
Abstract
Introduction
OMIM, Online Mendelian Inheritance in Man® available at:
There are also rare cases that are caused by accidental intake of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a prodrug that is converted to MPP+, an inhibitor of mitochondrial complex I. Therefore, although they represent a very small proportion of all cases, these examples support the idea that mitochondria might be important in pathogenesis in PD because in both cases, mitochondria are placed close to the cause of the disease.
Another possible clue to mechanism in PD considers the pathological events in the disease. Many types of neurons are damaged in PD, but prominent amongst them is the almost complete loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). These, and other neurons that are vulnerable to the disease process, tend to be projection neurons with long unmyelinated axons (12). In many cases, such neurons maintain a large synaptic field, requiring a substantial amount of energy in their axons. Therefore, it is possible that the types of neurons that are lost in PD have specific energetic requirements which, again, might implicate mitochondria in preferential vulnerability to disease.
However, it is not clear if mitochondrial dysfunction is a sufficient explanation for pathogenesis in PD. For example, although there is evidence of mitochondrial dysfunction in sporadic PD, this could easily be secondary to other events. The argument that energetics might be important in preferential vulnerability in PD is weakened by the fact that many neurons have high energy requirements. And while there are mitochondrial genes that cause parkinsonism, there are mutations in both mitochondrial and nuclear encoded mitochondrial genes that do not cause parkinsonism as a prominent symptom.
Therefore, mitochondria are theoretically relevant to some forms of PD, but it is not clear if mitochondrial dysfunction is either necessary for or sufficient to explain all forms of PD. In this review, we will discuss some of the ways in which PD genes impact mitochondria to try and understand whether there are specific aspects of mitochondrial function that may be important in this disease. Before doing so, we will first briefly review some specific aspects of mitochondrial biology that may be relevant to PD.
Mitochondria and Mitochondrial Dynamics
Mitochondria are double membrane organelles with an independent genome (mitochondrial DNA, or mtDNA) that have several functions, including supplying cells with energy through production of ATP, regulating local calcium levels, and participating in cellular signaling. ATP production is achieved through the movement of electrons through the complexes of the electron transport chain (ETC). During the process of electron transfer through the ETC, hydrogen ions are pumped across the inner mitochondrial membrane. Differences in H+ ion concentration across the inner mitochondrial membrane result in a mitochondrial membrane potential (MMP, or DYm). Also during ATP production, oxygen is consumed and a small number of electrons leak, which can react with oxygen locally to produce superoxide and lead to reactive oxygen species (ROS). Measuring mitochondrial function can therefore include measuring ATP production, the activities of the complexes (I–V) of the ETC, membrane potential, oxygen consumption, and/or ROS generation.
As well as being important by virtue of generating ATP, mitochondria are also highly dynamic organelles. They are trafficked throughout the cytoplasm and undergo fission and fusion events. Mitochondrial mobility is thought to be especially important in neuronal cells to deliver mitochondria to the site of synapses where they not only provide ATP for sustained synaptic transmission, but also probably help to buffer calcium changes (44, 101). A proper distribution of mitochondria is dependent on mitochondrial transport as well as fission and fusion processes. The fission and fusion of mitochondria is a highly regulated process. Some of the key events are mediated by dynamin-family GTPases, three of which, mitofusin 1 and 2 (Mfn1/2) and optic atrophy protein 1 (OPA1), promote fusion, and the pro-fission dynamin related protein (Drp1). We will discuss fusion and fission separately because of this distinction between the molecular machineries that mediate the processes, although it should be noted that there is evidence that the two processes are linked temporally with each other (98).
Mitochondrial Fusion
Mitochondrial fusion occurs when two distinct organelles fuse together and their contents, including matrix proteins and mitochondrial DNA, then mix (Fig. 1). The mitofusins are localized to the outer mitochondrial membrane by two transmembrane domains. Both the N-terminal GTPase domain and C-terminal two heptad repeat coiled-coil domains face the cytosol (86, 90). This orientation of the protein permits mitochondrial tethering by homotypic and heterotypic oligomerization of Mfn molecules to allow GTPase-dependent fusion of the outer membranes. The process of fusion occurs preferentially for mitochondria with an intact mitochondrial membrane potential (51, 56).
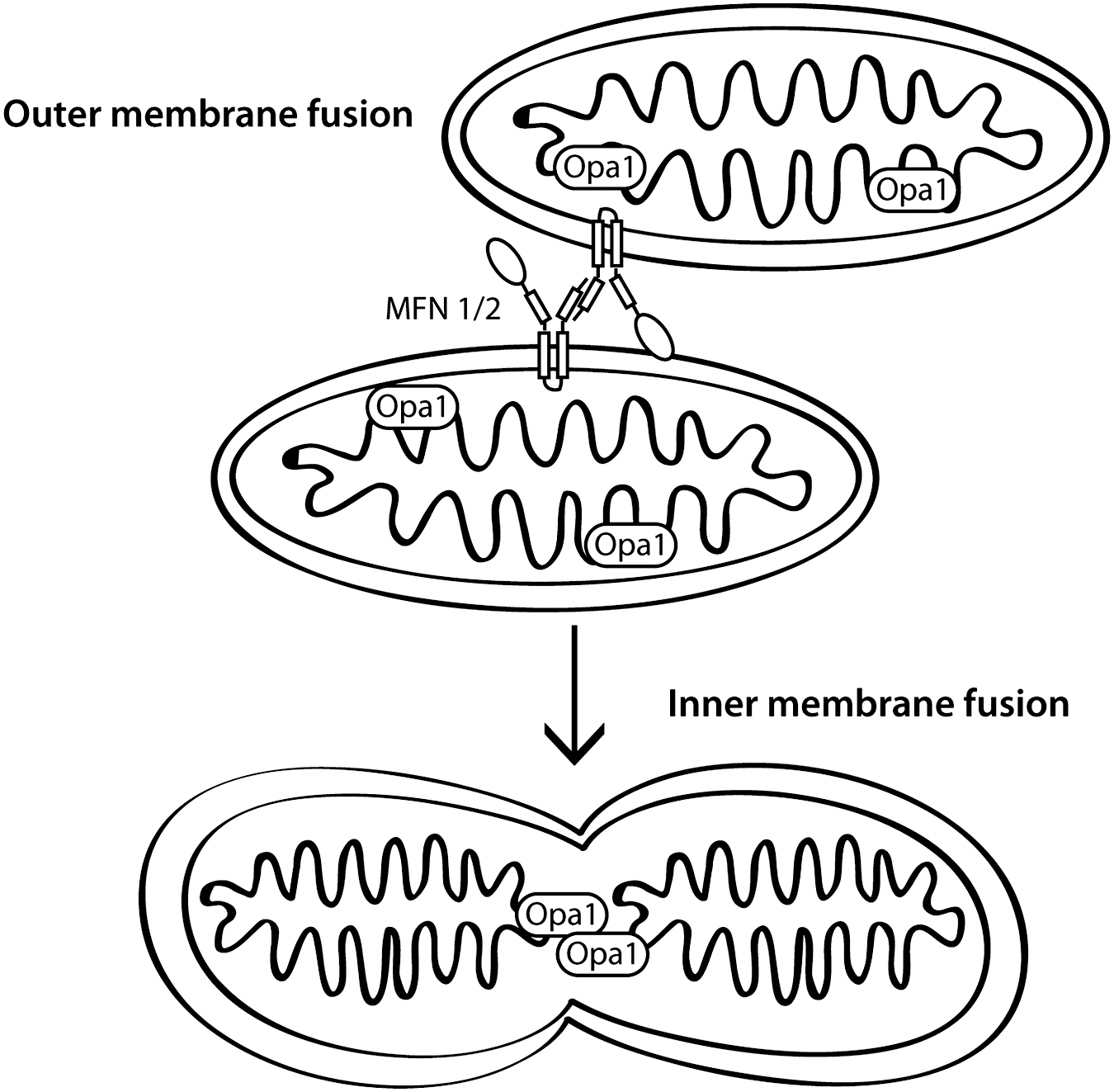
The same mitofusin molecules are also important for mitochondrial transport by kinesin motors through interaction with the atypical rho GTPase Miro and the adaptor protein Milton (42), and for regulating apoptosis through interaction with pro-apoptotic Bcl proteins (13). Although the functions of Mfn1 and 2 partially overlap, especially in that both proteins promote fusion, there is evidence suggesting that each has unique properties. The fusion activity of Mfn1 is higher than that of Mfn2, but its affinity for GTP is lower, resulting in more efficient mitochondrial fusion (47). Furthermore, in cells lacking Mfn proteins, defects in inner membrane fusion by Opa1 are rescued by restoration of Mfn1 alone, but not Mfn2 (22). Mfn2 is involved in several functions not directly associated with mitochondrial fusion, including mitochondrial and ER tethering to allow coupling of calcium homeostasis between the two organelles, as well as binding to signaling molecules including p21 Ras, in order to limit Erk activation, and anti-apoptotic Bcl-2 family members (reviewed in (27)).
OPA1 mediates fusion of the inner mitochondrial membrane, which requires oligomerization and GTPase activity of the Opa1 protein (2, 22, 29, 77, 91). Opa1 is located in the intermembrane space by virtue of a mitochondrial localization sequence and, in the long isoform a transmembrane domain that anchors it in the inner mitochondrial membrane. Knockdown of OPA1, in addition to leading to fragmented mitochondria, also results in a loss of membrane potential, disorganized cristae with widened junctions, and cytochrome c release, resulting in apoptosis (76). Proteolytic processing of OPA1 into soluble isoforms occurs after loss of membrane potential. The corollary reduction of membrane-associated isoforms interrupts the normal functions of OPA1 to inhibit mitochondrial fusion, leads to cristae remodeling, and also promotes cytochrome c release (6, 48). Therefore, loss of Opa1 is associated with both changes in mitochondrial dynamics and apoptosis.
Mitochondrial Fission
The fission proteins Drp1 and Fis1 promote mitochondrial fission, the process by which a single organelle is cleaved into two or more daughter mitochondria, in mammalian cells (Fig. 2). Drp1, another dynamin related GTPase, is usually present in the cytoplasm as oligomers. When the oligomers assemble into larger complexes on the mitochondrial outer membrane they promote GTPase-dependent changes in the membrane to mediate fission (reviewed in (16)). FIS1 is an outer mitochondrial membrane protein with a single C-terminal transmembrane domain that transiently interacts with Drp1 for recruitment of Drp1 oligomers to the membrane (112). Fis1 also promotes apoptosis independent of its effects on fission (3).
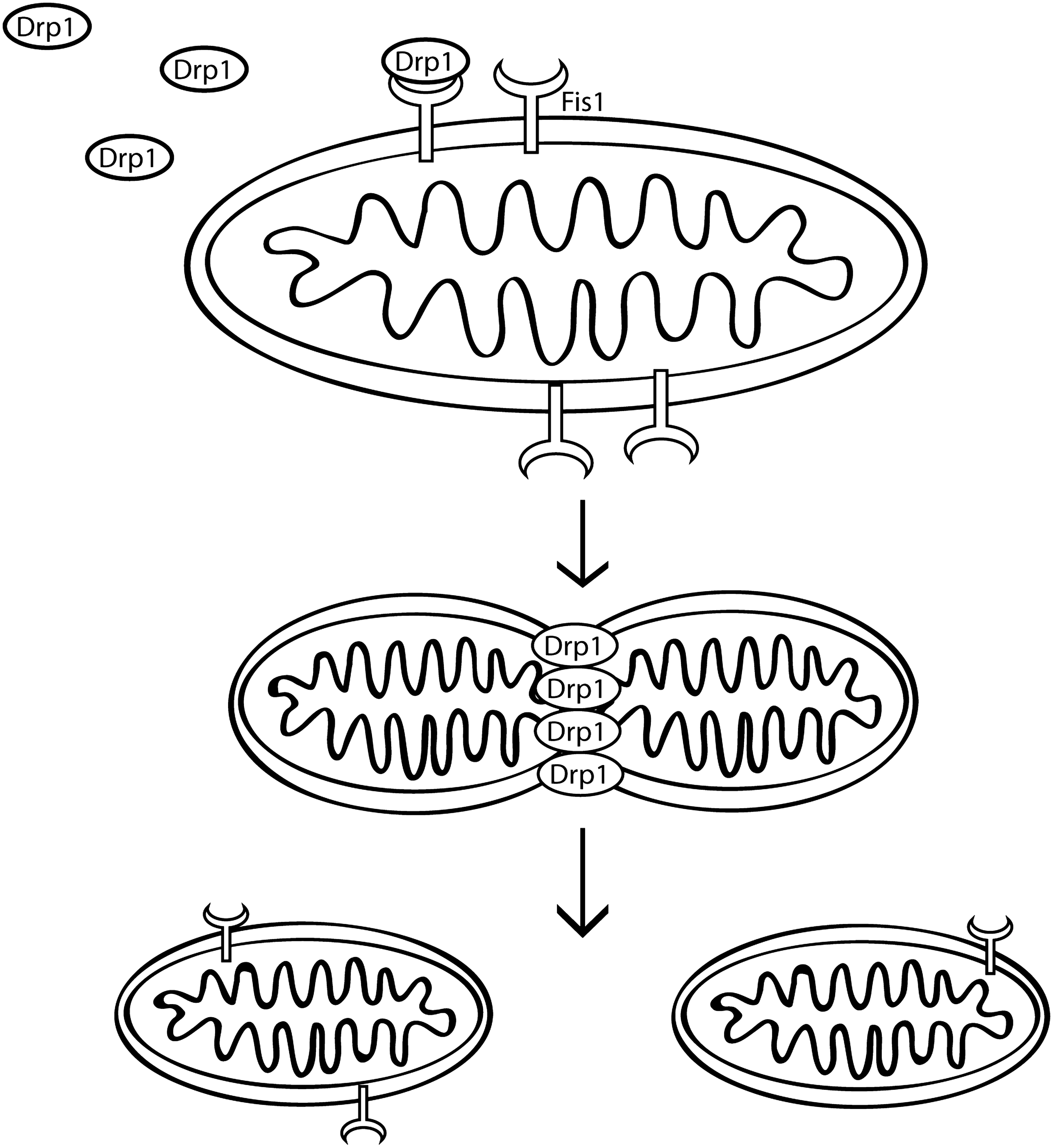
In addition to requiring interaction with Fis1 for mitochondrial localization, post-translational modifications regulate both mitochondrial recruitment and oligomerization of Drp1. Modifications include phosphorylation, which can inhibit or promote fission by regulating binding to other proteins or by directly affecting GTPase activity (reviewed in (16)). Less well-studied modifications include sumoylation (34), ubiquitination by MARCHV, an outer mitochondrial membrane ubiquitin ligase that ubiquitinates both Drp1 and Fis1 (111), and redox dependent S-nitrosylation of cysteine 644 by nitric oxide (NO) thought to promote fission (19). Therefore, mitochondrial fission is influenced by cellular signaling events in the cytosol by a number of signaling pathways.
Regulation of Mitophagy
The above discussion shows us that the mitochondrial network is subject to cycles of fission and fusion, the timing of which is often related to mitochondrial membrane potential. However, mitochondria that are significantly damaged need to be removed from the cycling pool of organelles to maintain the overall mitochondrial health of the cell. The process by which this occurs is a selective form of autophagy, called mitophagy.
Autophagy is a highly conserved process in which proteins and organelles are degraded either by nonselective engulfment of cytoplasmic contents or by selective removal of specific cargoes within a double-membrane structure, the phagophore, that eventually fuses with lysosomes to degrade the vesicle contents (reviewed in (113)).
Identification of mitochondria to be degraded is likely dictated by mitochondrial membrane potential whereby depolarized mitochondria are tagged for degradation. Mitophagy is also influenced by mitochondrial dynamics as when fission is blocked nonfunctional mitochondria accumulate independent of membrane potential (98). Several previous studies have suggested that fission of mitochondria occurs where nonfunctional and oxidized proteins are preferentially accumulated in a single daughter mitochondrion that does not undergo subsequent fusion and is degraded, whereas the other mitochondrion has higher membrane potential and undergoes further rounds of fission and fusion (98). This unequal mitochondrial division is thought to be important to maintain the population of highly functional mitochondria and to provide a mechanism by which nonfunctional mitochondrial components can be isolated for autophagic removal.
The cellular components required for yeast mitophagy are more fully characterized than the pathways needed for mitophagy in mammalian systems. Briefly, in yeast, Atg32 is the specific receptor protein needed for sequestration of mitochondria, in addition to the core autophagic proteins. Atg32 is then recognized by Atg11 and Atg8 and transported to the vacuole (reviewed in (103)). In mammalian systems, the homolog of Atg32 has not been identified although candidate proteins including Nix and p62 have been suggested (Fig. 3). Nix is an outer mitochondrial membrane protein that binds to LC3 (Atg8) and has been reported to mediate mitochondrial removal in the maturation of red blood cells, as well as to contribute to depolarization-induced mitophagy (31, 89, 114). A second depolarization-induced mitophagy pathway that will be discussed in further detail below utilizes p62 binding to mitochondrial substrates of parkin ubiquitination for mitochondrial transport to autophagosomes (39).

The regulation of mitophagy is not fully characterized. Several conditions, including starvation and increased ROS, have been shown to increase mitophagy. However, because mitochondria are critical for ATP production and also a significant source of intracellular ROS, they are themselves likely to significantly affect autophagic activity. The activation of autophagy observed with inhibition of the electron transport chain is ROS dependent (17). One example of how increased ROS levels interact with autophagy is by oxidation of ATG4 leading to its inactivation, which allows LC3 conjugation to autophagosomes (93). Therefore, the relationships between mitochondrial function, autophagy in general, and mitophagy in particular, are probably complex with several inter-related events mediating the overall identification of damaged mitochondria and their rates of removal from the mitochondrial pool.
Mitochondria in Parkinson's Disease
There is extensive evidence for mitochondrial involvement in dopaminergic neuron degeneration and Parkinson's disease. Inhibitors of complex I of the electron transport chain, including MPTP/MPP+ and rotenone, have been used to model the loss of dopamine neurons that characterize the disease (reviewed in (8)). Mitochondria from Parkinson's disease patients have defects in the activity of complex I of the ETC in many regions, including the substantia nigra, skeletal muscle, and platelets (10, 53, 70, 92), although not all studies have found significant decrement in this measure.
In several of the mitochondrial toxin models, there is evidence that changes in mitochondrial morphology may impact toxicity. For example, rotenone treatment of primary neurons causes rapid mitochondrial fission, and reducing fission or increasing fusion not only prevents mitochondrial fragmentation but also rescues cell death (7). 6-OHDA-induced Drp1-dependent mitochondrial fission occurs early, before loss of membrane potential, and is required for 6-OHDA-induced cell death (43). MPTP treatment leads to gross morphologic changes in mitochondria (21). There are a few reports of altered mitochondrial morphology in sporadic PD both in the brain, substantia nigra, and caudate nucleus, and in skeletal muscle and cybrid cell lines in which enlarged mitochondria with disrupted cristae can be observed (1, 5, 54, 97). Therefore, mitochondrial damage in PD may be associated with altered mitochondrial morphology that might, in turn, be associated with altered mitochondrial dynamics.
Also implicated in Parkinson's disease is an increased level of reactive oxygen species (ROS). As discussed above, cellular ROS is primarily generated in mitochondria (reviewed in (4)). This increase in oxidative stress can cause damage to complex 1, reducing its efficiency, and leads to modification of other cellular components. Whether ROS generation is an early or later event in PD is not clear, and whether oxidative stress is required for PD pathogenesis is not known.
Autosomal Recessive Parkinsonism Genes and Mitochondria
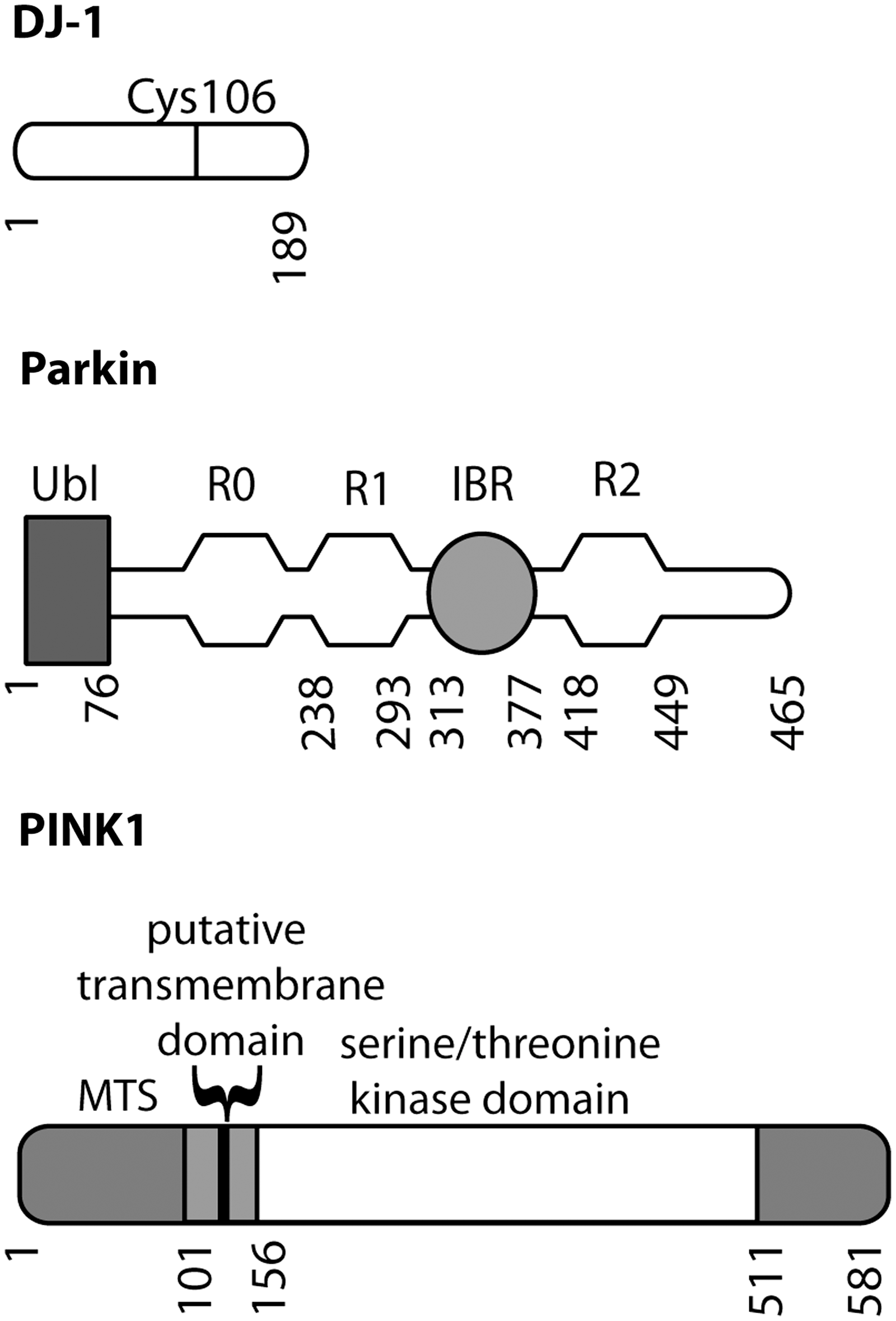
There are several genes for PD including three that are associated with autosomal recessive parkinsonism (Fig. 4). The three best characterized are parkin, PINK1 (Pten-induced novel kinase 1), and DJ-1, and there is evidence that each may have specific roles in the regulation of mitochondrial function. Importantly, these genetic forms are each recessive and include genomic deletions and whole exon duplications in the case of parkin (61). The pattern of inheritance and the types of mutations found implies that the disease is caused by loss of normal protein function.
The human phenotypes associated with mutations in any of these three genes are similar. Affected patients tend to have a relatively early onset of between 20 and 50 years of age, which is earlier than that seen for ‘typical’ sporadic PD (age at onset between 50 and 70 years of age). Recessive parkinsonism often has a relatively benign course, with mild disease over many years. Some cases have Lewy bodies, the a-synuclein positive inclusions that characterize sporadic PD, although not all cases do.
Parkin was the first of these genes to be identified. Parkin is an E3 protein–ubiquitin ligase with an N-terminal ubiquitin-like (ubl) domain and three RING (really interesting new gene) finger domains together with an IBR (in-between RING) domain. RING-type E3 ligases bind simultaneously to their protein substrates and to E2 enzymes that carry activated ubiquitin (Ub). The ubiquitylation reaction occurs because of the relative proximity of the E2–Ub complex and substrate; RING-type E3 ligases are therefore more akin to scaffolding proteins than to traditional enzymes that catalyze reactions. The addition of one or more ubiquitin molecules to the substrate can either be a signal for protein degradation by the proteasome, or can modify substrate function in other ways, for example, by changing cycling of the protein through different endocytic compartments. Several parkin substrates have been proposed, although none have been universally accepted.
DJ-1 was the second gene identified for recessive parkinsonism. DJ-1 is part of an evolutionarily ancient protein superfamily, and DJ-1 homologues are found in most aerobic species. Although the precise function of the protein is not known, DJ-1 appears to be important in regulation of cellular responses to oxidative stress. This is achieved in part by modification of a specific cysteine residue (Cys106 in human DJ-1) to form a cysteine-sulfinic acid (14).
PINK1 (PTEN-induced novel kinase 1) is the most recently discovered of the three genes and encodes a serine/threonine protein kinase. The full-length protein is localized to mitochondria, although some cleavage products are found in the cytosol (9, 99). Despite having measurable kinase activity using model substrates, the identification of authentic PINK1 substrates is still controversial, as none have been shown to show differential phosphorylation in vivo, such as in knockout brains.
One link between these three genes is that Drosophila models of both PINK1 and parkin deficiency have similar mitochondrial phenotypes in which large, highly fused mitochondria are found. In the same models, there is evidence of decreased ATP levels, degeneration of flight muscles, and loss of dopaminergic neurons (23, 80, 110). Parkin overexpression can complement PINK1 deficiency but the reverse is not true, suggesting a common pathway in which parkin is downstream of PINK1 (23, 80, 110). Several recent studies have suggested that PINK1 or parkin deficiency in flies causes excessive mitochondrial fusion, as overexpressing fission proteins or knocking down the fusion machinery can rescue the phenotypes. DJ-1 deficient flies also have a mitochondrial phenotype. However, the pathology in this model is more dependent on levels of oxidative stress and DJ-1 cannot rescue PINK1 or parkin deficiency (66, 67, 110). In this model, therefore, DJ-1 is likely in a parallel pathway to PINK1/parkin.
Mammalian PINK1 and parkin-deficient phenotypes are characterized by accumulation of fragmented mitochondria with lower membrane potential (26, 32, 88, 109). Subsequently, this deficit was shown to result from reduced mitochondrial mobility and dysregulation of fission and fusion proteins, leading to a net enhancement of fission processes (26, 88). The enhancement of fission in PINK1-deficient cells may be dependent on changes in calcium signaling, as loss of PINK1 expression leads to calcium release from the mitochondria through the Na(+)/Ca(2+) exchanger resulting in accumulation of calcium and increased ROS generation (36). Furthermore, Drp1 is dephosphorylated in PINK1-deficient cells by overactivation of the calcium-dependent phosphatase calcineurin and by blocking calcineurin activity in these cells mitochondrial fragmentation can be reversed (88). Therefore, some of the mechanisms involved in mitochondrial fragmentation in mammalian cells have been identified, although it should be noted that morphological alterations are not seen in all circumstances, such as in knockout mice (37, 41).
Therefore, in Drosophila models the net effect of PINK1/parkin deficiency is to shift the balance of mitochondrial dynamics towards fusion, whereas in mammalian cell models the bias is towards fission. Quite why the two models give different results is difficult to understand. Aspects of the PINK1/parkin pathway are conserved, as in both cases parkin can rescue the effects of PINK1 deficiency, arguing against there being a major evolutionary difference in the relationship between PINK1/parkin and mitochondria. Two other possibilities are likely, although there may be other explanations. One simple explanation is that the effects of PINK1/parkin deficiency on the balance between mitochondrial fusion and fission are indirect and, therefore, depending on the precise physiological state of the tissue, different mechanisms may predominate. An alternative possibility is that the relationships between morphology, fission/fusion, and functional effects on the overall health of the cell are complex. In this scenario, the amount of phenotypic rescue of the cell or tissue may not be strictly correlated with fission or fusion. It is worth noting that several of the fission and fusion GTPases also play roles in other aspects of mitochondrial function, such as transport within the cell.
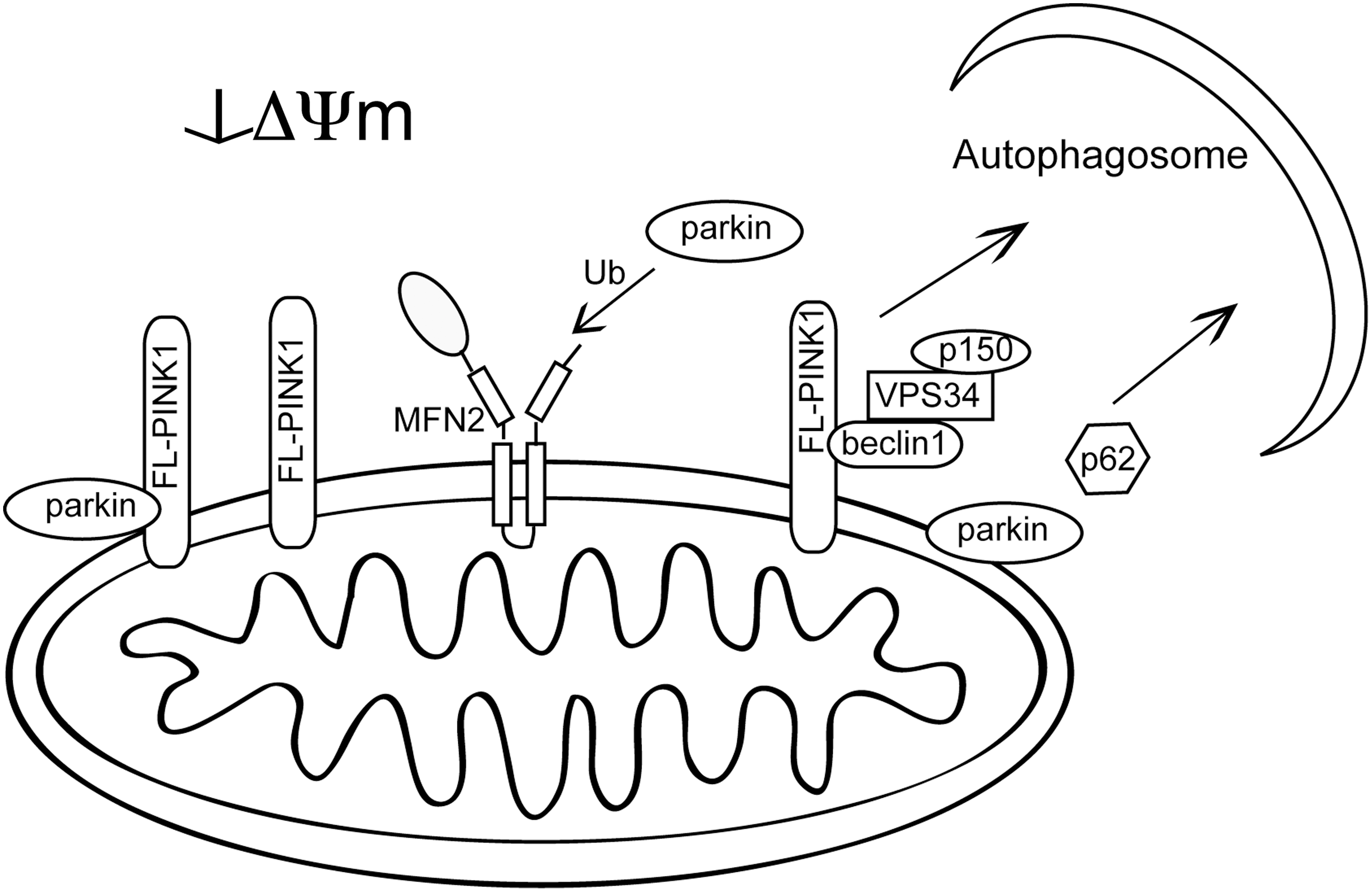
When it was originally reported that parkin deficiency was associated with mitochondrial phenotypes there was a conundrum, as parkin is generally present in the cytosol and nucleus, leading to an uncertainty as to how a mitochondrial effect might arise. The observation that parkin is recruited to CCCP-decoupled mitochondria to promote selective removal of depolarized mitochondria helped explain this apparent disconnect and also led to renewed interest in the stability and processing of PINK1. Under basal conditions, where mitochondrial membranes are highly polarized, PINK1 is constitutively synthesized but rapidly processed and degraded by voltage-sensitive proteolysis with very little full-length PINK1 being accumulated on the mitochondrial outer membrane (58, 94). However, if mitochondria become depolarized, full-length PINK1 is stabilized on the membrane. The increased level of PINK1 recruits parkin to the mitochondria and this relocalization of parkin subsequently leads to autophagic degradation of these depolarized mitochondria (28, 49, 74).
Although complete understanding of this pathway is still lacking, aspects of this process have been characterized. In Drosophila, the rhomboid-7 mitochondrial protease was shown to cleave dPINK1, and later the mammalian ortholog PARL was shown to mediate PINK1 N-terminal cleavage (28, 49, 107). Proteolysis of PINK1 still occurs in PARL-deficient cells, although with different products consistent with PARL being the preferred protease. In Drosophila there are no homologs of rhomboid-7, resulting in the disappearance of the shorter dPINK1 isoform when rhomboid-7 is absent (107). Several of the disease-linked mutations in PINK1 have altered processing that results in a greater ratio of full-length to cleaved isoforms of PINK1 (28). Mitochondrial accumulation of full-length PINK1 has been shown to increase autophagy and therefore mutations that preferentially increase full-length PINK1 may affect mitochondrial morphology and function through induction of mitophagy (28, 64, 102).
Parkin relocalization to mitochondria is fully dependent on PINK1 kinase activity and is interrupted by some, but not all, pathogenic PINK1 and parkin mutations (40, 64, 74, 75). Kinase-dead PINK1 accumulates on depolarized mitochondrial membranes similar to wild-type protein, but is unable to support parkin recruitment (40). Parkin expression is necessary to support depolarization-dependent K63 ubiquitination of mitochondria (39, 75), and mitochondria with parkin at the surface are sequestered and then degraded by autophagy. The perinuclear clustering of depolarized mitochondrial is dependent on microtubule polymerization and in some experimental settings also requires expression of the autophagy adaptor p62 (39, 72, 75, 102). A recent report has demonstrated the proteasome dependence of parkin-mediated mitophagy. Following mitochondrial translocation of parkin, K48- and K63-linked ubiquitination of mitochondrial substrates is increased and the proteasome is recruited to mitochondria, leading to degradation of multiple outer mitochondrial membrane proteins including Mfn1 and Mfn2, neither of which is necessary for mitophagy (15). Removal of outer mitochondrial membrane proteins was independent of and necessary for autophagy.
However, there are still some important questions remaining about the PINK1/parkin pathway and the relationship of these two proteins to mitochondria. Specifically, there is an apparent contradiction between the observation that parkin overexpression can rescue several aspects of PINK1 deficiency and the absolute requirement of PINK1 for parkin recruitment to depolarized mitochondria. It is possible that parkin is functional within the cell without requiring recruitment to mitochondria; the recruitment itself may be an epiphenomenon. However, the strong effect of PINK1 on parkin localization suggests that there is communication between the two proteins, which would be unusual if this had no functional significance. There are clearly some important further directions to be followed regarding the details of the relationship between these two proteins, and the nature of the signaling pathway between them, but we have to consider that parkin may have PINK1-dependent and -independent functions. Furthermore, PINK1 expression is not necessary for removal of mitochondria via mitophagy, as upregulation of autophagy genes suppresses PINK1-deficient phenotypes in Drosophila and autophagy is upregulated with transient knockdown of PINK1 in mammalian cells (26, 60).
Another concern about the phenomenon of parkin recruitment is that it has not yet been shown to occur in more intact settings that of cultured HeLa cells. The depolarization of whole populations of mitochondria within a cell triggers massive parkin recruitment that is unlikely to represent what happens in a neuron. More selective processes, such as the recruitment of parkin to a subset of depolarized mitochondria in Mfn1/2-deficient fibroblasts that have a population of mitochondria with variable membrane potentials is perhaps more realistic of what may occur in vivo (73). However, recent results have indicated that parkin recruitment may be less robust in neurons compared to other cell types, potentially depending on the metabolic state of the cell (100). Therefore, there is a need for a model for recruitment of parkin under pathological or physiological circumstances in neurons, preferably in vivo.
In addition to promoting autophagic removal of damaged mitochondria, PINK1 and parkin may also more directly regulate mitochondrial dynamics. As discussed above, parkin has recently been shown to ubiquitinate the mitochondrial fusion proteins Mfn1 and Mfn2, and depolarization-induced Mfn2 ubiquitination and removal by the proteasome is enhanced by the expression of parkin and PINK1 in both Drosophila and mammalian models (38, 83, 115). Removal of mitofusins from the mitochondrial surface prevents fusion with neighboring mitochondria and would allow for sequestration of damaged organelles from the mitochondrial network. Mfn2 not only regulates fusion, but has also been found to mediate mitochondrial transport along microtubules through its interaction with Miro and Milton complexes that have been shown to promote both anterograde and retrograde mitochondrial transport (69). Furthermore, overexpressed full-length and N-terminal cleaved PINK1 have been shown to interact with Miro2, and this interaction increases the proportion of full-length PINK1 associated with mitochondria (105). It is not clear whether the association of PINK1 with Miro/Milton complexes requires mitofusins. Therefore, it is likely that there are at least two pathways of PINK1 function, transport of mitochondria along microtubules, and mitochondrial quality control.
Thus, when mitochondrial membrane potential is intact, the cleaved isoform of PINK1 predominates and can still interact with and promote Mfn2-dependent mitochondrial transport. However, when mitochondrial membrane potential dissipates, parkin-mediated ubiquitination of Mfn2 would increase, leading to inhibited mitochondria transport (Fig. 5). In contrast, PINK1 involvement in mitophagy requires accumulation of full-length PINK1, which can both recruit parkin to mediate selective mitophagy and can also bind to beclin1 to promote autophagy (68) (Fig. 6). The beclin1/Vps34 interaction with PINK1, unlike parkin recruitment, does not depend on PINK1 kinase activity. Differences in PINK1 and parkin null phenotypes between Drosophila and mammalian models might, in part, be due to differences in redundant pathways for mitochondrial transport and removal.

DJ-1, also linked to recessive parkinsonism, functions to promote mitochondrial viability. Under standard conditions it is localized to the cytosol, but with oxidative stress DJ-1 itself becomes oxidized at cysteine 106 and translocates to the mitochondria. Mitochondrial localization and protection against oxidative stress both depend on oxidation of this cysteine residue (14). Cellular, Drosophila, and animal models of DJ-1 deficiency are all characterized by markedly enhanced sensitivity to oxidative stress with some, but not all, studies observing a higher proportion of depolarized and fragmented mitochondria (45, 46, 50, 52, 67, 79, 81, 95). Models in which DJ-1 deficiency results in fragmented, disconnected mitochondria have partial phenotype reversal by inhibition of Drp1 or by overexpression of PINK1 or parkin (46, 95). Enhanced autophagy with DJ-1 deficiency has been reported in some studies (46, 85, 95), though reduced autophagy has also been seen in some models (52). Mitochondrial effects of DJ-1 are rescued by antioxidant treatment, indicative of increased fission resulting from increased oxidation and not necessarily a direct effect on fission and fusion machinery (46, 95). Again, this supports the idea that DJ-1 plays a role in mitochondrial health that is independent of the PINK1/parkin pathway.
Kufor-Rakeb disease, caused by recessive mutations in the lysosomal 5 P-type ATPase ATP13A2, is characterized by widespread neurodegenerative features that include early onset parkinsonism and pyramidal degeneration (84). The precise function of ATP13A2 is unclear, but disease-associated mutations lead to mistargeting of ATP13A2 to the endoplasmic reticulum instead of the lysosomal membrane, which may impair protein degradation through insufficient acidification of lysosomes. Potentially supporting the idea that ATP13A2 mutations are associated with lysosomal function, Tibetan terriers with recessive ATP13A2 mutations have widespread neurodegeneration and ceroid lipofuscinosis (33). Although Kufor-Rakeb syndrome cases have not been autopsied, if the results in dogs are predictive of the human condition, then ATP13A2 mutations would be associated with a lysosomal storage disease.
Autosomal Dominant Parkinsonism Mutations and Mitochondria
The above discussion has centered on genes for autosomal recessive parkinsonism, where evidence of a role in mitochondrial function is reasonably well established. However, there are other genetic forms of PD, including two well-characterized genes for autosomal dominant disease, LRRK2 and α-synuclein (Fig. 7). Given the history of thinking about mitochondria in PD, effects of these two genes on mitochondrial function and morphology have been examined in several studies.
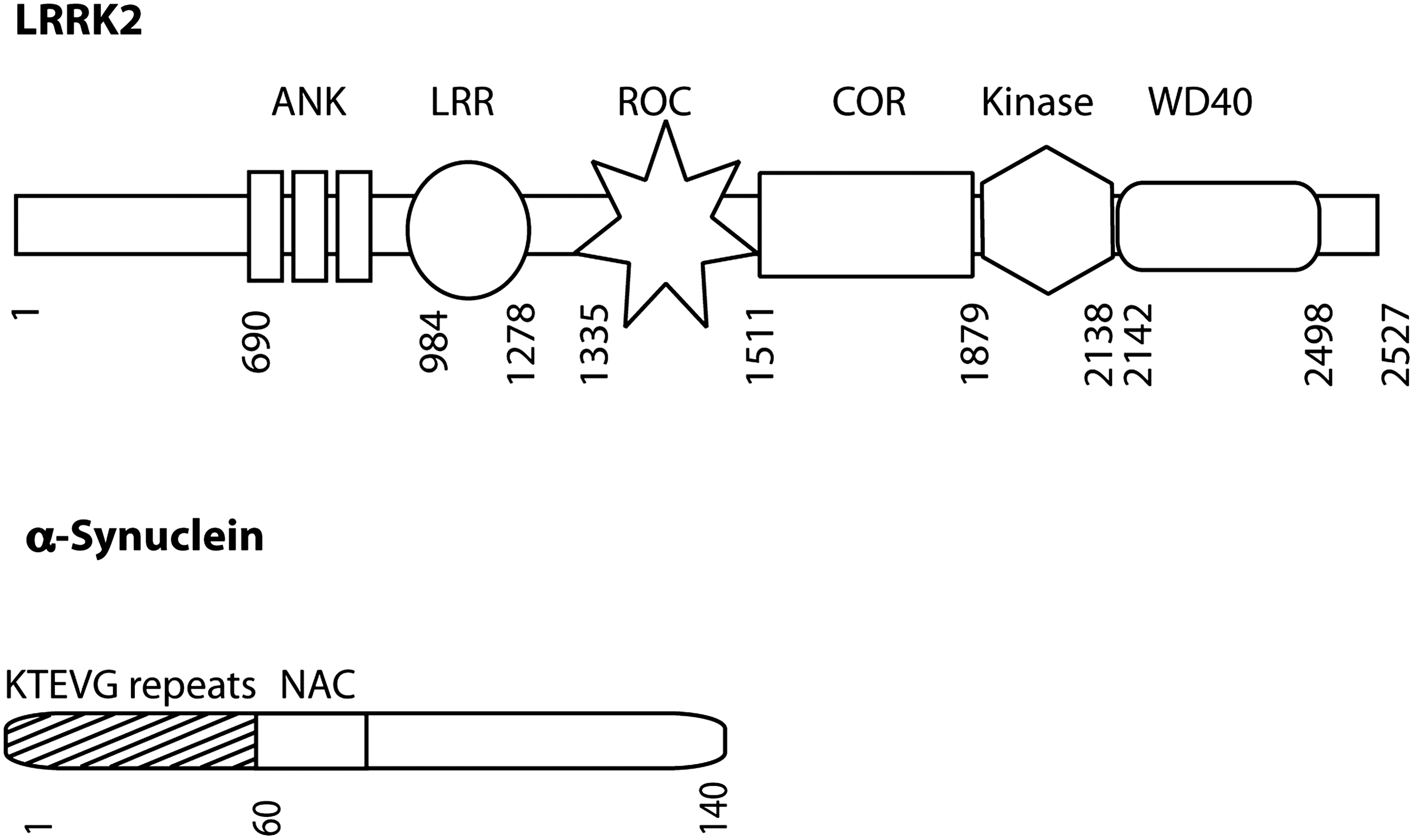
α-Synuclein, a component of Lewy bodies, was the first identified genetic cause of parkinsonism (82). It is located throughout the cytosol and it has been known since some of the early cloning papers that a fraction of the protein is membrane associated, being particularly concentrated around synaptic vesicles (63). More recently, α-synuclein was suggested to contain an N-terminal cryptic mitochondrial targeting signal that promotes mitochondrial import of α-synuclein, allowing its interaction with the inner membrane (30). Accumulation of wild-type and A53T α-synuclein in mitochondria led to decreased mitochondrial complex 1 activity and increased levels of ROS (18, 30).
In addition to inhibiting mitochondrial function, α-synuclein expression also inhibits autophagy, which might therefore indirectly change mitochondrial turnover rates. Increasing α-synuclein levels were shown to reduce LC3-II accumulation by inhibition of Rab1, a small GTPase that controls vesicle traffic from the ER to the Golgi (108). Decreasing Rab1 expression or increasing α-synuclein levels led to mislocalization of ATG9, a protein involved in early autophagosome generation from the ER-Golgi compartments (108). Thus α-synuclein inhibits autophagy at the earliest stages of vesicle formation rather than inhibiting mature or vesicle degradation.
Not only does α-synuclein modulate autophagy, but its removal, including of mutant forms, is in part achieved through both bulk and chaperone-mediated autophagy (CMA) (25, 104). Furthermore, disease-associated mutations in α-synuclein allow the protein to interact more strongly with the LAMP2A receptor, but internalization of mutant protein into the lysosome is less efficient, which inhibits degradation of both α-synuclein and other CMA substrates (25). A recent report shows increased autophagy may connect increased levels of α-synuclein with mitochondrial dysfunction (20). Fewer mitochondria are found in cultured cortical neurons expressing either wild-type or A53T α-synuclein and expression of A53T α-synuclein leads to lower ATP levels and neuron loss. Reduced mitochondria levels could be reversed by reducing parkin expression, inhibiting Drp1 activity, or increasing expression of Mfn2 as well as by reducing expression of necessary autophagy genes (20).
LRRK2 is a large multidomain protein with both GTPase and kinase activities that is localized primarily to the cytoplasm with an appreciable amount being membrane associated. Some reports have suggested that a portion of LRRK2 is present at the outer mitochondrial membrane (11, 106), although this is not seen in all models.
A more specific role for LRRK2 in mitochondrial function is supported by attenuation of peroxide toxicity by overexpression of wild-type, but not mutant, LRRK2 as well as the ability of human LRRK expression to protect against paraquat and rotenone toxicity in Caenorhabditis elegans (59, 87). A recent study reported mitochondrial phenotypes in fibroblasts taken from G2019S carriers that have lower mitochondrial membrane potential, and reduced levels of total intracellular ATP as a result of lower ATP production from substrates linked to complexes I, II, and IV (71). Mitochondria were longer and more interconnected in G2019S fibroblasts and although this morphology phenotype was mild, increased mitochondrial length correlated with reduced ATP (71).
Whether these mitochondrial phenotypes are a cause of mutant LRRK2 pathology, or simply result from other primary events, is unclear. One of the first phenotypes associated with overexpression of LRRK2 mutants in cell culture models was a reduction in neurite length and branching, with opposite effects from LRRK2 knockdown which results in increased neurite length (62). Changes in neurite length and mitochondrial morphology and function could both result from cytoskeletal changes mediated by LRRK2 expression. LRRK2 interaction with tubulin has been reported, and neurons from mice overexpressing G2019S LRRK2 have increased F-actin in filopodia, which correlates with reduced neurite length (35, 78). Given the necessity of microtubule transport in mitochondrial distribution, it is possible that cytoskeletal disruption might prevent effective mitochondrial transport.
Alternatively, LRRK2 regulation of mitochondrial morphology could lead to reduced neurite length as mitochondrial fission is known to promote synaptosomal distribution of mitochondria, and synaptic mitochondria control synaptic strength and synaptic transmission which regulate dendritic length (57, 101). Consistent with synaptic changes are that G2019S and wild-type LRRK BAC (bacterial artificial chromosome) transgenic mice exhibit deficiencies in striatal dopamine release (65, 96), and overexpression of human LRRK2 in Drosophila leads to a reduction of mitochondria in terminal boutons with overexpression of wild-type or G2019S mutant LRRK2, resulting in fewer type 1 boutons and reduced length of the neuromuscular junction (55).
Overall, there are several pieces of data that link a-synuclein and LRRK2 to mitochondria. However, these are somewhat indirect and there is equal data suggesting that either protein might be associated with other membranous structures in cells. It is therefore not clear if mitochondrial association is either necessary or sufficient to explain the toxic effects of dominant mutations, although given the interest in this organelle in PD, mitochondrial damage is an important theory to explain damage to neurons caused by mutations in LRRK2 or a-synuclein.
Conclusion
Despite the recognition that mitochondrial function, dynamics, and quality control are likely to contribute significantly to neuronal loss in Parkinson's disease, a better understanding of what defects are primary in pathogenesis and how environmental and genetic causes of mitochondrial dysfunction interact to cause disease is needed. Available data indicate that genetic and suspected environmental causes of PD impact mitochondrial function, dynamics, and removal at different stages. Inhibition of complex 1 activity by environmental toxins, ROS accumulation, or dominant mutations may initiate disease, whereas recessive mutations, specifically DJ-1, may function to limit mitochondrial damage in conditions of increased oxidative stress. Subsequently, if mitochondria become depolarized or excessively fragmented, PINK1 and parkin-regulated pathways may sense these impaired mitochondria and respond by limiting fusion and eliminating nonfunctional proteins.
However, a substantial amount of work is required to understand the molecular details of PINK1/parkin related pathways, as well as the effects of DJ-1 or the dominant mutations in a-synuclein and LRRK2. A particularly difficult question is to understand why mutations in these genes cause parkinsonian phenotypes and not neuronal damage more generally. It is possible that dopamine neurons in the substantia nigra have specific energetic requirements that render them susceptible to mitochondrial damage. However, mutations in mitochondrial genes tend to be associated with damage to peripheral nerves (e.g., Charcot-Marie Tooth syndrome associated with mutations in mitofusin) or optic atrophy (e.g., mutations in Opa1). Therefore, there is probably something specific about the roles of the PD genes and mitochondria. One possibility is that the genes for PD play more regulatory roles in mitochondrial function and, perhaps, these roles are more important in situations such as oxidative stress. We propose here that focusing on regulatory aspects may be a critical direction for the field in the future.
Footnotes
Acknowledgments
This research was supported entirely by the Intramural Research Program of the NIH, National Institute on Aging.