
Abstract
Introduction
Pater and Loeb (181) were among the first to show that perceived fatigue is an independent predictor of QOL and survival in cancer patients. Numerous other studies have followed confirming their results (14, 20, 38, 202). A component of perceived fatigue could be a decline in cognitive function. Evaluation of cognitive impairment involves functionality of multiple domains, which include visuospatial skill, memory, language, and motor function (258).
Over half of patients undergoing chemotherapy exhibit cognitive impairment (109), which is associated with patients perceived fatigue (18). This sense of tiredness can persist from 6 months to 2 years following remission, providing insight into the debilitating, and sometimes long-term side effects of cancer and its treatment (120, 132, 149).
While documenting perceived fatigue is useful clinically, it is difficult to discriminate between a sense of tiredness (i.e., perceived fatigue) and physiological fatigue. Physiological fatigue can be divided into two components, central and peripheral fatigue. Central fatigue involves the central nervous system and the inhibition of neurological reflexes. Central fatigue is a factor with cancer (275), however the interaction of central fatigue and chemotherapy is unknown, limiting the discussion of this component further. Peripheral fatigue is muscle specific and involves the loss of muscle function, divided into two components: muscle fatigue and muscle weakness. Muscle fatigue is defined as the loss of force that is reversible by rest, while muscle weakness is an impaired ability to generate force and is not relieved by rest (177a). Based on available data, this review will discuss the effects of chemotherapy on muscle weakness.
Previous studies have used performance assessments to document muscle weakness in cancer patients that have received chemotherapy. Compared to healthy controls, patients show a slower chair-rise time, indicating a decrease in muscle strength (25, 85). Hand-grip force, another measurement of muscle weakness, is decreased in cancer patients (25, 96, 230, 231, 269). These studies point to a prominent clinical problem of debilitating muscle weakness with cancer treatment.
One underlying mechanism of the muscle weakness experienced by chemotherapy-treated patients is a developed state of oxidative stress, defined in this review as a disruption of redox signaling and control (112). Numerous chemotherapeutic agents directly or indirectly produce a state of oxidative stress. Drugs that include a quinone moiety in their chemical structure can directly produce a state of oxidative stress by interacting with molecular oxygen and undergoing redox cycling, leading to the generation of reactive oxygen species (ROS) (40). Other chemotherapeutic agents can indirectly produce a state of oxidative stress by decreasing antioxidant levels, crippling the cell's defenses against elevated oxidants (7, 158).
Circulating biomarkers are a nonspecific systemic index of oxidative stress in the body. In cancer patients under going treatment, circulating markers of oxidative stress, in the form of lipid peroxidation and protein carbonyl content, are elevated (44, 91, 105). These markers reflect events of oxidative stress that may exist in various tissues, including skeletal muscle. No current data exist to describe the level of muscle-derived oxidants in cancer patients. However, circulating biomarkers serve as an index about the level of oxidative stress in the body and could signify an elevation in muscle-derived oxidants (201).
In skeletal muscle, exposure to elevated oxidants are known to cause muscle weakness and accelerate the rate of fatigue (191, 235). Antioxidant exposure delays the rate of fatigue, supporting this connection (118, 136, 165). Chemotherapy-induced oxidative stress in cancer patients could be a reflection of the elevated muscle-derived oxidants, an underlying mechanism for the muscle weakness experienced by patients. This article reviews how chemotherapy can affect striated muscle, increasing muscle-derived oxidants and leading to muscle weakness in patients.
Anthracycline Therapy
Numerous chemotherapy drugs have been approved by the Food and Drug Administration to treat patients in the clinic (97). For the purpose of this review we focused on a class of chemotherapy drugs called anthracyclines. For over 50 years, anthracyclines have been used widely in the clinic to treat multiple types of cancers (e.g., leukemia, lymphoma, breast, prostate, ovarian, lung) (see Table 1; (86, 209)). Due to the extensive literature available, the effects of chemotherapy on muscle function are best defined in this class and accessible for evaluation. The mechanisms by which anthracyclines kill tumor cells are various, including: inhibition of DNA replication and RNA transcription, free radical generation leading to DNA damage or lipid peroxidation, DNA alkylation, interference with DNA unwinding or DNA strand separation and helicase activity, and inhibition of topoisomerase II (209). Some or all of these effects are responsible for inhibiting tumor cell growth, preventing division and metastasis. Anthracyclines can also negatively affect noncancerous tissues, including striated muscle, which contributes to the fatigue and muscle weakness in patients treated with anthracycline-based chemotherapy. A comprehensive literature search over the past 5 years (2005–2010) was performed to document fatigue reports associated with each drug in the anthracycline group (Table 1). Two anthracycline drugs (teniposide and valrubicin) were not included in the table due to the absence of current data documenting associated fatigue with drug administration.
The grade refers to the severity of the adverse event with a numerical scale: (1) Mild, (2) Moderate, (3) Severe, and (4) Disabling. Abbreviations: NCI, National Cancer Institute; NSCL, non-small cell lung; QOL, quality of life; SCL, small cell lung.
Most studies observing fatigue with anthracycline chemotherapy use the National Cancer Institute Common Toxicity Criteria for Adverse Events (CTCAE) to grade the severity of patients perceived fatigue (173a). This method allows reporting of adverse events with descriptive terminology. The grade refers to the severity of the adverse event with a numerical scale: (1) Mild, (2) Moderate, (3) Severe, and (4) Disabling. The adverse event fatigue has specific descriptions associated with the numerical scale: (1) Mild fatigue relieved by rest, (2) Fatigue not relieved by rest—difficulty performing activities of daily life, (3) Fatigue not relieved by rest—interfering with activities of daily life, and (4) Disabling fatigue. In Table 1, each study lists the cancer population and the range of the grades of fatigue associated with the chemotherapy drug. A few studies used a standard QOL questionnaire the European Organization for Research and Treatment of Cancer QLQ-C30 (EORTC QLQ-C30) to report chemotherapy-associated fatigue. This questionnaire has three multi-item symptom scales measuring fatigue and global health status (3, 228). Studies that assessed fatigue using a QOL questionnaire are also included. Table 1 documents the grades of fatigue in cancer patients treated with a specific anthracycline. In studies assessing patient fatigue with anthracycline-based chemotherapy, 47% document grade 4 fatigue, categorized as disabling and effecting physical capabilities of the patients. This table also illustrates that fatigue is a common problem with anthracycline chemotherapy occurring independent of the drug type.
Aside from the antitumor effects, anthracyclines are known to have toxic side effects in normal tissue, including oxidative stress (86). Anthracyclines can generate oxidants through two mechanisms: interaction with the mitochondrial respiratory chain and through a nonenzymatic reaction with ferric iron (103). Numerous drugs in the anthracycline class cause oxidative stress in both humans and rodents.
Elevated oxidants in circulation have been reported in cancer patients following administration of the anthracycline epirubicin (28, 134, 150). Following epirubicin administration in rodents, oxidants are elevated and antioxidants are decreased in both cardiac (54) and hepatic tissue (117). Irinotecan is another example of an anthracycline that causes oxidative stress. Markers of lipid peroxidation are elevated in both plasma and intestines of rodents following irinotecan administration (259, 264). In human breast cancer cells, topotecan exposure causes a decrease in glutathione, along with an increase in lipid peroxidation (7, 243). This drug-induced oxidative stress is a potential mechanism underlying the documented fatigue experienced by cancer patients.
Chemotherapy is generally a combination of drugs administered in a standardized treatment regimen, specific for the cancer type. Various interactive effects of different drugs could occur, leading to collateral damage of nontargeted tissues. The current reductionist approach in the literature provides some clarity on the negative effects of a single chemotherapeutic agent on nontargeted tissues. The available studies offer valuable information on the drug's mechanism of action and potential interventions that could offset the negative side effects. The next step for the field is to move toward a more clinically relevant approach, in order to directly apply the findings to patients. The existing data focuses on a single chemotherapeutic agent, which provides foundational knowledge about the individual drug and lays the groundwork for future studies in patients. Based on available data involving chemotherapy effects on striated muscle, this review will focus on a single chemotherapy agent, doxorubicin, a representative anthracycline.
Doxorubicin as the Prototype
Doxorubicin is an antibiotic that exerts its antitumor activity by inhibiting DNA Topoisomerase II, thus preventing DNA replication (240). Other antitumor activities of doxorubicin include: generation of ROS leading to DNA damage and apoptotic cell death, stimulation of p53-DNA binding, activation of the caspase cascade, and DNA cross-linking (76, 155). The Farmitalia Research Laboratories of Milan discovered the drug in the early 1960s (11). Since its discovery, doxorubicin has been used widely in the clinic, seen as one of the most effective anticancer drugs (43, 263). Early on in the clinic, reports of doxorubicin causing severe fatigue (174, 175) and affecting cardiac muscle function (124) were documented. Further investigation into the mechanism of the drug revealed elevated oxidants in noncancerous tissues were a likely cause of the cardiotoxicity (40, 49, 60).
Cardiac muscle
Doxorubicin-induced cardiotoxicity is well known in the field of chemotherapy drug research, and has been reviewed widely in the literature (39, 40, 155, 162, 237, 244). The first observations of doxorubicin cardiotoxicity were by Lefrak and colleagues (124). They documented the deterioration of cardiac muscle function by echocardiography, and assessed postmortem cardiac muscle tissue of two patients after doxorubicin administration. Since that first report, numerous studies over the past 30 years have investigated the mechanisms behind doxorubicin-induced cardiotoxicity (237).
The principal mechanism of doxorubicin cardiotoxicity is an increase in oxidative stress, manifested through elevated oxidants, markers of protein oxidation, and decreased antioxidant activity (39). Elevated oxidant activity is observed in cardiomyocytes following doxorubicin exposure (52, 208, 224, 242, 274). A similar response is observed in rodent cardiac tissue, localized to the mitochondria and sarcoplasmic reticulum (59). This elevation in oxidants comes from multiple sources in the cell. The mitochondria are thought to be the primary source of doxorubicin-induced oxidants (34, 37, 48, 60, 261). Complex I (NADH dehydrogenase) of the electron transport chain is the specific site of doxorubicin reduction, forming an unstable semiquinone (49). The doxorubicin semiquinone is then oxidized to the stable quinone form, transferring an electron to oxygen to produce the superoxide anion (O2 -) (162).
The elevation of oxidants caused by doxorubicin is protected with the overexpression of mitochondrial specific antioxidants. Figure 1 illustrates how overexpression of manganese superoxide dismutase protects against mitochondrial and myofilament damage caused by doxorubicin (276). Overexpression of other antioxidants such as glutaredoxin 2 (57) and glutathione peroxidase (GpX) (271) also protect against doxorubicin-induced cardiotoxicity, pointing to the involvement of mitochondria.

Other secondary sources of oxidants include NADPH oxidase and altered Fenton chemistry. Doxorubicin-induced oxidant activity was blunted in cardiomyocytes treated with an inhibitor of NADPH oxidase (223). Mice deficient in gp91, a required subunit for NADPH oxidase activity (123), are protected from doxorubicin-induced cardiotoxicity (268). Altered Fenton chemistry occurs during the redox cycling of doxorubicin. During this process aglycone metabolites are produced that alter iron homeostasis, leading to elevated oxidants (155).
Based on its chemical structure, doxorubicin can directly stimulate an increase in oxidants by undergoing redox cycling. However, a secondary, indirect method for doxorubicin to stimulate oxidants is via an inflammatory response. One potential secondary mediator of the inflammatory response is tumor necrosis factor alpha (TNF), a pro-inflammatory cytokine produced by many cell types, including cardiac and skeletal myocytes. Circulating levels of TNF are elevated in cancer, with chemotherapy exacerbating this response (24). Doxorubicin-treated animals and patients exhibit a stress response, characterized by an increase in serum levels of inflammatory cytokines, especially TNF (87, 167, 238, 250). Doxorubicin not only stimulates an increase in circulating TNF, but also increases the production of TNF by cardiac muscle (169) and upregulates the TNF receptor subtype 1 (TNFR1) in cardiomyocytes (41). Inhibition of TNF prevents doxorubicin-induced cardiotoxicty and diminishes glutathione depletion and lipid peroxidation (158). TNF is known to elevate ROS in striated muscle (95, 135), mediated through TNFR1 signaling (95). Elevated levels of TNF caused by doxorubicin are an indirect method to increase oxidants and lead to cardiac muscle dysfunction.
This increase in oxidants can modify vital proteins, leading to cardiac muscle dysfunction. Markers of protein oxidation, such as nitrotyrosine and protein carbonylation, are increased in cardiac muscle with doxorubicin exposure (34, 129, 217). Alterations of vital proteins can alter cardiac structure, leading to impaired contractile function. Cardiac tissue exposed to hydrogen peroxide results in altered myofibrillar proteins: troponin I, tropomyosin, actin, and myosin-binding protein-C (16, 30). The post-translational modifications of myofibrillar proteins leads to a decrease in maximal force and compromises cardiac function (16). Oxidative modifications of myofibrillar proteins can alter myofilament structure, resulting in dysfunction of striated muscle.
Doxorubicin can also cause oxidative stress by altering cellular antioxidant expression and activity. In the literature, discrepancies exist between whether doxorubicin inhibits inherent antioxidants, or stimulates an increase in antioxidant activity. In rodent cardiac muscle, data exist in both groups. In vivo doxorubicin administration decreases the content of antioxidants: superoxide dismutase (SOD) (93, 158), catalase (CAT) (158), and glutathione (GSH) (106, 125, 158). Activity of SOD is also diminished with doxorubicin (67, 125, 154). These studies hypothesize one mechanism doxorubicin stimulates oxidants is by diminishing cellular antioxidants. However, other studies show an increase in SOD (4, 6, 73), CAT (6, 73, 93, 106, 158), and GpX (4) activities with doxorubicin treatment. These data support the hypothesis of a cellular adaptive response to doxorubicin, elevating antioxidant activity to combat the doxorubicin-induced increase in oxidants.
Studies involving the administration of antioxidants in combination with doxorubicin support oxidant involvement. Rodents administered N-acetylcysteine (NAC) before doxorubicin exposure were protected from doxorubicin-induced cardiotoxicity and myocardial lesions (61). The same results were observed in a canine model of doxorubicin treatment (97). NAC is a nonspecific reduced thiol donor that increases muscle cysteine and GSH availability (78). Doxorubicin is known to decrease GSH content (106, 125, 158), a vital antioxidant in striated muscle. Infusion of glutathione prevented the cardiac contractile impairment caused by doxorubicin (256). Other antioxidants also protect cardiac muscle function in doxorubicin-induced cardiotoxicity. Vitamin E, a lipid soluble antioxidant, protected against doxorubicin-induced left ventricular dysfunction (195) and prevented elevated oxidant activity (19).
Cardiotoxicity limits the amount of doxorubicin given in the clinic (43, 236). However, on this limited dose, numerous reports show patients receiving doxorubicin-based chemotherapy experience debilitating fatigue, often in the moderate to severe category (Table 1). This persistence of weakness indicates that other striated muscles, involved in exercise and daily activity, may be affected.
Skeletal muscle
Individual case reports document physician-observed lower extremity muscle weakness in patients undergoing doxorubicin-based chemotherapy (94, 110). Schwartz documented weakness and fatigue in breast cancer patients through four cycles of doxorubicin chemotherapy (213). Following doxorubicin exposure, patients exhibited a decline in functional ability (12 min walk test) and a rapid increase in fatigue (Visual analogue scale). Weakness and fatigue are evident 1–5 years after doxorubicin exposure in lymphoma and leukemia patients (68, 248, 255). These studies suggest skeletal muscle weakness caused by doxorubicin exposure.
Existing studies in both rodents and humans show the negative effects of doxorubicin on skeletal muscle. The toxicity of doxorubicin is used in the clinic for the treatment of blepharospasm and cervical dystonia, causing permanent muscle necrosis in patients. Direct injection of doxorubicin into skeletal muscle causes loss of muscle mass, altered myofilament structure and depressed force in rodents (45, 46, 74, 75, 139 –143, 145), nonhuman primates (138, 139, 144, 146), and patients (147, 267). Doxorubicin is also administered through isolated limb perfusion in patients with limb sarcoma tumors. This therapy leads to loss of limb muscle function and reduction in size of both Type 1 and Type 2 muscle fibers (21). Pfieffer and associates used a canine model of isolated limb perfusion with doxorubicin, observing a significant increase in doxorubicin concentrations in the quadriceps, along with muscle atrophy and weakness (187).
Rodent models of systemic doxorubicin treatment consistently reveal a negative affect of doxorubicin on skeletal muscle function. The few studies in the field use an intraperitoneal route of administration for doxorubicin, adapted from the cardiac literature and related to clinical treatment of advanced ovarian cancer or peritoneal carcinomatosis (234, 252). Doroshow and colleagues observed skeletal muscle loss of myofibrillar organization and interstitial edema following a single intraperitoneal injection of doxorubicin (62). This method results in skeletal muscle toxicity exhibited by nucleolar segregation and altered distribution of the perinucleolar chromatin in hindlimb skeletal muscle (151, 254).
Doxorubicin also causes a catabolic response leading to the loss of muscle mass. Patients undergoing doxorubicin-based chemotherapy show loss of muscle mass (21, 245). The skeletal muscle atrophy induced by doxorubicin is thought to occur through the upregulation of the E3 ubiquitin-ligase atrogin1/MAFbx, suggesting catabolism through the proteasome pathway (273). Doxorubicin is a known stimulator of apoptosis in cardiac myocytes (237), with possible similar effects on skeletal muscle that could contribute to catabolism. Apoptosis is induced in C2C12 myotubes following exposure to doxorubicin in vitro (98). Oxidant-mediated apoptosis is a common signaling pathway in skeletal muscle atrophy, leading to caspase activation and proteolysis (15). In striated muscle, doxorubicin stimulates both the formation of ROS (242) and activation of caspases (261), which could be linked to apoptotic signaling pathways. We have documented significant loss of muscle mass in both hindlimb (87) and respiratory muscle (88) following doxorubicin treatment.
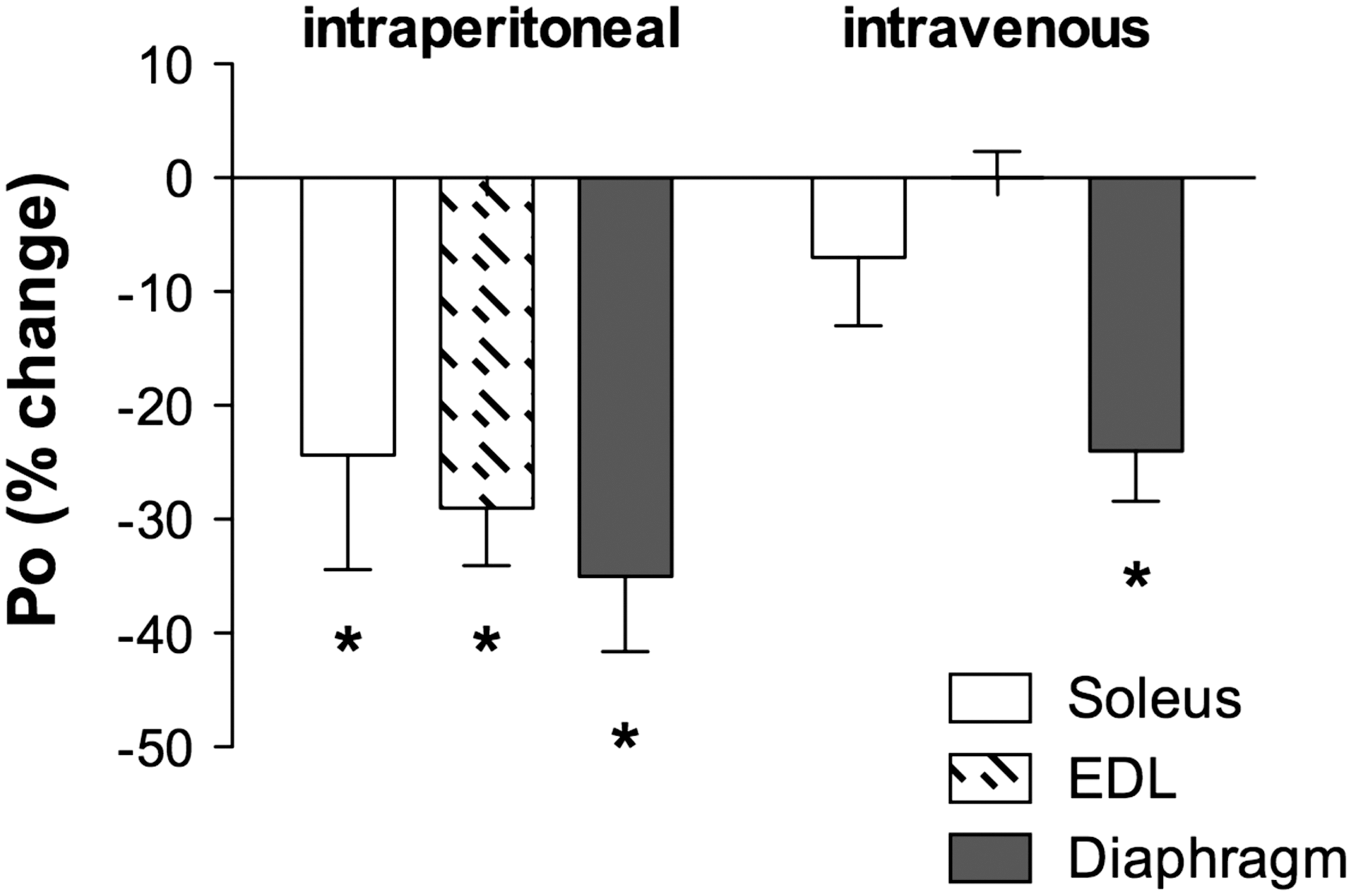
In vitro contractile function preparations have been used to determine the negative effects of doxorubicin on skeletal muscle function. Daily injections of a low dose of doxorubicin (1.15 mg/kg) depressed hindlimb muscle force, both fast-twitch and slow-twitch muscles, 4 weeks following exposure (71) (Fig. 2). The authors hypothesized the loss of force was associated with a decrease in the muscle-specific isoform of SERCA, an effect on calcium homeostasis (71). We have shown a single injection of doxorubicin depresses force of hindlimb and respiratory muscles (Fig. 3) (87). Most recently, we established a model of doxorubicin-chemotherapy using an intravenous injection, a method commonly used in the clinic (33, 43). Our studies conclude that doxorubicin causes respiratory muscle dysfunction, despite the route of administration (88).
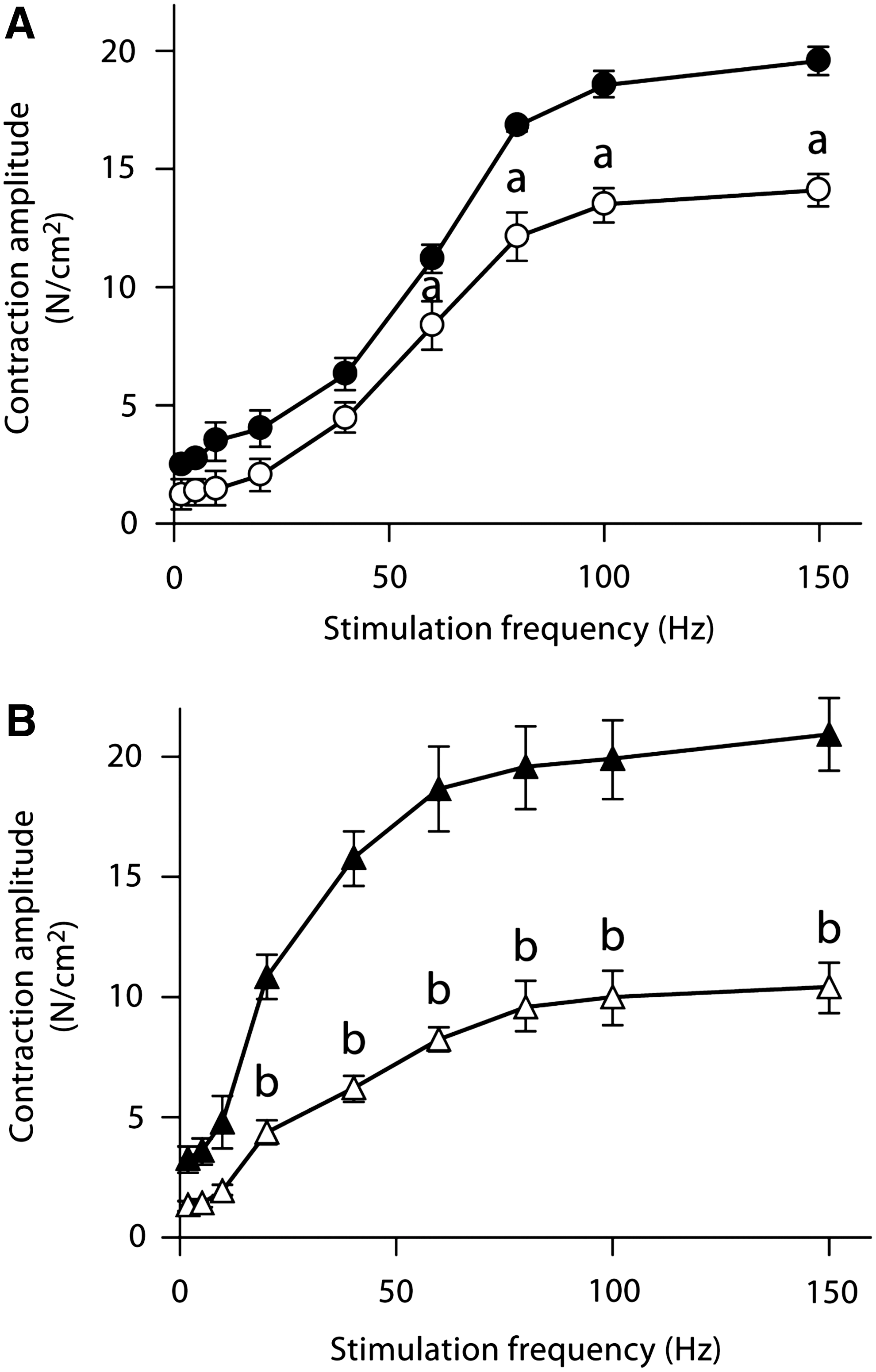
Studies have shown doxorubicin accumulates in skeletal muscle up to 24 h following administration (62, 187). This accumulation of doxorubicin in the muscle suggests a direct effect of doxorubicin on skeletal muscle function, however the data are divided. In vitro doxorubicin experiments have been conducted using varying concentrations of the drug (1–175 μM). van Norren and colleagues observed a depression in absolute force and an accelerated rate of fatigue in intact hindlimb muscles exposed to doxorubicin (253). They observed a dose-dependent depression in force, with impaired relaxation. Parallel experiments using lower doxorubicin bath concentrations (2 μM), similar to serum levels found in patients (55, 188), found no change in skeletal muscle force (87, 89).
Studies utilizing permeabilized skeletal muscle fibers have observed how direct doxorubicin exposure alters calcium homeostasis. Doxorubicin increases the rate of tension development in calcium-activated fibers (50, 282). The doxorubicin-induced tension development is blunted by ruthenium red, an inhibitor of the ryanodine receptor, suggesting doxorubicin alters calcium availability. Isolated skeletal muscle sarcoplasmic reticulums exposed to doxorubicin show increased calcium release (241, 282). This is in agreement with another study showing increased calcium influx in C2C12 myotubes exposed to doxorubicin (253). The data suggests doxorubicin acts similar to caffeine, sensitizing the ryanodine receptor to activation calcium, and stimulating calcium release from the SR (5, 282). Intact single fibers exposed to doxorubicin show an increase in tetanic intracellular calcium that does not alter tetanic force (87). These studies investigating doxorubicin-induced skeletal muscle dysfunction establish a negative effect at the molecular level that could be related to weakness in patients undergoing chemotherapy.
One potential mechanism of doxorubicin-induced skeletal muscle dysfunction is an induced state of oxidative stress, similar to cardiac muscle. Few studies exist that analyze doxorubicin-induced oxidative stress in skeletal muscle. We have shown doxorubicin increases oxidant activity in skeletal muscle, along with markers of protein oxidation (88). Multiple sources where oxidants are produced exist in skeletal muscle, including the mitochondrial electron transport chain, NADPH oxidase, and phospholipase A2 (78, 107, 191). Yamada and colleagues showed a decrease in complex I activity in isolated mitochondria from skeletal muscle following multiple low dose doxorubicin injections (272).
Similar to cardiac muscle, elevated TNF levels caused by doxorubicin can lead to increased oxidants and muscle weakness in skeletal muscle. Exposure to TNF depresses both respiratory and hindlimb skeletal muscle force (95, 198), shown to be mediated through TNFR1 (95). The accepted mechanism by which TNF mediates contractile dysfunction is through elevated ROS (95). Pretreating with Trolox, a hydrophilic antioxidant, can prevent TNF-induced contractile dysfunction (95). Taken together, these results suggest that elevated levels of TNF caused by doxorubicin could exert an additive oxidant effect, leading to skeletal muscle dysfunction. We have shown TNFR1 signaling is required for doxorubicin-induced muscle weakness in both respiratory and hindlimb muscle (87, 89). TNF mediates the majority of signaling through TNFR1, leading to the activation of cytotoxic cascades and elevated ROS (95, 260). Increased levels of TNF following doxorubicin exposure lead to an additive oxidant effect, contributing to doxorubicin-induced muscle weakness.
In skeletal muscle, elevated oxidants are known to cause muscle weakness and accelerate fatigue, reviewed previously (191, 218, 235). Exposure to high concentrations of exogenous oxidants results in muscle weakness (10, 29, 122, 173, 189, 190). The rate of fatigue is slowed with antioxidant exposure, suggesting a prominent role for oxidants (119, 164, 165, 197). In a state of oxidative stress, redox-sensitive proteins can be modified, altering signaling and contractile function. Exposing myofibrillar proteins, such as myosin and actin, to oxidants can result in modifications that alter protein structure and formation, and decrease force generation (17, 115, 192).
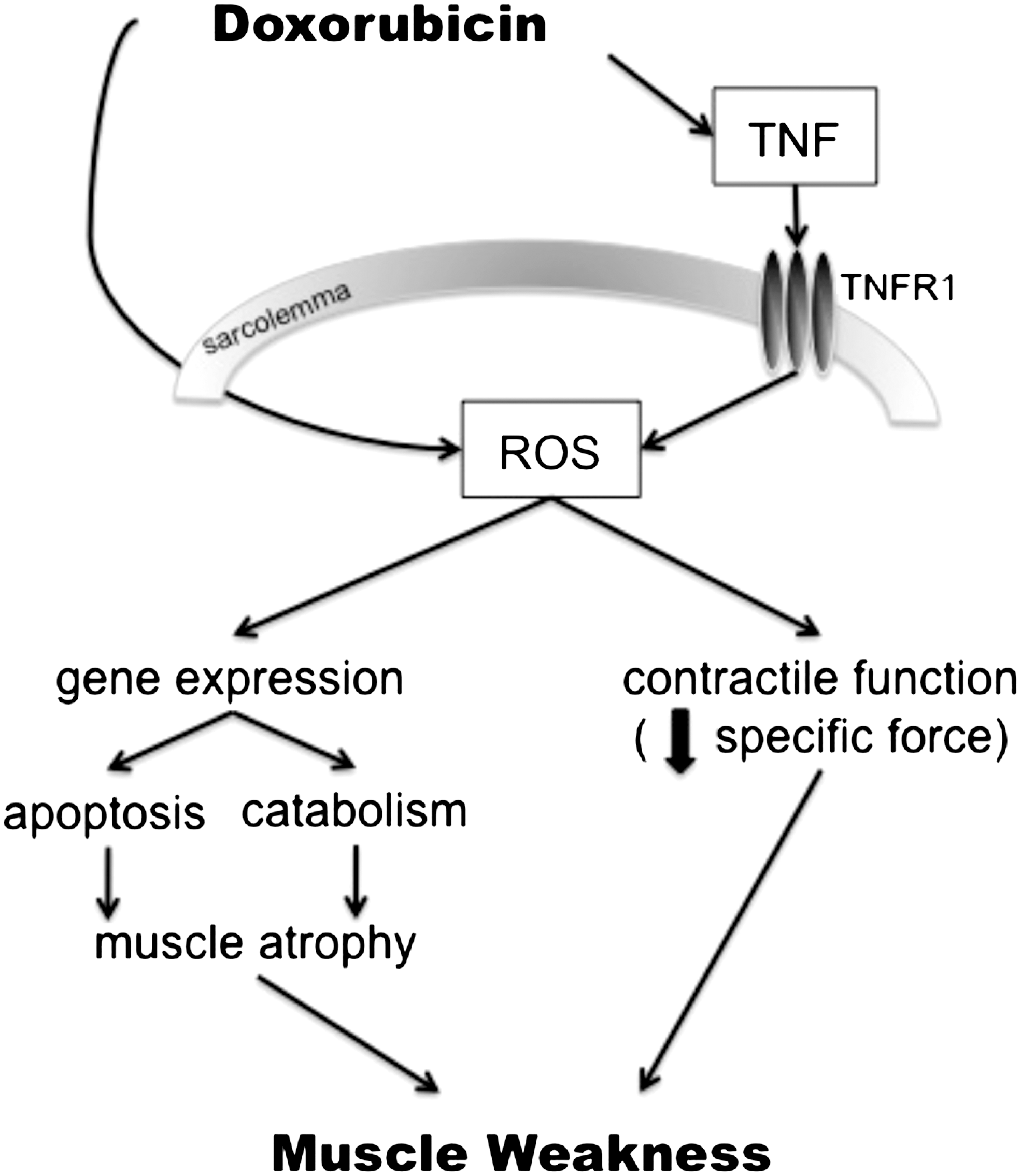
How doxorubicin effects skeletal muscle function is an emerging field. Based on the cardiac literature and current pool of data, the expected underlying mechanism is oxidative stress, occurring via a two-fold pathway (Fig. 4). Doxorubicin can directly stimulate ROS production through redox cycling, or indirectly via TNF-signaling. These two mediators, ROS and TNF, can then lead to the negative effects of doxorubicin on skeletal muscle function. The documented weakness in cancer patients undergoing chemotherapy is a significant clinical problem, welcoming studies for interventions to alleviate these severe side effects.
Future Studies
The emerging field of skeletal muscle dysfunction with chemotherapy warrants further investigation. The majority of reports in the literature document perceived fatigue through patient self-report or physician observations. Only a few studies use quantitative measures to show a decrease in muscle strength (21, 142, 230, 267). The process by which chemotherapy drugs elevate oxidants and cause weakness is also undefined. Documentation of skeletal muscle weakness, along with the mechanism involved, are required.
Translational studies are needed to investigate interventions that would prevent the debilitating weakness and fatigue with chemotherapy. The majority of chemotherapy drugs administered cause oxidative stress in noncancerous tissues, making antioxidant interventions attractive. A recent study published in this journal showed that a cysteine-rich protein diet increased muscle strength (hand-grip force) and quality of life in patients undergoing chemotherapy (245). Control comparisons were made with patients receiving an alternative protein diet that was equal in protein content with only minimal quantities of cysteine, indicating a possible antioxidant effect. Cysteine availability is vital in maintaining adequate glutathione levels, an important cellular antioxidant. Cysteine is the rate-limiting step in glutathione synthesis, an amino acid that can replenish loss of glutathione caused by doxorubicin (4, 106, 113). Rodent studies utilizing the antioxidant NAC also support antioxidant therapy. Pretreatment with NAC prevented loss of body weight and cardiotoxicity caused by doxorubicin (61, 97). NAC could also benefit skeletal muscle dysfunction caused by chemotherapy. In healthy individuals, NAC promotes endurance, improving exercise performance, and slowing the rate of fatigue (136, 137, 148, 199). A few studies published in the early 1980s assessed NAC protection against doxorubicin-induced cardiomyopathy in patients. Dresdale and associates administered NAC to disease-free cancer patients with documented doxorubicin-induced cardiomyopathy (63). NAC did not reverse the abnormal cardiac function in patients, administered over 2 years following chemotherapy. The acute and chronic cardiotoxicity caused by doxorubicin, evident by an increase in tubular area and mitochondrial damage, was not protected with NAC (172, 249). These early studies did not address the timing or dose dependency of NAC, necessitating further studies of the drug, possibly with interventions given pre-chemotherapy exposure.
NAC is not the only antioxidant available to use as a research tool. Further research investigating antioxidants already approved for human use could provide relief for cancer patients from debilitating muscle weakness. An emerging field is developing that requires more systematic testing. As shown in Table 1, patients report fatigue while exposed to multiple different chemotherapeutic agents within the anthracycline class. Fatigue and weakness are common side effects in cancer treatment. These debilitating side effects not only compromise quality of life in patients, but also limit the effectiveness of the treatment. Studies investigating the mechanistic link between chemotherapy-induced oxidative stress and muscle dysfunction lay the groundwork for the development of novel therapies that can lead to improved quality of life and increased physical activity in patients.
Footnotes
Acknowledgments
The authors thank Erin Wolf for editorial assistance. Our work in this area is supported by the National Institutes of Health Grant R01CA139843 (to D.K. St. Clair) and by the American Heart Association Predoctoral Fellowship 09PRE2020088 (to L.A.A. Gilliam).