies have found increased numbers of mtDNA deletions in the substantia nigra neurons of deceased PD subjects (8, 63). Surveys of low abundance heteroplasmic point mutations have also been performed. In these studies methodologic approaches and in some cases interpretations have varied. These studies have generally concluded that low abundance heteroplasmic point mutations are common in the brains of aging individuals, and between PD and control subjects point mutation burdens are equivalent (4, 95, 121, 125). In terms of mutation patterns or localizations, some have found no differences between PD and control subjects (4, 121), whereas others have claimed characteristic mtDNA signatures that distinguish PD subject brains from those of control subjects. For example, Smigrodzki et al. and Parker et al. have reported low-abundance heteroplasmic mutations in a defined region of the ND5 gene are found in most PD subject brains but rarely in the brains of persons who died without PD (95, 125).
Are PD-Relevant mtDNA Signatures Inherited or Acquired?
MtDNA mutations accumulate with advancing age. It has been postulated that these mutations are acquired or “somatic” mutations, and that they may contribute to common age-related disorders such as Alzheimer's disease, PD, and type II diabetes (156). Empiric evidence further supports the possibility that age-associated increases in mtDNA mutation may drive aging-related phenotypes (66, 144). Whether the responsible mtDNA mutations are point mutations or deletions is a topic of debate (29, 64, 154). Interestingly, humans with mtDNA polymerase gamma variants or mutations that could potentially accelerate mtDNA mutation accumulation can present with parkinsonism or Parkinsons's disease (26, 48, 71, 72).
Transgenic mouse models have leveraged defective mtDNA polymerase gamma proofreading to accelerate formation of mtDNA somatic mutations (66, 144). Free radical species are also known to mutagenize DNA, with mtDNA being affected more than nuclear DNA (79). Somatic DNA mutation rates are presumably higher in the mitochondria than the nucleus because mitochondria are the main source of oxygen radical production in eukaryotic cells (117). In one common mutagenic reaction, a cytosine nucleotide is oxidatively deaminated, and the lost amino group is replaced by an oxygen double bond. This reaction converts the cytosine to uracil. When DNA replication occurs, the uracil of the replicating strand is read as a thymine, and an adenine is placed in the daughter strand at a position where there previously was a guanine. In the next replication cycle, the uracil is replaced by thymine. The end result is substitution of an A-T pair for a G-C pair (82, 92, 98). Imbalances in mitochondrial purine and pyrimidine pools also appear to facilitate the relatively high rate of both point and deletion-type mtDNA somatic mutations (126, 127).
As was discussed in the previous section, mtDNA point mutations and deletions do accumulate in the aging substantia nigra. Deletion accumulation in PD subjects exceeds that of non-PD subjects (8, 63). Point mutation burden is comparable between groups, although certain point mutations seem to be found only in PD subjects (95, 121, 125). It could be argued these findings imply a role for somatic mtDNA mutation in PD, but this is a somewhat complicated subject. MtDNA heteroplasmic ratios can drift over time, so the fact that mtDNA mutational burdens increase during aging does not prove that those mutations were not inherited. Also, it has been shown that mtDNA point mutations may themselves induce mtDNA depletion and potentially, therefore, deletion (45, 149). This raises the possibility that increased levels of substantia nigra mtDNA deletions in PD subjects could actually serve as a marker for potentially inherited mtDNA point mutations. Also, it is important to keep in mind that the PD complex I defect is seen in multiple tissues throughout the body, which would mean an acquired mutation would likely have to arise very early during development, and probably during the oogonia gamete stage (24, 36).
Parker et al. hypothesized that inherited mtDNA mutations are potentially more relevant to sporadic PD than acquired mutations (94, 97). This view is perhaps more consistent with the multi-tissue complex I defect seen in sporadic subjects. Parker has further hypothesized that, in general, the inheritance of low abundance heteroplasmic mutations could play a role in a variety of sporadic diseases because diseases arising from the inheritance of low abundance, heteroplasmic mutations would be expected to show neither Mendelian nor consistent maternal epidemiology (93) (Fig. 8).
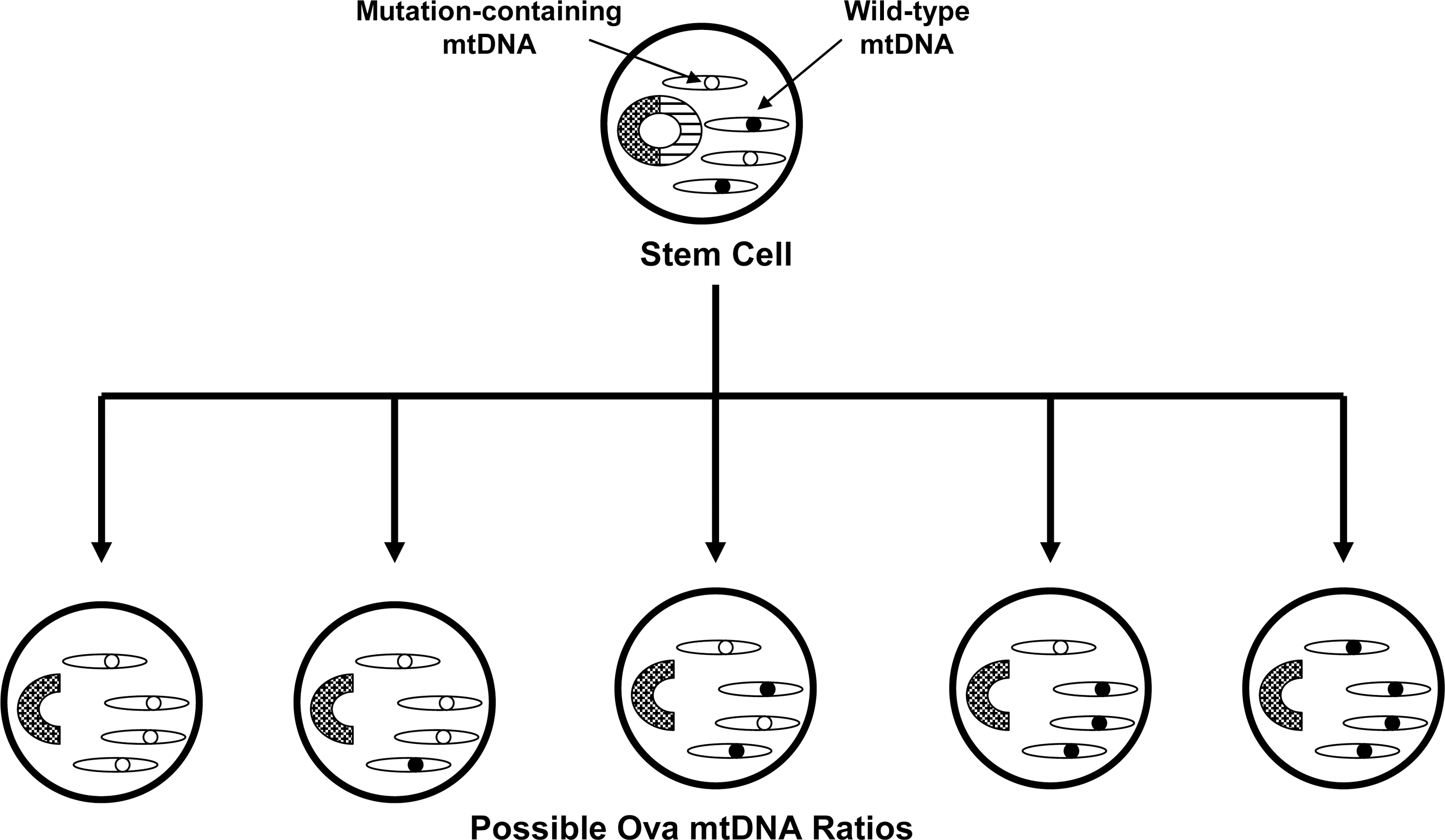
It is increasingly recognized that gene inheritance may contribute to the development of sporadic diseases. In sporadic PD, this is reflected by the finding that even though Mendelian inheritance patterns are not clearly demonstrated sporadic PD subjects are more likely to have a PD-affected parent than are non-PD subjects (97, 140). When data on such parent–child relationships are closely examined subtle evidence of maternal inheritance bias does emerge. Wooten et al. found that among PD clusters in which there are two affected siblings and an affected parent, the affected parent is more likely to be the mother than the father (161). Further, an analysis of multiple PD subject databases found sex ratio differences between PD probands and the PD-affected parents of those probands (134). While the proband sex ratio showed more men than women, the PD-affected parental generation showed more women than men. This finding is consistent with a maternal inheritance bias.
Lastly, inherited homoplasmic mtDNA polymorphisms either in isolation or as part of haplogroups may influence PD risk (5, 38, 102, 151). It increasingly appears these polymorphisms may subtly influence mitochondrial function (40, 54, 99). This may apply to even extremely common synonymous mtDNA polymorphisms that do not cause amino acid substitutions (70). It has been observed that inherited mtDNA sequence variations may influence the acquisition of somatic mutations (45). It is therefore worth considering the possibility that both inherited and acquired mtDNA variation contributes to the development of sporadic PD.
Implications and Conclusions
The perceived relevance of mitochondria to PD has certainly evolved over the past three decades. During the 1980s and early 1990s, the MPTP story and subsequent demonstration of complex I dysfunction in idiopathic PD subjects catapulted mitochondria to the forefront of the PD research community. During the late 1990s this interest waned as investigators began to identify mutations in families with Mendelian PD subtypes. As the last decade progressed, though, an emerging impression that many genes implicated in Mendelian PD influence or are influenced by mitochondrial function restored the mitochondria to a position of research prominence (15, 142).
Advances in transgenic technology now make the development of disease models in which histological, biochemical, and clinical phenotypes are induced by nuclear gene mutations increasingly commonplace. On the other hand, modeling sporadic diseases that do not have identifiable Mendelian causes is conceptually challenging. Because of this, it is extremely tempting to want to extrapolate findings from Mendelian PD model studies to sporadic PD. While in some ways this will no doubt advance our knowledge of PD in general and especially our understanding of the Mendelian forms, over-generalizations could prove dangerous and an over-reliance on Mendelian models could impede PD therapeutic development as it has in other fields (129, 132, 133). In this respect PD cybrids currently provide one of the most straightforward ways to model the PD complex I defect. While their past contributions to the PD research field are well recognized, an active role for PD cybrids in ongoing PD research and especially PD therapeutic development requires consideration. In the meantime, some groups are currently developing new technologies that would facilitate the direct transfer of exogenous mtDNA to cells (56, 57). This could enable new iterations of the cybrid technique, such as the creation of PD cybrid cell lines that contain mtDNA from PD subject substantia nigra neurons.
Footnotes
Acknowledgments
This work was supported by grants from the Morgan Family Foundation and the PD Foundation of the Heartland.