
Abstract
Introduction: Canonical View of Sarcopenia and Clinical Ramifications
“
Unfortunately, health issues related to sarcopenia will continue to mount as U.S. and global populations are growing older. By the year 2040 up to 15 million Americans will be age 85 or older (117). Indeed, the number of persons aged >65 years is expected to double from approximately 35 million in 2000 to an estimated 71 million in 2030. Correspondingly, the population aged >80 years is expected to increase from 9.3 million in 2000 to 19.5 million in 2030 (Centers for Disease Control, MMWR, 2003).
Sarcopenia is traditionally defined as a degenerative loss of muscle mass and strength. Muscle atrophy is considered a central part of an age-related process of remodeling in skeletal muscle, whereby changes in muscle architecture occur. These include (a) loss in the number of muscle fibers via apoptosis and necrosis, (b) reduction in cross-sectional area of remaining muscle fibers, and (c) accumulation of fibrotic collagen (18, 102). Sarcopenia appears to be progressive, as beyond age 50 muscle mass declines at an average rate of 1%–2% per year, a 30%–40% reduction in both skeletal muscle mass, and fiber number is typical by age 75 (27, 55, 98, 102). Further, atrophy and loss of muscle fibers preferentially affects fast-twitch or type II muscle fibers. Contractile force generation erodes with aging due to the product of fiber atrophy coupled with a diminishment of muscle quality, or active tension expressed per cross-sectional area (99). Remodeling and fibrosis are thought to play a substantial role in the impairment of muscle quality in elderly populations. When muscle atrophy is coupled with a reduction in muscle quality, joint torque production by skeletal muscles through skeletal lever systems is lessened over 30% by age 75, and in excess of 50% by age 85 (102). Muscle atrophy has been consistently documented in the gerontology literature across human and gerontological models. In humans, fiber cross-sectional area of vastus lateralis (VL), a common target of sarcopenia studies, was up to 50% lower in septuagenarians than in young adults (84, 128). A ≥30% decrease in muscle mass by age 75 has been a common occurrence in sarcopenia studies (62).
The etiology of sarcopenia in human populations is considered to be multifactorial and includes (a) endocrine alterations, (b) loss of alpha motorneurons, (c) repeated damage/repair cycling, and (d) reduction in physical activity (97). Endocrine-related changes with aging include impaired release of androgens, growth hormone, thyroxine, elevated cortisol, and increased insulin resistance (33, 59, 114). Insufficiency of androgens and growth hormone/insulin-like growth factor-1 (IGF-1) signaling exacerbate wasting and impeded repair and growth (33, 34). In addition, poor nutritional status exacerbates skeletal muscle wasting and weakness in the elderly (20).
Progressive muscle wasting and remodeling has also been demonstrated in rodent and nonhuman primates. Gastrocnemius mass decreased by 23% between 6- and 27-month-old Fischer-344 rats (105). The Fischer-344X Brown Norway (FBN) hybrid rat exhibits a large reduction in muscle mass (−45%), fiber cross-sectional area (−30%), and muscle fiber number (−27%) between 18 and 38 months (77, 105). Muscles in older FBN rats also displayed a more heterogeneous muscle morphology, characterized by fibrosis and fat infiltration (105). In C57BL/6 mice, skeletal muscle mass also decreased with aging, with percent atrophy ranging from 15% to over 30% (10). Thus, the relative progress of sarcopenia in old rodent models appears to be comparable with aging human subjects (21).
Skeletal muscles in aging populations also are susceptible to injury as a result of repetitive eccentric contractions. Remodeling is a likely explanation for age-associated weakness and enhanced risk of damage induced as muscle fiber size diminishes, and accumulation of connective tissue and fat increases. Encroaching fibrosis increases internal work of the muscle and suppresses muscle quality (98). Therefore, mechanistic approaches designed to unravel the mysteries of sarcopenia must focus on the process of skeletal muscle remodeling, in addition to atrophy. A number of molecular and cellular candidates have been linked to sarcopenia and remodeling, including IGF-1 and its splice variant mechano-growth factor (MGF) (38), disrupted mechanotransduction (94), alterations in satellite cell signaling (39, 98), mitochondrial dysfunction (53), oxidative stress (118), apoptosis (68), autophagy (126), insulin resistance (121), and dysregulation of the ubiquitin/proteosome pathway (5). Satellite cells as pluripotent nuclei may differentiate into adipocytes or fibroblasts, particularly with advancing age or disease (98). In addition, underlying mechanisms contributing to fibrosis and remodeling may include elevation of inflammatory cytokines (22, 96).
Based upon cell signaling commonalities, in this review we will address the concept that a significant portion of age-related sarcopenia may be related to chronic inactivity and underlying chronic disease.
Living in a Box
The human genome supported a hunter-gatherer lifestyle and physiology 40,000 years ago and predominates our healthy phenotype, designed to cover large distances in acquiring and transporting food and water. Thus, it is designed to facilitate physical activity throughout the lifespan. Postmodern, Westernized societies from Japan to New York to Texas have become increasingly sedentary over the last century, “living in a box” at home, commuting, and in the office. Indeed, it has been argued that inactivity encompasses a discrete pathology that alters the expression of critical genes involved in pro-inflammatory signaling (14, 15). As a consequence, inactivity induces a phenotype that promotes the etiology and prevalence of 25 chronic diseases, including coronary artery disease, hypertension, type II diabetes, obesity, cancer, and Alzheimer's (14), all affected by pro-inflammatory signaling. Indeed, inactivity elevates risk of coronary artery disease (+45%), stroke (+60%), hypertension (+30%), type II diabetes (+50%), breast cancer (+31%), colon cancer (+41%), and osteoporosis (+59%), while contributing to 340,000 premature deaths, at a cost of $150 billion per year (15). Importantly, the chronic diseases above may also elicit muscle wasting. Indeed, emerging evidence indicates that aging, inactivity, and chronic diseases may regulate an overlapping set of genes and proteins involved in oxidative stress, inflammatory signaling, and stress protection (15, 16, 22, 95).
Although sarcopenia has been reported as a commonality among the aged since the Greek civilizations, the vast majority of inductive scientific studies involving human and mammalian subjects have been collected with sedentary, aging populations serving as the control group. Therefore, our understanding of true aging in mammalian skeletal muscle has been, unfortunately, limited by the focus on inactive human and caged animal models fed ad libitum. Logically, it could be argued that many mammalian studies involving rodents, primates, and humans have truly been evaluating the combination of aging and chronic inactivity. Further, the incidence of numerous pathologies that can elicit muscle wasting and/or weakness increases with age (37). These include insulin resistance, hypertension, coronary artery disease, peripheral vascular disease, infection, chronic obstructive pulmonary disease (COPD), and cancer that impair muscle function, many of which elicit muscle wasting, possibly distinct from the true aging process.
In addition to aging and chronic disease, disuse and inactivity in young or old animals also elicit substantial muscle atrophy (1, 56, 57). Disuse or mechanical unloading models are characterized by skeletal muscle atrophy and susceptibility to damage (57). Mounting evidence indicates that oxidative stress, NF-kappaB (NF-κB) activation, and impaired stress response are important driving mechanisms behind muscle wasting (1, 56, 57, 101). In addition, translocation of important signaling proteins such as neuronal nitric oxide synthase (nNOS), from the subsarcolemmal cytoskeleton, may elicit muscle wasting during disuse and Duchenne muscular dystrophy, possibly via activation of ubiquitin ligases (111, and [Kim, et al., unpublished observation]). In addition, there is accumulating evidence that unloading-induced myopathy is redox sensitive (1, 28, 56, 57, and [Kim, et al., unpublished observation]).
Skeletal muscles that experience mechanical unloading are also susceptible to damage upon reloading (57). Interestingly, many of the signaling pathways (insulin/IGF-1, nNOS, inflammatory cytokines, NAD(P)H oxidase, NF-κB activation, and apoptosis) contributing to muscle dysfunction during disuse and mechanical unloading are also shared with long-term physical inactivity (13), aging (94, 105, 106), and muscular dystrophies (36, 124). However, lifelong inactivity models will provide a more thorough understanding of muscle remodeling during aging than those of extreme disuse (casting, hindlimb unloading, denervation, and mechanical ventilation), as extreme disuse results in increased fast-twitch myosin isoforms and fiber-type, whereas aging is characterized by a shift from type I to type II fibers (103).
Upregulation of pro-inflammatory genes in aging skeletal muscle have been associated with sarcopenia and impaired muscle function (40). There is now substantial evidence that oxidative stress, mitochondrial function, and pro-inflammatory signaling are also central to insulin resistance (8). Mitochondria are an important source of hydroperoxides, which in turn interfere with insulin receptor substrate-1 (IRS-1) phosphorylation, and subsequent translocation of glucose transporter type 4 (GLUT4) receptors to the sarcolemma (8, 12). Disuse, sedentary lifestyle, and aging all increase the likelihood of insulin resistance, particularly as oxidative stress and pro-inflammatory signaling rise (7). Insulin resistance may exacerbate endothelial dysfunction and exert positive feedback on metabolism, thus elevating oxidative stress (116). Moreover, commonly used medications to increase insulin sensitivity such as angiotensin II receptor blockers, angiotensin-converting enzyme inhibitors, and peroxisome proliferator-activated receptor (PPAR) agonists reduce pro-oxidant genes such as Nox2 (NAD(P)H oxidase) in phagocytic and nonphagocytic cells (115). Therefore, anti-inflammatory therapy may be efficacious in attenuating sarcopenia in part by reducing insulin resistance.
Chronic Inactivity and Aging Skeletal Muscle
If many of the same genes and proteins expressed in disuse and chronic diseases (e.g., type II diabetes and cardiovascular disease) are also expressed with advancing age (13, 25), it is likely that at an interaction between aging and disuse exists. Elevated leakage of reactive oxygen species (ROS) from mitochondria, for example, is commonalities for disuse and aging (80, 103). Recent investigations that have combined aging and extreme disuse indicate that tumor protein53 (p53) and mitochondrial B-cell lymphoma 2 (Bcl-2) pathway signaling produce nonadditive effects (110). Interestingly, muscle atrophy with disuse or mechanical unloading is stimulated by increased oxidative stress and impairment of stress proteins, including heat-shock protein 70 (HSP70) and antioxidant enzymes (e.g., catalase and glutathione peroxidase) (101). Van Remmen and colleagues demonstrated that knockout of the copper, zinc isoform of superoxide dismutase (Cu,ZnSOD or SOD1) gene increased oxidative stress, while reducing muscles mass, and increasing damage in a manner mimicking accelerated sarcopenia (82). In addition, SOD1 knockouts displayed diminished contractile force, which was exacerbated with advancing aging (47, 82). It is uncertain whether short-term unloading or disuse is influence by inflammatory cytokines reported to be important in myopathy with muscular dystrophies, cachexia (COPD, cancer, chronic heart failure, and diabetes), and aging models of muscle wasting (69, 101). However, chronic inactivity may indeed elevate circulating and tissue-level inflammatory cytokines.
Underlying illness or pathology may exacerbate age-associated sarcopenia or could play a contributory role to muscle wasting associated with the aging process. The interactive nature of underlying disease and aging, particularly when coupled with chronic inactivity, is possible because of shared pro-inflammatory pathways. Indeed, Chung's Inflammatory Hypothesis of Aging, first articulated a decade ago (22), postulated that increasing loss of homeostasis at the tissue level was a function of increased pro-inflammatory signaling. Pro-inflammatory mediators proposed included inducible nitric oxide synthase (iNOS), NAD(P)H oxidase, pro-inflammatory cytokines (e.g., tumor necrosis factor-alpha [TNF-α] and interleukin-1beta), and NF-κB (22, 73, 75). In addition, caloric restriction (CR) reversed age-related increases in pro-inflammatory mediators in the kidney, heart, and brain (22). TNF-α contributes to IRS-1 phosphorylation and insulin resistance, possibly via AMP-activated protein kinase (AMPK) and p38 mitogen-activated protein kinase signaling (24, 35, 129). Chronic inflammation also increased tissue damage, and has been noted with disuse and aging (71).
Chronic elevation of pro-inflammatory as a purveyor of skeletal muscle wasting has significant global importance, as upregulation of inflammatory proteins such as TNF-α, iNOS, and NF-κB is reflective not only aging and disuse, but also disease and cachexia (2, 19, 37, 56, 57, 71 119). For example, in a series of experiments, Reid and colleagues demonstrated that elevation of TNF-α lead to mitochondrial dysfunction as well as exacerbating release of ROS (63, 64, 81). Further, TNF-α stimulated ubiquitin-driven proteolysis via Forkhead box protein O4 (FoxO4) and impaired contractile function in skeletal muscle (63, 64, 81).
While the etiology of cachexia has been categorized as metabolic and inflammatory in nature (96), increasing recognition of shared pathways between bedrest (disuse) and sarcopenia exists (34). Skeletal muscle dysfunction and weakness during chronic heart failure has also been attributed to TNF-α, NF-κB, iNOS, and oxidative stress (3, 42). In addition, recent data indicate that sepsis causes insulin resistance via iNOS-induced nitration of IRS-1 (89). Indeed, disuse, cachexia, and sarcopenia can all be argued as metabolic disorders where inflammatory signaling, mitochondrial dysfunction function, endocrine regulation, and protein turnover are shared (34), possibly distinguished by FoxO isoform activation and alterations in mechanotransduction (86).
Support for pro-apoptotic signaling as a central mediator of muscle wasting can be found in a host of pathologic conditions, including chronic heart failure, mechanical ventilation, muscle denervation, COPD, and muscular dystrophy (2, 19, 74, 100, 104, 114). Oxidative stress, disruption of Ca2+ homeostasis, and insufficient stress response can activate pro-apoptotic signaling (104, 105), a consistent outcome of aging, inactivity, muscular dystrophy, as well as wasting observed with chronic disease (104). For example, activation of apoptosis with disuse is greatest in aging skeletal muscle (104), such that a ≥25% decline in skeletal muscle mass is correlated with elevated terminal deoxynucleotidyl transferase-mediated dUTP Nick End Labeling (TUNEL)–positive nuclei and DNA fragmentation.
Apoptosis is accomplished in skeletal muscle via caspase-dependent and independent signaling (6). Pro-apoptotic signaling (e.g., caspase-3 activation) may contribute to sarcopenia not only by reducing the number of myonuclei and satellite cells, but also by increasing proteolysis (6). There are 3 caspase-dependent pathways: the mitochondrial Bcl-2 pathway dependent on caspase-9 activation, the caspase-8-dependent fas ligand/cytokine pathway, and the Ca2+/endoplasmic reticulum (ER) stress pathway that activates caspase-12. Caspase-3 cleavage serves as an integrating center for the caspase-8, -9, and -12, pathways that ultimately induce caspase-3 activation clearly respond to aging (67, 68). Cleavage of caspase-3 is elevated with aging in limb muscles, associated with a 35% reduction in mean fiber cross-sectional area (105). Consistent with the “constant domain” model for myonuclei, where each myonuclei governs its own cellular volume or domain (4), apoptosis and removal of myonuclei and satellite cells in a postmitotic tissue-like skeletal muscle can result in fiber atrophy as well as fiber loss (4, 74).
Leeuwenburgh and colleagues suggested that mitochondrial damage and oxidative stress contribute to apoptosis and sarcopenia (46). Pro-apoptotic Bcl-2-associated X protein (Bax) was elevated with aging, whereas anti-apoptotic Bcl-2 protein expression declined in fast and slow-twitch skeletal muscle (105). Mitochondrial-dependent and caspase-independent signaling in skeletal muscle was activated as well. Translocation of caspase-independent endonuclease G (EndoG) and apoptosis-inducing factor (AIF) from the mitochondria to the nucleus was also present in old, but not in young animals (31, 60, and [Kim, et al., unpublished observation]). ER stress is also elevated with aging, and associated impairment of calcium homeostasis activated caspase-12 and thus caspase-3 (54, 92). ER stress may further promote cytochrome c release from the mitochondria (27). Alway and colleagues reported that aging and disuse may act in an interactive manner, with noncaspase-dependent apoptosis (AIF) amplified with aging (104).
Receptor-mediated signaling, including TNF-α, induces apoptosis by the activation of Fas-associated death domain receptors (FADD), caspase-8, and thus caspase-3 activation (90). TNF-α-driven apoptosis is believed to be critical in atrophy. Indeed, Degens and Alway proposed that low-grade inflammation is the primary contributor to skeletal muscle wasting with aging and chronic disease (25, 26). While human data remain limited, an age-related increase in apoptosis, coupled with a 60% decrease muscle fiber amounts, has been documented (109, 125). TUNEL-positive nuclei in the VL muscle stained for DNA fragmentation were 87% higher compared with muscle in young adults (125).
It is probable that some fiber loss with aging occurs in relation to α-motor neuron denervation or mitochondrial dysfunction, whereby both mechanisms may lead to elevated apoptosis and muscle dysfunction (18, 53, 104). Elevated oxidative stress results in early degeneration of skeletal muscle, producing a muscular dystrophy or amyotrophic lateral sclerosis-like phenotype (94). For example, knockout of the Cu,ZnSOD results in significant damage and atrophy of skeletal muscle reminiscent of myopathy (82). In summary, distinguishing the effects of aging on skeletal muscle function from underlying chronic inactivity and chronic disease may be challenging given their shared pro-inflammatory signaling, (114).
Exercise as a Sarcopenia Intervention in Aging Populations
Conversely, long-term exercise training appears to reduce the pro-inflammatory phenotype and reduce apoptosis (105, 106). While the mechanisms remain to be elucidated, exercise training reduces iNOS and oxidative stress, while increasing stress protective HSP70 in skeletal muscles in heart failure, coronary ischemia, and aging models (37, 56, 106). In combating sarcopenia, short- and long-term studies have largely focused on the benefits of resistive training or moderate to higher intensity level weight-bearing exercises. Indeed, the human literature suggests that resistive training is the most consistent method to combat sarcopenia in an aging population with adequate protein intake. In 10-week to 12-month intervention studies in rat and mouse models, endurance training appears to positively impact apoptosis, muscle mass, and muscle fiber cross-sectional area. Alway and colleagues have shown that treadmill exercise training elevates anti-apoptotic markers, including Bcl-2, X-chromosome-linked inhibitor of apoptosis, and apoptosis repressor with a caspase recruitment domain protein (ARC) (104). In concert, treadmill exercise reduced DNA fragmentation and caspase-3 activation (104, 105). Recent findings from Rasmussen and colleagues indicate the importance of adequate protein intake and mitigation of age-related changes in micro-RNA involved in translation repression and gene silencing (29, 30).
Sirtuin and PPAR-γ coactivator 1-alpha (PGC-1α) signaling has recently been linked to improving insulin signaling and slowing of the aging phenotype. Further, SIRT (Sirtuin) family signaling may be inducible with exercise. New data indicate that exercise training normalizes silent information regulator-6 (SIRT-6) in skeletal muscles from old rats (52). In addition, NAD-dependent deacetylase sirtuin-1 (SIRT-1) levels were upregulated in old rats by a single bout of exercise, and corresponded to an improvement in phosphorylation of IRS-1, tyrosine protein phosphatase nonreceptor type-1, and insulin signaling (87). Upregulation of SIRT family proteins may in turn elevate PGC-1α and thus may reduce oxidative stress and apoptosis (87). For example, a recent study (85) reported that knockout of NAD-dependent deacetylase sirtuin-3 (SIRT-3) prevented exercise-induced upregulation of AMPK and PGC-1α. Conversely, high fat meals reduced SIRT-3 and AMPK phosphorylation, whereas CR or fasting elevated SIRT-3 and p-AMPK. In addition, Wenz et al. (122) recently showed that overexpression of PGC-1α reduced markers of insulin resistance and sarcopenia.
When begun in middle age or later, treadmill exercise training appears to have a modest effect on reduction in muscle mass and fiber cross-sectional area (49, 105). McArdle and Jackson conducted a series of studies that demonstrated the importance of HSP70 in reducing oxidative stress and sarcopenia in aging muscles (5, 71). Starnes et al. also demonstrated that HSP70 was inducible with treadmill exercise training is retained in old age (108). While significant protection against muscle fiber atrophy has been noted in some studies, muscle mass and strength loss may persist even with maintenance of HSP70 (49).
Tarpenning et al. (112) concluded that endurance training reduced age-related decline in muscle strength and sarcopenia in human subjects. However, exercise intensity may be a critical factor when combating sarcopenia in the elderly (78). Indeed, Evans and colleagues showed that resistive training was most effective in alleviating muscle atrophy when used in aging populations, and this is still the consensus today (32, 78). Interestingly, it is thought that anti-inflammatory drugs may promote hypertrophy and strength gains in response to resistance training in older populations (40). In contrast, chronic use of anti-inflammatories blunt hypertrophy in response to resistive training in young populations (40). Additional research shows that weight training in combination with supplementation of branch-chain amino acids may be more efficacious in older populations at risk of sarcopenia than resistive training alone (99). In summary, while exercise training in an older population appears to partially reduce apoptosis and augment stress response proteins (e.g., HSP70 and SIRT-1), functional effectiveness of these interventions has been equivocal with lower-intensity exercise, unless begun at a young age.
Lifelong voluntary exercise in ameliorating sarcopenia
While exercise interventions in aging populations yield positive, modest results in alleviating sarcopenia, emerging data suggest that lifelong voluntary exercise provides substantially greater protection against age-associated apoptosis, muscle fiber atrophy, and remodeling (51, and [Kim, et al., unpublished observation]). Further, some of the cell signaling pathways modulated by lifelong activity and CR may be shared (55). Lifelong wheel running significantly protected against age-associated reduction in fiber cross-sectional area (51) (Figs. 1 and 2). In addition, lifelong wheel running also reduced remodeling, as visualized by attenuation of accumulation of extracellular space and collagen I (51). Wheel running also ameliorated age-associated elevation of oxidative stress. However, protection against fiber atrophy and remodeling was not related to any changes in either Cu,ZnSOD or manganese isoform of superoxide dismutase (MnSOD) protein expression. While IGF-1 levels were lowest in old, sedentary rats fed ad libitum, the most effective protection against age-related suppression of IGF-1 was provided by wheel running (Fig. 3). This is in agreement with lifelong protection of skeletal muscle mass by IGF-1 studies by Rosenthal and colleagues using mice that overexpress the IGF-1 gene (120). In addition, Goldspink presented evidence that MGF, the splice variant of IGF-1 specific for skeletal muscle, is critical in satellite cell activation and preservation of muscle mass with aging (38).
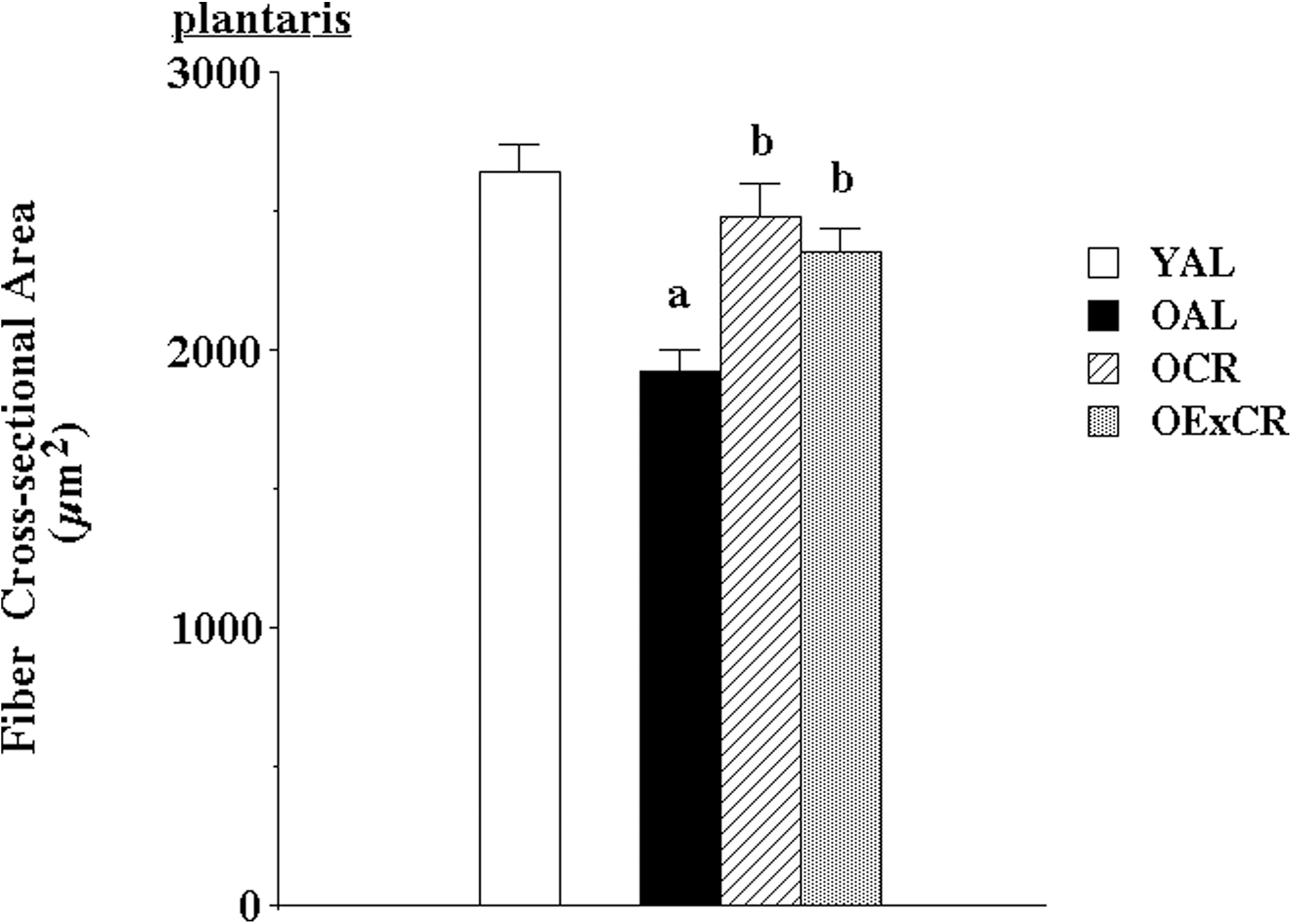


We recently demonstrated that lifelong wheel running ameliorates age-related elevation and translocation of pro-apoptotic EndoG to the nucleus (Kim, et al., unpublished observation). In contrast, wheel running had little effect on elevation of AIF. Further, lifelong wheel running resulted in higher levels of HSP25 and phosphorylation of HSP25 at Ser82 in the plantaris muscle of old rats that participate in lifelong, voluntary exercise. HSP25 may protect against apoptosis via death receptor pathways. However lifelong wheel running did not have a protective effect for HSP70. Lack of HSP70 response may be related to low-intensity exercise levels. Interestingly, Murlasits et al. reported that resistive training upregulated HSP25, but not HSP70 in rats (83). In contrast, Jackson and colleagues demonstrated that lifelong overexpression of HSP70 reduces muscle damage and myopathy (17, 70). Therefore, lifelong upregulation of HSPs may be a critical mechanism in reducing age-associated sarcopenia via HSP25 and HSP70.
Lifelong wheel running also reduced apoptosis and improved autophagy signaling (microtubule-associated protein 1 light chain 3, lysosomal membrane protein-1) in old Fischer-344 rats (126). In the heart long-term wheel running reduced apoptosis mediators, including cell-cycle-checkpoint kinase 2 (Chk2) and p53 via Chk2, p53, IGF-1, and endothelial nitric oxide synthase (123). Recently, Leick et al. (61) demonstrated that PGC-1α was necessary to retain exercise protection in skeletal muscle against (a) downregulation of MnSOD and citrate synthase and (b) attenuation of elevated Bcl-2 pathway signaling. Reynolds et al. (93) reported that wheel running might protect the Akt/mTOR (mammalian target of rapamycin) pathway of protein synthesis against age-related decline. In summary, lifelong exercise may limit sarcopenia and apoptosis, mediated by preservation of key stress proteins, including HSP25, IGF-1, and PGC-1α. While the efficacy of and mechanisms underlying protection by lifelong, habitual exercise are beginning to emerge, exercise may be very effective in reducing the risks of sarcopenia regardless of intensity levels when practiced over significant portions of the lifespan.
CR Delays Skeletal Muscle Dysfunction
Prolonged, moderate CR of 30%–40% has been the most consistent rodent and primate model used to increase longevity (67). Moderate (30%–40%) and mild (8%–10%) CR have also been efficacious in preserving skeletal muscle function (51, 67, and [Kim, et al., unpublished observation]). Although moderate CR may initially elicit a small reduction in muscle mass, further reduction in mass and fiber CSA with advancing age are substantially attenuated by CR (27, 76, and [Kim, et al., unpublished observation]). Further, CR may also alleviate the age-related decline in contractile function (9, 88). Interestingly, CR attenuates pro-inflammatory genes associated with oxidative stress in muscles from old and young animals (23). Kim et al. (51) found that mild CR reduced muscle fiber atrophy and partially limited reduction in IGF-1. CR was also effective in attenuating loss in muscle fiber number, oxidative stress, and mitochondria abnormalities (67). Indeed, CR significantly reduced mitochondrial proton leak and ROS generation, lipid peroxidation, and protein damage in skeletal muscle (67) and increased the expression of genes involved in ROS scavenging (107). CR also reduced the incidence of age-related abnormalities in the electron transport chain, and accumulation of mtDNA deletions in mitochondria (9, 58). Recently, CR has been found to elevate SIRT-3/PGC-1s signaling, and thus enhancing mitochondrial function and muscle mass (85).
CR may limit sarcopenia by reducing age-associated apoptosis. Activation of caspase-3 and fragmentation of DNA were significantly reduced by CR (27, 88). Specifically, CR attenuated fas/cytokine pathway signaling, reduced age-associated elevations of TNF-α, FADD, and caspase-8 cleavage (88). Further, the ER/Ca2+ stress apoptotic pathway was also downregulated by CR with a reduction in procaspase-12 and AIF (27).
Aging Mammals in Natural Habitat and Skeletal Muscle Senescence
Unlike rodent models of aging housed in laboratory animal facilities, wild mammals typically remain highly active and in mild caloric deficit over much of their lifespan. Free-living animals are susceptible to stressors such as predators, altered environmental conditions, conflict-inflicted wounds, and infectious disease. Therefore, stress protection and antioxidant strategies are paramount in skeletal muscle integrity and function. However, there is a paucity of data that characterize the existence of a senescent phenotype in skeletal muscles of aging mammals in their natural habitat.
We initially chose mammalian species of disparate size and longevity to examine naturally occurring senescent phenotoypes. Two were diving mammals, and thus experience repeated hypoxia-reoxygenation during breath-hold over the course of their lifespan. One such species was the Weddell seal (Leptonychotes weddelli). In the Ross Sea region of Antarctica, a local population of seals has been under scientific investigation since the 1980s, resulting in a high percentage of known-aged, old (20+ years) individuals (113). We thought that these initial experiments might shed light on the influence of chronic inactivity, and ad libitum feeding on skeletal muscle senescence, including sarcopenia. Seals were captured, sedated, and biopsies taken (43). A second breath-holding mammal, the diminutive water shrew (Sorex palustris), was obtained in woodlands of Manitoba, Canada. The third species, the short-tailed terrestrial shrew (Blarina brevicauda), is also a carnivore subjected largely to cold stress and altered ambient gas partial pressures associated with underground burrows, rather than breath-holding during hunting.
In the long-lived Weddell seal, muscle fiber cross-sectional areas of longissimus dorsi and pectoralis major showed either an increase or no change compared with muscle samples from young adults (43) (Table 1). Collagen content increased by 115% in longissimus dorsi and 65% in pectoralis. In addition, a shift of the collagen isoform profile from type III to the stiffer type I also occurred with age, with an 79% increase of the collagen type I/III ratio in pectoralis and 49% in longissimus dorsi (Fig. 4). In older adult water and terrestrial shrews, muscle fiber cross-sectional areas increased, rather than losing size, whereas increases in extracellular space and collagen I staining were observed (44). The ratio of type I/type III collagen also was elevated with age in both species of shrews. However, the ratio of muscle fiber to collagen levels was lower in the terrestrial shrew compared with its aquatic sympatric species. Thus, aging mammals that are active in their natural habitat may exhibit fibrosis as a primary characteristic of the aging phenotype in skeletal muscle.
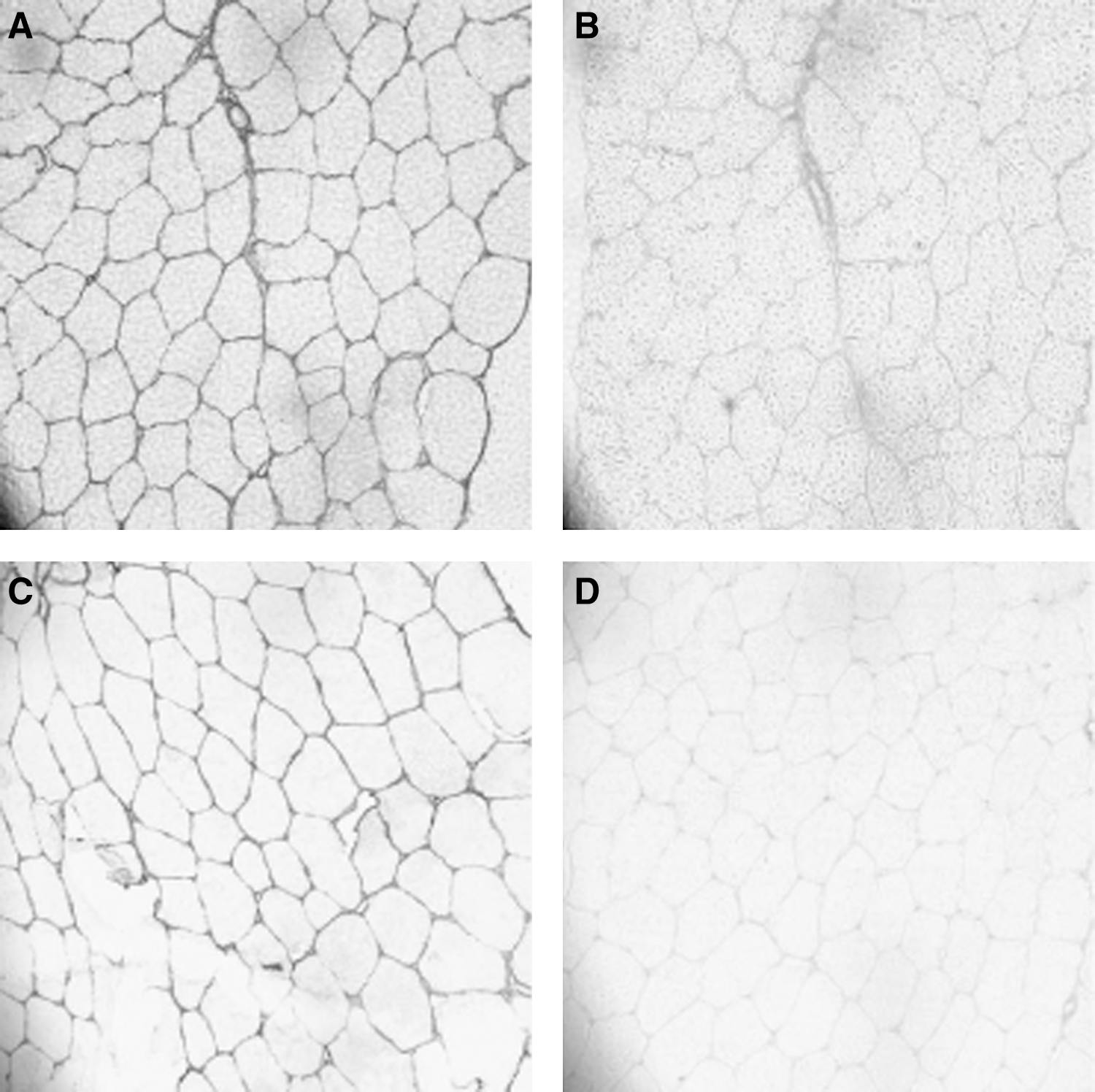
Muscle was transversely sectioned at 7–9 μm and stained with hematoxylin before analyses. A single asterisk denotes significant differences between species (α=0.05); double asterisks indicate significant differences between age classes. Values are means±standard error.
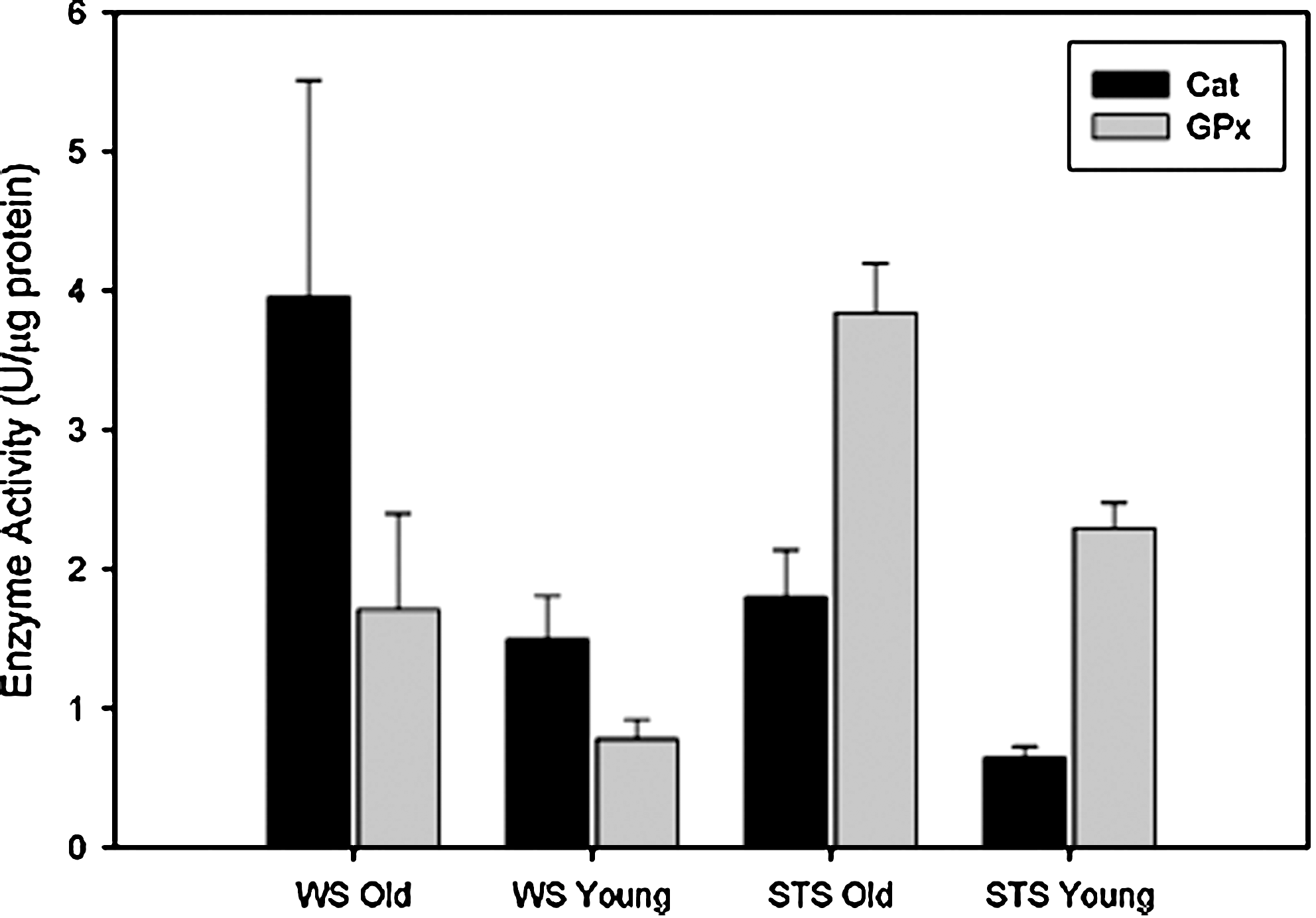
Although hydroperoxide levels increased, markers of oxidative damage (4-hydroynonenal) showed no increase with aging in any shrew species (44). There was also no increase in DNA fragmentation or TUNEL+ nuclei in old shrews (44). Interestingly, Cu,ZnSOD was dramatically upregulated with age, with particularly high levels discovered in the water shrew (60%) compared with terrestrial shrew (25%) (45). Protection against hydroperoxides was thus augmented with age. Catalase levels doubled in the old water shrews, whereas glutathione peroxidase was elevated by 120% in its terrestrial counterpart with age (Fig. 5). It is possible that the remarkable upregulation of stress protection and antioxidant enzymes in the shrews with aging may limit oxidative damage and apoptosis, reflective of lifelong activity and/or adaptation to their unique environments. In short, these data suggest that senescence in muscles of old mammals living in their natural habitats may not be characterized by atrophy and apoptosis as much as by fibrosis. Fibrosis elevates internal work of aging muscles, reducing force production and increasing susceptibility to damage. Because of the early stage of these studies and potential influence of natural selection and removal of weaker senescent animals by predators or disease, extrapolation to senior human patients is cautioned at this time.
Conclusion
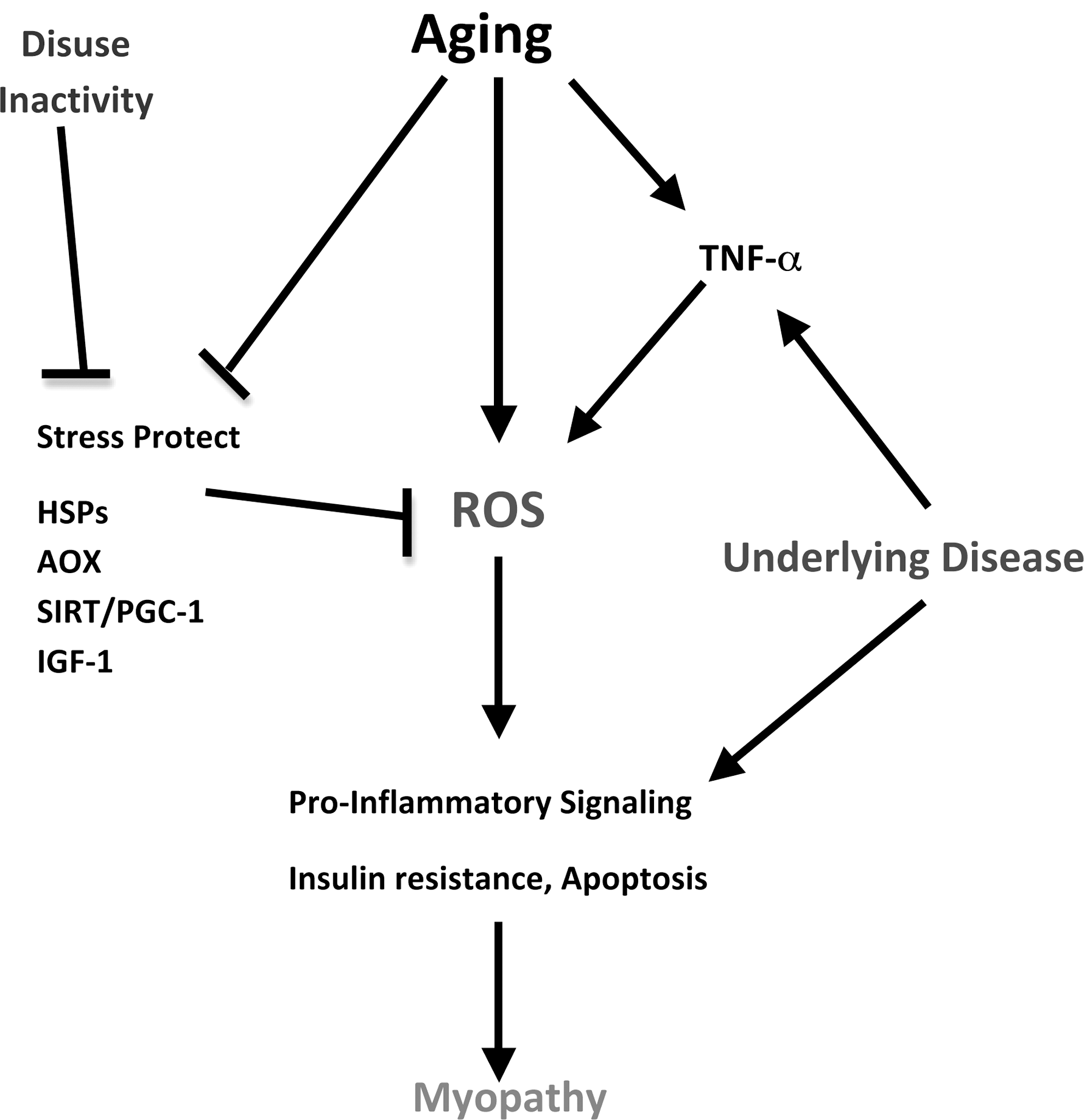
We have taken an integrative approach to re-examining the notion of understanding age-related sarcopenia in mammals by using sedentary, caged rodents. CR, short-term and lifelong exercise, human patients, and aging mammals in the natural habitat were used as models to provide a cell signaling perspective on the interactive nature of aging and chronic inactivity. Given that much of the cage-restricted rodent and human aging literature includes inactivity as part of the aging approach, the use of such subjects as control groups is cautioned. In addition to inactivity, underlying disease (e.g., cardiovascular disease and insulin resistance) may also impact the expression of the aging phenotype observed in skeletal muscle. Indeed, models that involved maintenance of lifelong activity expressed less muscle atrophy and apoptosis into old age, consistent with an ability to maintain or increase key stress-protective proteins (e.g., HSPs, IGF-1, SIRT-3, PGC-1α, Cu,ZnSOD, and catalase). The literature also supports the notion that lifelong activity is far more effective in mitigating the effects of sarcopenia than treatments commencing in middle age or beyond. A prospective integrative model is found in Figure 6.
Footnotes
Acknowledgments
This work has been supported by grants from NIH (AR054084), NSF (055185F), AHA (0555064Y, 0855158F), and The Sydney and J.L. Huffines Institute at Texas A&M University.
Author Disclosure Statement
There are no existing financial conflicts.