
Abstract
Pancreatitis: Incidence and Problem
The exocrine pancreas is composed primarily of pancreatic acinar cells, which comprise over 90% of the gland and are considered the primary site of local injury in AP. Over a decade ago, we proposed that disruption of acinar cell Ca2+ homeostasis underlies the development of pancreatic injury (167). Subsequent studies have confirmed the central role of Ca2+ in the pathogenesis of AP as induced by various precipitants, including caerulein hyperstimulation (82, 133), bile salts (78, 164), nonoxidative alcohol metabolites (fatty acid ethyl esters [FAEEs]), and fatty acids (FAs) (27, 28). Sustained, global elevations of Ca2+ induced by these diverse agents led to premature Ca2+-dependent activation of zymogen granules (ZG), vacuole formation, and cell death (82, 133).
Under conditions in which tissue antioxidant levels are low, or the production of reactive oxygen species (ROS) exceeds the normal range of antioxidant capacity, oxidative stress develops (59). Several decades ago a role for ROS was proposed in AP, based on results derived from the application of various precipitants, including infusion of oleic acid and ductal obstruction, in a canine model (139). Subsequent studies have demonstrated increased oxidative stress linked to AP, including that induced by ductal obstruction (160) and taurocholate (134), the latter study concluding that ROS were involved in mediating tissue damage but were not a trigger for development of AP per se. Further, scavenging of ROS by antioxidants did not reduce acinar cell damage in two models of AP, with only a reduction of oedema observed (150). Thus, the precise involvement of ROS in the pathogenesis of AP has remained unclear.
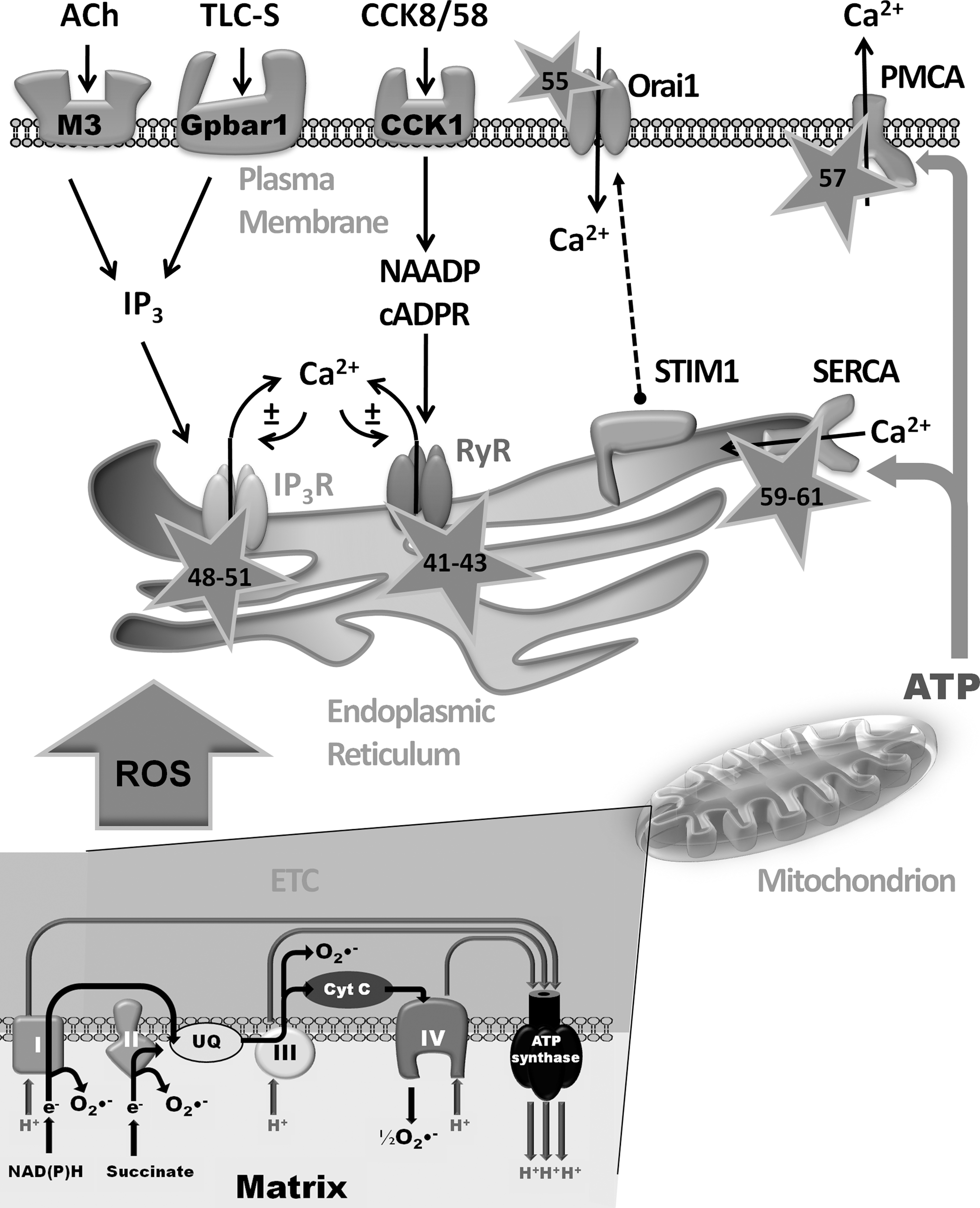
Interestingly, extensive evidence suggests that Ca2+ signaling and ROS are intimately linked (16), with many facets of Ca2+ homeostasis sensitive to the cellular redox status (Fig. 1). In view of the fundamental role that Ca2+ overload plays in the development of AP, this review will discuss the roles of Ca2+ and ROS in the pathophysiology of the exocrine pancreas, their interaction to modulate cell death pathways, and potential for therapeutic manipulation.
Ca2+ Signaling in Health and Disease
Physiological Ca2+ signaling
The pancreatic acinar cell is a polarized, nonexcitable secretory unit that displays perfectly organized morphology so that spatiotemporally defined Ca2+ signals are generated in response to appropriate physiological neuro-hormonal stimulation (Fig. 1) (121, 122). Thus, cholecystokinin (CCK) and acetylcholine (ACh) stimulate their respective membrane receptors on human (104) and rodent acinar cells (24) to cause discrete oscillatory elevations of Ca2+ that are restricted to microdomains (123), which lead to fusion of ZG membranes with the apical membrane, (41) resulting in the exocytosis of inactive digestive enzyme precursors into the pancreatic duct. Recent evidence, however, suggests that transient global Ca2+ signals may be required for physiological enzyme secretion, whereas local signals generate primarily fluid secretion (89). A combination of type 2 and 3 inositol 1, 4, 5 trisphosphate (IP3) receptors (IP3R) may be essential for Ca2+ signals linked to exocrine secretion in pancreatic acinar cells, since responses to CCK and ACh were absent in double knockout mice, but not in single knockouts for each individual IP3R subtype (44).
Physiological Ca2+ signals are generally confined to the apical pole by a buffer barrier of perigranular mitochondria. These organelles temporarily uptake Ca2+ released from the endoplasmic reticulum (ER), upregulating ATP production, and inhibiting global waves from permeating to the basolateral area (157, 165). When stimulation reaches sufficient intensity, or when mitochondria are inhibited, this barrier is overwhelmed and local repetitive Ca2+ responses in the granular area are transformed into global Ca2+ waves, underlining the complex role of mitochondria in physiological acinar cell function.
It is paramount that homeostatic mechanisms are maintained to prevent sustained global Ca2+ elevations and preserve mitochondrial function. Therefore, Ca2+ must be cleared continuously from the cytosol to restore resting state. Thus, Ca2+ is returned to the ER stores by the sarcoendoplasmic Ca2+-ATPase (SERCA) or extruded by the plasma membrane Ca2+-ATPase (PMCA), processes that consume ATP and are dependent upon fully functioning mitochondria (Fig. 1). Mitochondrial ATP production and bioenergetic regulation of the cell are influenced by Ca2+ continually leaked from the IP3R into the mitochondria (19) and the resting Ca2+ is dictated by a balance between leak, extrusion/reuptake mechanisms, and Ca2+ entry. Ca2+ must periodically be allowed into the cell to refill intracellular stores via store-operated Ca2+ entry (SOCE), which has recently been shown to occur by the interaction of the ER sensor protein stromal interaction molecule 1 (STIM1) and the plasma membrane channel Orai1, which form a complex at ribosome-free ER plasma membrane junctions in acinar cells (90) (Fig. 1).
Abnormal Ca2+ signaling
Disruption of normal Ca2+ signaling was postulated as a trigger for pancreatitis over a decade ago (167). Hyperstimulation of acinar cells with CCK-8 caused large sustained cytosolic Ca2+ elevations, premature intracellular digestive enzyme activation, and necrosis (133). Subsequently, it was shown that major precipitants of AP, such as bile salts (Fig. 1) (78, 164) and nonoxidative metabolites of ethanol (27, 28), also generated toxic elevations of Ca2+ that resulted in cellular necrosis.
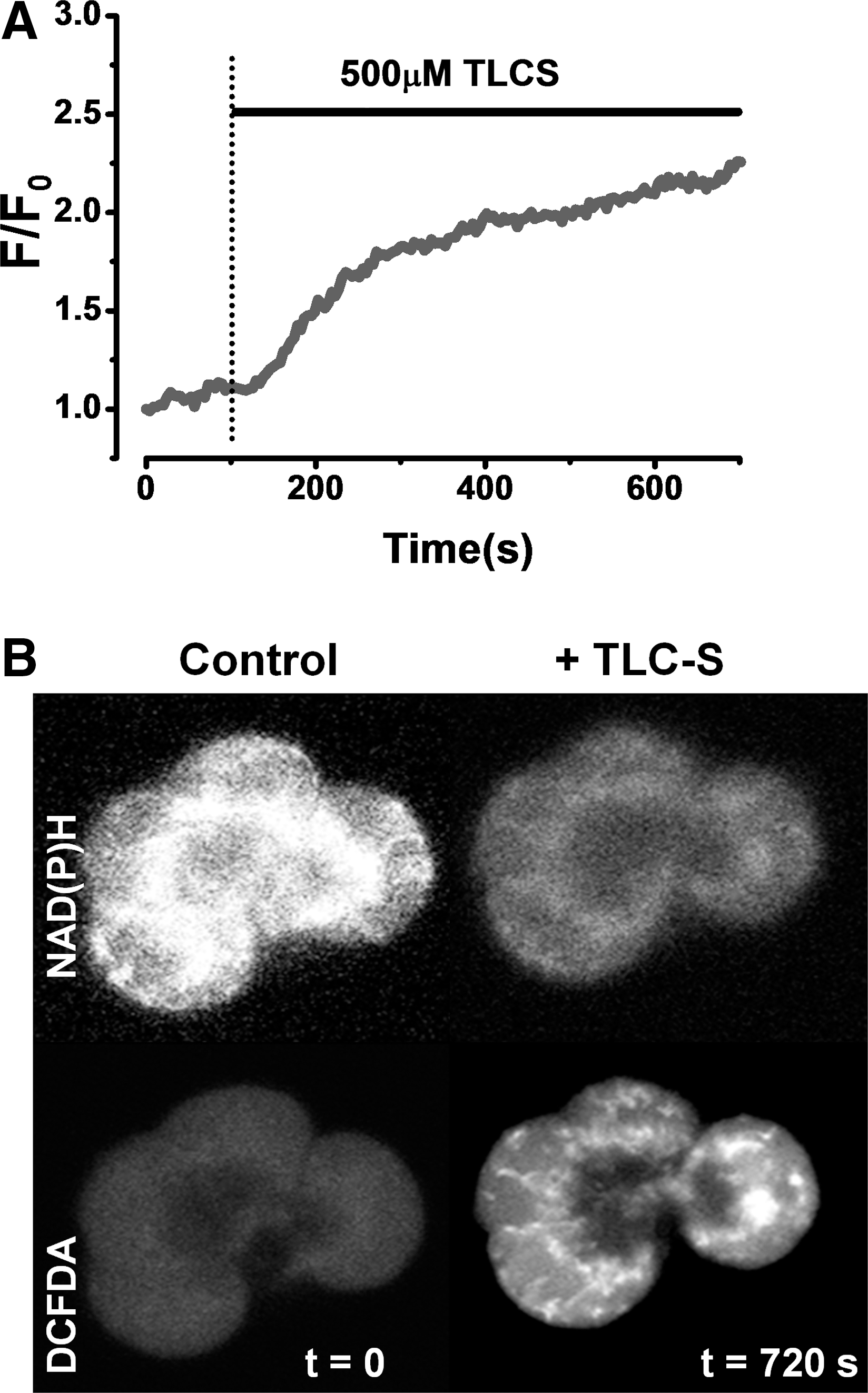
Ca2+ release from internal stores in response to these precipitants was dependent on IP3R opening (27, 46, 164). However, sustained Ca2+ entry triggered by Ca2+ store depletion was required to raise cytosolic Ca2+ to detrimental levels; abrogation of the cytosolic Ca2+ rise with intracellular Ca2+ chelators prevented deleterious changes such as trypsinogen activation, vacuolization, and necrosis (29, 78, 133). The mechanism of cellular necrosis was further defined with respect to nonoxidative ethanol metabolites, which produced a Ca2+-dependant inhibition of mitochondrial function, resulting in loss of membrane potential (ΔΨm), NAD(P)H, and ATP production (29). Experiments using mitochondrially targeted luciferase in pancreatic acinar cells have demonstrated that bile acids and nonoxidative ethanol metabolites cause a profound decrease of mitochondrial ATP, in addition to cytosolic reductions (29, 166). Recently, we have demonstrated that the bile acid taurolithocholic acid 3-sulfate (TLC-S) induced sustained cytosolic Ca2+ increases (Fig. 2) accompanied by an initial rise in NAD(P)H autofluorescence as previously described (120). However, the stimulatory effect on mitochondrial function was quickly lost in the presence of sustained cytosolic Ca2+ elevation, with NAD(P)H autofluorescence rapidly decreased, an action accompanied by a large, sustained increase in mitochondrial Ca2+ ([Ca2+]m) (14) (Fig. 2); such signals are detrimental to mitochondrial function, thereby inhibiting ATP production. A reduction in ATP output would modify IP3R function (7) and inhibit ATP-driven Ca2+ pumps, thereby impeding clearance of elevated cytosolic Ca2+ from the cell (Fig. 1) (123). In accord with this hypothesis, supply of intracellular ATP via a patch pipette abrogated the development of sustained Ca2+ elevations and consequent necrosis in response to nonoxidative ethanol metabolites (27, 29) and TLC-S (14). Evidence derived from such experiments highlights the fundamental importance of normal mitochondrial function for Ca2+ homeostasis and cell integrity under conditions of pancreatic insult (102).

Influence of ROS on Ca2+ signaling
Given the importance of restriction of cytosolic Ca2+ levels within physiological limits and discrete microdomains, the cell must finely coordinate Ca2+ entry, release, and exit mechanisms to ensure that mitochondrial dysfunction does not ensue. Studies in other cell types suggest that many of these Ca2+ homeostatic processes are sensitive to the intracellular redox environment. For example, both IP3R and ryanodine receptor (RyR) contain multiple cysteine residues that are sensitive to ROS, implying a modulatory role for free radicals on Ca2+ release channels (Fig. 1) (40, 96, 152), potentially influencing events such as deleterious intracellular activation of trypsinogen (68). Further, oxidation of thiols prevents negative regulation of the IP3R by calmodulin by inhibiting its binding to the receptor (42, 60, 175). The general effect of RyR oxidation is to increase channel activity, a process mediated by enhanced subunit assembly and inhibition of calmodulin binding, a negative regulator of the receptor (60). The sensitivity of the IP3R appears more complex, since ROS may sensitize the IP3R to activation by IP3 but inhibit channel function per se (73, 74). It has been shown that thiol oxidizing agents sensitize Ca2+-release channels in a manner sufficient to induce cytosolic oscillations in the absence of physiological stimulation (15, 100) and thus function to preserve IP3R-linked Ca2+ oscillations necessary for exocrine secretion (18). However, the situation is likely to be complex. A study in vascular smooth muscle cells has demonstrated that while exogenously applied ROS enhanced the release of IP3-mediated Ca2+, this did not occur when nonhydrolysable analogs of IP3 were used instead of IP3, potentially suggesting an additional role for ROS in the breakdown of IP3 (155).
Important recent progress has been made with respect to elucidation of the vital Ca2+ entry mechanism in pancreatic acinar cells, which involves formation of an STIM1-Orai complex (90). Under conditions of store depletion, STIM1 senses Ca2+ changes within the ER and translocates to the plasma membrane forming a functional complex with Orai1 (98). Interestingly, the Ca2+ release (IP3R) and Ca2+ entry mechanisms appear spatially distinct in the acinar cell, with ribosome-free portions of ER forming close associations with the plasma membrane (90). Recently, redox regulation of Orai1 has been reported in T-cells that may function to tune cellular Ca2+ signaling (12). This study showed that Orai1, which possesses an external cysteine residue, was sensitive to H2O2, whereas Orai3, in which this residue is absent, was insensitive. T-cells became progressively resistant to ROS after differentiation into effector cells, which correlated with an increased Orai3 expression. This might enable cells to proliferate, differentiate, and secrete cytokines when present in oxidizing environments such as the inflamed pancreas. Whether ROS sensitivity plays a role in Orai-mediated Ca2+-entry in cells of the pancreas has yet to be determined.
Despite the lack of voltage-sensitive Ca2+ channels in the pancreatic acinar cell, there is evidence to suggest that Ca2+ channel blockers may decrease the severity of experimental pancreatitis (65, 149) although postendoscopic retrograde cholangiopancreatography pancreatitis was unaffected by nifedipine treatment (125). The development of selective inhibitors of SOCE in pancreatic acinar cells, however, may provide an attractive proposition for amelioration of AP, and progress in this field is eagerly awaited.
Ca2+-ATPase pumping mechanisms that lower the concentration of cytosolic Ca2+ play a critical role in homeostasis of the pancreatic acinar cell. Their importance is underscored by the fact that Na+/Ca2+ exchangers, which have been shown to exert a homeostatic influence in other cell types, appear to be nonfunctional or absent. These energy-requiring pumps are also sensitive to the redox environment (Fig. 1). For example, H2O2 applied to pancreatic acinar cells profoundly altered the normal pattern of CCK or ACh evoked Ca2+ signals, increasing the proportion of global, sustained responses as the concentration of H2O2 increased (17), potentially via inhibition of the PMCA, thereby decreasing Ca2+ clearance from the cytosol through the plasma membrane. This effect appears linked to mitochondrial function, since mitochondrial depolarization and consequent ATP depletion correlated with a fall in PMCA activity (4). The SERCA pumps are subject to oxidative modification, possessing as many as six redox-sensitive cysteine residues (145). Current evidence suggests that mild oxidative conditions lead to oxidation of Cys674, upregulating pump activity (1), whereas prolonged exposure can cause suphonylation of the same residue and oxidation of other residues, eliciting irreversible SERCA inhibition and exacerbating Ca2+-overload (1, 49, 154). Such divergent effects highlight the enormous complexity of the yet partially understood relationships between Ca2+ signaling and ROS; fine-tuning of the levels of localized, subcellular ROS may affect Ca2+ homeostasis positively or negatively.
Control of Redox Status
ROS generation
Before discussion of the involvement of ROS in cellular toxicity, it is pertinent to briefly highlight where and how ROS are produced, and what endogenous protective mechanisms are in place to deal with their excessive formation. In this respect, it should be emphasized that ROS are both generated under physiological conditions as an integral part of the mitochondrial ATP production and exert functions as signaling molecules in their own right, rather than simply being the widely reported mediators of undesirable toxicity. Growing evidence suggests that the balance between ROS-producing and ROS-scavenging systems underpins the proper functioning of the cell.
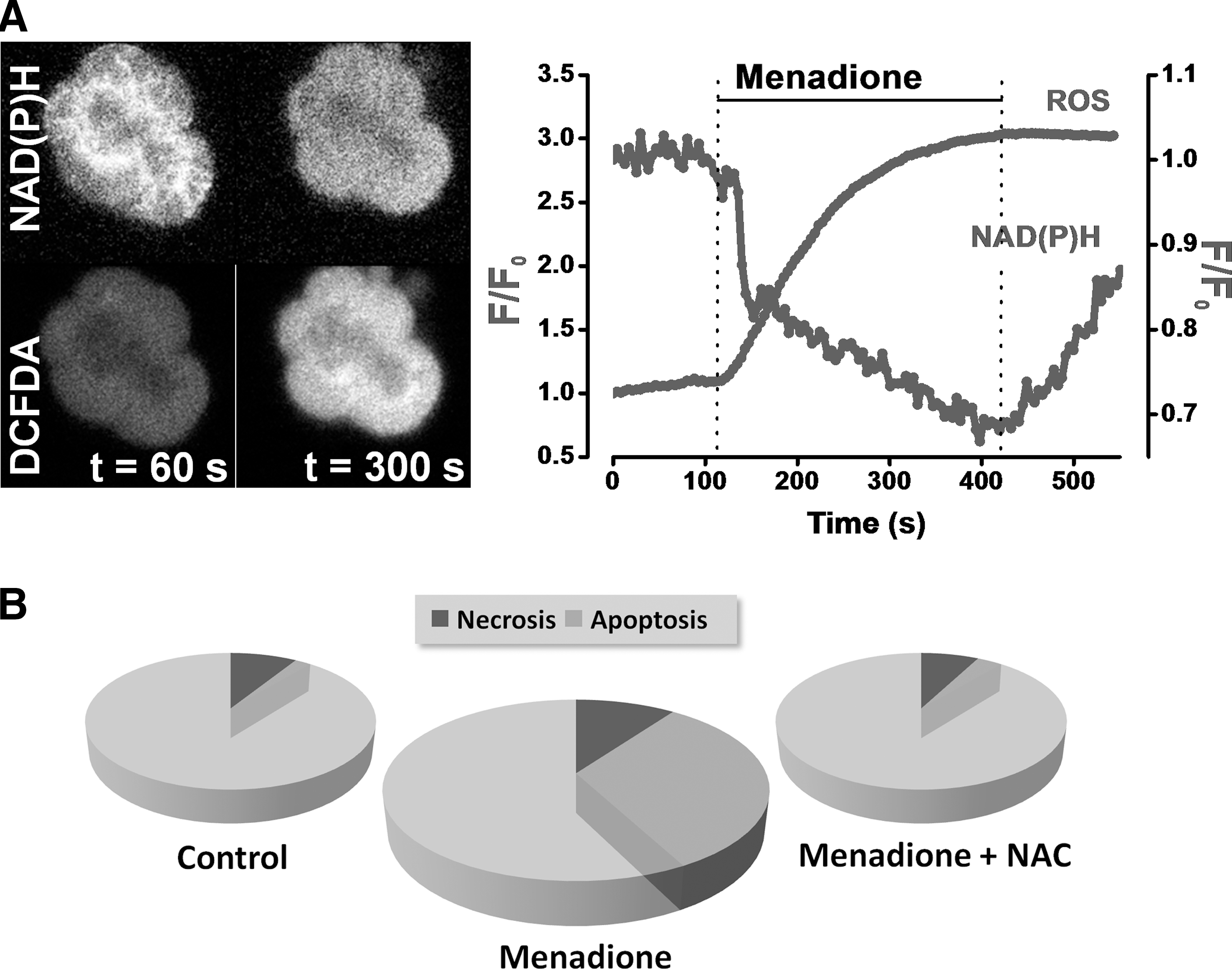
Mitochondria account for 90% of the total cellular oxygen consumption and are the principal source of physiological ROS (64). An estimated 1%–2% of all O2 consumed is linked to production of radicals. To generate ATP, mitochondria oxidize NADH and FADH2 produced by the tricarboxylic acid (TCA) cycle or via β-oxidation of FAs, in a series of reactions catalyzed by enzyme complexes I–IV located on the inner mitochondrial membrane (IMM), which constitute the electron transport chain (ETC). It is thought that 10 or more components of mitochondria produce ROS (2), but the main sources of superoxide (O2•−) are complexes I and III of the ETC (Fig. 1). Nonmitochondrial sources of ROS are perhaps less well characterized; however, they may be prodigious in their ROS production. Specific ROS generation by NADPH oxidase and myeloperoxidase are critical to the immune response and are certainly involved in the development of the systemic complications of AP. We have previously measured substantial Ca2+-independent ROS generation in freshly isolated murine pancreatic acinar cells in response to menadione (vitamin K3) (Fig. 3A) (26). Quinones such as menadione enter rapid redox cycles within the cell, consuming NAD(P)H and producing superoxide (Fig. 3A). Importantly, our findings indicated that the consequence of this acute ROS generation was promotion of acinar cell apoptosis, rather than necrosis; scavenging of radicals with N-acetyl-L-cysteine (NAC) prevented apoptosis (Fig. 3B).
We have recently shown that application of a high concentration of the bile salt TLC-S caused a significant increase in ROS generation in human and murine pancreatic acinar cells (Fig. 4) (14). Mitochondria were the site of TLC-S-induced ROS production; thin confocal sections showed a mitochondrial distribution of 2,7-dichlorodihydrofluorescein diacetate acetyl ester (DCFDA) fluorescence (Fig. 4B), whereas distribution of ROS-sensitive indicators colocalized with mitochondrial markers, and the inhibition of ETC with antimycin A and rotenone abolished all detectible ROS (Fig. 5) (14). The generation of ROS elicited by the bile acid was Ca2+-dependent since pretreatment of cells with 1, 2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) abolished this effect, and thus likely to result from the overload of mitochondria due to sustained cytosolic Ca2+ elevations. The membrane-bound enzyme NADPH oxidase has been implicated in the generation of ROS by bile acids in AR42J cells, derived from a rat acinar cell tumor (172). In this study, caerulein stimulated the activation of NADPH oxidase, expression of pro-apoptotic factors, and subsequent apoptosis, effects that were Ca2+-dependent and sensitive to diphenyliodonium (DPI) implicating NADPH oxidase in the production of ROS within AR42J cells (171). In freshly dispersed murine pancreatic acinar cells, however, we found that NADPH oxidase was not the source of bile acid- (14) or menadione-induced (26) ROS production since these effects were insensitive to DPI. Further, while AR42J cells express functional NADPH oxidase, the presence of this enzyme could not be confirmed by immunohistochemistry in primary pancreatic acinar cells and may even be absent (53).
During the course of AP there is significant recruitment of immune cells to the organ. Importantly, ROS induce both activation and proliferation of immune cells (70, 151, 153). Moreover, the recruited cells, particularly neutrophills, monocytes, and macrophages, generate copious ROS via the NOX family of NAD(P)H oxidases (6), which consist of flavoprotein, cytochrome b, and regulatory subunits (83). Upon activation, NAD(P)H oxidase assembles in the plasma membrane and reduces molecular oxygen to superoxide. Phagocytes employ NAD(P)H oxidase 2 to destroy pathogens within the phagosomal space (107). Within neutrophils, H2O2, formed from superoxide, is further converted to the extremely reactive hypochlorous acid by myeloperoxidase (169), a common marker of pancreatitis severity (10). While the activity of NAD(P)H oxidase and myeloperoxidase in the production of powerful oxidants is vital in the role of immune cells as host defense, under conditions in which these oxidants are directed against host tissue, worsening of inflammatory diseases such as pancreatitis results (126). ROS production is enhanced in neutrophils obtained from patients with AP (159) and ROS derived from neutrophil NADPH oxidase has been shown to implement damage in experimental AP (53).
ROS defense mechanisms
To regulate the concentration and localization of ROS, numerous antioxidant strategies have been developed by the cell. The proximal ROS generated within mitochondria is the superoxide anion (Fig. 1), which at neutral pH is a moderately stable radical generally confined to the mitochondrial interior provided organellar membrane integrity is uncompromised. Toxicity is reliant upon onward generation of further reactive species that may react directly with biomolecules such as proteins, lipids, and DNA (75, 86, 117). The deleterious effects of mitochondrial ROS are prevented by various antioxidant systems. For example, O2•− is enzymatically converted to H2O2 by a family of metaloenzymes, the superoxide dismutases (SOD) (43). While O2•− may dismutate to H2O2 spontaneously, the rate is slow and O2•− may either reduce transition metals (particularly Fe2+), which in turn react with H2O2 to produce fiercely reactive NO•, or spontaneously react with HO• to produce peroxynitrite. Therefore, it is important that the cell maintains O2•− at the lowest concentration possible and it has been reported that SOD levels are decreased in cerulein-induced pancreatitis (30, 39, 110), suggesting compromise in the disease state. The role of SOD enzymes is to restrict dismutation to a rate limited only by the diffusion of the O2•− radical. The majority of O2•− is produced and released into the matrix that contains a specific form of SOD with manganese at its active site (MnSOD), which eliminates O2•− formed by the components of the ETC located on the IMM leaflet or within the matrix space itself (43). Expression of MnSOD can be upregulated by oxidative activation of pro-survival nuclear factor kappa B (NF-κB), thus limiting further ROS production.
Like the cytosol, mitochondria possess small molecule antioxidants such as ascorbate, glutathione (GSH), and vitamin E (α-tocopherol), which are concentrated at levels above those of the cytosol (77, 162). Both ascorbate and GSH are actively transported into mitochondria, ascorbate in its oxidized form via Glut1 where it is recycled by the ETC and prevents oxidative damage to the mitochondrial DNA (77). A number of lipophillic antioxidants, such as α-tocopherol (Vitamin E), polyphenols, and carotenoids, are present within cellular membranes. Under physiological conditions polyphenols and carotinoids may perhaps be discounted as major sinks of radical species, since their concentration in plasma is rarely above 1% of that of α-tocopherol (45). The hydrophobic α-tocopherol radical is scavenged by ascorbate (84).
The oxidation of GSH to glutathione disulphide (GSSG) is an important step in the channeling of potentially harmful radicals (168). GSH is synthesized from the precursor NAC and consists of three amino acid residues, glutamate, cysteine, and glycine, in which the central thiol group accounts for the reducing activity of the molecule. The pancreas contains high levels of GSH, ∼2 μmol/g tissue, which represents the fourth highest among the visceral organs (61, 108). Further, the rate of metabolic GSH turnover is high in the pancreas, with only the liver and kidneys possessing higher activities (61). The pancreas therefore appears well adapted to deal with oxidative stress. The actions of GSH are dependent upon turnover between reduced (GSH) and oxidized (GSSG) forms; GSSG is created and can be shuttled back to GSH via GSH reductase. Crucially, this requires the donation of an electron from NAD(P)H primarily generated within the TCA cycle. However, although GSH is present in millimolar concentrations the rate constant for reaction with H2O2 is negligible, and therefore intracellular diffusion of H2O2 per se would not be markedly affected by GSH (22). It is the presence of the selenium-containing enzyme GSH peroxidase (GPx), which catalyzes the reduction of peroxides to water (or related alcohols) and the concomitant oxidation of GSH, that makes this a favorable reaction.
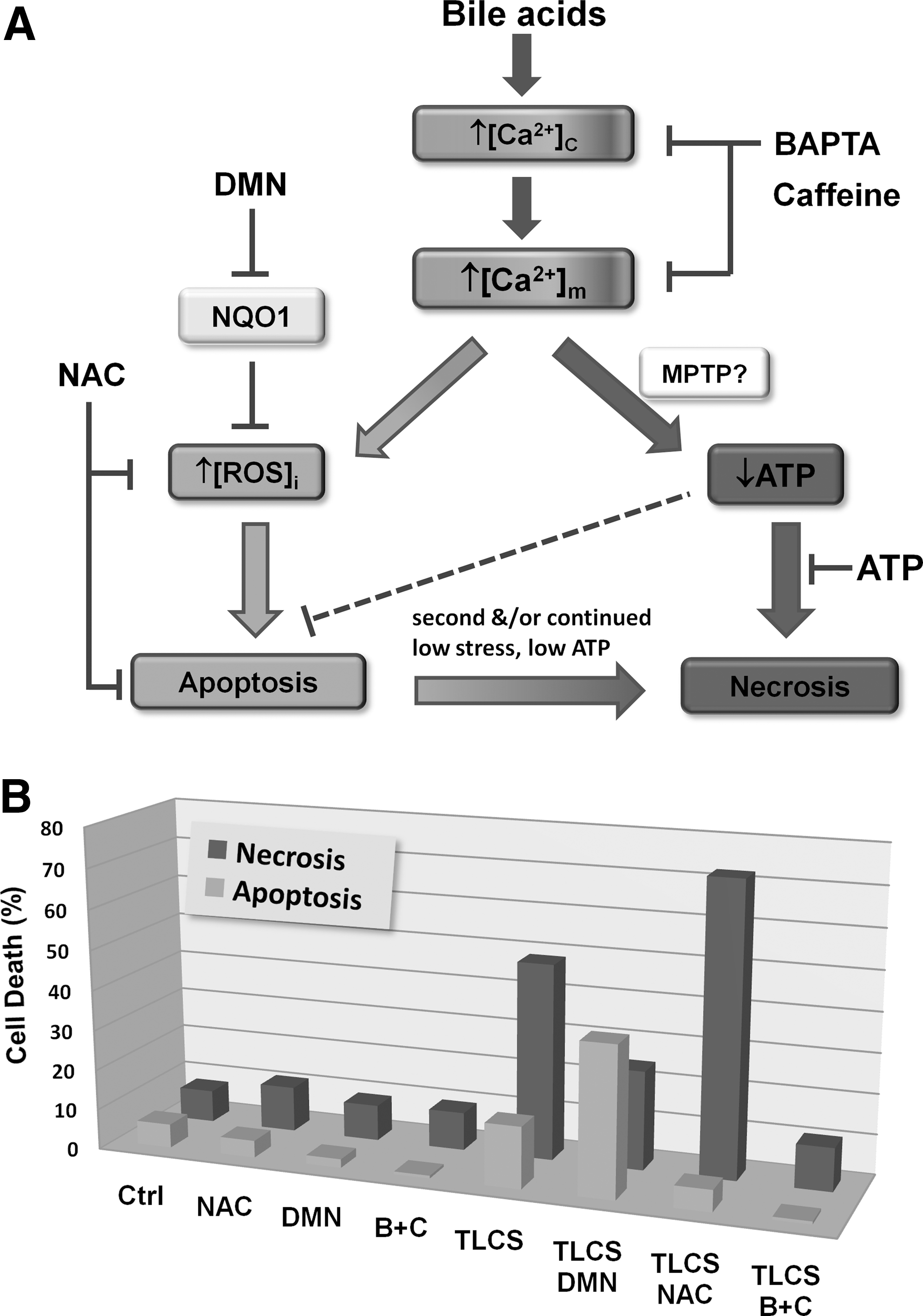
In addition to GPx, there are complementary enzyme systems that perform important antioxidant defense roles. Of particular relevance is NAD(P)H quinone oxidoreductase 1 (NQO1) an FAD-containing (flavoprotein) obligate two-electron reductase that functions as a cellular detoxifying mechanism (34, 136). It is ubiquitously expressed in all tissues and its expression is upregulated in both AP and pancreatic adenocarcinoma (61, 88, 91), suggesting a function to limit oxidative stress in disease states. NQO1 uses NAD(P)H derived from the TCA cycle as a reducing cofactor in multiple reactions such as the production of relatively stable hydroquinones from endogenously generated quinone species, for example, tocopherol and coenzyme Q10 (8, 146). NQO1 performs many protective functions, including direct scavenging of superoxide (147), maintenance of the reduced, antioxidant form of coenzyme Q10 (8), and stabilization of p53 (3), which controls the transcription of multiple genes involved in oxidative stress and cancer. We have recently highlighted the important influence of NQO1 in the pancreatic acinar cell to modulate the internal redox environment. Inhibition of this enzyme with the inhibitor 2,4-dimethoxy-2-methylnaphthalene (DMN) dramatically increased the generation of menadione-induced ROS (26), since one-electron reduction directly to the relatively stable hydroquinone was prevented, thereby shunting the quinone into two-electron reductive pathways that generate unstable semiquinone intermediates and superoxide within redox cycles. Further, DMN also potentiated bile acid-induced ROS production (14), an action that promoted apoptosis (Fig. 6) and consistent with previous observations underlining the importance of ROS in this modality of cell death.
Ca2+, ROS, and Cell Death
Cell death mechanisms in the exocrine pancreas
The severity and/or duration of stress applied to pancreatic acinar cells is a major influence on cell death modality (25). Apoptosis and necrosis represent distinct ends of the cell death spectrum, believed to be composed of up to 11 types (97). Apoptosis is genetically regulated (116), whereas necrosis is largely considered an uncontrolled default mechanism of cell death. More recently, autophagy has become of interest to the field of pancreatology. Classically viewed as a major intracellular degradation mechanism for both long-lived proteins and whole organelles, autophagy operates under stress conditions to promote survival under starvation conditions or cell death when apoptosis is inhibited (173). Degradation within autolysosomes is catalyzed by hydrolases, specifically cathepsins L and B (13), and functions to recycle vital nutrients such as amino acids (101). Although the role and significance of autophagy in disease states is incompletely understood, recent pioneering work has shown that mice deficient in Atg5, a protein central to autophagy, exhibited reduced trypsinogen activation and almost no AP with the exception of mild oedema (62). Subsequent work has reported impaired autophagic flux to mediate vacuolization and trypsinogen activation in rodent models of AP that may be due to an imbalance between cathepsins L & B (92). Future studies are required to elucidate the relative importance of autophagy in severe pancreatitis disease models that are characterized by significant necrosis.
The principal mechanism of pancreatic acinar cell death is necrosis (25, 80), the extent of which determines the severity of the disease. Whereas necrosis elicits inflammation, apoptosis involves a regulated cascade of signaling events that result in the clean removal of the dead cell from the tissue (97). These major forms of cell death coexist to differing extents in established models of AP induced by caerulein hyperstimulation, bile acids, choline-deficient ethionine-supplemented (CDE) diet, and pancreatic ductal obstruction (38, 76, 119). Induction of apoptosis reduces the severity of caerulein-induced pancreatitis (9), whereas inhibition of caspase activity (therefore, apoptosis) leads to severe necrotizing pancreatitis (93). In experimental AP at least, the severity correlates directly with the extent of measured necrosis and crucially bears an inverse correlation with apoptosis reviewed in (52). Thus clinically, the balance of cell death between apoptosis and necrosis may be critical for the outcome of the disease, a hypothesis that requires thorough testing.
Apoptosis: The role of ROS
The manner in which ROS mediate apoptosis is incompletely understood; however, there are direct interactions between radical species and apoptotic machinery. For example, H2O2 can mediate cytochrome c release, caspase-3 activation, and DNA fragmentation in a manner dependent upon translocation of Bax/Bak proteins, a crucial step in the initiation of apoptosis (23, 137, 141). Generation of ROS by the glucose oxidase system has also been demonstrated to induce apoptotic changes such as increasing the Bax/Bcl-2 ratio and subsequent DNA fragmentation.
In rat, the antioxidant melatonin prevented much of the apoptosis produced in an ischemia/reperfusion experimental pancreatitis model demonstrating the fundamental relationship between ROS and apoptosis (103). However, perhaps counter-intuitively, ROS and other oxidants may inactivate caspases (138) or activate pro-survival NF-κB, the latter shown in taurocholate (161), ethanol (51), and caerulein (54) models of pancreatitis. Thus, the participation of ROS in disease models appears multifactorial and complex. Importantly, when release of cytochrome c occurs, this interrupts the ETC, an action that causes further generation of ROS, damage to mitochondrial components, and augmentation of the oxidative burden (21, 174). Thus, once apoptosis is initiated, mitochondria become a persistent source of ROS but not ATP. When excessively generated, the unstable nature of radicals means that cell injury by direct oxidative damage to subcellular components may occur. Lipid peroxidation is often used as a biomarker of oxidative stress and there are components of every cellular membrane that are extremely vulnerable. Polyunsaturated FAs are found abundantly throughout the ER, mitochondrial, and plasma membranes and are freely peroxidated by ROS, primarily hydroxyl free radicals. This may lead to disruption of membrane integrity, loss of secretory function, and necrosis (142).
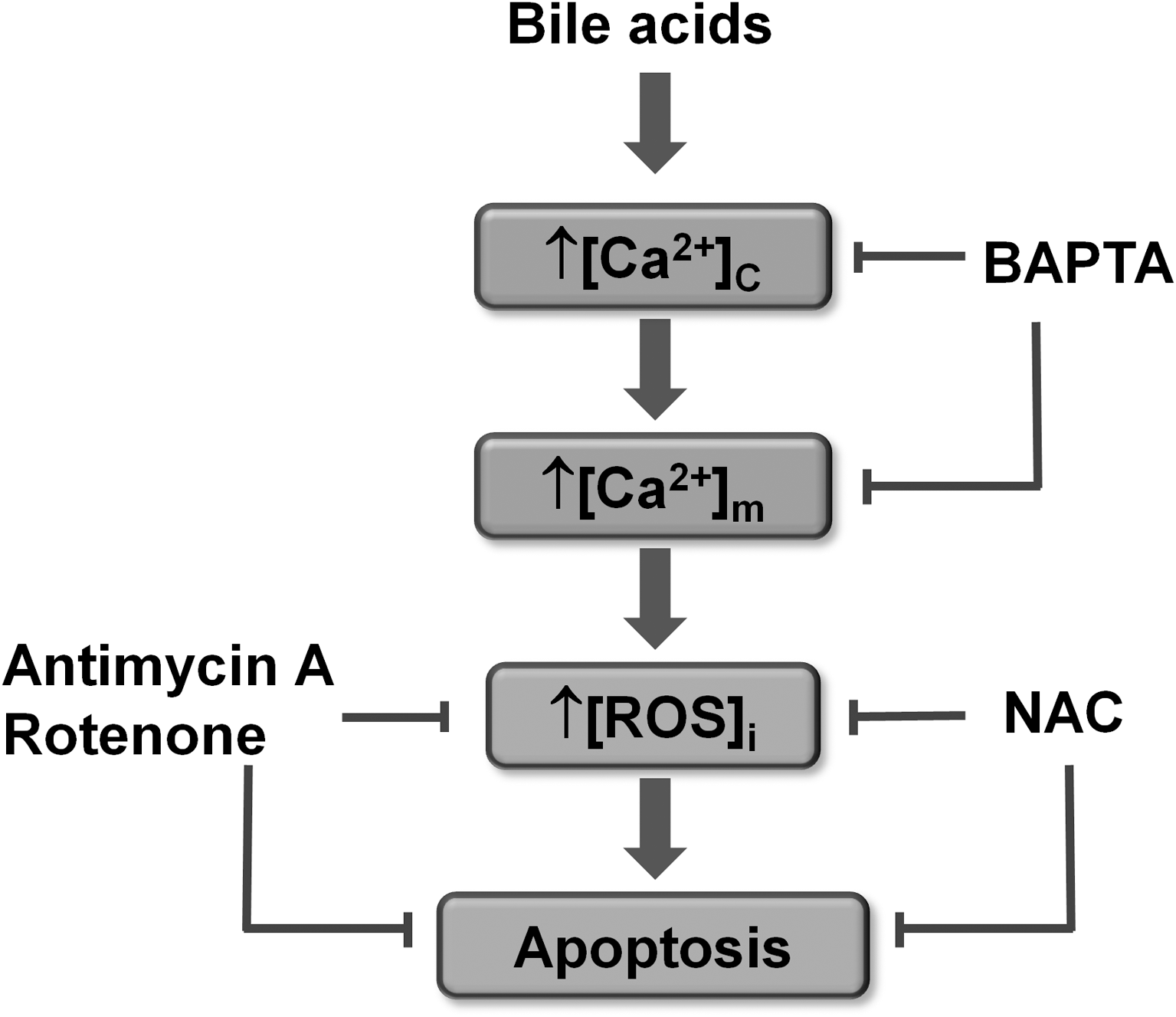
The interplay between ROS and apoptotic signaling is intriguing. Pancreatic acinar cells generate significant levels of ROS via redox cycling when exposed to the oxidant menadione (Fig. 3A) (26). This ROS production induced apoptosis (measured by both caspase activation and phosphatidylserine translocation) in a manner completely abolished by the antioxidant NAC, intimately linking ROS and apoptosis within the pancreatic acinar cell (Fig. 3B). Recently, the role of ROS in modulating cytochrome C release and subsequent apoptosis has been elegantly demonstrated in isolated mitochondria and intact pancreatic acinar cells (114). The pathophysiological relevance of acute ROS production was further strengthened by recent findings that ROS were generated by the bile acid TLC-S (Fig. 4) and which also promoted apoptosis (14). Our findings suggest that [Ca2+]c elevations induced by bile acids elicit Ca2+ uptake into the mitochondrial matrix increasing [Ca2+]m. This rise generates mitochondrial ROS with subsequent caspase activation and apoptosis; when mitochondrial ROS were decreased by NAC or by inhibition of the ETC with antimycin-A and rotenone, apoptosis was prevented. In support of the involvement of Ca2+ in the route to eventual apoptosis, preincubation with the Ca2+ chelator BAPTA-AM significantly dampened [Ca2+]c elevations, abolishing both ROS production and apoptosis. The actions of bile acids were modulated by NQO1; NQO1 inhibition revealed ROS generation by TLC-S at concentrations that could not be detected under control conditions. Further, bile acid-induced apoptosis was potentiated when NQO1 was inhibited (Fig. 6) (26, 14), implicating NQO1 in the regulation of intracellular ROS and consequent apoptosis, a view consistent with current evidence in diverse cell types. For example, overexpression of NQO1 has been shown to decrease cellular ROS production (33), whereas the absence of NQO1 in KO mice has been shown to potentiate menadione-induced toxicity (132).
Importantly, when manipulation of bile acid-induced ROS was performed, the balance of cell death modality was affected. For example, potentiation of TLC-S-induced ROS by DMN augmented apoptosis while concomitantly reducing necrosis. Conversely, NAC treatment abolished apoptosis but potentiated necrosis (Fig. 6). Our current view is that ROS generation constitutes a crucial determinant of pancreatic acinar cell death, primarily in promotion of apoptosis, an action that may serve as a protective mechanism under conditions of acute cellular stress. Whether such an effect of ROS would be beneficial or detrimental in an in vivo setting, by shifting the apoptosis/necrosis balance in pancreatic acinar cells, has not yet been established in models of AP.
Necrosis: The importance of Ca2+
Early studies have demonstrated the crucial part played by cytosolic Ca2+ in the induction of apoptosis (71, 170). Indeed, Ca2+ overload may be the common pathway that links all forms of cell death (135). However, it has been recognized that pathophysiological stimulation of pancreatic acinar cells with CCK, bile acids, or nonoxidative ethanol metabolites causes sustained elevations of cytosolic Ca2+ that primarily trigger necrosis (Fig. 6) (27, 78, 133). Coapplication of BAPTA-AM and caffeine, an IP3R blocker, completely prevented the necrosis induced by the bile acid TLC-S in an in vitro toxicity model (Fig. 6) (14). A common feature of Ca2+ overload in pancreatic acinar cells is that mitochondrial function becomes severely inhibited, leading to loss of ATP production (27, 102, 166). The actions of AP precipitants at the cellular level are mirrored within in vivo models of pancreatitis, which have demonstrated reductions of cellular ATP (113), and ATP depletion has been proposed as a master switch for a transition from apoptosis to necrosis (85).
How might sustained elevations of cytosolic Ca2+ trigger eventual necrosis? It is known that as cytosolic Ca2+ rises, mitochondrial uptake of Ca2+ occurs via an electrochemical gradient driven by the ETC. To achieve this, movement across both mitochondrial membranes must occur. Originally, the outer mitochondrial membrane was thought freely permeable to small ions such as Ca2+. However, this is likely mediated by the voltage-dependent anion channel (VDAC), a pore that regulates Ca2+ movement into the inter-membrane space (47). Transport across the IMM is primarily achieved via the mitochondrial uniporter, a channel currently poorly characterized (79, 127). Crucially, it is highly Ca2+-selective and allows its entry into the matrix to stimulate ATP production. This is achieved by a number of synergistic mechanisms, including allosteric activation of three Ca2+-dependent enzymes within the TCA cycle, α-ketoglutarate dehydrogenase, isocitrate dehydrogenase, and pyruvate dehydrogenase (94, 95), which elevate reductive intermediates such as NAD(P)H, feeding the ETC. The adenine nucleotide translocase (ANT), which transports ADP, and ATP synthase (Complex V) are both involved in ATP production and are regulated by Ca2+ (32, 99). Overall, the effect is to increase the supply of substrates to the ETC, allowing for a faster rate of oxidative phosphorylation and ATP production. This is highlighted by simultaneous measurements of cytosolic Ca2+ and mitochondrial NAD(P)H autofluorescence in human and rodent pancreatic acinar cells, in which physiological stimulation with CCK-8 and CCK-58 caused oscillatory Ca2+ rises that triggered associated oscillatory NAD(P)H elevations (24, 104, 165). Importantly, however, the movement of Ca2+ into the matrix is at the expense of ΔΨ m and large sustained Ca2+ elevations may completely collapse the proton motive force ceasing the production of ATP. Such transition from low permeability to high is likely to occur via the formation of a mega-channel, the mitochondrial permeability transition pore (MPTP).
Involvement of the MPTP in necrosis
Mitochondrial permeability transition was observed over 30 years ago as the formation of a nonspecific pore enabling the passage of low-molecular-weight solutes (63, 66, 67). This supramolecular complex, formed from pre-existing components assembled at the contact sites between the inner and outer membranes of the mitochondria, contributes to cell fate and may play a role in determining the severity of AP (102). The formation of the MPTP in response to sustained cytosolic Ca2+ and ROS breaches the permeability barrier of the IMM, allowing free movement of all solutes <1500 Da into and out of the mitochondrial matrix. This includes proton movement into the matrix and thus dissipates the proton gradient and prevents further oxidative phosphorylation and mitochondrial ATP generation. In addition, ROS generated by the ETC, usually confined within membrane structures, may exit. The exact regulation and composition of this pore are still under vigorous debate; however, current evidence suggests that the core machinery consists of the ANT and phosphate carrier, which together form a pore modulated by cyclophilin D (57). Short-term flickering of the MPTP may be a physiological phenomenon for the removal of Ca2+ from the matrix (69, 115) and function to match mitochondrial Ca2+ to specific substrates (37). However, sustained opening is triggered by high levels of mitochondrial Ca2+ taken up from the cytosol and other stimulants, principally ROS.
Interestingly, a recent study suggests that cyclophilin D may have a more wide-reaching role as a redox sensor within the mitochondria, with oxidation of the protein influencing its conformation and activity (87). Formation of the MPTP has been noted in a number of disease states, particularly in cardiac and neurological ischemia/reperfusion injury but awaits characterization in the pancreas.
Both Ca2+ and ROS appear central to the opening of the MPTP. Previous studies in hepatocytes have shown that MPTP opening occurs via menadione-induced oxidative stress (124), consistent with a model of ROS-sensitized Ca2+-dependent MPTP induction (20, 156). Animals lacking the VDAC and ANT, apparent components of the MPTP, may still be able to form the MPTP (55, 58, 81), whereas those lacking cyclophilin D are markedly protected from necrosis but have normal apoptotic responses (106). Elevation of mitochondrial Ca2+ is a required step in the opening of the MPTP and subsequent loss of membrane potential and ATP synthesis (56). However, both Ca2+ and ROS exist in a positive feedback loop to uncouple ΔΨm. ROS were postulated to uncouple membrane potential over a decade ago (72) and subsequent evidence has demonstrated the involvement of specific mitochondrial uncoupling proteins in this process (36). This uncoupling is consistent with a model in which ROS is generated in an uncontrolled manner via a positive feed-forward loop (105).
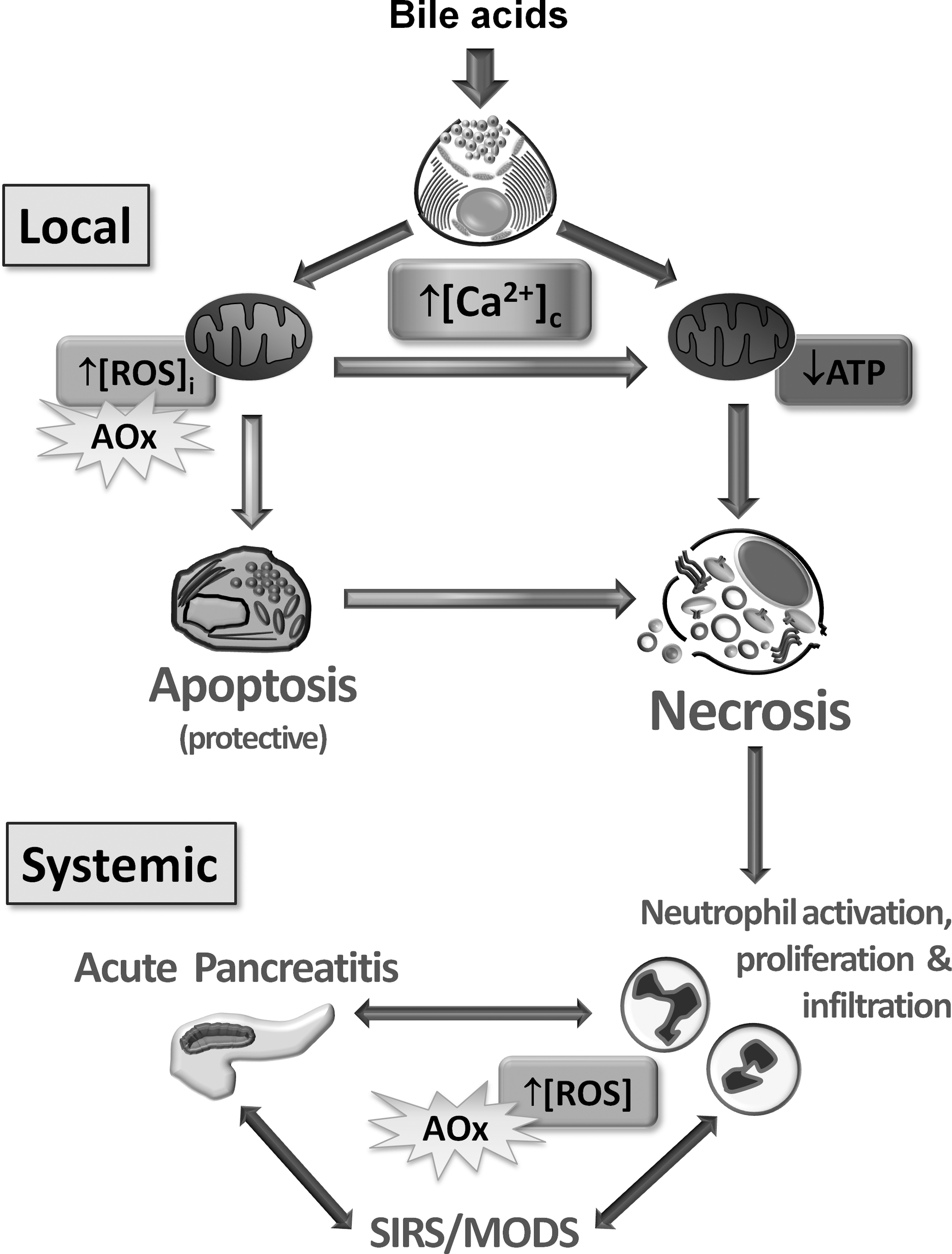
Importantly, when considering the role of ROS in the pathophysiology of the pancreas, it should be appreciated that multiple targets of ROS exist. Interventions designed to inhibit radical generation or limit their effects by scavenging will necessarily affect diverse systems in vivo. In AP, localized inflammation can escalate to a full-blown systemic inflammatory response syndrome (SIRS) involving activation and infiltration of neutrophils, the latter ROS-producing cells exacerbate the progression of experimental pancreatitis (53). ROS also activates NF-κB, leading to an influx of neutrophils via chemokine and cytokine transcription; the neutrophils release further ROS and reactive nitrogen species during the respiratory burst (142). Thus, oxidative stress amplifies the initial tissue damage experienced in AP (Fig. 7).
Since a role for ROS was first suggested in AP (139, 140), studies have produced conflicting findings. Antioxidants were shown to be beneficial in ameliorating experimental AP induced by caerulein (50), pancreatic ductal obstruction (144), and taurocholate (48). However, reduction of ROS by scavengers did not decrease acinar cell damage in two models of AP (150). Further, Ebselen, a GPx analog, has shown beneficial effects in caerulein and CDE mouse models but was without effect in Na-taurocholate rat experimental pancreatitis (109). There may be a number of explanations for these disparate results and conflicting findings with other antioxidant treatments, including differences between combinations of agents, doses, species, and types of model. Importantly, antioxidant treatments have frequently been given simultaneously, or before, AP induction in animal models from which it would prove difficult to draw a human clinical parallel. The main focus of animal studies has tended toward proving the involvement of oxidative stress and not the standardized testing of the efficacy of specific antioxidant therapies. Thus, the present preclinical evidence evaluating a role for oxidative stress in pancreatitis models is complex and most likely subject to variation depending on the precise experimental conditions employed. The importance of ROS in the disease state should be placed firmly in the context of evidence obtained from human studies, specifically the assessment of antioxidant treatments for AP.
Clinical experience with ROS
Antioxidants are grouped into agents that are free radical-scavenging enzymes (e.g., SOD), enzyme cofactors (e.g., selenium), enzyme substrates (e.g., vitamins A, C, and E), or nonenzymatic antioxidants (e.g., Ebselen and CV-3611). The rationale for the use of such therapy in the treatment of AP is essentially twofold. First, an increase in oxidative stress has been implicated in the pathogenesis of many systemic phenomena, including the SIRS and sequelae, such as acute respiratory distress syndrome (ARDS) and multiple organ dysfunction syndrome (MODS), all of which are major features of AP. Greater oxidative stress has been observed in patients with severe and mild AP compared to healthy volunteers (158). In remote organ injury, oxidative damage to plasma constituents such as proteins and lipids is a mortality predictor in patients with established ARDS (128 –131). Second, a decrease in antioxidant levels has been demonstrated in a variety of animal AP models, including caerulein hyperstimulation, CDE diet, and Na-taurocholate (31, 111, 112, 143). Thus, a reduction in oxidative stress by antioxidant treatment should theoretically be beneficial in AP patients.
Unfortunately, the promise of antioxidant therapy has not been borne out by human clinical trials that have produced a range of conflicting findings. Most trials have concentrated on scavenging free radicals via the GSH pathway. Selenium, a cofactor required for efficient GPx function, and NAC have both featured in these studies. Other studies have used vitamins A, C, and E, to augment the antioxidant defense. Unlike animal studies, enzymes have not been directly administered and also such studies frequently used combinations of antioxidants, making it difficult to determine effects of individual compounds. A combination of antioxidants, including NAC and selenium, was assessed using an observational study on patients (163), showing that antioxidants were safe, with no reported side effects, and restored antioxidant levels toward normal, though only vitamin C and selenium were significantly improved. However, the authors failed to demonstrate a significant impact on mortality in severe AP. A further prospective study assessed efficacy of high dose vitamin C treatment in patients with AP (35). The high-dose group demonstrated greater levels of plasma antioxidants, and reduced levels of lipid peroxidation, and recovery from clinical symptoms was significantly quicker. This study demonstrated promising results; however, the fate of the patients with severe AP was not documented adequately and this must be a crucial endpoint for any potential treatment. A further major criticism of these trials and others is a lack of randomization and blinding, impeding objective, accurate conclusions regarding the potential benefits of antioxidant therapy.
Importantly, a recent single-center study in the United Kingdom has addressed the criticisms leveled at previous studies, including the lack of patient randomization. The first reported randomized, controlled trial of anti-oxidant therapy in AP (148) evaluated the effects of administration of a combination of NAC, selenium, and vitamin C to AP patients over 3 years. Relative serum levels of antioxidants rose, whereas markers of oxidative stress fell in the active treatment group during the course of the trial. However, at 7 days, there was no significant difference in the primary end point, organ dysfunction, between test and control groups or for any secondary end-point of organ dysfunction or patient outcome. Further, this study highlighted a trend toward a more deleterious outcome in patients given antioxidant therapy and the potential benefit to manipulate ROS in the treatment of AP remains far from clear. In accord, a recent meta-analysis of randomized clinical trials suggested that antioxidant supplements may increase mortality in several diseases (11). Our findings on the role of ROS in pancreatic acinar cells (14) are consistent with the lack of effect of antioxidant therapy in clinical AP and, furthermore, provide an explanation for the potential of antioxidant treatment to worsen AP since we found that ROS-scavenging shifted pancreatic acinar cell death from apoptosis to necrosis.
Conclusions
Tightly controlled, transient Ca2+ signals are integral to the transduction of neurohormonal stimulation that leads to exocrine secretion. Ca2+-release channels, Ca2+-entry mechanisms, and energy-dependent Ca2+-pumps are sensitive to the cellular redox environment, and generation of ROS, principally by mitochondria, is likely to profoundly influence Ca2+ homeostasis. However, when sustained, global elevations of Ca2+ are generated by precipitants of AP, mitochondrial function is compromised. The acute Ca2+-dependent generation of ROS in the pancreatic acinar cell promotes apoptosis, a process that allows for removal of dead cells without eliciting inflammation, and may constitute a protective mechanism; the level of stimulation is dependent on the balance between ROS-producing systems and the activity of multiple antioxidant defense mechanisms present in the cell.
A sustained mitochondrial Ca2+ increase, however, eventually causes rundown of ATP production since ΔΨm is lost, probably via MPTP formation, with dramatic consequences for the cell. Inhibition of mitochondrial energy production leads to failure of homeostatic ATP-dependant Ca2+ pumps that normally return cytosolic Ca2+ to basal levels. Further, apoptosis is compromised since energy-dependant caspase activation is inhibited and cells are shunted into necrosis; abrogation of deleterious Ca2+ elevations or supply of intracellular ATP prevent necrosis induced by bile acids and FAEEs, and such approaches may prove promising for amelioration of AP. Necrosis triggers the SIRS that recruits neutrophils to the site of inflammation, causing further release of ROS and damage may escalate in a vicious cycle resulting in MODS (Fig. 7). Application of antioxidants may thus reduce systemic inflammation. However, concurrent inhibition of ROS-mediated apoptosis in the exocrine pancreas may also exacerbate organ injury by increasing necrotic cell death. In accord, recent clinical findings have shown that not only is antioxidant therapy ineffective in the treatment of AP, but also it may actually worsen the outcome, a profile that may be common to other diseases. Thus, ROS cannot be simply viewed as villains of the piece, but are likely to exert far more subtle and complex actions in the body.
Footnotes
Acknowledgments
The authors are grateful for the valuable support of the Medical Research Council (United Kingdom) and the National Institute for Health Research (United Kingdom).