
Abstract
Introduction
With respect to mitochondria and cancer, initial observations were made at the beginning of the third decade of the twentieth century by Otto Warburg, who noticed that cancer cells consume more glucose and produce more lactate than normal cells (a comprehensive review on the life and work of Otto Warburg in the context of modern knowledge can be found in Ref. 108). Confusingly, this was later misinterpreted by Warburg and others, contending that all cancer cells must have damaged respiration, when in fact the changes in most types of cancer may be more accurately explained in terms of regulation of both glycolysis and respiration (206). It should be noted, however, that in a certain (probably minor) subset of cancers, this envisaged damage to mitochondria or respiration actually occurs in a form of relatively rare mutations in nuclear genes encoding essential components of oxidative phosphorylation, such as complex II (CII) in paraganglioma and pheochromocytoma (83), or other metabolic pathways, exemplified by the enzyme isocitrate dehydrogenase in gliomas (46). In addition, mutations directly in mitochondrial DNA occur in up to 60% of cancers (116). Even though most of these mitochondrial mutations are silent, about one quarter of them have a potential to alter mitochondrial function, which may contribute to the phenomenon described by Warburg.
Mitochondria are central to the phenotype characteristic of cancer cells, as mitochondria from cancer cells increase the glycolytic rate in the cytosol of nonmalignant cells, whereas mitochondria from nonmalignant cells decrease the rate of glycolysis in the cytosol of malignant cells (28). It therefore appears that cancer cell mitochondria are different from the mitochondria in normal cells. The characteristic alterations of cancer cell mitochondria include the association of upregulated hexokine II with the porin-like voltage-dependent anionic channel (VDAC) in the mitochondrial outer membrane (MOM) (130), attenuated entry of pyruvate into the tricarboxylic acid cycle (TCA) in response to hypoxia-inducible factor 1 (HIF1), and Myc upregulation and corresponding increase in glutamine metabolism (45, 108, 206), downregulation of complex V (CV) function (43), elevated mitochondrial membrane potential (11, 49, 84, 138, 190), and increased propensity to generate reactive oxygen species (ROS) (196).
Considering the mitochondrial characteristics listed above that are typical of many different cancer types, the possibility of selective targeting of cancer cell mitochondria represents an enticing option for anticancer therapy (79, 170). Mitochondria, responsible not only for the maintenance and regulation of cellular metabolism but also for the execution of apoptosis (109), may be effectively coerced by pharmacological interventions into expediting the demise of cancer cells using an array of compounds we proposed to name ‘mitocans' (a word derived from ‘mitochondria’ and ‘cancer’) (149, 153). Mitocans induce cancer cell death by directly targeting various sites in and around the mitochondrion, thereby bypassing the frequent defects in apoptotic machinery upstream of mitochondria. In the present review, we will provide an overview of the mitocans (some of them are well known whereas others are quite novel) and divide them into classes based on their mitochondrial target. We will highlight the role of mitocans targeting the electron transport chain (ETC), which comprise a number of intriguing compounds often highly selective for cancer cells.
Mitocan Classes
In this section we divide the mitocans into eight distinct classes of compounds with defined modes of action, stemming from their molecular targets around or inside the mitochondria (149, 153). The individual classes of mitocans are listed in Table 1, graphically depicted in Figure 1, and are numbered based on the location of their respective targets from outside of MOM to inside the mitochondrial matrix. Each of the classes is represented by a number of compounds, some of which are covered in the next paragraphs.
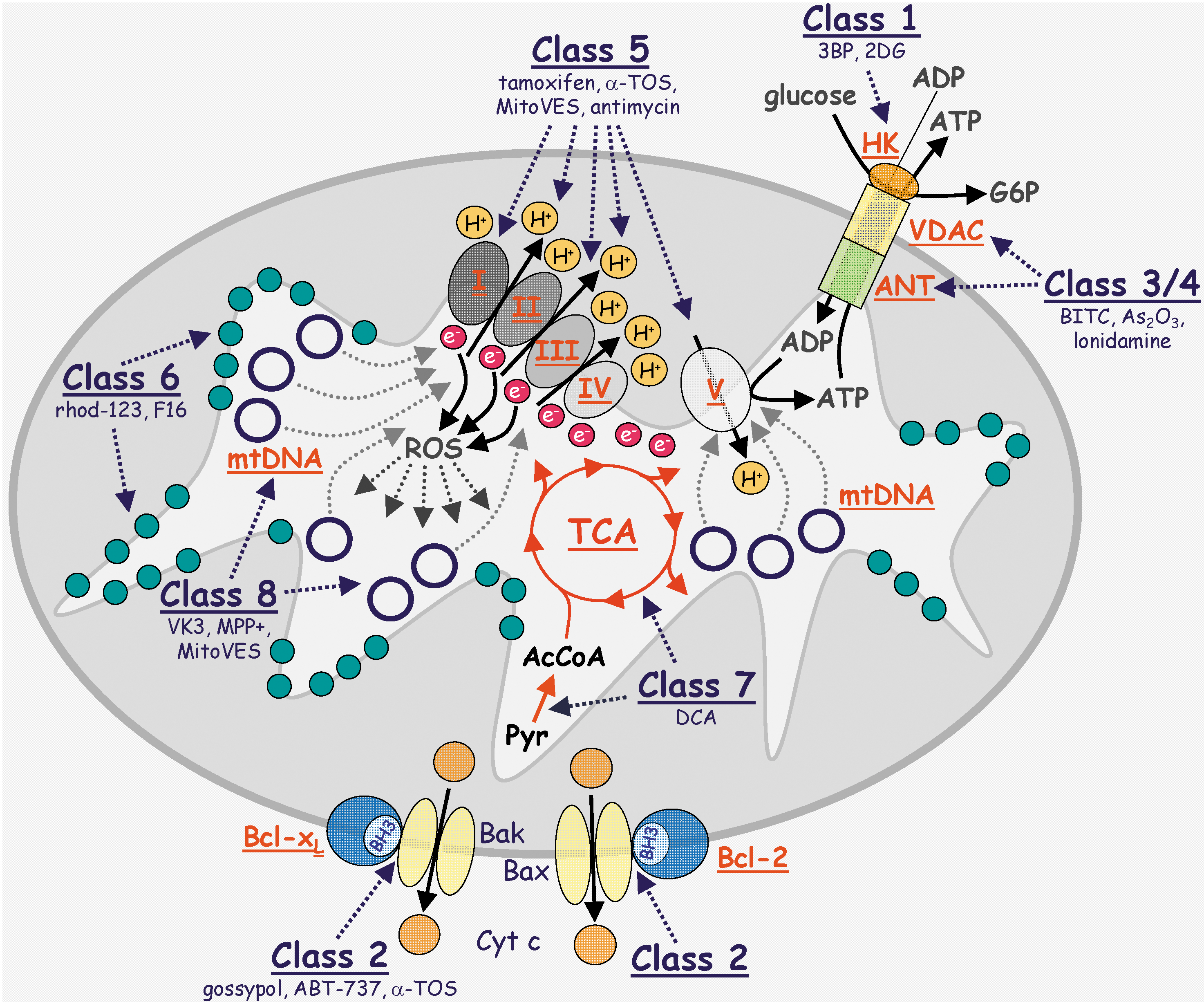
Class 1: Hexokinase inhibitors
Hexokinase (HK) is an enzyme, whose main role is to phosphorylate glucose, converting it to glucose-6-phosphate (G6P), which is a substrate for metabolic pathways ultimately coupled with ATP generation. HK has a very important function in cancer. Besides converting glucose to G6P, which can then enter the metabolic machinery to, ultimately, yield ATP, HK is associated with the cytosolic side of VDAC, a transmembrane protein in the MOM. The isotype HK-II is overexpressed in cancer cells in response to hypoxia, being transcriptionally regulated by HIF1. A channel is formed by VDAC and the adenine nucleotide transporter (ANT) extending across the MOM and the mitochondrial inner membrane (MIM), respectively, interconnecting the mitochondrial matrix to the cytosol. The function of the channel is to transport outside mitochondria the ATP made by the electron transport chain (ETC) of the MIM. When expressed at higher levels, as in cancer cells, HK-II binds both ATP and glucose, resulting in the production of G6P. A direct correlation has been established between the growth of carcinomas and the levels of HK activity (28). According to Koobs (107), mitochondrial-bound HK limits respiration when tumor cells are utilizing glucose (known as the Crabtree effect), even though large amounts of ADP continue to be produced. The continuous phosphorylation of glucose by ATP (proceeding by the mitochondrial-bound HK) reduces the level of phosphate available for oxidative phosphorylation, and thereby prevents attaining maximal rates for state 3 respiration (8). Hence, HKII via its mitochondrial localization also helps to stabilize mitochondria, suppressing apoptotic death of cancer cells and promoting their survival (130).
Several hexokinase inhibitors have been found to inhibit cancer growth and of these, considerable focus has been on 2-deoxyglucose (2DG) and 3-bromopyruvate (3BP). The basis for the action of 2DG is to inhibit HK activity and hence, glycolysis, with the result that the binding of HK to VDAC is prevented. By this mechanism, 2DG promotes the susceptibility of malignant cells to other forms of treatment (184). This finding, as well as recent reports that 2DG promotes cancer cell apoptosis when used in combination with another mitocan, the anti-diabetic drug, metformin, provides the basis for testing these compounds in the clinical setting (13). 3BP is an alkylating agent that inhibits both HK activity and the mitochondrial complex II (CII) and consequently is included in the mitocan class 2 and 5 (Fig. 1). Recent data indicate that 3BP acts by binding covalently to HKII, causing its dissociation from VDAC (36). 3BP causes cancer cell death due to the rapid depletion of ATP and suppresses tumor growth considerably in animal models. For these reasons, 3BP is another candidate for cancer clinical trials (106, 130).
Class 2: Compounds targeting Bcl-2 family proteins
Bcl-2 family proteins comprise a class of polypeptides whose state of relative balance in levels of the family members decides the cell's ‘fate’. When the levels of expression of the pro-apoptotic members are greater, then it will result in the demise of the cell, whereas greater levels of expression of the anti-apoptotic Bcl-2 family proteins will provide a pro-survival ‘environment’. Although recent findings revealed novel functions for the Bcl-2 family members, including a role in the biogenesis of mitochondria (189), we will only briefly discuss these proteins as targets for class 2 mitocans, resulting in promoting apoptosis of cancer cells. The basis for the action of this mitocan class stems from the finding that the anti-apoptotic and pro-apoptotic Bcl-2 family proteins interact via their Bcl-2 homology-3 (BH3) domains, thereby preventing the BH3 domains from forming large channels or pores in the MOM, a prerequisite for apoptotic signaling downstream of mitochondria (221). Since the MOM channel is made of oligomers of the pro-apoptotic Bax or Bak protein BH3 domains increased expression of the anti-apoptotic BH3 interacting proteins Bcl-2, Bcl-xL, or Mcl-1 will protect cancer cells from apoptosis. Moreover, the anti-apoptotic Bcl-2 family proteins are often overexpressed in cancer cells (122). Therefore, small molecules or BH3 mimetics, targeting the interaction between the anti- and pro-apoptotic Bcl-2 protein members is of clinical importance (102, 224).
The BH3 mimetics include the natural polyphenolic compound gossypol. This agent has been shown to interact with BH3 binding domains, thereby interfering with the interaction between Bcl-2, Bcl-xL, or Mcl-1 and the pro-apoptotic proteins, Bax or Bad. The result is the oligomerization of Bax or Bad to form channels and activation of the post-mitochondrial apoptotic signaling (160). Gossypol has served as a structure for developing more efficient BH3 mimetics, such as the highly intriguing compound, ABT-737 (Abbot Laboratories, ref. 205). ABT-737, as well as an oral version, ABT-263 (199), are now being tested in clinical trials. The apoptogenic compound α-tocopheryl succinate (α-TOS) has been reported to interact with the BH3 binding domain of Bcl-2 and Bcl-xL, which suppressed their binding interaction with the pro-apoptotic protein Bak, arresting proliferation of prostate cancer cells and inducing their death by apoptosis (181). This activity of α-TOS is interesting in that it complements a second apoptogenic activity due to its ability to also interact directly with the ubiquinone sites of the mitochondrial CII (see below for details). In addition, the mitochondrial complex III (CIII) Qi site inhibitor, antimycin A, also acts as a BH3 mimetic (201), suggesting that on a more general level, compounds that interact with ubiquinone binding sites in the mitochondrial electron transport chain may have a tendency to be BH3 mimetics as well, consistent with the possibility that the BH3 binding site in Bcl2 family members might also be a ubiquinone binding site (153).
Class 3 and 4: Thiol redox inhibitors and VDAC/ANT targeting drugs
The redox environment of cancer cells is distinctly different from that of normal cells in that cancer cells show much higher intrinsic levels of ROS and also, as a result, appear more vulnerable to agents that induce further elevations in oxidative stress, since their antioxidant capacity is relatively inferior (93, 193). Therefore, compounds that oxidize thiol group and/or deplete the mitochondrial glutathione pool will cause substantial apoptosis of cancer cells (72, 196). Agents like arsenic trioxide (134, 166) or isothiocyanates, represented by phenylethyl isothiocyanate (PEITC) (198, 217) have been shown to possess relative selectivity in killing cancer cells by upsetting the normal homeostasis in the cellular redox environment. Intriguingly, PEITC has been reported to efficiently kill resistant leukemia cells (197).
The permeability transition pore complex (PTPC) forms as a super channel comprising the VDAC/ANT system of proteins embedded in the MOM and MIM, respectively, interconnecting the mitochondrial matrix with the cytosol, and serves as a mode of transport for a variety of solutes and small molecules including ATP and ADP (228). Deregulation of the VDAC/ANT complex results in apoptosis induction in cancer cells. Compounds that modulate the PTPC include lonidamine, arsenites, and steroid analogues (represented by CD437) (12). Interestingly, an arsenite analogue 4-(N-(S-glutathionylacetyl)amino) phenylarsineoxide (GSAO) was shown to inhibit the function of ANT by crosslinking its cysteine residues. This was followed by generation of oxidative stress and induction of apoptosis, which was selective for proliferating, angiogenic endothelial cells while being nontoxic to growth-arrested endothelial cells (54). These results indicate that GSAO can selectively kill endothelial cells in the context of a growing tumor, acting in an anti-angiogenic manner. Similar findings were reported for the mitocan, α-TOS (see more on its molecular mechanism below).
Class 5: Electron transport chain targeting drugs
Members of this class are discussed in more detail in a separate section below.
Class 6: Lipophilic cations targeting the inner mitochondrial membrane
The molecular target of this group of compounds is given by the relatively high transmembrane potential that exists across the MIM (Δψm,i). In fact, it has been documented that cancer cells have a considerably higher Δψm,i than that in nonmalignant cells due to altered mitochondrial bioenergetics (138). This feature will dictate the intracellular targeting of lipophilic cations which, as a result of the increased Δψm,i in cancer cells, will make these mitocans relatively more selective for cancer cells, since the Nernst law defines that with each increase of Δψm,i by −60 mV, a corresponding 10-fold increase occurs in the accumulation of cationic compounds in the MIM (139, 207).
A prime example of this mitocan class targeting the inner membrane is rhodamine-123, which was reported to accumulate in mitochondria of src-transformed cells as early as in the 1980s (98), showing selectivity for cancer cells (114). Following from this, the selective toxicity of a number of delocalized lipophilic cationic agents, including the peptide (KLAKKLAK)2 towards cancer cells was found (64). A compound termed F16, with a delocalized positive charge, was identified by high-throughput screening and was found to be selective and effective against breast carcinomas with high levels of HER2 expression (66).
Class 7: Drugs targeting the tricarboxylic acid cycle
The TCA cycle, also referred to as the citric acid cycle or Krebs cycle, is a source of electrons that are fed into the ETC that are used to drive the electrochemical proton gradient required for the generation of ATP. The TCA cycle is based on the addition of acetyl-CoA (formed in the mitochondrial matrix by conversion of pyruvate, catalyzed by pyruvate dehydrogenase) to oxaloacetate to form citrate. Citric acid is then, in a series of reactions, converted to oxaloacetate, which then again adds a molecule of acetyl-CoA. During this process, electrons are released to drive the proton gradient that is coupled to the generation of ATP. An interesting step in the TCA cycle involves succinyl dehydrogenase (SDH), also known as the mitochondrial respiratory complex II, which converts succinate to fumarate, releasing two electrons that are used to reduce ubiquinone (UbQ) to ubiquinol (UbQH2), which is then carried to CIII of the ETC.
A number of compounds exist that target the TCA cycle as well as the reaction converting pyruvate to acetyl-CoA, a prerequisite for pyruvate to enter the mitochondria and the TCA cycle. The enzyme pyruvate dehydrogenase, catalyzing the reaction, is negatively regulated by phosphorylation via the pyruvate dehydrogenase kinase (PDK), which is often upregulated in cancer by HIF1 and Myc transcription factors (45). This skews the balance towards the predominantly glycolytic phenotype typical of cancer cells. Dichloroacetate (DCA), a relatively basic compound, is selective for killing cancer cells by suppressing the activity of PDK (22), thereby promoting the activity of its target, pyruvate dehydrogenase. In this way, DCA increases the supply of pyruvate into the TCA cycle and causes a shift from anaerobic glycolysis to oxidative glycolytic metabolism accompanied by dissipation of Δψm,i, generation of ROS, and activation of the K+ channel, events that typify cancer cells (22). DCA is already in clinical use to treat patients with mitochondrial deficiencies, and therefore, its development as an anticancer drug should be less complicated than with a completely novel agent.
Class 8: Drugs targeting mtDNA
Mitochondria are unique organelles because they carry their own genetic information encoded on a small circular genome, referred to as mitochondrial DNA (mtDNA). The mammalian mitochondrial genome has a size of over 16 kB, and encodes 13 subunits of the mitochondrial complexes I, III, IV, and V, 24 tRNAs, 12S, and 16S rRNA, and also contains a region called the D-loop sequence, which is important in the regulation of mtDNA replication (5).
To date, several compounds have been reported that interfere with the function and stability of mtDNA and other drugs that affect the activity of the mitochondrial DNA polymerase-γ. For example, vitamin K3 (menadione) targets mtDNA by inhibiting the activity of DNA polymerase γ that is specific for replication of mtDNA, with ensuing induction of apoptosis (177). Similar effects were reported for fialuridine which induces mitochondrial structural defects (123). The Parkinsonian toxin 1-methyl-4-phenylpyridinium causes a reduction in the copy number of mtDNA by destabilizing the structure of the mitochondrial D-loop (137,203).
We have been studying vitamin E analogues as anticancer drugs, epitomized by the redox-silent α-TOS (see Chapters 3.2 and 4 for details on the apoptogenic signaling induced by α-TOS and similar agents). To enhance the toxicity of this drug, we modified it by adding a triphenylphosphonium (TPP+) group onto it, targeting the drug to the mitochondrial inner membrane (MIM) (57, 58), as has been done previously for a variety of antioxidants (103, 145). This mitochondrially targeted vitamin E succinate (MitoVES) was found to be superior to the untargeted α-TOS in its apoptogenic activity. Interestingly, we found that MitoVES modulates mtDNA and more specifically, at subapoptotic doses, suppressed the D-loop derived transcript levels in a range of cancer cells, a phenomenon that was not observed for α-TOS. This was accompanied by inhibition of cancer cell proliferation (Neuzil et al., unpublished). It is not clear at this stage, whether modulation of mtDNA by MitoVES is a direct effect on the mitochondrial genome or whether it is mediated by ROS generated in the MitoVES-treated cells (see Chapter 4 for details). Nevertheless, MitoVES provides an intriguing possibility for interfering with tumour progression by means of suppressing the proliferation of cancer cells without necessarily inducing apoptosis.
Class 9: Drugs targeting other sites
This group comprises a number of agents that affect mitochondria but their targets have not yet been fully elucidated. While effects on specific components of mitochondria or mitochondrial function have been reported for many such compounds, in these cases the molecular target has not yet been defined. A prime example of such compounds as is betulinic acid (73) of potential clinical relevance.
Anticancer Drugs That Target the Electron Transport Chain
Mitocans comprise a large group of different compounds, many of which were discussed above and in recent reviews (72, 75, 80, 169, 207). The following sections focus in more detail on the class 5 of mitocans, which target the mitochondrial ETC, the central system of mitochondrial energy production and the process of oxidative phosphorylation (176, 178).
The production of energy is achieved by transporting electrons in a coordinated manner from NADH or FADH2 (generated from substrates in the TCA cycle) to the final acceptor, molecular oxygen, to produce water (Fig. 2). The four protein complexes of the mitochondrial ETC are embedded in the mitochondrial inner membrane, and, by passing electrons from complexes I and II via complex III to complex IV (abbreviated as CI etc.) with the help of electron carriers, UbQ and cytochrome c, generate energy that is maintained as an electrochemical proton gradient across the mitochondrial inner membrane (MIM). CV, the F1F0-ATPase, then uses the energy from the proton gradient to generate ATP from ADP and inorganic phosphate, which provides the cell with its fundamental energy substrate. ETC, due to the large electron flow, is the major source of mitochondrial ROS, with CI and CIII identified as prime superoxide-generating sites (1, 144). In addition, recent evidence also points to a role CII in ROS formation (120, 121). The superoxide by-product is not harmful when ROS production is controlled within the constraints of the cellular redox system, as the cell must maintain moderate ROS levels necessary to sustain the cellular signaling processes. However, deregulation leading to permanently higher levels of ROS may predispose, over time, to carcinogenesis (70). By contrast, an acute and substantial increase in ROS levels may have a much more immediate effect and commit the cell to undergo apoptosis (76, 128).

Cancer cells often show higher levels of oxidative stress and higher saturation of their antioxidant defenses, and consequently may be more sensitive to apoptosis induction by compounds disrupting the cellular redox balance (196). In addition, the ETC itself plays a role in apoptosis, because defects in the ETC are associated with abnormalities in apoptosis induction (112, 120). Furthermore, inhibition of ETC complexes results in increased ROS production, such that targeting the ETC complexes with compounds from the class 5 of mitocans provides a possible strategy for eliminating cancer cells with relatively high specificity. The next section provides an overview of the apoptosis-inducing class 5 mitocans (Table 2) targeted to the individual macromolecular complexes of the mitochondrial ETC and their anticancer potential. The list is not exhaustive, and further research will very likely identify additional candidates.
Compounds in clinical use or undergoing clinical trials are in bold.
Complex I
The first and largest complex of the ETC, NADH:UbQ oxidoreductase or CI, functions by transferring electrons from NADH derived from the TCA cycle to UbQ, which passes them on to CIII. CI contributes to the maintenance of the proton gradient on the MIM, pumping four protons into the intermembrane space for each NADH oxidized. In most mammalian cells, CI comprises 45 subunits, including seven that are coded by the mitochondrial genome, and has a total molecular mass of about 1,000 kDa (178). The crystal structure of CI provided insight into the mechanism of proton pumping and electron transfer revealing that the electrons are removed from NADH by a flavin group, relayed to a series of iron–sulfur clusters, and finally donated to UbQ bound within CI (17, 62).
CI is proposed as one of the sites for superoxide formation within the ETC. It is not known precisely how the superoxide is formed, but the flavin group, the N2 iron sulfur cluster and the UbQ binding sites have each been suggested to participate in the process (17, 68, 86, 110, 111, 113). The basal level of superoxide production from CI is dramatically increased either due to retrograde electron transport or inhibition of CI. With regard to inhibition, more then 60 classes of compounds are known to inhibit the function of CI (50). Most of these inhibitors are specific for overlapping binding sites within the UbQ-binding region (159, 195), but despite this, substantial differences were seen with respect to the ability of individual compounds to induce superoxide production (67, 113). Some well-known CI inhibitors are considered neurotoxic and are often used as pesticides, and as such, have been implicated in the development of neurological disorders such as Parkinson's disease, inducing similar symptoms in experimental animals (18, 53, 60, 162). Recent observation suggests that the neurotoxicity of rotenone and-methyl-4-phenylpyridinium (MPP+) is unrelated to the inhibition of CI, and instead results from their ability to destabilize microtubules (37). The equally potent CI inhibitor piericidin, which does not disrupt microtubules, is not neurotoxic. For this reason, rotenone, even though it efficiently induces apoptosis in cancer cells (124, 166, 214), is not considered for cancer treatment and piericidin could be a better candidate.
In the context of anticancer applications, a well-studied analogue of rotenone is deguelin, an inhibitor of the Akt and nuclear factor-κB (NF-κB) signaling pathways. Deguelin shows chemopreventive anti-tumor activity in experimental cancer and xenograph models without major toxic effects and also inhibits angiogenesis (39, 51, 117, 147, 156, 202). This anti-tumor activity is at least in part related to its interaction with heat shock protein HSP90, but direct inhibition of CI also plays a role, because cells with defective respiration are insensitive to deguelin-induced apoptosis (87, 104, 157). The lower affinity for CI (60 times lower for deguelin compared to rotenone) and the lack of the microtubule-related side effects may possibly contribute to the more favorable toxicity profile of deguelin (30,50).
Interestingly, the anti-breast cancer drug, tamoxifen, routinely used in the clinic as an antagonist of the estrogen receptor in the treatment of hormone-dependent breast cancer was also reported to inhibit CI at the flavin site. This inhibition was accompanied by increased cellular hydrogen peroxide production, which was synergistically enhanced by estradiol (142). This observation could potentially extend the use of tamoxifen to other cancer types besides hormone-dependent breast cancers, and its prior history as a drug already clinically approved may make its application to other cancers more straightforward. Indeed, some anti-proliferative effects of tamoxifen against cells lacking the estrogen receptor, as well as against prostate cancer have been observed and the combination of tamoxifen with estradiol can induce apoptosis of tamoxifen-resistant breast cancer (14, 41, 161). Targeting the ETC may therefore be an important additional component of tamoxifen's anticancer activity, despite some doubt remaining as to whether CI is the only target of tamoxifen in the ETC (146).
Finally, the biguanidine, metformin, an AMP-activated protein kinase (AMPK) activator used in the clinic to treat type-2 diabetes also inhibits CI and blocks mitochondrial respiration, readily inducing apoptosis in p53 deficient cancer cells, and prevents the growth of p53-deficient xenographs (29). It also displays anti-tumor effects in vivo in pancreatic cancer and kills breast cancer cells as well as breast cancer stem-like cells; patients given metformin show a reduced incidence of cancer and increased effectiveness of chemotherapy to treat their tumors (reviewed in Ref. 38). Thus, metformin may be a promising option for cancer treatment when used either alone or in combination with other anticancer drugs (13).
Complex II
CII or succinate dehydrogenase (SDH), is the smallest of the respiratory complexes, and in mammals, all four of its subunits are encoded by nuclear genes (178). Unlike Complex I, III, and IV, CII does not directly participate in the maintenance of the proton gradient. It is also unique in that it forms a direct link between the TCA cycle, where it catalyzes the oxidation of succinate to fumarate, and the mitochondrial ETC. CII transfers two electrons removed from succinate to oxidized UbQ to form reduced UbQH2, which then transports the electrons onto CIII, before UbQ is recycled. In this way, CII can feed electrons into the UbQ pool, contributing to the electron flow that exists between CI and CIII, thereby indirectly helping to maintain mitochondrial membrane potential by proton pumping from respiratory complexes, CIII and CIV.
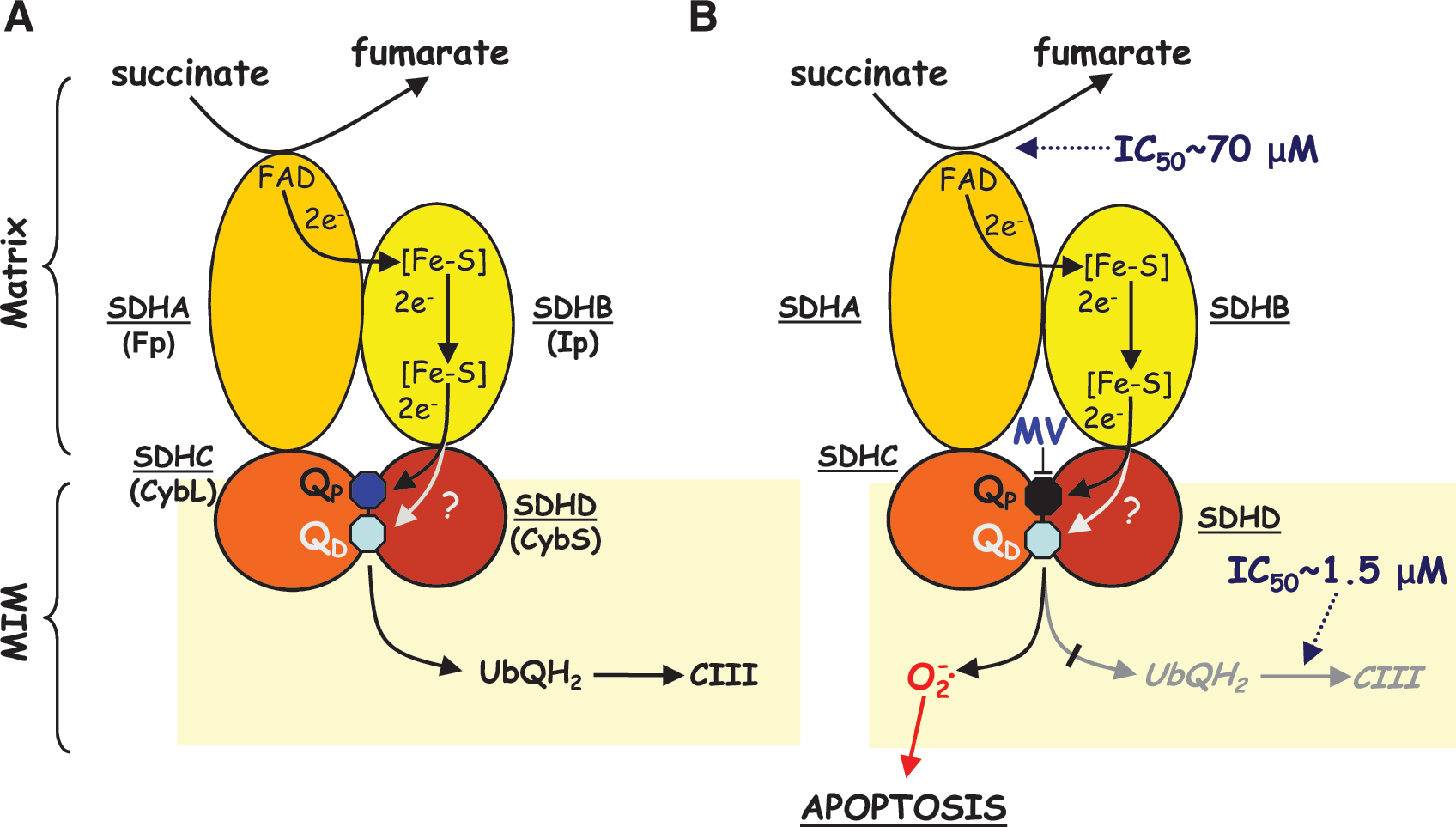
Unlike CI, the complete crystal structures of both prokaryotic and eukaryotic CII have been solved, providing insight into its architecture and the association of the four subunits, SDHA, SDHB, SDHC, and SDHD, helping to explain its mechanism of action (191, 219). The SDHA subunit, exposed to the mitochondrial matrix, comprises the active site of the SDH enzyme for converting succinate to fumarate. Electrons generated during this process are channeled via the iron–sulfur (Fe:S) clusters of the SDHB subunit to the UbQ-binding sites within subunits SDHC and SDHD embedded in the MIM. Of the two UbQ-binding sites suggested by the crystallographic studies, the proximal one (QP; closer to the matrix) is probably fully functional, whereas the distal site (QD) may be a nonfunctional ‘pseudo-site’ (129). Although CII has often not been considered a major source of mitochondrial superoxide generation, blocking the UbQ-binding sites with specific inhibitors nevertheless induces substantial ROS generation (see below).
One compound that triggers high levels of ROS generation from CII is the emerging anticancer drug, α-TOS (56, 57, 59), which belongs to a group of ‘redox-silent’ analogues of vitamin E (α-tocopherol). α-TOS has shown significant anticancer potential in various preclinical cancer models, including the difficult-to-treat mesotheliomas and HER2-positive breast carcinomas, without adverse effects or toxicity (154, 155, 187, 208). The UbQ-binding sites on CII appear necessary for the mechanisms of α-TOS anticancer activity, involving superoxide production, increased calcium levels, and the induction of apoptosis and its anticancer activity can be inhibited by pre-treating with antioxidants, supporting α-TOS working as a pro-oxidant (59, 78). α-TOS treatment of cancer cells sets in motion a number of intracellular processes, but the precise sequence and mutual relationship of each step have yet to be fully elucidated.
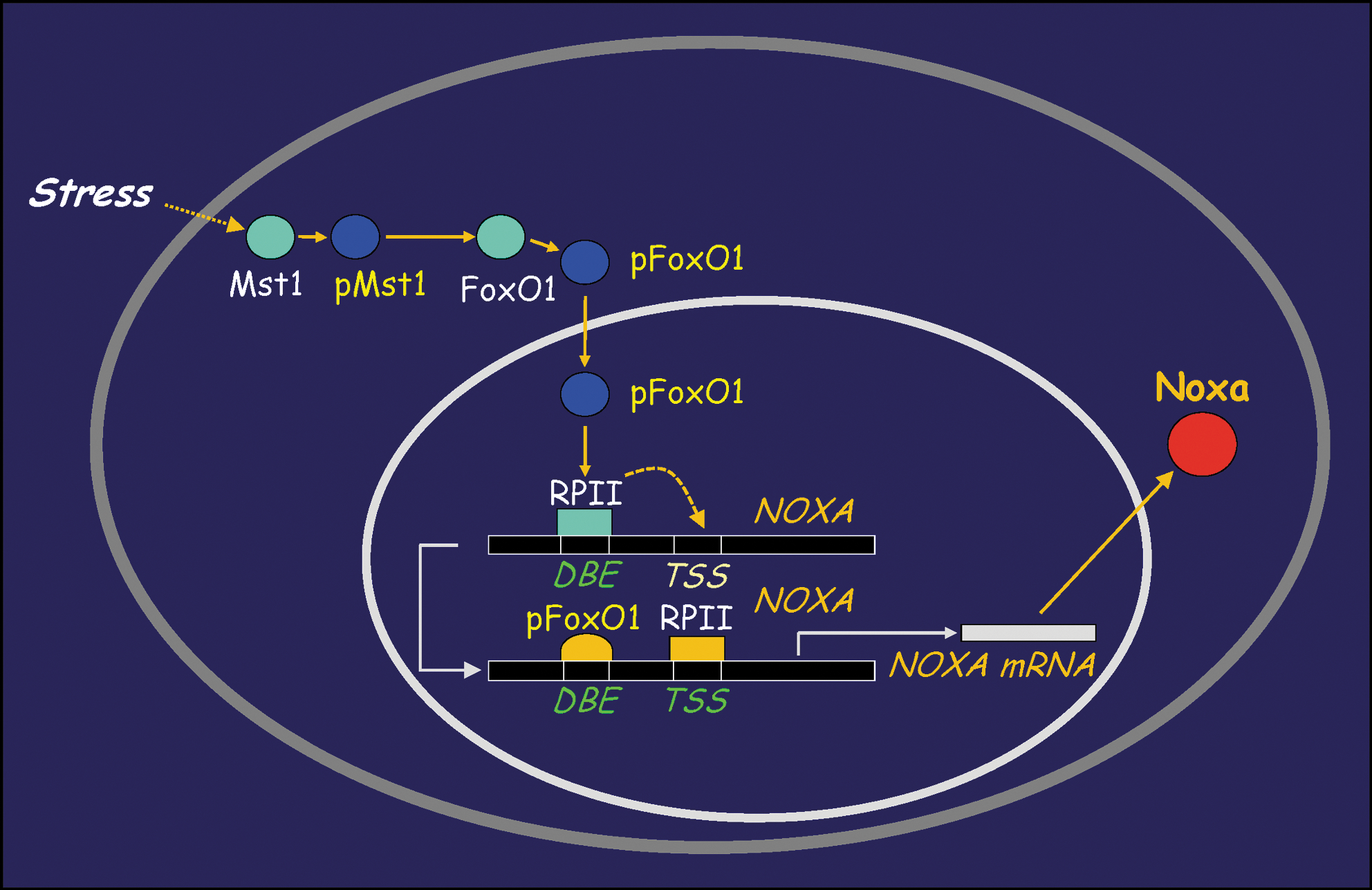
α-TOS treatment of cancer cells results in upregulation of the pro-apoptotic protein NOXA, in a p53-independent manner, an event that is regulated by the Mst1-FoxO1 axis (204) (Fig. 3). NOXA induction suppresses the anti-apoptotic function of Mcl-1, thereby promoting Bak/Bax polymerization and mitochondrial permeabilization (167, 211). At the same time, α-TOS promotes mitochondrial apoptosis by acting as a BH3 mimetic (181). These processes are depicted in Figure 4, which also indicates that the ROS generated in cells from targeting of CII by α-TOS may also cause dimerization of Bax and its ensuing mitochondrial translocation (44). Furthermore, α-TOS activates sphingomyelinase, resulting in the formation of the pro-apoptogenic product, ceramide (211). Ceramide production is an early event detectable within the first 15–20 min following α-TOS addition, well before the other drug-induced effects are observed. Another pro-apoptotic effect of α-TOS and the related compound, α-tocopheryloxybutyric acid (2) is to inhibit the Akt pro-survival signaling pathway. The mechanism of Akt inhibition has not yet been fully clarified, but downregulation of Ras signaling and inhibition/downregulation of epidermal growth factor receptors have been implicated (55, 182). Finally, α-TOS can also inhibit cell cycle progression by reducing expression of the transcription factor E2F1 (4).
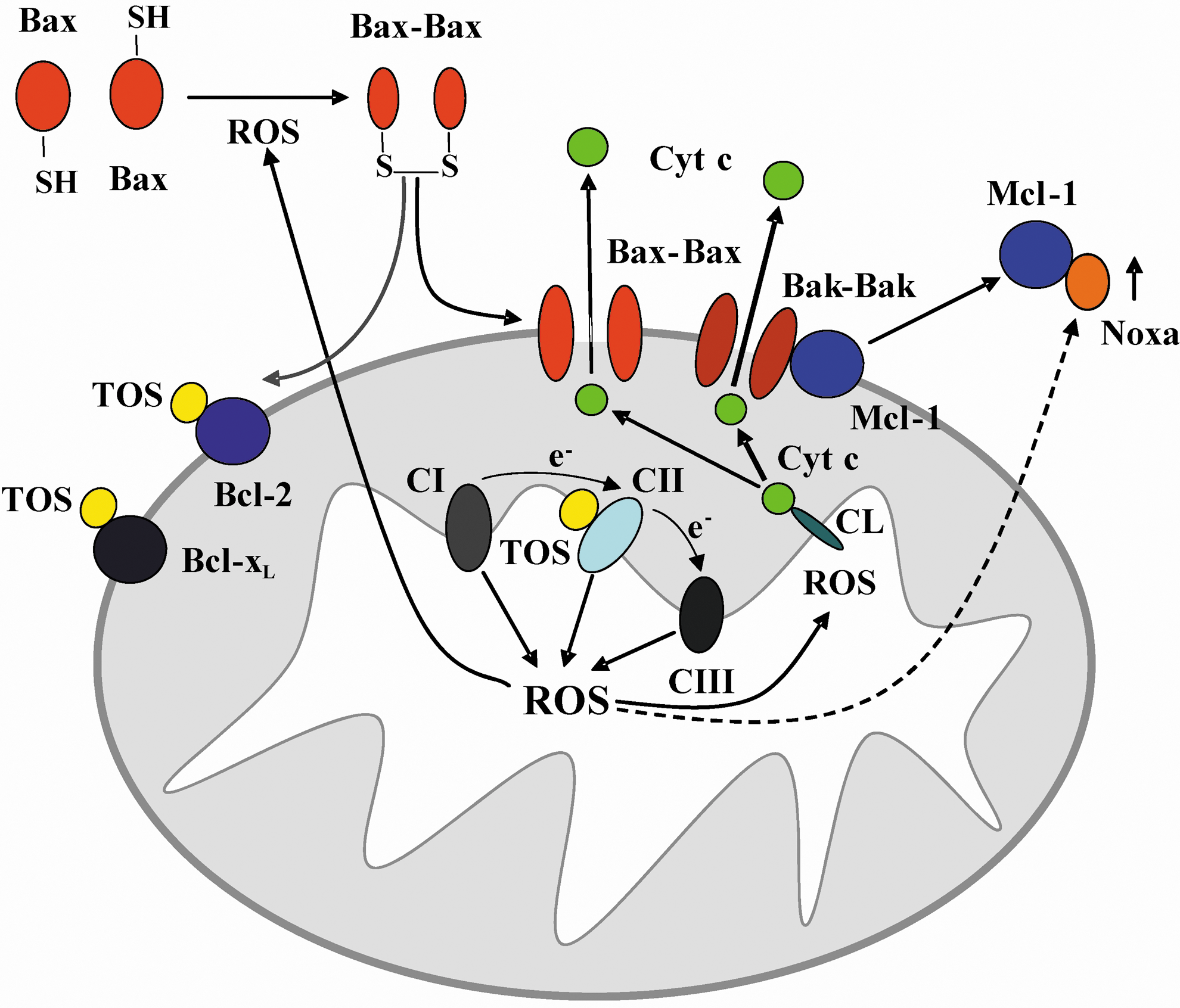
α-TOS has other indirect anticancer effects by attenuating angiogenesis as a result of the direct elimination of proliferating endothelial cells and by disrupting autocrine as well as paracrine signaling via the FGF2 pathway (151, 187). In this way, α-TOS eliminates tumor cells not only directly by inducing apoptosis but also indirectly by inhibiting angiogenesis. Furthermore, the relative specificity of α-TOS for targeting cancer cells is enhanced by the higher esterase activity in normal cells cleaving it to form vitamin E (α-tocopherol), which has antioxidant properties. This has been clearly shown in rat hepatocytes, where α-TOS protects against oxidative stress (150, 152, 225).
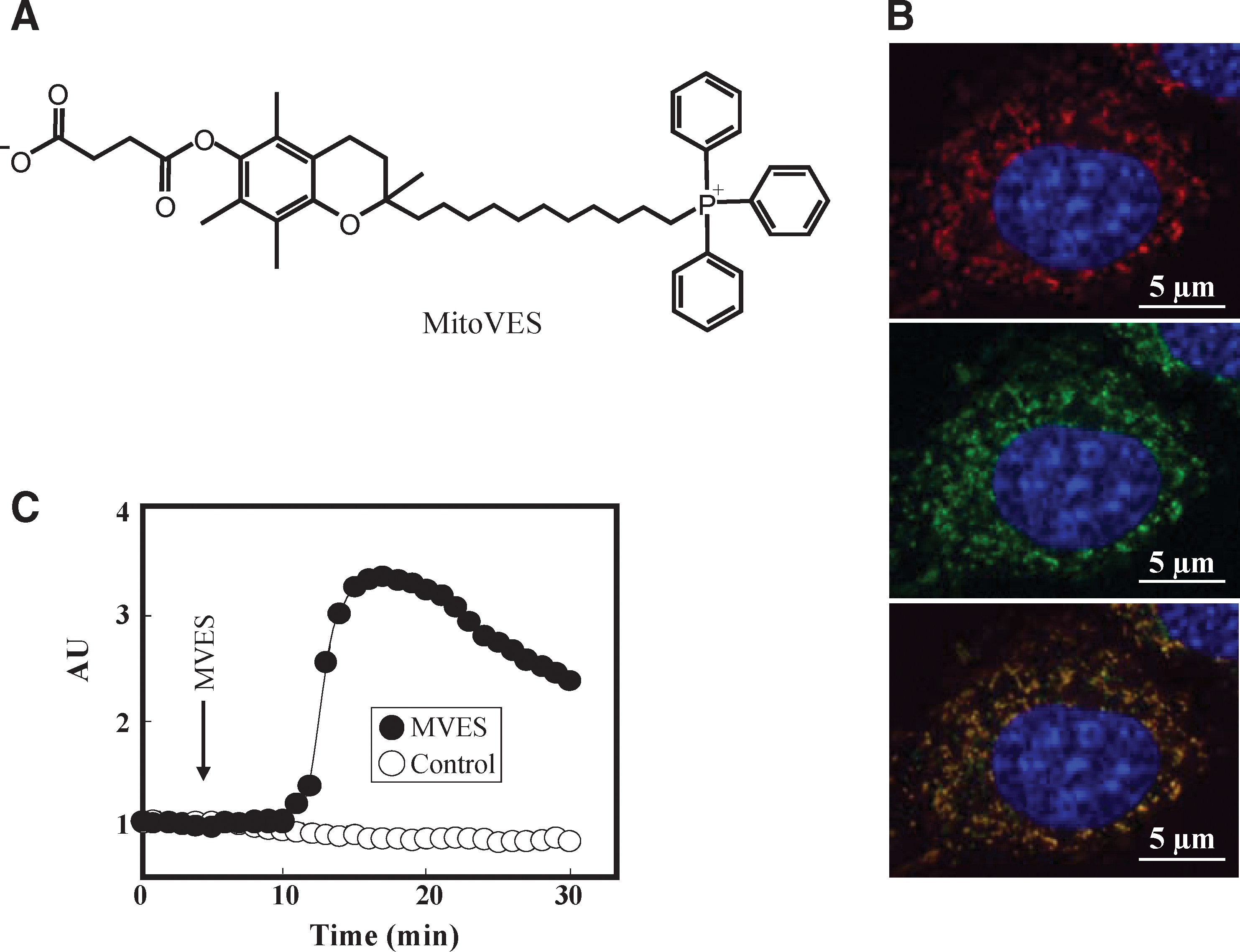
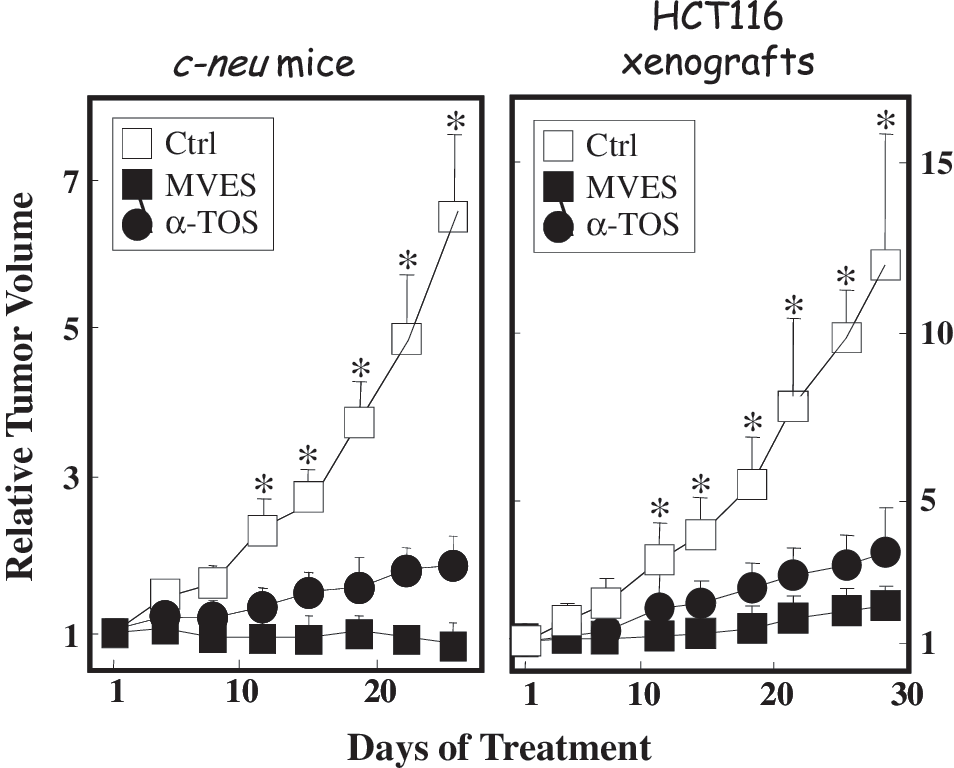
The knowledge that α-TOS targeted CII's UbQ-binding sites led us to design a more specific mitochondrially targeted analogue, MitoVES. The structure of this compound (Fig. 5A) causes it to compartmentalize to mitochondria (Fig. 5B), particularly to the surface of the MIM facing the matrix (57, 58). Consequently, cells treated with MitoVES respond rapidly by generating ROS within several minutes (Fig. 5C) which translates into much greater apoptogenic activity in cancer cells and superior anticancer efficacy in animal models (Fig. 6). MitoVES features physicochemical features endowing it with intriguing properties, as depicted in Figure 7. Thus, MitoVES selectively targets the Qp site of CII such that the IC50 value for electron transfer from CII to CIII was found to be 1.5 μM, whereas the IC50 value for conversion of succinate to fumarate was much greater at 70 μM.
Thenoyltrifluoroacetone (TTFA), the archetypal inhibitor of the UbQ binding sites of CII, also induces an increase in oxidative stress and calcium levels (133). However, in contrast to α-TOS, its practical application as an anticancer agent is restricted, as it is also very toxic to noncancerous cells, such as hepatocytes (226). Interestingly, the anti-diabetic and anti-inflammatory drug, troglitazone, which contains the α-tocopheryl moiety similar to α-TOS, also interferes with CII activity (186). The drug was registered, but was subsequently withdrawn from the clinic due to liver toxicity (85). Another group of compounds targeting the UbQ site(s) of CII are atpenins, natural products isolated from bacteria, structurally similar to UbQ (136). The potential use of these agents as anticancer drugs has yet to be established.
Other compounds, including malonate and 3-nitropropionic acid (3NP), that specifically inhibit the succinyl dehydrogenase activity in the SDHA subunit of CII are also able to stimulate ROS production and induce cell death. Nevertheless, their use for cancer treatment may be problematic, because they exhibit neurotoxic properties, and in experimental animals they produce symptoms similar to Huntington disease, a severe human neurodegenerative disorder (23, 81, 92, 125). Hence, with regard to future anticancer drug development, the UbQ-binding sites in subunits SDHC and SDHD may prove to be more ideal targets than sites in the SDHA catalytic domain, given the associated problem of neurotoxicity. Inhibiting SDH activity also blocks the TCA cycle as well as the transfer of electrons to the ubiquinone pool and hence, the metabolic TCA cycle arrest could have much more serious consequences for the cell than the selective inhibition of electron transfer at the UbQ sites. In the latter case, the TCA cycle would not become fully inhibited, but the disruption of the electron flow at the UbQ sites would still allow for efficient superoxide formation.
The eligibility of CII as a target for anticancer drugs is not compromised by the occurrence of mutations in subunits SDHB, SDHC, and SDHD in cancer. These mutations are associated only with relatively infrequent and nonaggressive familiar paragangliomas and pheochromocytomas, and in most common cancers such as breast cancer, only one in a million patients carries a mutation in CII (165, 179). Therefore, CII represents an invariant target in most cancers.
Complex III
The electron flows from CI and CII are integrated at CIII, also known as the cytochrome bc1 complex or ubiquinone-cytochrome c oxidoreductase, which mediates transfer of electrons to another electron carrier, cytochrome c. CIII, similarly to CI, pumps protons across the MIM, contributing to the proton gradient that forms the mitochondrial membrane potential. Along with CI, CIII is considered to be a major source of superoxide radicals due to electron leakage, which is further increased either by the inhibition of the UbQ-binding sites or by an increase in the mitochondrial membrane potential (35, 61). The crystal structure of mammalian CIII containing 11 protein subunits (including one mitochondrially encoded) has been solved (94, 215) and the mechanisms for proton pumping and electron transfer have been determined. CIII contains two binding sites for UbQ involved in the ‘Q cycle’ in which two molecules of UbQH2 are oxidized at the Qo site per one molecule of UbQ reduced at the Qi site, allowing CIII to transfer protons across the MIM (40). Specific inhibitors of the Qi site such as antimycin A induce superoxide production at the Qo site, but Qo site inhibitors themselves do not have this effect (61). Antimycin A effectively eliminates cancer cells by apoptosis (214), although antimycin A is also a BH3 mimetic (201). Interference with the anti-apoptotic proteins of the Bcl-2 family could make antimycin A an interesting option for targeting cancers resistant to apoptosis due to upregulation of Bcl-2/Bcl-xL.
Another compound, adaphostin, similarly to antimycin A, also targets the Qi site of CIII to induce oxidative stress and apoptosis in cancer cells (115). Originally, adaphostin, a dihydroquinone derivative, was identified as an inhibitor of the Bcr-Abl kinase in chronic myeloid leukemia, but turned out to be also highly effective against Bcr-Abl-negative cancer cells as well as against cells with a mutated Bcr-Abl, resistant to the Bcr-Abl kinase inhibitor drug, imatinib mesylate. The action of adaphostin depends on stimulating superoxide production and appears to be relatively cancer cell-specific (31, 32, 47). Caspase-independent inhibition of the Raf1/MEK/ERK, Akt pathways and activation of the JNK and p38 MAP kinases have commonly been shown to occur during adaphostin-induced apoptosis (222) and the activation of p38 MAP kinase also results in cell cycle arrest in G1 (143).
Plant-derived polyphenols, represented by resveratrol (trans-3,4',5-trihydroxystilbene), are also efficient respiratory chain inhibitors of several ETC complexes, especially CIII, and induce mitochondria-mediated apoptosis (194, 229). The dichotomy of cytoprotective and cytotoxic effects associated with resveratrol (also linked with the beneficial health effects of red wine) may be concentration dependent and related to the widespread action on multiple cellular processes. In any case, the chemoprotective activities of resveratrol have been observed in various preclinical cancer models (7, 88, 229), and the anticancer potential of resveratrol is currently being investigated in several ongoing clinical trials, primarily in colorectal cancer (e.g., NCT00256334, NCT00920803, NCT00433576). Another representative from the pholyphenol group is xanthohumol, derived from hops, which also effectively induces mitochondrial superoxide generation and apoptosis by CI and CIII inhibition associated with cytochrome c release from mitochondria (188). Xanthohumol shows other activities as well, including inhibiting the NFκB and Akt pathways, stimulating the apoptotic arm of the unfolded protein response, suppressing angiogenesis, and repressing tumor growth in experimental cancer models (3, 89, 127, 135, 141).
Isothiocyanates are natural compounds found in cruciferous vegetables such as broccoli, watercress, or cabbage, and like the polyphenols, they provide beneficial effects for preventing cancer. For example, benzyl isothiocyanate (BITC) inhibits CIII in cancer cells, initiating ROS-mediated apoptosis followed by translocation of Bax into the mitochondria in a JNK and p38 MAP kinase-dependent manner (148, 173, 216). These effects were specific for cancer cells with minimal effects on normal cells (216), a beneficial and advantageous property for any anticancer drug. BITC suppressed angiogenesis and xenograft tumor growth in vivo in experimental cancer models and was shown to be an efficient chemopreventative (209, 210, 218). Interestingly, phenethyl isothiocyanate (PEITC), a related compound, also selectively induced apoptosis in cancer cells by ROS accumulation, even though this drug mainly appeared to deplete the glutathione pool by inhibiting glutathione peroxidase (198). Given the similarity between both compounds, it is likely that PEITC will also directly induce the production of ROS from the mitochondrial ETC (26).
Finally, CIII is also inhibited by lamellarin D, a hexacyclic pyrrol alkaloid derived from marine invertebrates and has been shown to act as a topoisomerase I inhibitor (48, 65). Mitochondrial respiration was disrupted at the level of CIII by lamellarin D treatment, which was accompanied by the loss of the transmembrane potential, calcium release, and induction of mitochondria-dependent apoptosis, all of which were relatively specific for cancer cells (9, 10, 105). Surprisingly, mitochondrial ROS generation was not stimulated by lamellarin D, suggesting a mechanism different from that of the CIII-targeting compounds discussed above. Interestingly, treating cancer cells with a modified lamellarin D derivative, PM031379, induced mitochondrial ROS generation and successfully induced apoptosis in a non-small cell lung carcinoma cell line, U1810, that was chemoresistant to the parental compound, lamellarin D (74). The difference between these two compounds was that whereas both induced the initial mitochondrial-specific apoptotic steps, only PM031379 induced the nuclear translocation of the apoptosis-inducing factor (AIF) in a ROS-dependent manner.
Complex IV
The final step in electron transport via the ETC is performed by CIV, also known as cytochrome c oxidase. This complex removes electrons from cytochrome c and donates them to molecular oxygen, producing water while, at the same time, transporting protons across the MIM. Along with CI and CIII, CIV directly contributes to the maintenance of the proton gradient and the mitochondrial membrane potential. Of the 13 subunits that form CIV in mammals, three are encoded by the mitochondrial genome and 10 by the nuclear DNA (178). As the final element in the ETC, CIV has a physiologically important role in regulating ETC activity. For example, CIV is regulated by the HIF1 transcription factor in hypoxia, which results in an isoform switch increasing CIV activity (71). Furthermore, CIV is also regulated by nitric oxide, which competes for oxygen binding and inhibits mitochondrial respiration (140).
Surprisingly, there are not many anticancer agents that directly target CIV, in spite of the important role of this complex in regulating ETC activity. In fact, many of the compounds mentioned in connection with CIV actually modulate the expression of its various subunits at the level of mRNA expression and thereby indirectly modulate CIV mediated apoptosis. For example, N-(4-hydroxyphenyl) retinamide (4HPR), also known as fenretinide, downregulates CIV by destabilizing the mRNA transcript for subunit III of CIV, inducing ROS-mediated apoptosis in cancer cells (220). A number of clinical trials are currently underway to evaluate the merits of this compound for treating different types of cancer. Anticancer drugs from the anthracycline family, such as doxorubicin, also downregulate the expression of CIV subunits II and V at the level of mRNA. However, in this case, the CIV downregulation was associated with cardiotoxic side effects of these drugs and did not appear to relate to their anticancer activity (33). Interestingly, direct interaction with CIV and inhibition of its activity by doxorubicin and daunomycin has also been reported (163).
Induction of apoptosis in a leukemic cell line with the antioxidant and lipoxygenase inhibitor 3-tert-butyl-4-hydroxyanisole (BHA) was shown to also target and inhibit CIV directly at relatively high concentrations (185) but these effects could not be replicated with mitochondria derived from murine skeletal muscle (69, 158). Similarly, inhibition of CIV activity and apoptosis induction was also observed for the lipophilic cationic compound N-retinyl-N-retinylidene ethanolamine (A2E) in mammalian retinal pigment epithelia, as well as in other cells. However, A2E is associated with age-related macular degeneration and its potential use in cancer has not been explored (180, 192). With respect to CIV function in cancer cells, perhaps the most relevant compounds for current cancer therapy could be the porphyrin photosensitizers, such as Photofrin II, which in photodynamic therapy inhibit CIV, resulting in ROS production required for successful treatment (91). Similarly to the antioxidant MitoQ (103), the mitocan MitoVES (57, 58), polyphenols (19, 132), and manganese porphyrin superoxide dismutase mimics (6), photosensitizers were successfully delivered into mitochondria using cationic tags or peptide conjugates, resulting in increased phototoxicity (42, 118, 183).
Complex V
The proton gradient maintained by complexes I–IV drives the conversion of ATP by CV—the ATP synthase (F1FO-ATPase), which attaches inorganic phosphate to ADP and thereby supplies the cell with essential energy in a form utilized in many cellular processes. CV functions as a miniature molecular engine with two parts. Controlled dissipation of the proton gradient by the transmembrane FO portion provides torque that drives the rotational movement of the F1 portion of the enzyme, resulting in the synthesis of three ATP molecules per one 360° turn of the enzyme (52, 101).
Inhibiting the FO portion of CV with oligomycin or its derivatives can induce apoptosis, but its selectivity for cancer-derived cell lines is limited, indicating that targeting CV in this manner may not be a very effective intervention for cancer (174, 175). Interestingly, oligomycin treatment can reduce the availability of ATP for caspase activation or changes in intracellular pH which will under certain conditions prevent apoptosis and/or switch to necrotic cell death. (63, 119, 126, 223).
CV is also a molecular target of resveratrol and related compounds such as piceatannol. Crystal structures of the F1 subcomplex bound to resveratrol and piceatannol have been solved (77, 227) and this adds CV to the growing number of intracellular targets for resveratrol, providing the basis for the complexity of effects associated with this polyphenol. Resveratrol binding to CV may partly explain how this drug can promote the induction of apoptosis in cancer cells (194).
CV can also be inhibited by the metabolite 3,30-diindolylmethane (DIM) derived from the compound indole-3-carbinol (I3C) found in cruciferous vegetables. DIM stimulates ROS formation and induces apoptosis in a JNK and p38 kinase-dependent manner, as well as inhibiting the cell cycle by upregulating the p21 cell division kinase inhibitor (82). In addition, DIM modulates the aryl hydrocarbon receptor and displays anti-proliferative, pro-apoptotic, and anti-angiogenic activities in a number of cancer cell lines and experimental tumor models (172). Clinical testing of DIM verified its safety in human subjects, and it is used for treatment of recurrent respiratory papillomatosis, a benign noninvasive neoplasia caused by infection with the human papilloma virus (171, 212). Clinical trials in cancer patients are ongoing, including trial numbers NCT00212381 in cervical dysplasia and NCT00888654 in prostate cancer.
Bz-423, a 1,4-benzodiazepine derivative, is another inhibitor of CV activity which acts in a noncompetitive manner to induce superoxide production, albeit associated with less ATP depletion than for oligomycin (97). Bz-423 is effective against a number of cancer cell lines, inducing cell cycle arrest and apoptosis in a Bax- and Bak-dependent manner, associated with superoxide-dependent apoptosis signal-regulating kinase (ASK1) release from thioredoxin and JNK activation (20, 21). It is also currently being considered for the treatment of psoriasis. Last but not least, CV is targeted by lipophilic cationic dyes such as rhodamine 123 (27, 138) with advantages that they are more readily accumulated by cancer cell compared to normal cell mitochondria (49, 138, 190), which should provide greater anticancer specificity. Indeed, selective elimination of cancer cells and prevention of tumor growth has been observed for rhodamine 123 (15, 16, 90) which in phase I clinical trials of patients with hormone-refractory prostate cancer showed good retention in the prostatic tissue. However, rhodamine 123 did not show therapeutic benefits because its effects were not statistically significant at the maximum tolerated dose (99). A rhodamine analogue, MK-077, has entered into phase I clinical trials, but its administration was associated with hypomagnesaemia and renal toxicity (25, 168).
Mechanism of Action and General Consideration for ETC Targeting Mitocans
Mitocans, compounds targeting cancer cell mitochondria and in particular, the mitochondrial ETC, constitute a promising group of cancer therapeutics. The advantage of the ETC in this context is that it is still fully or partially active in most tumors and that the mutation rate in the ETC complexes is relatively low. The only ETC complex that shows nuclear-coded mutations with links to cancer is CII, giving rise to relatively benign and/or infrequent neoplasias such as pheochromocytomas and paragangliomas (83). Certain subsets of cancers (about 15%) will also harbor nonsilent mitochondrial DNA mutations (116), and these mutations may compromise the induction of apoptosis due to nonfunctional ETC complexes that contain mitochondrially encoded subunits (112, 120). Even in this situation, the targeting of unaffected complexes (such as CII, with all subunits coded by the nucleus) may be plausible, as demonstrated by efficient killing of cells without functional CI by the CII inhibitor α-TOS (56, 59). It should be noted, however, that the efficiency of apoptosis induction by CII-interfering agents in cells with ETC compromised downstream of CII (e.g., in CIII, CIV) or in CV has not yet been examined. It can be anticipated, though, that targeting CII will induce apoptosis in cancer cells regardless of the potential mutation in the downstream components of the ETC, since targeting CII causes generation of ROS that, in turn, induce apoptosis (56, 57, 59, 197, 204).
One of the pivotal events in apoptosis induction by ETC-targeting mitocans is the induction of ROS. This is particularly apparent for mitocans directed at CI–CIII, and is exemplified by numerous studies that show the inhibition of apoptosis induction by antioxidants. The mechanism of ROS generation (mostly in the form of superoxide) from the ETC complexes is highly complex and is not yet fully clarified for isolated mitochondria, and only vaguely inferred for mitochondria in cells (24). Similar lack of clarity persists concerning the exact sequence of events after the initiation of ROS formation. An attractive option is that ROS trigger general mechanisms common to many mitocans targeting the ETC complexes. One of these mechanisms could be direct opening of the permeability transition pore, followed by apoptosis (128). Besides this direct effect, ROS may also diffuse into the cytoplasm in the form of hydrogen peroxide and activate signaling processes via the ASK1- and or MST1/Hippo-controlled pathways. While the ASK1 pathway, activated in response to ROS by the release of the ASK1 kinase from thioredoxin, has been known for some time (131), ROS-mediated apoptosis induction via the Mst1/Hippo-FoxO1-NOXA signaling pathway has only recently been described for the CII inhibitor α-TOS (204). This pathway is conserved in higher organisms and is activated by the addition of hydrogen peroxide. It may thus represent, similarly to the ASK1 pathway, a general mechanism of cellular response to sudden increase of intracellular oxidative stress with Mst1/Hippo auto-activation in the presence of ROS. However, the involvement of this pathway in apoptosis induction has as yet not been replicated for other compounds targeting the ETC.
Despite the possible common mechanism, a number of mitocans discussed here also possess other activities unrelated to the ETC. For example, antimycin A and α-TOS function as BH3 ‘mimetics'(181, 201), deguelin inhibits HSP90 (104, 157), tamoxifen is an estrogen receptor modulator (213), metformin stimulates AMPK (29), and resveratrol exerts a variety of effects (88). Even though some of these activities may result in apoptosis induction, others may be harmful, as demonstrated for the CI inhibitors rotenone and MPP+, which also disrupt microtubules. This interference with microtubules results in undesirable neurotoxicity and Parkinson's-like syndromes (37). Targeting of mitocans directly to mitochondria using lipophilic cations or other methods, as recently demonstrated for vitamin E analogues (57, 58), polyphenols (19, 132), and porphyrins (42, 118, 183), may be a route to avoid some of the unwanted side-effects unrelated to mitochondrial activity of the compounds. This represents an important development in the field, a paradigm shift in cancer therapy.
Conclusions
Mitochondria represent, due to their distinctive nature in a number of cancers, intriguing therapeutic targets across the landscape of cancers, and mitocans may thus provide a valuable therapeutic option for the future. Although some mitocans, in particular those targeting CI or CII, exert undesirable neurotoxic side effects, a number of these compounds are selective for malignant cells. The selectivity can be further improved by mitochondrial targeting, as exemplified, for example, by MitoVES. Moreover, the availability of invariant targets combined with the preference most mitocans show for cancer cells provides an incentive for further development of these exciting compounds, which may become an essential component of our anticancer ‘tool box’ of the 21st century.
Footnotes
Acknowledgments
This work was supported in part by grants from the Grant Agency of the Academy of Sciences of the Czech Republic (KAN200520703) and the Grant Agency of the Czech Republic to JN.