
Abstract
Oxidoreductases and metalloproteins, representing more than one third of all known proteins, serve as significant catalysts for numerous biological processes that involve electron transfers such as photosynthesis, respiration, metabolism, and molecular signaling. The functional properties of the oxidoreductases/metalloproteins are determined by the nature of their redox centers. Protein engineering is a powerful approach that is used to incorporate biological and abiological redox cofactors as well as novel enzymes and redox proteins with predictable structures and desirable functions for important biological and chemical applications. The methods of protein engineering, mainly rational design, directed evolution, protein surface modifications, and domain shuffling, have allowed the creation and study of a number of redox proteins. This review presents a selection of engineered redox proteins achieved through these methods, resulting in a manipulation in redox potentials, an increase in electron-transfer efficiency, and an expansion of native proteins by de novo design. Such engineered/modified redox proteins with desired properties have led to a broad spectrum of practical applications, ranging from biosensors, biofuel cells, to pharmaceuticals and hybrid catalysis. Glucose biosensors are one of the most successful products in enzyme electrochemistry, with reconstituted glucose oxidase achieving effective electrical communication with the sensor electrode; direct electron-transfer-type biofuel cells are developed to avoid thermodynamic loss and mediator leakage; and fusion proteins of P450s and redox partners make the biocatalytic generation of drug metabolites possible. In summary, this review includes the properties and applications of the engineered redox proteins as well as their significance and great potential in the exploration of bioelectrochemical sensing devices. Antioxid. Redox Signal. 17, 1796–1822.
I. Introduction
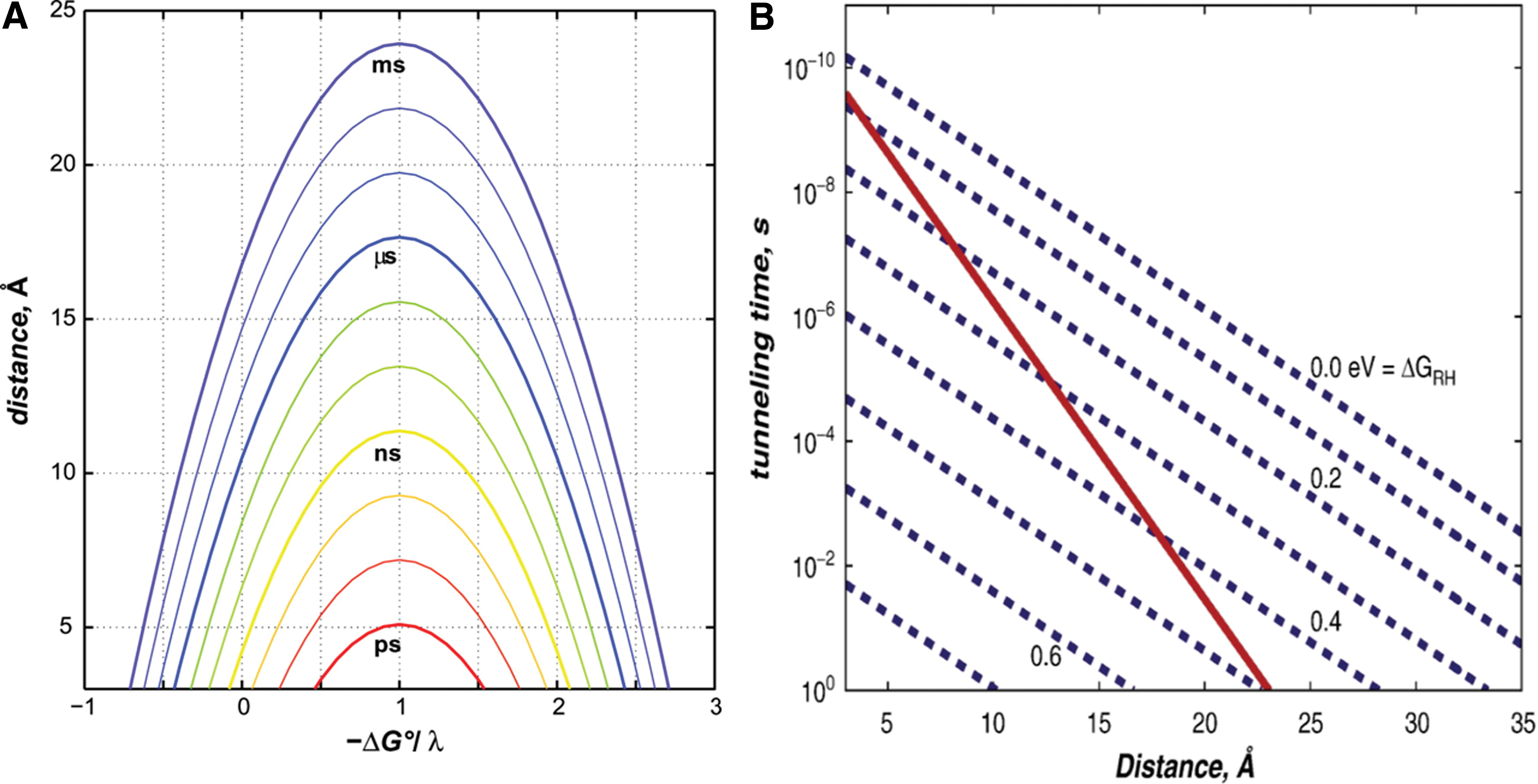
When proteins need to transfer electrons with high directional specificity over distances>14 Å, multistep tunneling through chains of redox centers occurs. Chains are defined as groups of single-electron redox centers that are more widely separated, having fewer neighbors. Redox-active amino acids such as tyrosine (Tyr) or tryptophan (Trp) can form chain elements, but usually a chain contains nonamino-acid cofactors, such as a planar heme or a cubic iron–sulfur (FeS) center. In some proteins, folding patterns may allow two catalytic sites to be placed sufficiently close to each other that no connecting chain is needed to assure physiologically competent tunneling rates. Although the chemistry at catalytic sites can be complex and the speed of catalytic turnover is difficult to predict, the intermediate chains appear rather simple. A free electron localized on a redox center has a wavefunction that extends beyond the cofactor in all directions, decaying exponentially into the electrically insulating amino-acid medium. Since the tails of these wavefunctions encounter other nearby redox centers, the electron can tunnel from one center to another, with a rate that decreases exponentially with distance (106, 107).
A. Redox properties of naturally occurring/wild-type proteins
Simple electron transferases include cytochromes (Cyt), blue-copper proteins, FeS proteins, and flavodoxins (Fld). Tyr and Trp radicals may serve as electron-transfer intermediates, as the redox window is not sufficiently oxidizing or reducing to react with the peptide backbone and the other side chains. Therefore, redox-active proteins can be compared with wide band-gap semiconductors with “dopants” (redox active species) that provide electron localizing sites. We briefly describe some of the different classes of naturally occurring or wild-type redox proteins.
1. Metalloproteins
In metalloproteins, which contain metal ion cofactors, metal-binding sites are responsible for catalyzing important biological processes, such as photosynthesis, respiration, water oxidation, molecular oxygen reduction, and nitrogen fixation. The most biologically relevant metals are the first-row transition metals, namely, iron, nickel, copper, vanadium, and manganese. Occasionally, transition metals combine with second-row transition elements such as molybdenum to form heterogeneous clusters.
a. Iron/heme proteins
Iron is abundant in biomolecules, and exists in the ferrous Fe(II) or ferric Fe(III) oxidation states. Iron-porphyrin complexes, known as hemes, form the prosthetic groups of a number of proteins. These hemoproteins exhibit an impressive range of biological functions, which include simple electron-transfer reactions, oxygen transport and storage, oxygen reduction to hydrogen peroxide or water, oxygenations of organic substrates, and the reduction of peroxides. This diversity of function is often extended further by combining heme groups with other cofactors, for example, flavins and/or metal ions. Such combinations frequently allow heme cofactors to couple electron transfers with other processes, such as the translocation of protons or the reduction/oxidation of other molecules. This versatility in function is made possible by a combination of differences in both the polypeptide and heme constituents of the various hemoproteins.
In heme proteins, such as hemoglobin, the oxidation state determines the ligands that bind to the iron of the active center heme: O2, CO, and NO bind to the ferrous form, whereas H2O, OH−, and CN− are ligands of the ferric state.
Myoglobin (Mb), one of the most widely studied proteins, is a heme protein that is found in the muscle tissue of most mammals and vertebrates, and is responsible for oxygen storage and transportation. The iron of the heme co-ordinates to four nitrogen atoms of the porphyrin ring and to the protein via the nitrogen from a histidine (His) residue. The sixth co-ordination site of the heme iron is available to bind exogenous molecules such as oxygen, carbon monoxide, cyanide, or water depending on the valence state of the iron (132). Although Mb does not function biologically or physiologically as an electron carrier, it has been frequently used as a model system that studies the electron-transfer properties of proteins.
Cyt P450, a superfamily of heme-containing monooxygenases, serve as versatile biocatalysts by converting a broad variety of substrates and catalyzing a great number of important chemical reactions in both biological systems and industrial applications. P450s are ubiquitously found in all domains of life, in many bacteria and all archaea, fungi, and many higher eukaryotes. In mammalian tissues, the P450 family plays essential metabolic roles by taking part in the detoxification of xenobiotics, biotransformation of drugs, biosynthesis of critical signaling molecules such as steroid hormones, fatty acids, eicosanoids, and catabolism of exogenous compounds as a source of energy (17, 61). The exposure to certain xenobiotics leads to the induction of specific P450 proteins that maintain cellular homeostasis in vivo (126). In catalyzing monooxygenation reactions, P450s generally require two electrons from either nicotinamide adenine dinucleotide (NADH) or nicotinamide adenine dinucleotide phosphate [NAD(P)H] to reduce O2 (Eq. 1), where the electrons are transferred to the heme of P450 via a protein electron transport system, usually a reductase and a FeS protein, depending on the type of P450s.
The spin state and electronic structure of the heme prosthetic group are determinant in the catalytic processes, and the redox potential of the Fe(III)/Fe(II) equilibrium can be perturbed by the changes in the molecular environment and the axial ligation of the iron in the course of oxidation and reduction (39).
In the Cyt family, four major groups of Cyt are currently recognized:
Cyt a.: Cyt, in which the heme prosthetic group is heme a, that is, the iron chelate of Cytoporphyrin IX.
Cyt b.: Cyt with protoheme (the iron chelate of protoporphyrin IX), as the prosthetic group but lacking a covalent bond between the porphyrin and the protein.
Cyt c.: Cyt with covalent thioether linkages between either or both of the vinyl side chains of protoheme side chains and the protein.
Cyt d.: Cyt with a tetrapyrrolic chelate of iron as the prosthetic group in which the degree of conjugation of double bonds is less than in porphyrin, for example, dihydroporphyrin (chlorin; heme d), tetrahydroporphyrin (isobacteriochlorins; heme d 1, siroheme). Heme, d has also been known as heme a 2.
Most peroxidases are heme-containing enzymes that oxidize a variety of xenobiotics by hydrogen peroxide. Although peroxidases also include nonheme-containing enzymes such as glutathione peroxidases and peroxiredoxins, only heme-containing peroxidases are considered in this review. The native enzyme contains a heme, usually ferriprotoporphyrin IX, with four pyrrole nitrogens bound to the Fe(III). The fifth co-ordination position on the proximal side of the heme is usually the imidazole side chain of a His residue. The sixth co-ordination position is vacant in the native enzyme on the distal side of the heme (30).
The following cycles of reactions are involved in the oxidation of xenobiotics:
Peroxidase+ROOH→Compound I+ROH
Compound I+XOH→Compound II+XO•
Compound II+XOH→Peroxidase+XO•+H2O
b. Copper metalloproteins
There are several different co-ordination modes for copper in metalloproteins, and the metal centers in the copper proteins are labeled as Type 1, Type 2, and Type 3 based on the spectroscopic properties of the proteins. Type 1 copper sites are characterized by an intense blue color, due to a strong absorption around 600 nm, and, hence, proteins containing Type 1 copper are often referred to as “blue” copper proteins, whereas Type 2 copper sites have spectroscopic properties similar to those found for square-planar co-ordination complexes. Type 3 copper sites contain two interacting copper centers, each co-ordinated by three His residues.
The blue copper proteins, also known as single-copper cupredoxins, are a different class of electron transfer proteins from heme proteins, as there is no prosthetic group associated with the copper. The copper center is directly co-ordinated with two His residues and one cysteine (Cys) residue in a trigonal planar structure, and a variable axial ligand such as methionine (Met) or glutamine (66). Azurin (Az), an arsenate reductase, is a redox-active cupredoxins and plays crucial roles in many important biological processes such as photosynthesis, respiration, cell signaling, and many other reactions in oxidoreductases (52, 125). Az has been widely studied in its electrotransfer properties and tuning its reduction potentials (18, 48, 88).
c. Zinc-finger proteins
After iron, zinc is the second most abundant transition metal in living organisms. Due to its fully occupied d shell, zinc is always present as Zn2+ in biological systems and does not change its oxidative state. In proteins, zinc ions play valuable roles in both enzyme catalysis and maintaining structure. The Zn2+ ion can stabilize the negative charges in the reaction intermediate, as in both carboxypeptidase and alcohol dehydrogenase. In catalytic sites, the zinc ion is usually exposed and bound to a solvent molecule (2). Zinc also acts as a strong Lewis acid, and can exist in several co-ordination geometries. This stability is due to zinc-finger proteins, relatively short lengths of peptide chains that fold around a central Zn2+ ion producing a compact domain (79). In many zinc-finger proteins, two zinc ions organize the structure of the protein domain; and in the most common configuration, Zn2+ is at the center of a tetrahedral co-ordination motif formed by two Cys and two His residues, creating a two-stranded antiparallel β-sheet and a short α-helix. Additionally, zinc can have one to four Cys ligands with His ligands completing a tetrahedral co-ordination; also, the zinc/thiolate clusters exist with one, two, three, and five ligand bridges (92). Zinc-finger proteins act as redox-sensitive molecular switches that affect intracellular signaling cascades (80).
d. Other metal-based (nickel, vanadium, and molybdenum) redox proteins
First-row transition metals such as nickel, vanadium, and molybdenum are required for many biochemical processes, particularly catalysis, electron transfer, and gene regulation. Transition metals are mainly useful because of their redox activity, their capability to bind and exchange ligands, and their high-charge density, which allows the polarization of substrates as well as the stabilization of transition-state intermediates. Transition metals can occur in mononuclear, dinuclear, and multiple nuclear centers. Their most frequent ligands are the nitrogen, oxygen, and sulfur atoms of the polypeptide chain; but the cofactors, such as tetrapyrroles, molybdopterins, small inorganic compounds such as carbon monoxide, and modified amino acids, are also used for both metal binding and modulating specific metal properties (45).
2. Flavoproteins
Flavoproteins are enzymes that catalyze oxidation-reduction reactions using either flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD) as a coenzyme, and they are involved in a wide range of biological processes, including photosynthesis, DNA repair, apoptosis, and bioluminescence. The flavin nucleotides are derived from the vitamin riboflavin. The fused ring structure of flavin nucleotides undergoes reversible reduction, accepting either one or two electrons in the form of one or two hydrogen atoms from a reduced substrate. When a fully oxidized flavin nucleotide accepts only one electron, then the semiquinone form of the isoalloxazine ring is produced, known as FADH
3. Quinoproteins
Quinoproteins are a class of oxidoreductase containing an amino acid-derived o-quinone cofactor, and the most typical member is pyrroloquinoline quinone (PQQ). Quinohemoproteins are quinoproteins that include a heme c moiety in addition to the quinone. PQQ-containing quinoproteins and quinohemoproteins have a common structural basis, in which PQQ is deeply embedded at the center of the unique superbarrel structure. Increased evidence for the structure and function of quinoproteins has revealed their unique position within the redox enzymes with regard to catalytic and electron-transfer properties, and also to physiological and energetic functions. The peculiarities of the quinoproteins, along with their unique substrate specificity, have encouraged their biotechnological applications in the fields of biosensing and bioconversion of useful compounds, as well as environmental treatment (93).
4. Redox-active amino-acid residue proteins
The four main electroactive amino acids, Tyr, Trp, Cys, and glycine (Gly), have been shown to form catalytically active, one-electron oxidized radicals (158). The proteins containing amino-acid redox cofactors are involved in a broad range of biochemical processes such as carbohydrate metabolism, DNA repair, nucleic acid biosynthesis, and energy transduction in photosynthesis and respiration. These enzymes typically operate at high oxidizing potentials, but despite this, several of the amino-acid radicals are remarkably stable. When incorporated into enzymes, redox features and reversibility of the catalytic protein radicals are improved relative to the properties of these species in solution (146).
B. Electron transfer in proteins
The understanding of the charge transport phenomenon through biological matrices such as proteins and DNA has generated intensive theoretical and experimental work in the past few decades. The seminal contributions of the Marcus theory, the superexchange charge transfer theory, and the definition of superior tunneling paths in proteins have had a tremendous impact on understanding biological processes in redox proteins involving electron transfer in photosynthetic reaction centers and charge transport (20, 91).
Electron transfer in proteins typically occurs between protein-bound cofactors, which include an electron donor and an electron acceptor. In the nonadiabatic theory, the electron is considered to switch between two states via a process called electron tunneling. The nonadiabatic regime is feasible for electron transfer only if there is a small overlap of the electron donating orbital and the electron accepting orbital. In an electron-transfer reaction, energy is placed in an orbital of a donor cofactor and is transferred to the acceptor via coupling with the nuclear vibrational modes of both the surrounding protein and the solvent. These nuclear vibrational modes are known as the “bath.” The final result is that the electron moves from being localized on the donor to being localized on the acceptor with some energy lost to the bath.
Single-electron transfer reactions can be found at the core of most biological redox processes. Most of these reactions involve quantum-mechanical electron tunneling between redox cofactors embedded in protein matrices separated by large molecular distances (>10 Å). The semiclassical theory of electron transfer provides a basic framework for understanding the specific rates of reaction (kET) between an electron donor (D) and an acceptor (A) held at a fixed distance and orientation. These rates depend on three critical parameters: (i) the driving force for the electron transfer (−ΔG°); (ii) the extent of nuclear reorientation (λ) in D, A, and the solvent that accompanies the formation of D+ and A−; and (iii) the electronic coupling (HAB) between the reactants [D, A] and products [D+, A−] at the transition state. The first two parameters largely depend on the chemical composition and environments of the redox centers, whereas the third is a function of the D–A distance and the structure of the intervening medium. In the case of biomolecules, the donor and acceptor cofactors are separated by large distances; thus, there is a negligible probability of the electron directly tunneling between the two orbitals. Electron tunneling is facilitated by a bridge medium which lowers the effective barrier height between the donor and acceptor orbitals by providing a series of virtual orbitals that the electron can transiently populate. The intervening protein medium between the donor and acceptor cofactors is considered to be the bridge.
The electron flow through proteins and protein assemblies in the photosynthetic and respiratory machinery commonly occurs between metal centers and other redox cofactors that are separated by relatively large molecular distances, often in the 10–20 Å range. Electron tunneling times should be in the millisecond to microsecond range for biological redox machines to function properly. Therefore, the maximum center-to-center distance for single-step tunneling through proteins should be no more than 20 Å, as shown in Figure 1A. The study of many redox protein assemblies suggests that charge transport needs to occur over distances that far exceed this single-step limit. In order to accomplish this long-distance tunneling, a process known as multistep electron tunneling (hopping) may be utilized. It shows that coupled reactions, particularly those with endergonic steps, can deliver electrons or holes rapidly to very distant sites. Requirements for functional hopping include the optimal positioning of redox centers and the fine tuning of reaction-driving forces. Modeling the kinetics of electron hopping is a straightforward problem that can be analytically solved without employing simplifying approximations (53). Let us consider the two-step tunneling reaction defined as
II. Engineering Redox-Active Proteins
Much effort has been made over the past several decades to understand the structure and function of redox-active proteins and to replicate or modify these properties through protein engineering to yield enhanced structural stability and functional capabilities. Nowadays, it is feasible to generate combinations of amino acids with any sequence, one amino acid at a time, due to the progress made in molecular biology and synthetic chemistry.
This de novo or “bottom-up” approach of protein design can be used to elucidate structural features that may remain hidden in biochemical and biophysical studies. The challenge lies in selecting appropriate sequences that will be the conformational free-energy minimum for the polypeptide chain and favor soluble, well-folded, and globular structures that can perform specific functions. Computational and experimental strategies are used to narrow in on specific sequence modifications that will fold into conformations with the lowest free energy. An examination of natural proteins has helped us discern two universal features. First, proteins fold in a manner that there is a simultaneous burial of hydrophobic residues and an exposure of hydrophilic residues from solvents. Second, there are abundant secondary structures such as α-helices and β-sheets. Both these characteristics play a crucial role in protein stability and, thus, they need to be considered during the development of de novo proteins (13). The de novo method is applied to redox proteins in order to better understand and improve the role of specific motifs in redox activity.
In redox proteins, there are different types of redox centers composed of metal atoms such as iron or copper or organic moieties such as FMN or FAD, which are capable of accepting and donating electrons. Nowadays, there is a broad spectrum of knowledge available about the catalytic mechanisms of metalloproteins and redox-active enzymes. The protein environment plays a critical role in controlling the redox properties and in the studies of native metalloproteins, and their variants have provided a great deal of insights into the fine tuning of the protein reactivity via both genetic and chemical approaches. These studies have also laid the groundwork for rationally modulating metal atoms or organic moiety reactivity toward the goal of providing novel chemical and biological catalysts. Progress in functional studies on designed redox protein active sites has demonstrated a remarkable control of electron-transfer kinetics and thermodynamics. In addition, advances in protein-ligand co-rdination chemistry show the practical approaches to produce complex metalloproteins containing more than one redox cofactor. Manipulating electron transfer between such recombinant proteins, with the aim of controlling the activity of the redox-active elements beyond their cellular physiological function, will allow us to engineer the protein-protein and protein-electrode interactions for the successful design of catalytic, electro-active surfaces for sensor construction (143). Furthermore, it is possible to design metalloproteins and redox proteins with unprecedented structural and functional properties (89). Equipped with insights from design processes, it may be possible to design new metalloproteins with improved properties such as higher stability and greater efficiency, or to impart them with functions not found in nature for use in an even wider range of biotechnological and pharmaceutical applications.
A. Methods available for designing artificial redox proteins
Several complementary strategies have been implemented for the creation or optimization of artificial proteins (82). We have aimed at providing an account of the genetic and chemical tools available for protein engineering with specific applications toward redox protein engineering.
1. Rational design
Rational design has been exploited as a powerful approach for unveiling the basic principles of redox-active mechanisms. The rational design of proteins is driven by the hypotheses of researchers and computer modeling programs based on available crystal structures of the proteins of interest to create new molecules with a certain functionality.
Rational design can also be accomplished with computational chemistry. The goal of in silico or computational design is to achieve the unbiased, quantitative design of protein structure and stability that does not depend on system-specific heuristics or subjective considerations. The approach is universally applicable and does not limit design efforts to specific protein folds or motifs. The use of new computer algorithms and ever-faster computers allows the rapid screening of vast combinatorial libraries without laborious modeling of individual structures (33). When there is no structural homology between the template protein and the target redox protein, a computer-aided design using programs such as Metal-Search and Dezymer has become indispensable toward designing novel redox functionalities (15, 37).
In the applications of rational design, site-directed mutagenesis is a commonly used biochemical technique that can be employed to study the function of engineered proteins. From a technical point of view, directed mutagenesis allows the production of any variation in a protein sequence in a fast and accurate manner. The loss of function accompanied by certain mutations (usually to highly conserved residues) allows the identification of residues that are essential for function. There are numerous reports of the site-directed modification of redox-related residues, such as Cys, Met, and His, in order to characterize the effect of the side-chain modification on protein behavior. Although serving a different purpose, the same mutagenesis techniques can be used in protein design to impart new functions into a protein scaffold by introducing residues that bind redox active ions, cofactors, or residues.
2. Directed evolution
Directed evolution is commonly used in protein engineering to harness the power of Darwinian selection in the evolution of proteins with desirable properties that are not found in nature, which can be performed both in vivo and in vitro (96). This method has been typically contrasted with rational design with parallel significance. The advantage of this technique is that the knowledge of structural data and the relationship between sequence, structure, and mechanism is not required for a fast generation of a huge number of mutants by mimicking Darwinian evolution (68). Directed evolution is very valuable in discovering the fundamental design principles of electron transfer in redox proteins when applied to proteins with known structures. In a directed evolution experiment, a pool of mutated enzymes is created via DNA modification, and the resulting proteins with beneficial properties are identified by selection or screening. This iterative process is repeated until the desired trait is improved. Several redox-active proteins have been submitted to directed evolution protocols for the selection of diverse phenotypes, frequently for industrial and environmental applications (6, 81).
The polymerase chain reaction technique used to amplify the DNA of the redox-producing mutants is error prone, and, thus, the availability of a suitable detection method is paramount. The direct estimation of product is the most reliable way of assessing the success of an evolved enzyme. However, this process is time consuming, labor intensive, and expensive due to the cost of reagents needed. For redox-active enzymes, the most frequent alternative methods are based on the estimation of the cofactors, cosubstrates, and surrogate substrates used (135). A more systematic technique based on the combinatorial mutagenesis is the so called combinatorial active-site saturation test (CAST), which was successfully applied to expand the substrate specificity of lipases (112). Although CAST requires the sampling of a large number of libraries, the process can be improved by the use of intelligent mutagenic DNA oligonucleotides. In a directed evolution experiment, a rapid screen or selection that reflects desired functions is the key to success.
3. Domain shuffling/molecular Lego
Domain shuffling has been used as a genetic engineering technique to swap or fuse functional modules among closely related proteins to mimic the natural molecular evolution in redox protein engineering. Domains of proteins with desired properties, such as electron transfer, are chosen as building blocks or modules to be fused at a genetic level, as a so-called molecular “Lego.” The novel molecular “Lego” approach makes it feasible to generate functional multidomain chimeric proteins with designed properties, beyond the restrictions imposed by the naturally occurring proteins. It serves as a prototype for engineering systems in the bioelectrochemistry exploitations. The link between the protein domains is generally achieved either by an engineered disulfide bridge or by a genetically engineered peptide linker between two proteins. The aim is to engineer electron transfer between recombinant proteins and to control the activity of the redox-active elements beyond their cellular physiological functions (119).
4. Catalytic antibodies
Antibodies are selected on the basis of their affinity toward stable antigens, whereas enzymes generally evolve maximum affinity for high-energy, transient transition states, thus stabilizing the transition state and catalyzing the reaction. By providing the immune system rate-limiting transition state of a particular reaction, it is possible to develop antibodies that behave as enzymes by binding to the substrate and promoting its transition state. One method by which this can be accomplished involves the use of a stable analog of the transition state as the antigen (108). It effectively directs immunological evolution along the same pathway as enzymatic evolution; the ultimate result is an antibody with catalytic activity. Catalytic antibodies can also be discovered by screening libraries of not only antibodies but also Fab fragments, RNAs, peptides, and synthetic organic molecules for enzymatic activities (121).
5. Protein reconstitution
Protein reconstitution is a well-practiced methodology that examines the structure-function relationship of proteins. The reconstitution method involves the elimination of the native redox-active center from the protein to yield the respective apoprotein or sometimes holoprotein. The next step is followed by the implantation of a structurally related cofactor or ion that generates the semisynthetic reconstituted protein that might exhibit new tailored functions and regulated electron-transfer properties which are absent in the native protein. The reconstitution process has been extensively used to study the mechanistic aspects of photoinduced electron transfer in proteins. The reconstitution of hemoproteins with synthetically modified heme units or metal photoporphyrin analogs enabled the synthesis of proteins with new designed photochemical, binding, and catalytic properties.
6. Incorporation of non-natural redox residues/metallocofactors
The possibility of expanding the natural repertoire of redox proteins and enhancing their properties has been pursued by protein engineers. Novel amino acids can be introduced into recombinant proteins in either a residue-specific or a site-specific manner. The most common approach that is used to incorporate amino acids into a protein relies on reading through amber stop codons (UAG/UAA/UGA) in mRNAs by a suppressor tRNA that is aminoacylated with the desired non-natural amino acid (101). The method is feasible both in vivo where systems rely on the misacylation activity of a mutant aminoacyl-tRNA synthetase, and in vitro where a suppressor tRNA is chemically aminoacylated.
Chemical synthesis allows the adept introduction of non-natural redox structures into proteins as compared with biological synthesis, which limits the components to α-amino acids (75, 76). Several reviews have documented the field of chemical synthesis of proteins and the different methods for the generation of peptide backbones and chemical ligation strategies (36, 59, 60, 100). Currently, a method known as semisynthesis is used, in which techniques originating from complementary fields such as physical biochemistry, chemical, and molecular biology are combined for protein modification. Semisynthesis is based on the chemical manipulation of the native protein to prepare homogeneous defined analogs using a limited number of steps and synthetic intermediates (43, 102, 103).
Unnatural metal cofactors and materials have been designed for introduction into small protein cavities as well as large protein cages such as viruses and ferritin (Fr). By the introduction of cofactors, the original functions of these proteins were dramatically changed, and they could be utilized for biosensing and catalysis. The method has been applied to protein–protein electron transfer systems and the utilization of a nanospace consisting of a protein assembly for the activation and concentration of metal complexes. We believe that such a study will provide intriguing implications on their applicable use as catalysts, sensors, metal drugs, and for other potential uses.
B. Electrochemical methods that characterize engineered redox proteins
Electrochemical and spectroelectrochemical methods can assist in understanding the structure-function relationships of mutated, as well as de novo metalloproteins and enzymes. Engineered redox-active proteins offer interesting opportunities for electrochemical studies of protein structure and function. Such characterization techniques would facilitate the potential use of engineered proteins in nanobiotechnological applications.
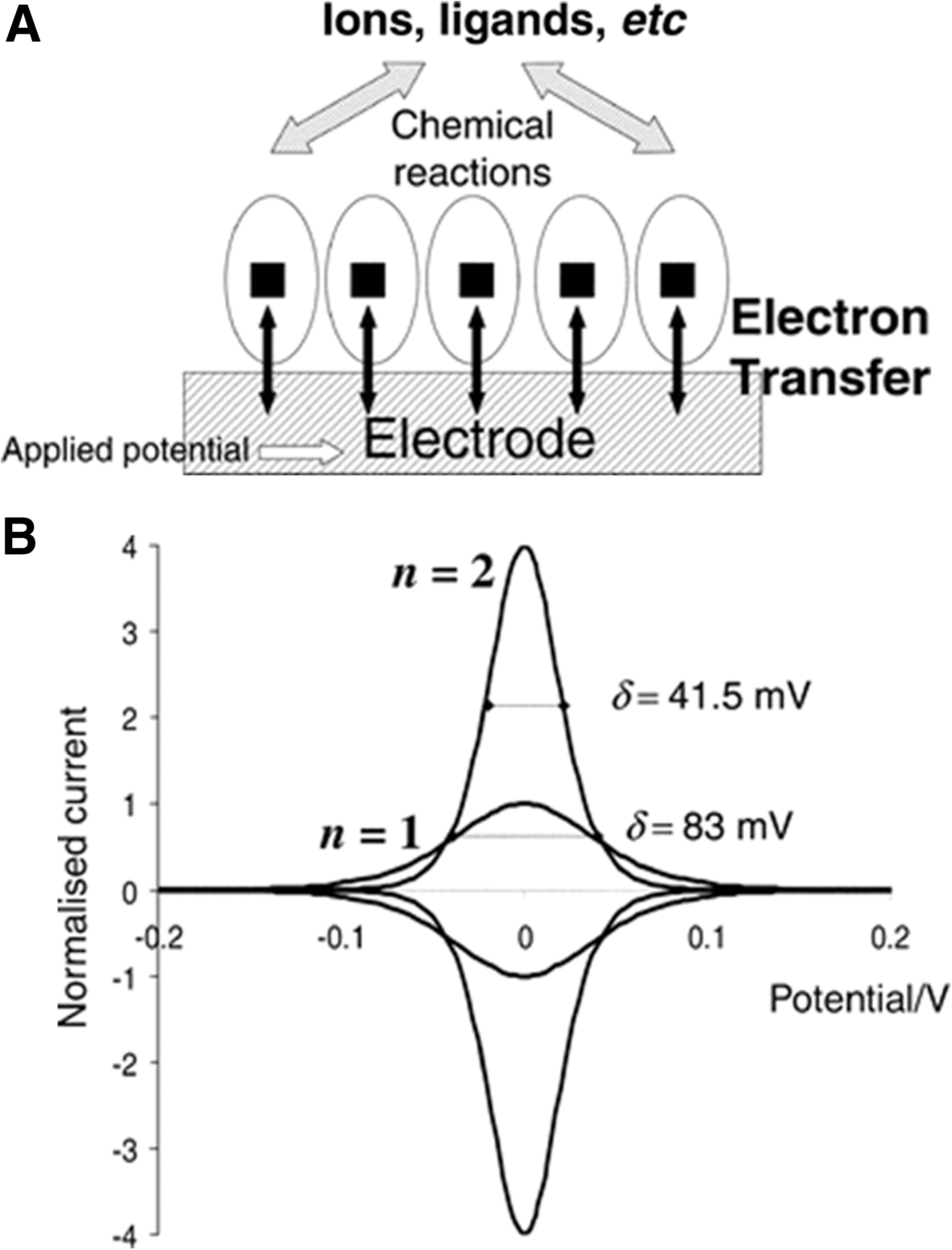
Since the past 20 years, electrochemistry has proved to be a useful means for understanding the biochemistry of metalloproteins and enzymes. It is also a powerful method for the exploitation of these proteins in biosensor and bioelectronic applications. Voltammetry can be briefly defined as an electrochemical technique in which current produced at an electrode is measured as a function of voltage (8). A typical voltammetric cell consists of three electrodes, namely working (where a redox reaction occurs), counter (inert material, places no part in a redox reaction but completes the circuit), and reference (provides a constant potential reference) electrodes and a supporting electrolyte. The redox properties of a certain sample can be evaluated by either dissolving it in the electrolyte solution or by immobilizing it on the working electrode surface. Net electron exchange between the protein and an electrode produces an electrical current that simultaneously quantifies and characterizes the underlying redox event(s). Schnepf et al. engineered four-helix bundle metalloproteins using combinatorial synthesis attached to a cellulose membrane via a cleavable linker and assembled on a cyclic peptide (120). Variations of hydrophobic amino acids and a few polar ones led to 462 distinct modular proteins, a number of which were found to bind heme-b and exhibited variations of redox potentials from −90 to −150 mV (111). Willner et al. covalently attached four-helix bundles containing two heme cofactors to gold electrodes. Cyclic voltammetry and differential pulse voltammetry (DPV) revealed redox potentials of −430 and −360 mV for the two heme cofactors and unidirectional electron transfer (151).
Direct electrochemistry of Cyt c was carried out as early as the 1970s using gold or various graphite electrodes. During the past two decades, direct electrochemistry has been developed into the method of choice for the analysis of soluble redox proteins and enzymes, a key element in protein design. Recently, this method has also been extended to a few challenging membrane proteins. Rusling and coworkers demonstrated direct reversible electron transfer for the photosynthetic reaction center from Rhodobacter sphaeroides, a purple bacteria found in fresh water or marine environments that can obtain energy through photosynthesis. In this case, the reconstituted photosynthetic reaction center in a polycation-sandwiched monolayer film (dimethyldiallylammonium chloride or ethylenimine) had a redox potential of 460 mV for its first primary donor, as measured by cyclic voltammetry. The polycations act as electrostatic glue, with their positive charges attracting the negatively charged reaction center to the similarly charged modified gold electrode (78). In another example, Baymann et al. investigated the Cyt bc1 complex from Rhodobacter capsulatus, a similar photosynthetic organism, by electrochemistry and visible/IR spectroscopy. They determined the midpoint potential of the [2Fe–2S] protein for the first time to be 290 mV at a physiological temperature (12).
Protein film voltammetry (PFV) has evolved as a very successful method in studying redox proteins, especially metalloproteins, which have been arranged as a layer on an electrode surface as illustrated in Figure 2A. PFV enables the protein to maintain its biological/catalytic functionality while undergoing fast interfacial electron exchange. This method provides an alternative perspective on complex redox enzymes that complements and extends conventional chemical studies. Figure 2B depicts the cyclic voltammetric signal that should be obtained for a simple reversible electron transfer reaction, in which the redox couple is immobilized on the electrode. Armstrong and colleagues have used PFV to elucidate the redox properties of several metalloproteins on the surface of pyrolytic graphite electrodes (4, 22, 139). An example of their work was the membrane-bound Escherichia coli dimethyl sulfoxide reductase, where it was shown that the catalytic activity of this complex Mo–pterin/FeS enzyme is optimized within a narrow window of electrode potential. The authors developed a full profile of the activity of this protein as a function of potential and found that there is an unusual potential dependence: in both directions, the activity of the enzyme reaches a maximum, beyond which the rate drops. Critical potentials were then assigned by the authors to the processes that switched the enzyme off in each direction. Finally, with the help of electron paramagnetic resonance (EPR) spectroscopy, the switch mechanism was rationalized in terms of a reaction pathway within this complex enzyme that was directed through Mo(V) (64).
Spectroelectrochemistry provides insights into the redox properties of novel metal sites with enzymatic activities engineered in existing nonredox proteins. Spectroelectrochemical titrations were also used by Springs and coworkers to investigate a library of random mutants at positions F61 and F65 of Cyt b562, one of the cofactors of Cyt c reductase participating in oxidative phosphorylation. This study showed a variability of redox potentials over a range of 105 mV, from 68 to 173 mV, interpreted as being due to the differing stability of the redox states of the mutants (162). Hellinga and coworkers used this approach to create new redox sites in thioredoxin. The structure of native thioredoxin, which does not contain metal centers, was submitted to the automated design software DEZYMER, which is able to identify regions of the protein where binding sites for nonheme iron and oxygen can be introduced by site-directed mutagenesis. Six mutants were designed to give various degrees of oxygen chemistry catalyzed by the engineered iron center (15).
III. Properties of Engineered Redox Proteins
Using the synthesis and characterization techniques just mentioned, researchers have engineered redox proteins with peculiar characteristics. As depicted in Table 1, we attempt to summarize the most relevant and interesting engineered proteins showing unique redox characteristics.
Az, azurin; Fd, ferredoxin; FeS, iron–sulfur; SCADS, statistical computationally assisted design strategy; Trp, tryptophan; Tyr, tyrosine.
A. Engineered copper proteins
Copper proteins are involved in numerous processes that impact life and the environment. The copper sites within this large class of metallobiomolecules function in a variety of ways, including transferring electrons over variable distances at divergent rates and potentials, binding and activating dioxygen for respiration and oxidation of substrates in metabolically important reactions, and reducing nitrogen oxides that are critical in the function of the global nitrogen cycle (136). The functional variability of this class of proteins is reflected in a structural diversity stemming from the variety of geometric, electronic structural, and spectroscopic features. Despite extensive research, the understanding of structure–function relationships at the molecular level is incomplete, and many questions remain unanswered concerning the detailed mechanisms and fundamental chemistry underlying copper-mediated processes in biology. One example is the reduction potential of blue copper proteins, where metal-ligand interactions in the equatorial plane and in the axial positions, geometry of the metal center, hydrophobicity around the metal center, the environment at the secondary co-ordination sphere (such as hydrogen-bonding patterns), and solvent exposure of the site are all expected to contribute to the structure-function relationship (18, 27, 52, 90).
We have previously mentioned that the blue copper Az is a 128-amino-acid protein with copper co-ordinating residues Cys112, His117, and Met121, which are located in the final 17 amino acids. With the moderate redox potential range of 265±19 mV, Az serves as an ideal system for testing the rational tuning of cupredoxin redox potentials. It has been previously demonstrated that the replacement of the axial ligand (Met121) contributes the largest changes to the redox potential (52, 125, 138). Berry et al. designed a series of unnatural amino-acid mutations to systematically probe the role of the axial hydrophobic Met ligand in the electron transfer kinetics of Az. The expressed protein ligation was used to replace the axial Met of Az with unnatural amino acids, as depicted in Figure 3. The highly conserved Met residue was replaced with the isostructural amino acids norleucine (Nle) and selenomethionine (SeM), allowing the deconvolution of different factors that affect the reduction potentials of the blue copper center. A careful analysis of the wild-type Az (315 mV vs. normal hydrogen electrode [NHE]) and its variants (M121Nle [455 mV vs. NHE] and M121SeM [340 mV vs. NHE]) obtained in this work showed that the large reduction potential variation was linearly correlated with the hydrophobicity of the axial ligand side chains. The plot shown in Figure 3 affirms that hydrophobicity is the dominant factor in tuning the reduction potentials of blue copper centers by axial ligands (18, 48).
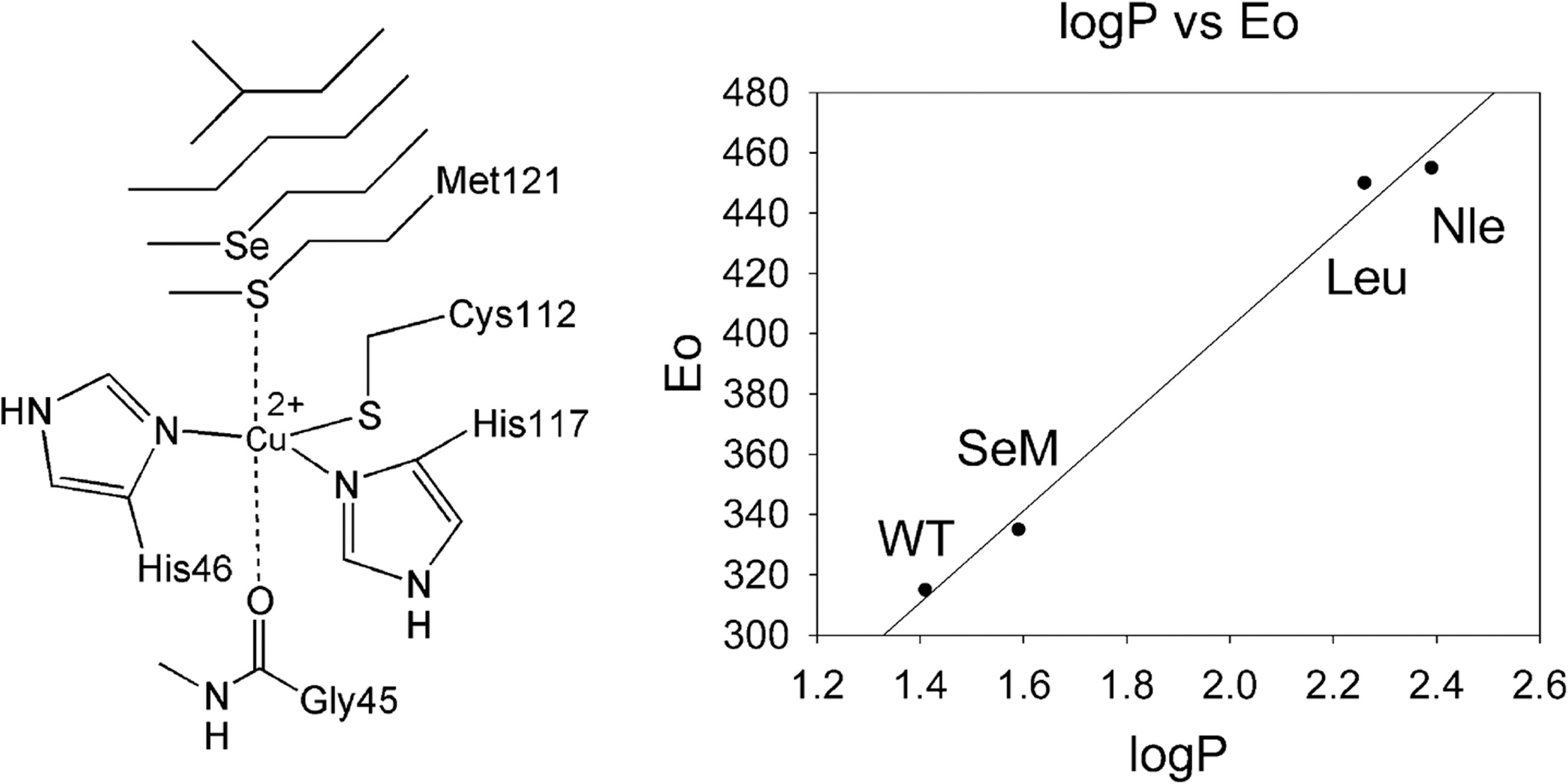
In another study, rational design was used to tune the reduction potential of Az by replacing Asn47 with Ser in rusticyanin to strengthen the hydrogen-bonding interactions between secondary structures and by additionally substituting Phe114 with Pro to affect the influence of the backbone carbonyl oxygen. The redox potentials of the Az proteins and their variants were measured by absorbing the copper-incorporated protein onto graphite electrodes and scanning for redox reactions using cyclic voltammetry (88). The results showed that the important secondary co-ordination sphere interactions were capable of tuning the redox potential of Az over a dramatic range without perturbing the metal-binding site. This study showed that it is possible to predictably control the redox potential of Az, an electron transfer protein, across the full range of cupredoxin potentials and may apply to other redox proteins.
Heteronuclear bimetallic centers, which perform important biological functions such as oxygen reduction, ligand degradation, and nitric oxide reduction, are found in many natural proteins. Designing artificial proteins that mimic the enzymes capable of complex reactions is even more challenging than designing homonuclear metal-binding sites due to the need to selectively place and bind two or more different metal centers (89).
One example is the heme-copper oxidases, which are not fully understood with regard to their structure and function, such as the role of the CuB center, the origin of spin coupling between CuB and heme, and the exact nature of the reaction intermediates. Sigman et al. designed and engineered a heme-copper (heme-CuB) center using sperm whale Mb as a substrate. This is necessary, because it is difficult to obtain large membrane-bound heme-copper oxidases in a single homogeneous form, partly because spectroscopic signals from the heme-copper center are often masked by signals from other metal-binding sites (126, 127). The design and engineering of a CuB center into Mb allows for a direct comparison of Mb and heme-copper oxidase in the same protein framework. Computer-aided designs using heme copper oxidases as a model showed which distal residues are needed to be mutated to His in order to allow the binding of CuB. Further UV-vis, elemental analysis, and EPR studies confirmed the binding of the CuB center on the protein and showed the importance of the CuB site in O2 binding and reduction as well as the significance of a proton network (123, 124).
In addition to the replacement or mutation of specific residues, organometallic compounds known to be effective in redox and catalytic reactions can be incorporated into proteins to control their redox activities. The ferrocene derivative (2-[(methylsulfonyl)thio]ethylferrocene) (Fc) is a redox active cofactor that has very low inner-sphere reorganization energy in electron transfer (0.3 kcal/mol). Az free of copper (Apo-Az), which can fold without copper, was used as the host for Fc, as it contains a highly conserved Cys residue (key entry/exit point for electron transfer) which was used to covalently attach a Fc cofactor. Hwang et al. discovered that when Fc was conjugated with Apo-Az (FcAz), it displayed improved solubility in water and higher stability of the ferrocenium ion. The incorporation of Fc increased the reduction potential of the complex from 402 to 579 mV (vs. NHE), and raising the pH from 4 to 9 resulted in a >80 mV decrease in the reduction potential of the protein-bound ferrocene (from 579 to 495 mV). Modification of the amino-acid residues present in close proximity to Fc allowed further fine tuning of the redox activity of FcAz, allowing its use as a redox agent for biomolecules to conduct electron transfer studies and in the design of new biosensors (67).
B. Engineered heme proteins
Heme proteins are one of the most diverse classes of proteins, with functions ranging from electron transfer to small-molecule transport, sensing, and activation. The redox potential of heme proteins is variable over a wide range of +400 mV (Cyt b559) to −400 mV (Cyt c3) (122). By systematically altering the characteristics of heme proteins, researchers have been able to convert one type of heme protein into another and to introduce new functions or substrate specificity (111, 141, 157). Additionally, the biological functions of the heme proteins involved in redox transfer reactions can be tuned by the protein environment through factors such as axial ligation, hydrogen bonding, out-of-plane distortion, and covalent attachment.
Heme-containing enzymes are appealing candidates for electron-transfer studies of engineered proteins with different redox properties due to the large catalytic versatility of the heme group. Cyt c is an essential component of the electron transport chain in mitochondria, which is highly conserved across the spectrum of the species. Iso-1-Cyt c from Saccharomyces cerevisiae, a species of yeast, is a member of the Cyt c family and is generally considered a suitable candidate for bioelectronic applications, because it contains a single surface Cys 102, which can be exploited for specific tethering and protein orientation, as well as a large positive charge, allowing electrostatic binding to negatively charged self-assembled monolayers (11, 23, 28). Casalini et al. studied the Met80Ala mutant of yeast Iso-1-Cyt c, in which the axial heme iron Met ligand is substituted by a nonco-ordinating Ala residue as an electrocatalyst for the amperometric biosensing of dioxygen and nitrite ions (29). This protein variant features an axial heme iron co-ordination position available for exogenous ligand binding. The Cyt c variant, as a small, engineered electron-transfer protein, was immobilized on a gold electrode, and it was found to exchange electrons efficiently, providing robust and persistent catalytic currents for dioxygen and nitrite ion reduction at different pH and temperatures (9, 109). In the cyclic voltammetry experiments, a polycrystalline gold wire was used as the working electrode, and a Pt sheet or wire and a saturated calomel electrode were used as a counter and reference electrode, respectively. The mutant protein was absorbed on the gold electrode coated with 4-mercaptopyridine by covalent protein linkage through gold wire dipping.
Cyt b562 is a small, soluble, four-helix-bundle protein with a single b-heme prosthetic group that is not covalently attached to the protein and could easily be removed and replaced by nonheme porphyrins, such as the chlorophyll analogue zinc chlorin e6 (Zn-Ce6). Cyt b562 was modified to mimic the central photochemical reaction in a photosystem of green algae and plants: one-electron transfer from a light-activated chlorin complex to a bound quinone molecule. The H63N mutant was first created to prevent the adventitious ligantion of porphyrins, such as Zn-Ce6, on the reconstitution of the apoprotein in this work. The unique quinone-binding site was introduced in the Cyt b562 protein by replacing a hydrophobic inner isoleucine with a Cys residue that could covalently attach to various quinone molecules. The bound quinones remain redox active and are able to be oxidized and re-reduced in a two-electron process. By binding different quinines to the Cyt b562 variant, the midpoint potential of semiquinone generation by one-electron reduction can be adjusted by about +500 mV. In the presented chlorin-protein-quinone complexes, light-induced electron transfer can occur from the chlorin to the bound oxidized quinone with electron transfer rates in the order of 108 s−1 (62).
As an example of molecular “Lego,” Valetti et al. used nonphysiological electron transfer partners, Fld, Cyt c553 (c553), and the heme domain of Cyt P450 BM3 (BMP) as building blocks to provide a controlled microenvironment that is capable of tuning the redox properties of the resulting complex (143). Fld is a bacterial protein including FMN, while Cyt c553 is an oridereductase dehydrogenating D-lactate. Fld was shown as a module for transferring electrons to c553 and BMP. The covalently linked assemblies of Fld-c553 and Fld-BMP complexes were constructed via engineered disulfide bridges or by fusion of the relevant genes via an engineered loop, as shown in Figure 4A. S35C and S64C mutants of Fld and M23C and G51C mutants of c553 were constructed to achieve such assemblies. The fusion proteins were found to be correctly folded and function by having efficient electron transfer from the FMN to the heme domain of BMP and c553, as shown in Figure 4B (119). This work has demonstrated the feasibility of the novel molecular “Lego” approach that engineers functional multidomain proteins with designed redox properties, with promising applications in the field of biosensors and bioelectronics.

Nanda et al. have established the de novo design of a β-sheet-metalloprotein called RMI, which mimics native rubredoxin. The RM1 β-hairpin structure was computationally designed by constraining the positions of the peptide that were strategic in mimicking and/or stabilizing the β-structure and by subsequently using another computer program, statistical computationally assisted design strategy, to select the most probable amino acids for the remaining positions. The redox-potential measurements of RM1 show a midpoint potential of 55 mV versus a standard hydrogen electrode at pH 7.5. Natural rubredoxins fall between −90 and +40 mV (vs. NHE). The single-chain RMI design showed activity for 16 cycles, which is thus far the longest, demonstrated redox cycling of a de novo designed rubredoxin protein mimic (100).
The expansion in amino acids by introducing unnatural amino acids contributes to the systematic determination of the role of each amino acid in tuning the activity of the metal center as well as in increasing the breadth of chemical transformations made by the proteins. Privett et al. introduced a non-natural amino acid called 4-β-(pyridyl)-L-alanine (Pal) as a heme-binding ligand in a four-α-helix bundle using solid-phase peptide synthesis methodologies. The effects of bis-pyridine were compared with the popular axial bis-His ligands used to design heme proteins. However, the bis-pyridine binds only a single oxidized heme some 60,000-fold weaker than the bis-His analog, inducing a 287 mV higher equilibrium midpoint reduction potential. This shows that switching the His amino acid for Pal on each helix at position 10 results in a major effect on the heme electrochemistry (287 mV, 6.8 kcal/mol) due to the alteration of the Fe(III) and Fe(II) co-ordination equilibrium (110).
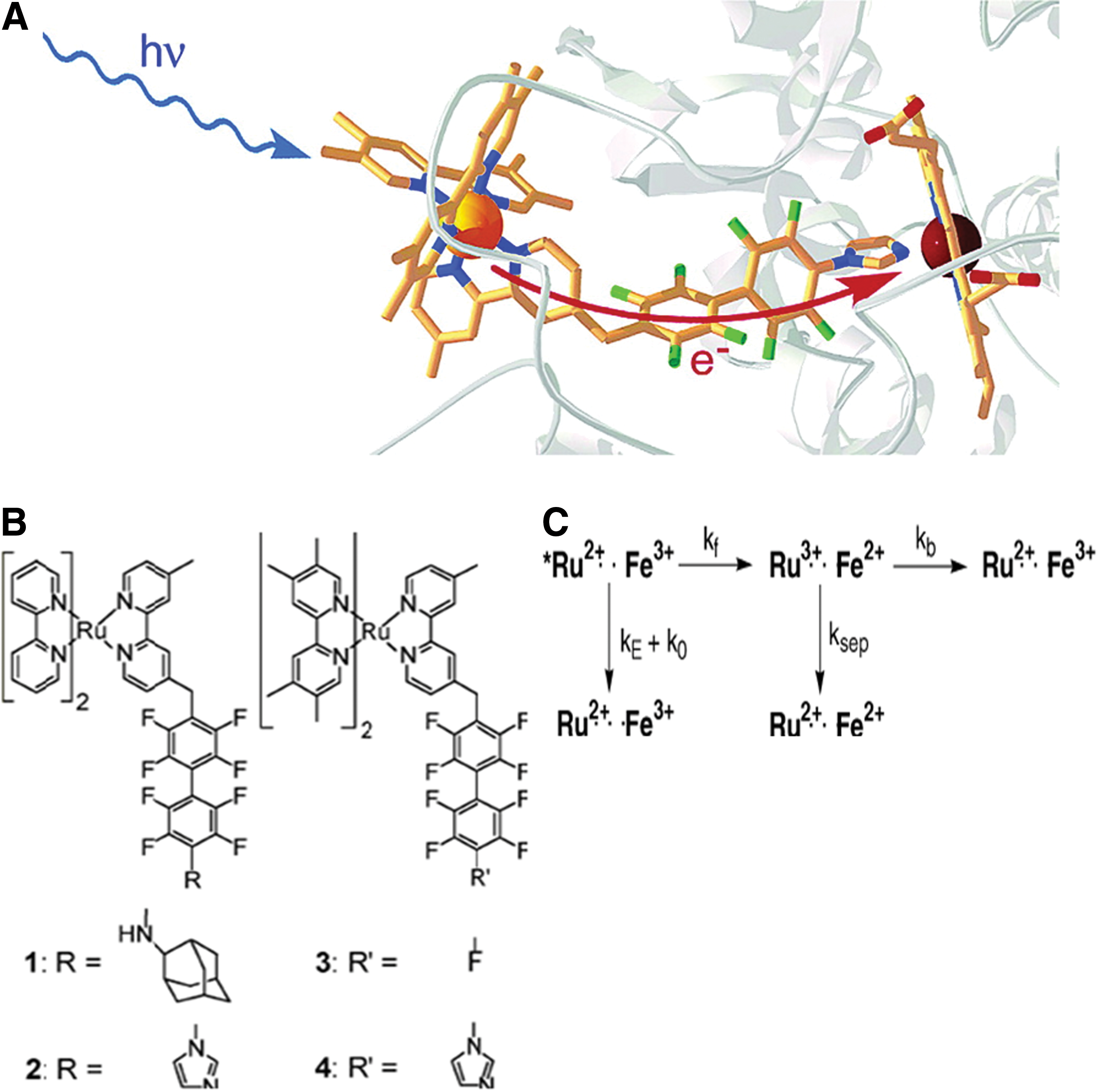
In another example, four ruthenium (Ru)-diimine complexes were synthesized to bind Cyt P450cam, in order to replace slow biological reduction (due to putidaredoxin) with a rapid optical redox trigger. In these Ru-diimine:P450 conjugates, Ru acts as the electron donor, and ferriheme acts as the electron acceptor. The Ru-diimine sensitizers were noncovalently coupled to a terminal functionality on the P450 via a perflurobiphenyl group. The four complexes all tightly bind P450cam: Ru–F8bp-Ad (
C. Engineered FeS cluster proteins
FeS proteins are a family of electron transfer proteins whose structures range from 2Fe-2S diamonds, intermediate 3Fe-4S butterfly-shaped clusters, to 4Fe-4S cluster cubes. The cluster formations of FeS are known to illicit a wide range of reduction potentials, that is, +450 to −700 mV (129). The factors that govern the reduction potentials of FeS clusters have been postulated to include the number of hydrogen bonds contained in the cluster (NH or OH to S), the proximity of charged residues, and the exposure to solvents. These factors have been implicated based on site-directed mutants, unnatural backbone substitutions, and theoretical calculations.
Ferredoxins (Fd) and high-potential iron proteins (HiPIPs), with both tetranuclear FeS cofactors and functioning as electron-transfer proteins, have Fe4S4 cluster active sites with nearly identical metrical parameters (25, 134). The Fds cycle between [Fe4S4]1+ and [Fe4S4]2+ states with potentials between −400 and −600 mV (vs. NHE), while HiPIPs utilize [Fe4S4]2+ and [Fe4S4]3+ cluster oxidation states operating at significantly higher reduction potentials, from approximately +200 to +400 mV (1). A comparison of Fd and HiPIP protein structures reveals a greater number of conserved active-site hydrogen bonds in Fd than in HiPIPs. This difference has been proposed to contribute to the striking difference in redox thermodynamics (7, 63). Low et al. synthesized two backbone-engineered analogu0 of HiPIP, [O]Val42 HiPIP and [O]Ala57 HiPIP, in which the H bonds were removed and replaced by ester linkages at positions 42 and 57, respectively, as shown in Figure 6A. Direct electrochemical measurements were conducted on the wild-type HiPIP and analogs to probe the functional effects of backbone NH
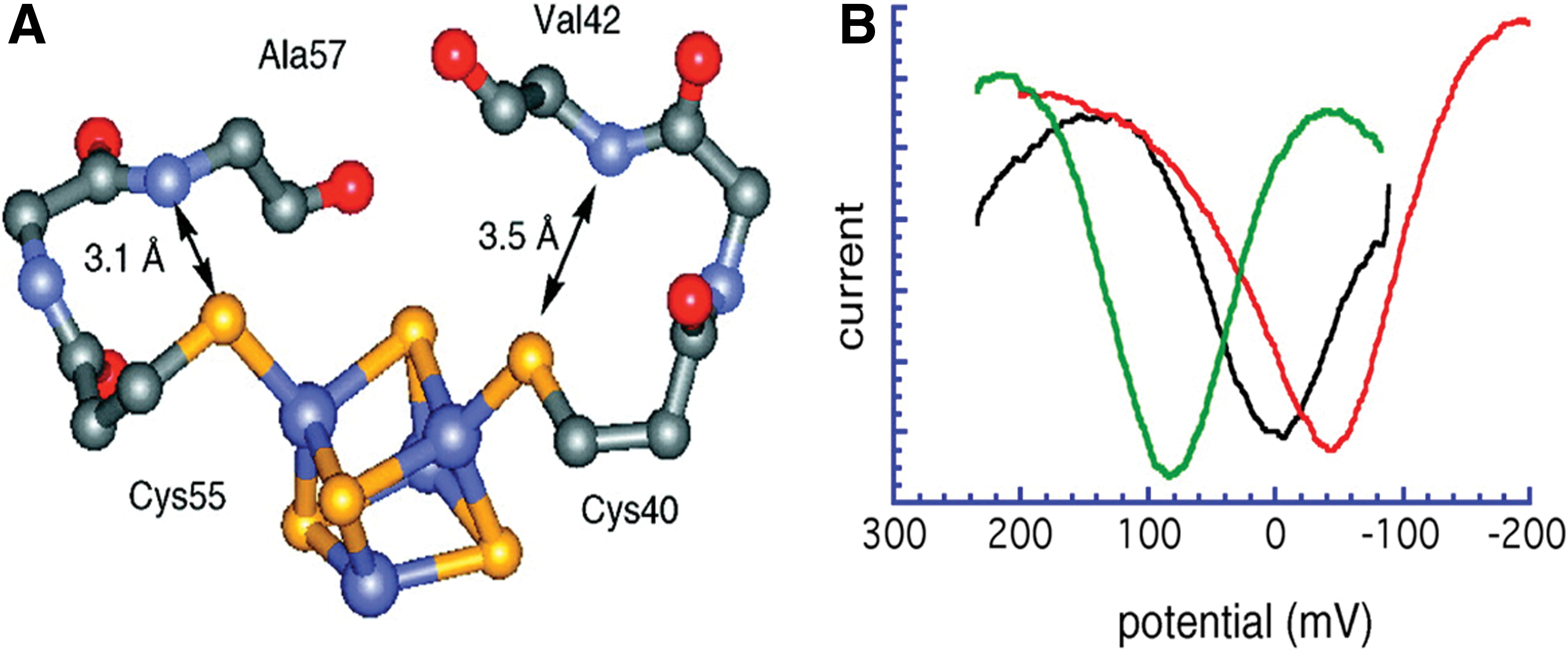
In another attempt of FeS cluster protein engineering, Sow et al. successfully achieved the total synthesis of a mini-ferridoxin (Fd) using a 58 amino acid Fd II from Desulfobivrio gigas, a sulfite reducing bacterium, as the template for the design process. They designed a 31-amino-acid polypeptide based on the sequence of D. gigas FdII, and synthesized it by solid-phase peptide synthesis. An FeS cluster was then incorporated into the polypeptide, and the miniFd was characterized in terms of its redox potential and spectroscopic properties (nuclear magnetic resonance [NMR], UV-visible, and EPR spectroscopies). The electrochemical characterization showed a quasi-reversible couple with a midpoint of −370 mV (vs. NHE), indicative of a Fe4S4 cluster. Although the immediate protein environment of the two FeS clusters in the miniFd and native Fd was not identical, the synthetic protein was found to be redox active, and its spectral properties were consistent with those of the native holoprotein (128).
D. Engineered proteins containing redox active residues
The role of redox active amino acids such as Tyr, Trp, Cys, and Gly in biological electron transfer and proton-coupled electron transfer continues to excite both experimental and theoretical interests. The study and characterization of these amino-acid radicals in nature can be challenging due to the sheer size and complexity of many amino-acid radical enzymes, combined with the often highly oxidizing nature of their radical cofactors. To study these redox active amino acids, Tommos and coworkers developed two synthetic de novo engineered model proteins known as α3W and α4W, depicted in Figure 7A. The aim of the de novo radical maquettes is to introduce the radical properties of interest as well as to build features that will facilitate the biophysical characterization of the system. Trp and Tyr are known as the catalytic species in the majority of amino-acid radical enzymes. α3Wand α4W were designed to contain the following key features: (i) the protein should be a monomeric three-helix (α3W) or four helix (α4W) bundle containing a single Trp or Tyr; (ii) the aromatic residue should be buried, as opposed to located in a solvent-exposed position where solution conditions are expected to dominate its chemistry; and (iii) the remaining residues should be redox inert in order to isolate the radical chemistry to a single site (146). The fluorescence properties of the α4W de novo designed protein contained a single Trp, suggesting that the residue was located in a sequestered position. Figure 7B depicts the DPV characterization, which indicates an elevated peak potential for the Trp in the folded form of the protein (pH range 7.0–8.5) relative to a partly unfolded form (pH range 11.4–13.2). Oxidation of the Trp is coupled to proton release, as indicated by a 53±3 mV/pH unit dependence of the peak potential of the 7.0–8.5 pH range (147).

IV. Applications of Engineered Redox Proteins
The engineered redox proteins just mentioned are primarily used for studying the basic mechanisms of electron transfer in proteins. In addition, they are used to investigate the effects of external factors such as pH, temperature, solvent effects, and internal factors, including electrostatics, amino-acid sequence, and reaction center distance. The knowledge acquired by studying various protein-engineered mutants can then be used to create proteins with desired properties in the applications of biosensors, biofuel cells, catalysis, and pharmaceuticals, as summarized in Table 2. We attempt to explain a few key applications of engineered redox proteins in each of these fields.
Cyt, cytochrome; ELISA, enzyme-linked immunosorbent assay; Fr, ferritin; HRP, horseradish peroxide; Ru, ruthenium.
A. Biosensors
Many native redox proteins have been utilized as the biorecognition components in the development of biosensor assemblies. Biosensors are powerful tools for detecting a wide range of analytes such as disease biomarkers, pollutants, toxins, and food contaminants. These analytical devices are based on the combination of a biorecognition element and a transducer element that converts the biorecognition event into a measurable signal. Electrochemical biosensors are emerging as key players due to their high sensitivity, ease of miniaturization, and low cost. Electrochemical biosensors often utilize natural or engineered proteins as their biorecognition elements. It is very difficult to find wild-type proteins with all the features required to construct the ideal biosensor, as they are not evolved or optimized for those applications, but are rather fine tuned to operate efficiently under the conditions of their native environment. Natural selection prevents random electron transfer in living organisms and ensures that valuable energy resources are not wasted and that toxic metabolites are not produced such as hydrogen peroxide (153). Nevertheless, protein engineering helps researchers manipulate biomolecules in order to produce tailor-designed biorecognition molecules for integration into biosensing platforms. The careful selection of the location and type of mutations gives rise to enzymes with enhanced properties, such as a higher affinity toward specific analytes, higher stability, higher electron transfer rates, and increased stability even after immobilization. These improvements have already resulted in biosensors with an enhanced performance (26).
In the previous section, we not only discussed theelectrode immobilization of redox proteins for providing insights into the mechanisms of electron transfer between protein partners, but also considered it a key step in the assembly of biomolecular electronic devices such as biosensors and biotransistors (114). Redox proteins incorporated on solid electrodes in an electrochemical environment may convert an unmediated transduction of a chemical reaction into electric signals. In addition, the exploration of functionally versatile and catalytically tunable molecular systems would allow the improvement of both the heterogeneous protein-electrode electron transfer rates and the stability of the hybrid interface in harsh conditions.
One of the examples of the biosensors utilizing redox proteins is the “lactate sensor,” which includes four different enzymes: lactate dehydrogenase, lactate oxidase, lactate monooxidase, and Cyt b 2 (44). Lactate, a salt or ester of lactic acid, is a product of fermentation and a metabolite from glucose during cellular respiration. The lactate sensor is used in sports medicine and other medical applications, such as measuring the concentration of lactate in blood to monitor oxygen deprivation and to indicate acidosis or bacterial meningitis (115).
Acetylcholinesterases (AChEs) have been used as the recognition elements for the detection of pesticides such as organophosphate and carbamate insecticides. They are mainly found at neuromuscular junctions, in the cholinergic nervous system, and in the red blood cell membranes. Since the active site of AChE from Drosophila melanogaster (Dm), the common fruit fly, is buried 20 Å inside the protein and the entrance to the active site is very narrow, Boublik et al. added mutations to alter some residues of that region (24). The replacement of glutamic acid, located at the rim of the active site gorge, by amino acids with bulky side chains, such as Trp or Tyr, has been demonstrated as red blood cell membranes increasing the inhibition constant for dichlorvos significantly. Double mutations, for example, replacing Tyr71 by aspartic acid (Asp) in addition to the Glu69 substitution, have led to even more sensitive enzymes. The double-mutant Dm-AChE was immobilized on microporous-activated conductive carbon and integrated into a flow system; the biosensor showed a limit of detection of 10−17 M. The mutant enzyme was immobilized on the carbon paste of screen-printed electrodes, and the enzymatic product thiocholine was detected via the mediator Co (phthalocyanine) at +100 mV versus Ag/AgCl (127).
In order to develop highly sensitive biosensors, proteins have been modified to enhance their electron transfer kinetics. Gorton and coworkers have widely studied the effect of mutations on the electron transfer pathways, using recombinant forms of horseradish peroxide (HRP) (46). Although the direct electrochemical reduction of wild-type HRP adsorbed on electrodes has been demonstrated, the electron-transfer kinetics was low, likely due to the long distance between the active site of the glycosylated native enzyme and/or unfavorable orientation (117, 118). The use of a nonglycosylated recombinant HRP containing a hexa-His tag accelerated the electron-transfer kinetics by improving the sensitivity of the gold electrodes toward hydrogen peroxide, and provided more stable biosensors due to the higher binding strength of the enzyme with gold (46).
Another way of facilitating electron transfer is by eliminating the amino acids that are not essential for enzyme functionality, thus making the active site more accessible. In order to prove this, microperoxidase-11, an undecapeptide portion of Cyt c that retains peroxidase activity, was linked to a heme c group obtained by HRP digestion to mimic the catalytic functions of HRP. The so-called “minizyme” has been observed to be very efficient not only as a biorecognition molecule in peroxide sensors (86, 87) but also as a bioelectrocatalytic label in enzyme (13, 73) and immunosensors (74) as well as a bioelectrocatalytic cathode in biofuel cells (149). Another example is the truncated laccase (LAC), a copper-containing oxidase enzyme found in many plants, fungi, and microorganisms. The truncated protein was generated using site-directed mutagenesis described by Gelo-Pujic et al. (163) The truncation enhanced the rate of electron transfer between the copper-containing active site and the electrode.
Oxidoreductases catalyze single- or multielectron redox reactions of many small organic or inorganic substrates. Often, the redox enzymes require redox partners in catalysis to balance the substrate/product redox half reaction. The redox partners, ranging from electron-transfer proteins to small-molecule cosubstrates, can be replaced by electrochemical electrodes to systematically study the redox transformation mechanisms and coupled chemical reactions. Nonetheless, most proteins lack direct electron transfer (DET) communication with substrate electrodes. This lack of communication presents one of the most fundamental obstacles in the development of bioelectronic systems. Ingenious methods have been developed to facilitate the electrical contact between biomolecular assemblies and electronic units (152). A few examples of these methods are the (i) structural engineering of proteins with electron relays (113); (ii) immobilization of redox enzymes in conductive polymers or redox-active polymers (65); and (iii) steric alignment of proteins on electron relays associated with electrodes (150).
1. Glucose biosensors
The glucose oxidase (GOx) enzyme electrode was one of the very first commercially successful biosensors and is believed to be the most frequently used analytical device in the world. These sensors take advantage of the high specificity of GOx for the catalytic oxidation of glucose to gluconolactone, a reaction that produces electroactive hydrogen peroxide as a by-product. Amperometry is used to oxidize H2O2 produced at the electrode/enzyme/sample interface, and the resulting current is proportional to the concentration of glucose (31). These measurements can be performed in a number of bodily fluids, using small sample sizes. For these purposes, a GOx sensor requires a rapid and linear response with a range of approximately 2–30 mM glucose (72, 85, 160). The enzyme electrode is generally disposable, and devices of this kind are now used by patients worldwide for the daily monitoring of blood glucose in the management of diabetes. Some other amperometric enzyme biosensors are also commercially available, including alcohol dehydrogenase, lactate oxidase, and glutamate oxidase.
The main barrier involved in the construction of glucose biosensors is the lack of direct communication between GOx and the electrode. Most enzyme-based electrodes require the presence of redox mediators that tunnel the electrons from the protein to the electrode surface. Reconstituted apoenzymes on relay-cofactor monolayers associated with the electrodes have been used to achieve efficient electrical communication. For example, apo-GOx was reconstituted on a PQQ-modified polyaniline film associated with the electrode. Effective electrical communication between the redox centers of the biocatalysts was observed and attributed to the alignment of the proteins on the electrode surface (150).
The emergence of metal and semiconductor nanoparticles with unique electronic and optical properties has enabled the generation of nanoparticle-enzyme hybrid systems for controlled electron transfer. The codeposition of gold nanoparticles with redox enzymes onto sensor surfaces was reported to operate without electron-transfer mediators, but the random and nonoptimized positioning of the enzymes did not allow efficient electron transfer. Highly efficient electrical contacting of a reconstituted GOx through a 1.4 nm single gold nanoparticle was accomplished by functionalizing the gold nanoparticles with N6-(2-aminoethyl) FAD, as depicted in Figure 8. The resulting nanoparticle-reconstituted enzyme electrodes stimulate mediatorless, bioelectrocatalyzed oxidation of glucose with a high electron transfer turnover rate of 5000 s−1. The mediation of electron transfer via the gold nanoparticles also minimized the interference of oxygen or ascorbic acid, the common electrochemical interferents present in biological samples (154).
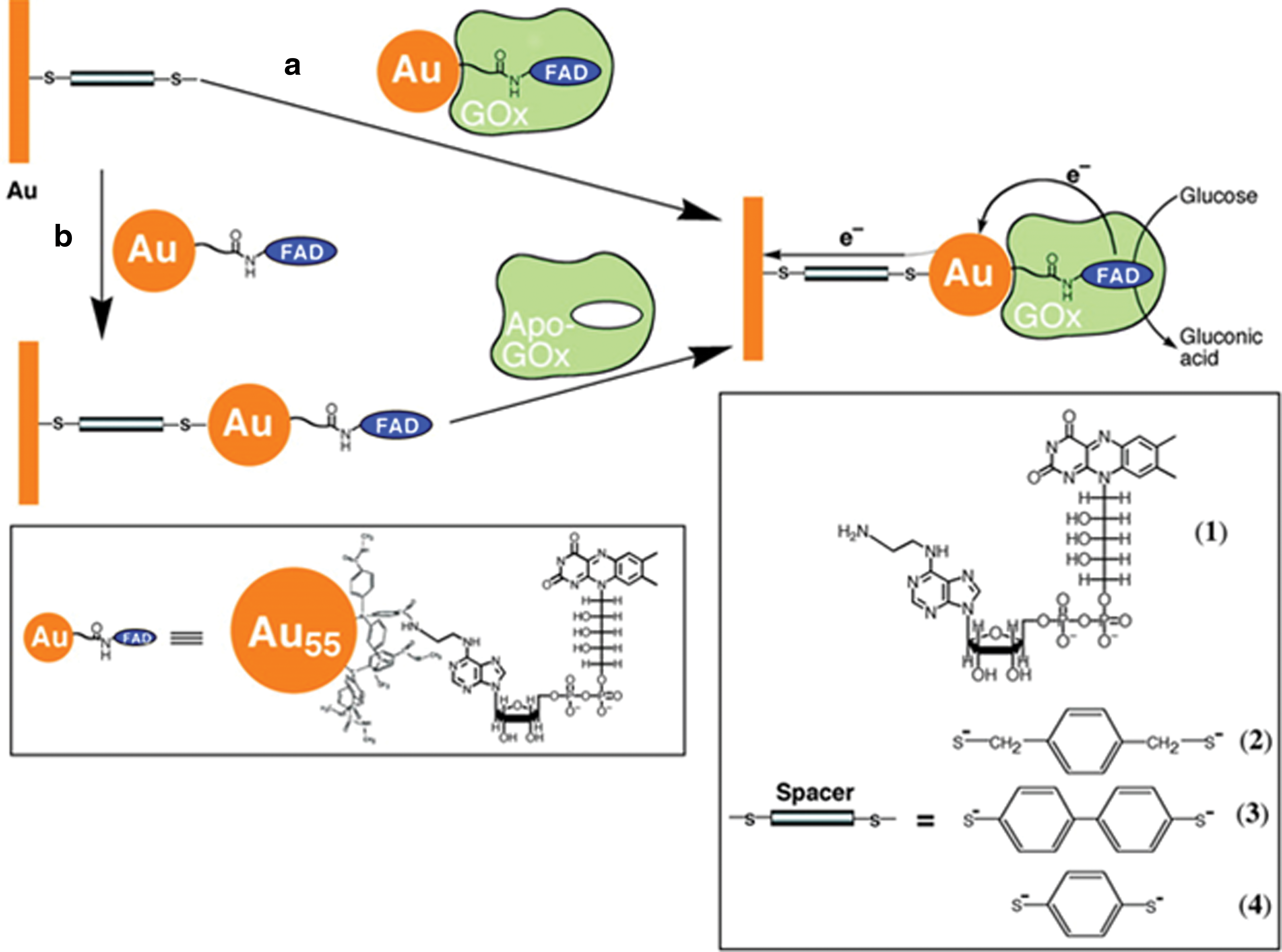
Yamazaki et al. used protein engineering as a tool to expand the dynamic range of glucose biosensors. Two mutants of PQQ-glucose dehydrogenase (GDH), His775Asp and His775Asn, having different Michaelis constant K m toward glucose, were produced. Their combination at a ratio of 1:1 in the construction of a biosensor provided a wider dynamic range (3–70 mM) compared with the wild-type enzyme-based biosensor. Moreover, the specificity of the biosensor for glucose was higher than for galactose, xylose, mannose, allose, and 2-deoxy-d-glucose (156).
2. Iron-based redox biosensors
Iron serves as an ideal redox-active cofactor for many biological processes because of its unique electrochemical properties. Being incorporated into many metalloproteins, iron plays important roles in metabolism, respiration, nitrogen fixation, and in sensing cellular redox status. Various iron cofactors are found at the active site in the iron-containing metalloproteins, and FeS cofactors can also been synthesized to form FeS clusters. Iron can be co-ordinated by amino acids in the polypeptide chain to form iron active centers, and based on the oxidation state, the ligands vary from His, Cys, Met, Glu, and Asp. The FeS cofactors can also been synthesized to form FeS clusters. In addition, iron could also be incorporated into porphyrin structures to form heme, and there are many different heme types giving different heme proteins.
Iron-based sensors act as switches that control protein activities in response to changes in cellular redox balance. The applications depend on the origin of iron in the sensor. The FeS clusters are able to serve as transcriptional and post-transcriptional regulators by poising their redox potentials to respond to specific oxidants or reductants. Iron is incorporated into porphyrin structures to form heme, with many different heme types forming a variety of heme proteins. Heme-based sensors are useful in small-molecule sensing such as O2, NO, and CO; some mononuclear iron proteins have been shown to be applied in redox sensing (such as the strong oxidant H2O2) and signal transduction.
In a study of Met80Ala Cyt c variants immobilized on solid electrode surfaces, a set of improved amperometric biosensors for molecular oxygen and nitrite ion were produced with more robust and stable reductive electrocatalytic performances at various conditions (28, 29). Utilizing these low-cost and stable engineered proteins made it possible for the exploitation of hemoproteins in the biochemical detection of chemical analytes of great environmental, clinical, and industrial significance, including CO, NO, NO2 −, and H2O2 (32). Furthermore, direct electrical communication between the redox centers and the metal electrodes yielded very good electrochemical performances without dramatically affecting the structural properties of the bound proteins, especially for the Cyt c variants whose redox potential could be reversibly modulated (23). This important achievement represented a series of possible suitable and appealing candidates for constituting the biocatalytic interface in biomolecular electronic applications, with more potential in nanostructured biosensing devices (3).
In protein design and molecular biology of redox proteins, molecular “Lego” has been developed to improve the electrochemical response of inaccessible metal centers in metalloproteins. By using enzymes with well-characterized electrochemical properties, this approach aims at building artificial redox chains that are able to communicate with the electrodes and facilitate efficient electron transfer (119). Several sets of molecular Lego based on engineered redox proteins have been reported to control the electrochemical activities of the redox active elements by linking electron transfer and the catalytic modules. P450 enzymes, highly relevant to the bioanalytical area, are attractive targets as the prototype of molecular Lego assemblies. The heme domain of the P450 BM3 gene-fused with Fld has been used as a module in the construction of an electrochemical assembly that solubilizes the core of human P450 2E1 with enhanced electrochemical properties (50). In another engineered artificial redox chain, Fld was adopted as a module for transferring electrons to Cyt c 553 and the heme domain of P450 BM3 by covalent linkage via engineered disulfide bridges with mutated Cys residues (119). The same approach was used with the copper-containing protein Az, in which a surface exposed Cys was engineered to provide the covalent bond for the linkage to the gold electrodes (35). The resulting molecule successfully maintained electrochemical redox activity.
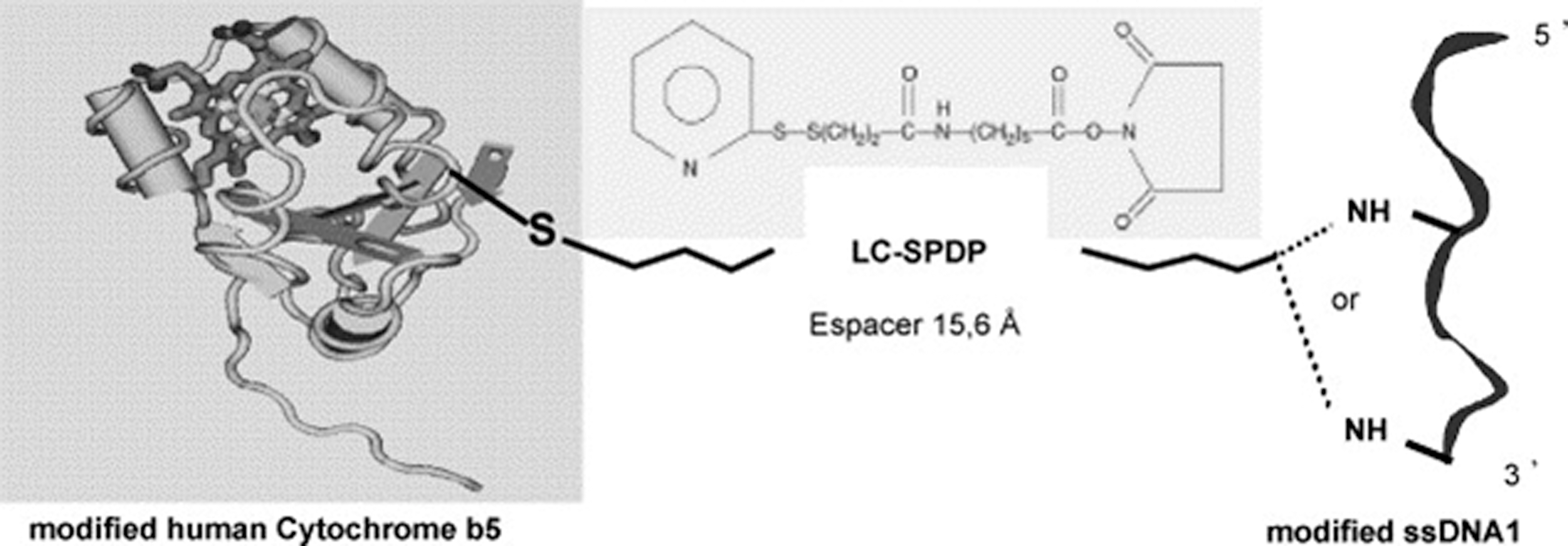
Molecular “Lego” need not only be applied for linking redox enzymes to other redox enzymes, but can also be used to design macromolecular assemblies with protein-DNA interactions for DNA sensors or bioelectronic devices. A highly controlled supramolecular assembly has been reported by Boireau et al., which contains an engineered redox protein, human Cyt b5, and modified oligonucleotides through a hetero bi-functional cross-linker (21). In this assembly, the DNA is placed in a biomimetic environment, and the Cyt b5-DNA complexes are able to bind to modified phospholipids that lead to a new generation of self-assembled dynamic DNA chips with low nonspecific interactions, a tunable immobilized probe density, and better hybridization efficiency. Figure 9 illustrates the covalent linkage of the genetically modified Cyt b5 to the modified oligonucleotide using a bifunctional linker. The hybridization of the DNA segment with a complementary oligonucleotide sequence can be easily monitored in real time by surface plasmon resonance analysis. There are a number of potential applications of this new assembly, including biochips with high sensitivity and selectivity, especially for the DNA-ligand interaction studies, as well as for highly organized and defined nanodevices in electronic and biomedical fields.
B. Biofuel cells
Enzymes or microbes incorporated in an electrical circuit produce a cascade of enzyme-catalyzed reactions, resulting in the production of electrons that can be harnessed as a biofuel cell. Among oxidoreductases, those that catalyze the reduction of oxygen, oxidation of hydrogen, or dismutation of peroxide are of particular interest. These molecules play important roles in signal generation in biosensors and current generation in biofuel cells. These systems have been designed to function on both the laboratory and industrial scales, and have great potential in being used as a sustainable biotechnological solution for alternative energy sources. There are many microbial fuel cells of different origins reported, such as sediment microbial fuel cells that harvest energy from the sediment-seawater interface, photoheterotrophic microbial fuel cells, and biomass-fueled ceramic fuel cells.
To operate the microbial fuel cells, electroactive metabolites and redox mediators shuttle electrons from the metabolic pathway of the microorganism to the electrodes. These bioelectrocatalysts are able to interact with the electrode surface and form a molecular transducer that directly converts chemical signals to electricity. In the previous sections, we have described the use of engineered redox proteins for biosensor applications. Further use of these engineered proteins can also be made toward the development of functional components of biofuel cell devices. The development of efficient biofuel cells that convert the chemical energy stored in abundant organic raw materials or biomass to electrical energy is a continuing challenge. Biofuel cells could provide cost-effective and environmental friendly solutions for the global energy crisis. A biobattery generates electricity from carbohydrates (sugar) utilizing enzymes as the catalyst, through the application of power-generation principles found in living organisms. Figure 10 illustrates a model biofuel cell consisting of an anode of sugar-digesting enzymes and a mediator, and a cathode comprising oxygen-reducing enzymes and a mediator, separated by a cellophane membrane (71).
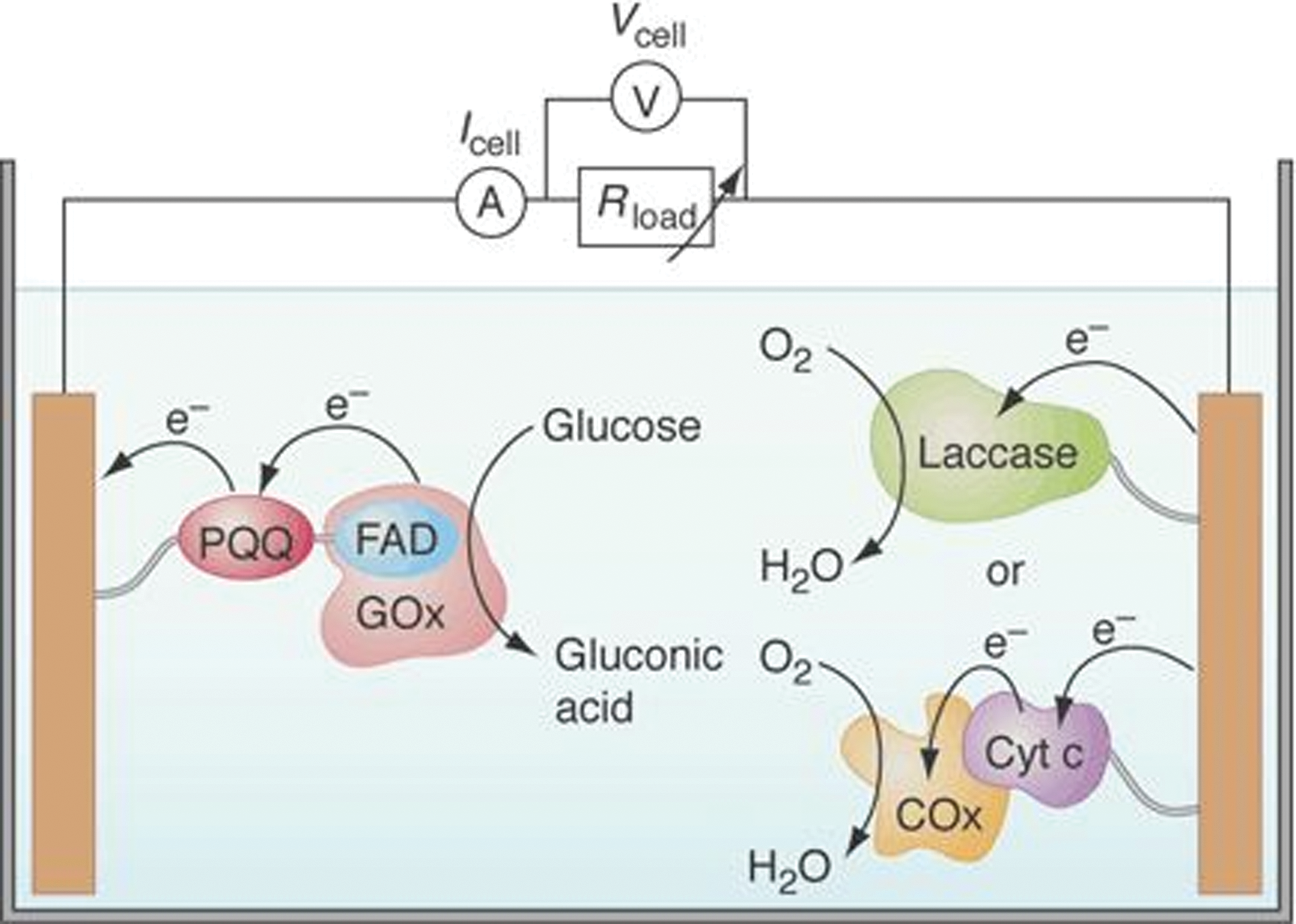
The use of body fluids such as blood (glucose) as a source of fuel toward the development of an implantable biofuel cell that powers prosthetics such as pacemakers or insulin pumps is also an attractive target. Pioneered by Adam Heller, miniaturized enzymatic biofuel cells (mBCs) convert blood sugars into electrical energy by employing GOx on the anode and bilirubin oxidase on the cathode (34). To match application demands, it is crucial to increase the lifetime and power output of mBCs. GOx is a stable enzyme with high substrate specificity and a relatively high K m value. In the field of self-blood glucose monitoring, GDH harboring PQQ as the prosthetic group is preferred owing to its oxygen insensitivity and high catalytic efficiency (79, 133). However, the application of PQQ-GDH for the bioanode is inherently limited because of its instability. In order to develop the ideal enzyme for glucose monitoring, Yuhashi et al. engaged in the protein engineering of PQQ-GDH. In these studies, one engineered enzyme, the Ser415Cys mutant (S415C) was obtained with enhanced stability using protein-engineering techniques. The S415C is stable even at 70°C. The alteration of the residue at position 415 did not affect the catalytic efficiency of this enzyme in glucose oxidation. They evaluated the potential application of the engineered PQQ-GDH, S415C, as the bioanode enzyme in glucose utilizing biofuel cell systems. The open circuit voltage for the system was 577 mV, and the maximum current generated was 4.3 μA (61.4 μA/cm2). The power generated from this cell was 1.23 μW (17.6 μW/cm2) at the external load of 200 kΩ. The biofuel cell employing wild-type GOx lost 6%–20% of its power per day, when they were operated continuously. The system employing the S415C maintained 80% of the initial response after 24 h of continuous operation (158, 161).
Most enzymatic biofuel cells utilize mediated electron transfer (MET)-type bioelectrocatalysis, as the MET-type system can be applied to most redox enzymes and is easily constructed (10, 158, 70). However, the MET system has several disadvantages such as thermodynamic loss and mediator leakage. To overcome these issues, another approach for bioelectrocatalysis is the utilization of DET-type fuel cells. Kamitaka et al. have developed one-compartment biofuel cells without separators, in which D-fructose dehydrogenase (FDH) and LAC work as catalysts of DET-type bioelectrocatalysis in the two-electron oxidation of D-fructose and four-electron reduction of dioxygen as fuels, respectively (70). FDH is a membrane-bound enzyme containing one flavin and one heme c as the prosthetic groups, and it can work as a catalyst of the electrochemical oxidation of D-fructose to 5-keto-D-fructose without electron-transfer mediators. This activity was maintained when FDH was adsorbed on Ketjen black (KB) particles immobilized on carbon papers to act as the anode. Multicopper oxidases such as plant and fungal LACs and bilirubin oxidase were chosen for DET-type bioelectrocatalytic reduction of dioxygen toward the development of the cathode. These enzymes catalyze the four-electron reduction of dioxygen to water through the oxidation of their specific substrates. Most of multicopper oxidases allow the electrodes to act as direct electron donors. Therefore, the thermodynamics, enzyme kinetics, and the electrode reaction kinetics of three kinds of multicopper oxidases were analyzed, and LAC from Trametes fungi was selected as the optimal enzyme for the cathode. The FDH-adsorbed KB-modified carbon paper electrodes and the LAC-adsorbed carbon aerogel-modified carbon paper electrodes have been combined to construct one-compartment biofuel cells without separators (70).
C. Photovoltaic cells
Recently, efforts have focused on the incorporation of redox proteins and enzymes into bioelectronics to exploit their efficient participation in electron transfer chains. Light-activated bioconjugate systems are being studied for their incorporation into practical devices to convert light energy into chemical energy. However, the few systems that have been investigated with an aim of practicing application energy conversion make use of reconstituted Mb or similar proteins that have little catalytic or biological activity. To address this issue, Peterson and coworkers have composed light-activated donor-acceptor bioconjugates via the covalent linkage of Ru(II) bisterpyridine chromophores and iso-1 Cyt c, as shown in Figure 11. Ru tpy chromophores were chosen due to their high stability and unique photophysical properties. The bioconjugates retain both their catalytic and biological activities after chromophore attachment. Photoinduced electron transfer was recorded between the Ru chromophores donor and Cyt acceptor. These light-activated conjugates can be used for numerous biosensing and biofuel cell devices (106).
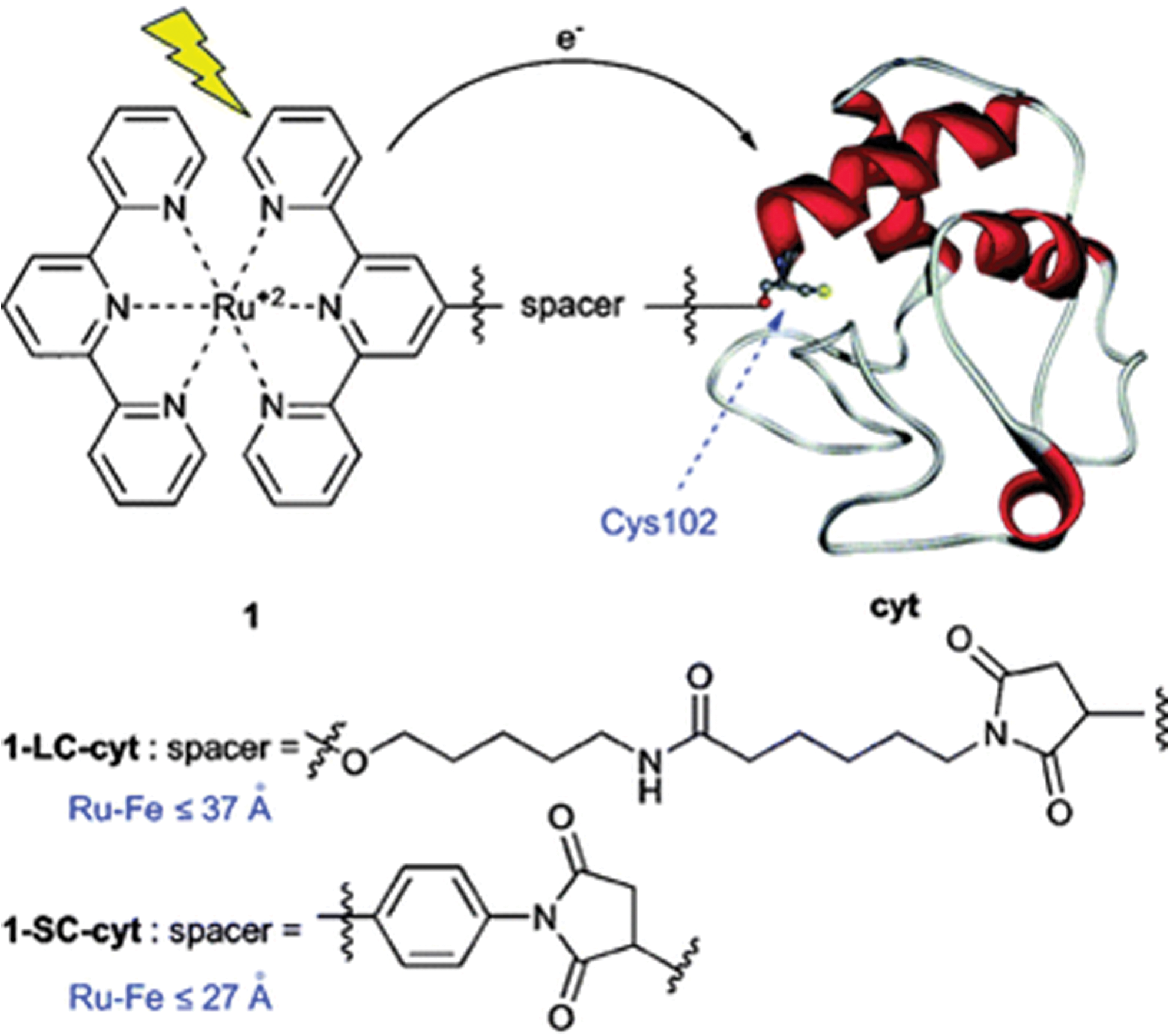
D. Electrochemical immunoassay
Enzyme-linked immunosorbent assays (ELISA) have been used for the detection of antibodies or antigens in immunological samples. Although there are many ways to perform these assays, in all cases, the enzyme acts as a signaling unit for the presence of either antigen or its antibody. The amperometric electrochemical ELISA method relies on the detection of a catalytic current due to electrochemically driven catalysis in the presence of a substrate. It has the ability to detect analytes in numerous matrices, ranging from animal tumors to environmental samples.
Peroxidases, such as HRP, are the most commonly utilized enzymes in electrochemical ELISA systems. They catalyze the H2O2-driven oxidation of their specific substrates, with the products of this chemical oxidation being electrochemically detected by applying a reducing potential. The other enzyme systems that have been used in electrochemical ELISA include GOx and choline oxidase. Several disadvantages are encountered on the chemical conjugation of peroxidases with antibodies or antigens, such as low reproducibility and undefined stoichiometry. Grigorenko et al. have developed a recombinant fusion of a protein analyte with HRP in Escherichia coli, where they employ the refolding of inclusion bodies and the reconstitution with heme. The genetic fusion approach enables the preparation of conjugates with a 1:1 stoichiometry and a defined structure (57).
E. Pharmaceutical applications of engineered redox proteins
From the very first studies on recombinant P450s, efforts were directed toward constructing fusions between P450s and redox partners in the hope of generating more efficient enzymes. The most important application of these enzymes in toxicology in the near future is likely to be the biocatalytic generation of drug metabolites for the pharmaceutical industry. Further tailoring will be necessary for specific toxicological applications, such as in bioremediation (51). Directed evolution has been used to good effect with bacterial P450s, particularly P450 BM3. There has been growing interest in engineering P450 BM3 to better metabolize druglike molecules (40, 105, 144, 159) for the generation of substantial amounts of authentic metabolites as well as in the optimization of lead compounds. The Arnold laboratory showed that P450 BM3 mutants could be used to generate metabolites from a drug (propranolol), a highly attractive goal for the pharmaceutical industry. Rounds of saturation and random mutagenesis led to modest improvements in the peroxide-supported activity of a mutant P450 BM3 heme domain toward the ring hydroxylation of propranolol (105). Yields obtained were comparable to those produced by recombinant human P450s in bacterial or baculovirus bioreactors (116).
One very specialized application of engineered P450s is in gene-directed enzyme prodrug therapy (GDEPT) (56). This is based on the ability of certain P450s to activate chemotherapeutic prodrugs such as cyclophosphamide, ifosfamide, and tamoxifen to their therapeutically active metabolites. The idea is that by the tumor-selective delivery of the gene for a highly active (engineered) catalyst, a greater therapeutic effect can be achieved with less systemic toxicity, especially if prodrug activation by liver P450s can be inhibited. This has been the motivation behind attempts to improve the activity and regioselectivity of P450 2B enzymes in the activation of cyclophosphamide and related drugs by Waxman and colleagues (81, 131). Since the enzymes are required to work in a physiological environment, the focus has been on improving activity and regioselectivity toward the desired biotransformation (as well as the gene delivery system), rather than on altering other properties of the P450 proteins. The provision of additional reductase may increase the efficacy of GDEPT, so tumor selective coexpression of P450 and reductase has been attempted (32, 69). A clear opportunity exists for the effective use of optimized P450-reductase fusion proteins (140). However, it is important to recognize that engineering P450s (with or without the reductase) that improve activity, regioselectivity, or electron transfer may alter the processing and the presentation of the mutant proteins by the immune system such that these “self” proteins appear foreign and may elicit an immune response in susceptible individuals.
Koder et al. have designed a di-heme-containing four-helix bundle with oxygen transport properties, whose O2 affinities and exchange timescales match those of natural globins. The engineering-based design of functional synthetic proteins progresses through four stages (Fig. 12): (i) the assembly of a simple, robust generic protein framework, such as a helical bundle, of an appropriate size to sustain eventual cofactor binding and catalytic function; (ii) insertion of cofactor-binding amino acids (His, phenylalanine to bind hemes), keeping the number of amino-acid changes low to control complexity; (iii) adjusting the sequence for improved structural resolution monitored using NMR spectroscopy and X-ray crystallography; and (iv) iteratively testing, redesigning, and adding engineering elements to refine function. This designed protein binds O2 even tighter (Fig. 12) than carbon monoxide, a desirable property that most native O2-binding heme proteins do not have. A key to its success is a reduction of helical-interface mobility to exclude water from the heme-binding site, thereby reducing heme oxidation and increasing O2 −binding stability (77).
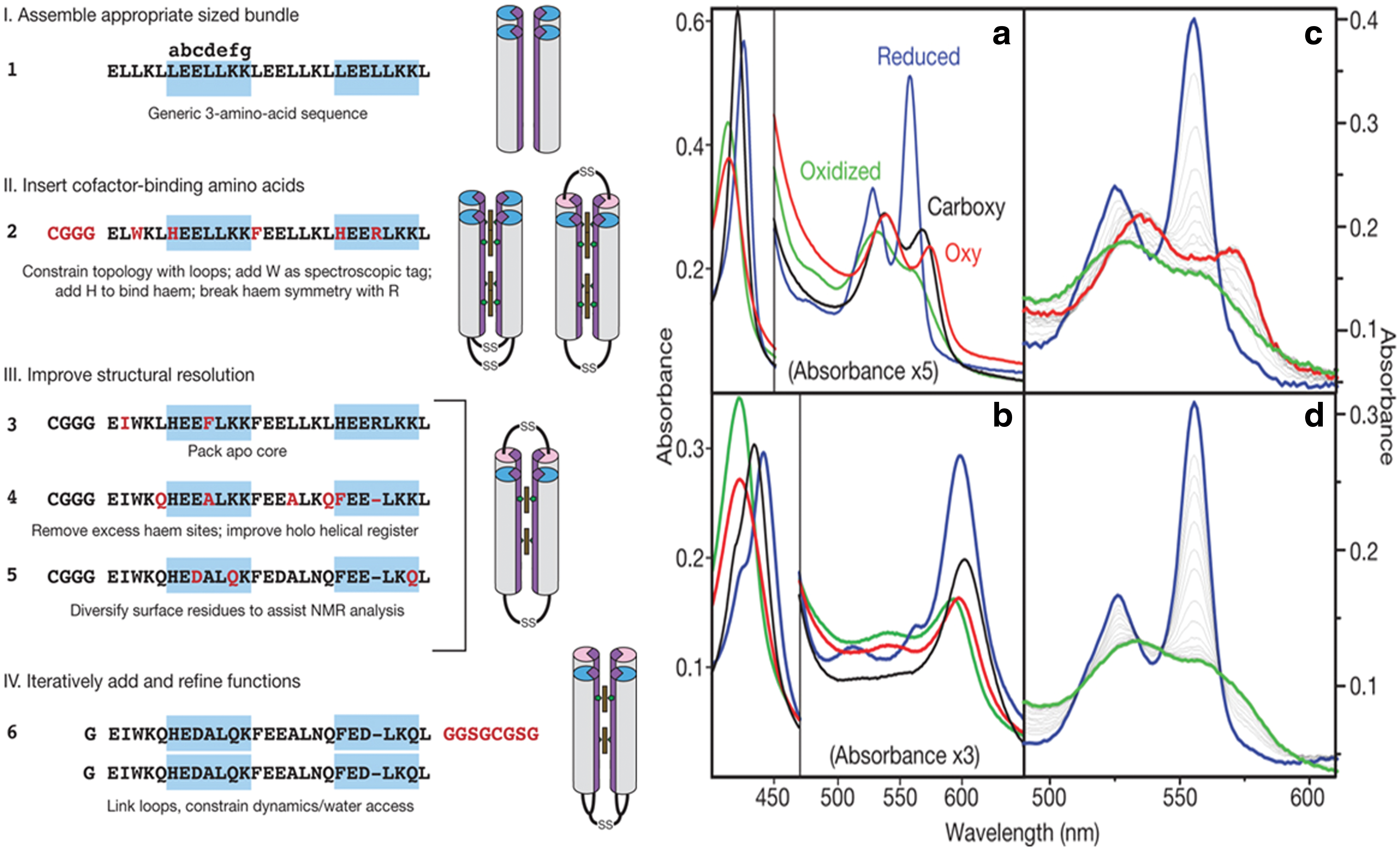
F. Engineered redox proteins in catalysis
The fusion of enzymatic proteins with metal-catalysts has recently drawn attention due to the combined advantages of both catalytic strategies. An increased understanding of chemical and enzymatic catalysis cumulated with the advent of genetic engineering and recombinant technology has led to the development of new biocatalysts with modified activities and specificities (5, 138, 148). Biomolecules such as protein, DNA, or antibodies have been used as hosts for the incorporation of metal complexes, resulting in the formation of hybrid catalysts with unique properties.
Harad and coworkers presented the first example of asymmetric hydrogenation of an amino-acid precursor catalyzed by the achiral complex of a transition metal rhodium combined with an antibody. The resulting hybrid antibody complex successfully reduces N-acetamido acrylic acid to N-acetyl-(S)-alanine with an efficiency >98%. The rhodium antibody was found to be a stereoselective catalyst displaying high substrate specificity due to second-sphere interactions (155).
Proteins are very attractive building components for the construction of self-assembled cage structures. Fr, an iron-storage protein comprised of 24 subunits that can assemble to form hollow cage-like structures, has been used in the preparation of size-restricted metal oxides and sulfides (41, 94, 95). Based on a similar idea, Watanabe and coworkers demonstrated that Fr could also be used to form a palladium nanocluster encapsulated in the apo-Fr cavity. Size-selective catalytic hydrogenation of olefins by the Pd•apo-Fr was observed at improved rates as compared with the protein-free reaction (142).
Carbonic anhydrase (CA) is a zinc metalloenzyme present in red blood cells that catalyzes the hydration of carbondioxide to bicarbonate. Replacing the zinc active site with manganese yielded manganese-substituted carbonic anhydrase (CA[Mn]), which showed peroxidase activity with a bicarbonate-dependent mechanism. In the presence of bicarbonate and hydrogen peroxide, CA[Mn] catalyzed the efficient oxidation of o-dianisidine with kcat/KM=1.4×106 M −1s−1 (104). Thus, the creation of a new peroxidase with non-natural biocatalytic activities was achieved by replacing the zinc ion with manganese.
V. Future Trends
Since the past few decades, redox proteins and metalloproteins have been playing significant roles as the catalytic components in the development of bioelectrochemical sensing devices. Biosensors and bioelectronics are rapidly progressing fields at the junction of chemistry, biochemistry, and physics, and their success depends on a better understanding of the biological components. Such electrical devices with a high sensitivity and selectivity have shown great potential in a broad spectrum of practical applications of clinical, pharmaceutical, environmental, and industrial interests. Furthermore, the advantages of the electrochemical biosensors such as portability, rapid measurements, and small sample size make them very attractive for biomedical and biochemical studies, as well as the numerous commercial applications that profit from miniaturized electrochemical instrumentation.
Recent progress in redox protein design and engineering shows rapid advancement toward novel protein synthesis with predictable structures and functions through both the types of protein scaffolds and the range of metal ions and redox cofactors that can be incorporated. These successful approaches of engineering the biorecognition components with rational and combinatorial strategies have opened the door to widespread exploitations of redox proteins for sensing devices with desired electrochemical properties. Furthermore, the developments in nanobiotechnology and material sciences facilitate further development of useful and reliable biosensor devices.
Protein engineering has made valuable contributions toward the design of redox proteins with functional properties and interacting surfaces. In the development of engineered redox protein-based biosensors, one of the long-term goals is to fine tune the redox potentials and increased sensitivity and selectivity of the metalloproteins for practical applications without dramatically changing the redox-active sites or electron-transfer properties. By tailoring such enzymes with desired bioelectrocatalytic properties and modifying electrode surfaces with controlled and sensitive biolayers, it is possible to achieve a designed pseudo-biological recognition between proteins and electrodes (49). The demonstration of Gox biosensors as commercially successful products and their continued improvement through the integration of metal nanoparticles, modified redox proteins, and nanomaterials, such as carbon nanotubes, has attracted much interest in recent years. It has the great potential for further developing biosensors that detect certain species in real-time biological systems with a similar concept, such as the detection of toxic reactive oxygen species using biosensors constructed with modified superoxide dismutase (14, 19, 98, 137). Besides preserving biofunctionality, the protein-nanotube/nanoparticle connection could ensure appropriate molecular orientation, flexibility, and efficient, reproducible electrical conduction.
Although the electrochemical properties of many redox proteins have been improved by protein engineering, there are still quite a few limitations and drawbacks in their applications in electrochemical biosensing device development, such as limited shelf life, nonspecific binding and adsorption, as well as instability of the biorecognition components. In addition, environmental and health issues regarding nanodevices have been raised as well as the possibility of contamination caused during the manufacture process and application (145). Many practical strategies and environmental friendly protocols have been developed and investigated to overcome or minimize these problems. In spite of the numerous challenges, there is no doubt that electrochemical biosensors utilizing native and engineered redox proteins will continue to be a powerful tool for biomedical and clinical usage, disease diagnostics, food industry, and many other related important practical applications.
Footnotes
Acknowledgments
This work has been partially supported by the grant of NSF 1036579, Wallace H. Coulter Foundations, the grant of DoD W81XWH-10-1-0732, and FIU Doctoral Evidence Acquisition Fellowships. The authors also wish to express their gratitude to Dr. Jennifer McKenzie for editorial assistance.