
Abstract
Introduction
The biomedical relevance of mitochondria is due not only to the host of genetic diseases that interfere with their own and the cell's functions, or to their relevance for aging (206, 216, 217). Via production of reactive oxygen species (ROS) they can have a role in carcinogenesis (160, 210, 211) and degenerative processes (1, 27, 94). Their involvement in apoptosis and necrosis makes these organelles a focal point for therapeutic efforts to induce the death of unwanted cells (60, 145, 220). This review summarizes the major functions of mitochondria, and briefly describes how plant-made small molecules (also referred to as herbals, phytocompounds, phytomolecules, or phytochemicals) may affect them.
Compounds of vegetal origin, most commonly present in the diet or in herbal extracts, can either act directly on mitochondria, for example as inhibitors of the oxidative phosphorylation system (57, 244), or affect mitochondria indirectly, by modulating cellular signaling cascades that have an impact on the organelles (e.g., 99, 183). Many, the so-called antioxidants, may in principle safeguard mitochondria as well as the cell by mopping up excess radicals. The theoretical risk exists of overdoing it, depleting the cell of a vital second messenger, hydrogen peroxide. The research community now fully realizes that the same antioxidant compounds may, depending on context, also act as pro-oxidants, and that this may be a very relevant type of action in vivo (66). If exerted at a low level—as is expected for dietary compounds that cannot reach high concentrations in the body due to low bioavailability and to metabolism (121, 218, 219)—pro-oxidant action can give rise to the adaptive phenomenon of hormesis, resulting in long-term indirect protection from redox stress (129, 168). Under more drastic (therapeutic) circumstances, the abundant generation of radicals may instead be exploited to precipitate cell death.
The focus of the review is on mitochondrial pathophysiology. For reasons of space and tractability, the review only touches those aspects of mitochondrial physiology of greatest relevance for its topic (Fig. 1). The pervasive interconnections are schematized in Figure 2. Much remains to be discovered: the mitochondrial proteome still comprises many “orfan” proteins; as their roles are identified, new possibilities for functional interactions with natural compounds are likely to emerge.
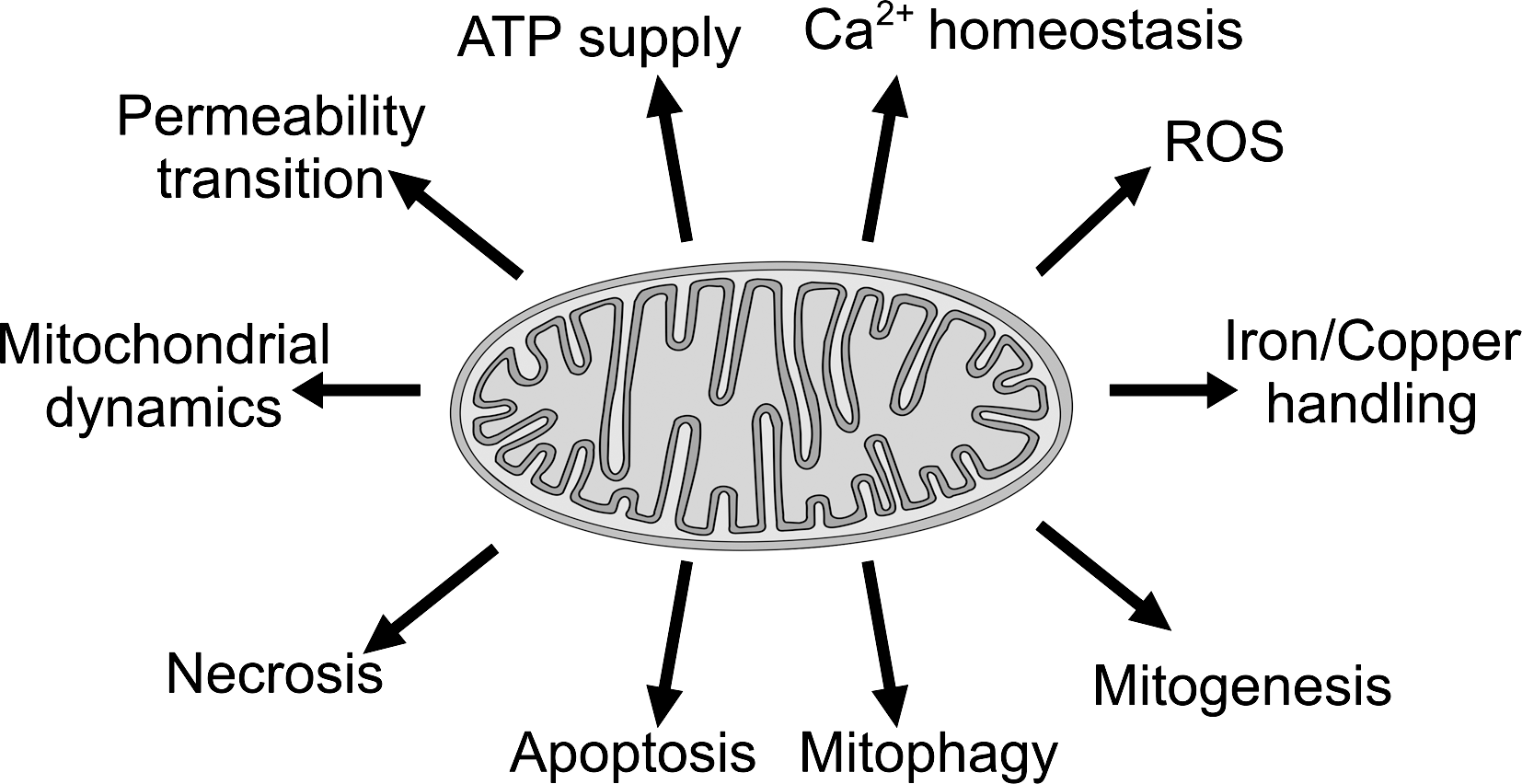

Only a small fraction of the innumerable articles in the field can be cited. We apologize to the many colleagues whose work is not adequately represented. Rather than producing detailed lists of plant-made molecules and their actions, we have tried to keep the discussion general, leaving room for the reader to make connections and extrapolations. In most cases a short overview of the relevant compounds and their possible mode of action follows that of each mitochondrial function or mito-related pathology. Particular attention has been devoted to resveratrol, possibly the most talked-about natural product. Phytocompounds act on cellular physiology in many ways, of which their functional interaction with mitochondria is but one. Several recent reviews together allow a comprehensive assessment of their pharmacology (e.g., 63, 76, 115, 182; resveratrol: 59, 96, 152, 181; flavonoids: 17, 72, 146, 154; catechins: 100, 104).
Mitochondrial Functions and Problems
ATP supply
The most obvious consequence of mitochondrial inadequacy is a deficit of cellular ATP, and the most obvious cause of mitochondrial inadequacy is genetic disease. The clinical relevance of mitochondria emerged with the identification of a variety of complex pathologies—metabolic disorders, myopathies, afflictions of the nervous system, etc.—originating from defects in mitochondrial function due to mutations or deletions of mitochondrial DNA (mtDNA). The mitochondrial genome, present in up to thousands of copies in a single cell, contains 37 genes, 13 of which encode subunits of the oxphosph apparatus. It suffers from a high mutation rate. Heteroplasmy prevails and the manifestation of clinical symptoms depends on the ratio of functional to nonfunctional copies. The origins, transmission, and evolution during lifetime of mtDNA alterations constitute a complex research area that cannot be tackled here but is covered by a number of recent reviews (e.g., 105, 206, 216).
Mitochondrial diseases arising from these alterations are heterogenous, reflecting the variety of possible mutations and deletions and the pecularities of mitochondrial genetics. mtDNA has been found in several cases to increase in pathologically affected regions, as if in an attempt to compensate dysfunction. Energy-intensive tissues such as the muscular and nervous systems are particularly affected. A broader definition of mitochondriopathy includes also defects of the estimated 1300–1500 components of the mitochondrial proteome that are encoded by nuclear DNA (10, 217). The MitoCarta database (144) has uncovered a large number of mitochondrial proteins with unknown function that may turn out to be targets for plant-made molecules. As the biochemical and functional characterization of these proteins progresses, more mitochondrial diseases come to be understood at the molecular level. One example of this type of progress is that of TMEM70, an ancillary factor of mitochondrial ATP synthase in higher eukaryotes (28).
Obviously, a shortage of ATP impacts on practically every aspect of cell life. Ca2+ homeostasis, for example, is deranged, in part because the sarco-endoplasmic reticulum calcium ATPases (SERCA) and plasma membrane calcium ATPases pumps cannot function optimally. Metabolic disorders are a common consequence of the impairment of oxidative phosphorylation, which forces cells to rely more heavily on glycolysis. The Warburg effect, that is, the reliance of cancer cells on aerobic glycolysis, was originally proposed to be a consequence of mitochondrial dysfunction and impairment of ATP production. Mitochondria have been found to be altered and functionally impaired in many types of cancer (47, 58). Glycolysis is now understood to be an important source of metabolic intermediates essential for the growth of any proliferating cells, and to offer other advantages to cancer cells, for example, resistance to hypoxia (58). Shifting the oxphosph/glycolysis ratio to favor the former antagonizes tumor cell growth (47), whereas an appropriate balance of the two pathways appears to be required for optimal tumor growth. These concepts are finding practical application with the development of anti-cancer drugs interfering with the aberrant metabolic flux of cancerous cells, such as dichloroacetate (131) and 3-bromopyruvate (92, 128).
Approaches to the management of mtDNA diseases are limited (177). A point of interest is whether the symptoms of mitochondrial myoencephalopathies might be reduced by an increase in mitochondrial biogenesis (177, 206). The notion derives from the prevalence of heteroplasmy, and posits that an increase in the number of WT mtDNA copies per cell might lead to an improvement in mitochondrial function even if accompanied by a corresponding increase in diseased copies. The idea seems negated by the often-observed failure of mitochondrial proliferation to remedy disease phenotypes, but has recently found some experimental support (139, 222). Phytochemicals may definitely help mitogenesis (see below), and may be useful to control other dysfunctions linked to mitochondrial genetic problems, such as excess ROS generation.
Ca2+ homeostasis
Mitochondria play an important role in the control of cellular Ca2+, since they can sequester this ion contributing to the shaping of the Ca2+ signal in space and time. In turn, they depend functionally on the external Ca2+ level, since physiological variations in this parameter are reflected in changes of matrix Ca2+ and in the activity of key matrix dehydrogenases, whereas pathogenic, prolonged, or exaggerated increases can induce the mitochondrial permeability transition (MPT; see below), dysfunction, and cell death (11). The mitochondrial Ca2+ uptake system predominantly displays a low affinity (μM range) for the ion (11). Such elevated concentrations are only reached transiently at Ca2+ hot spots close to plasma membrane Ca2+ channels or to sites of Ca2+ efflux from the endoplasmic reticulum (ER), which allow mitochondria to efficiently take in the ion as it is released from the stores (169) (Fig. 3). Mitochondrial Ca2+ uptake can also reportedly lead to increased ROS generation (2). Mitochondria in turn can have an impact on Ca2+ homeostasis also by influencing other components of the Ca2+-regulating system through their products, chiefly ATP, needed by Ca2+-transporting ATPases, and ROS, which target, among other relevant proteins, SERCA pumps, IP3 and ryanodine receptors, and several ion channels.

The interplay of Ca2+ signaling and mitochondria is highlighted by its role in pathology, in particular in ischemias, neurodegeneration, and excitotoxicity (see the sections on necrosis and on the mitochondrial permeability transition).
ROS and redox stress
Mitochondria are recognized to be the major source of ROS in most cells, under both normal and pathological circumstances (135). This results from the one-electron reduction of molecular oxygen to give superoxide anion, which can then be dismutated to O2 and H2O2 by superoxide dismutase (SOD). H2O2 can be eliminated by catalase or the thioredoxin/peroxiredoxin- and glutathione/glutathione peroxidase-centered redox defense systems (15, 30). For both systems reducing equivalents are provided by NADPH, linked to NADH by transhydrogenase (147). Imbalances in the levels of glutathione, the major cellular antioxidant, are critically linked to oxidative stress-induced apoptosis induction (27) and to major human pathologies, from aging and neurodegeneration on one side to cancer chemoresistance on the other. Mitochondrial glutathione has a direct, fundamental role in regulating ROS production by the organelles (124).
ROS production by healthy mitochondria is favored by a high transmembrane potential and a reduced state of the respiratory chain and of the NAD(P)H pool, which promote the leakage of electrons at complexes I and III of the respiratory chain. Conversely, depolarization has been reported to act in the opposite direction or to have no effect on ROS production. Other important mitochondrial sources of ROS are monoaminoxidases of the outer mitochondrial membrane (OMM) and p66shc (39). P66shc, a splice variant of adaptor proteins involved in Ras signaling, relocalizes to mitochondria upon phosphorylation by redox-sensitive kinases (c-Jun N-terminal kinase [JNK], PKCβ, and extracellular signal regulated kinases [ERK]) and produces ROS by catalyzing the direct transfer of electrons from cytochrome c to O2 (55). Thus, a positive feedback loop of ROS production is set up, which leads to mitochondrial dysfunction and dysregulation of Ca2+ homeostasis and apoptosis. P66shc is also intrinsically redox-sensitive, being activated by S-S bond formation and tetramerization. While its physiological roles are not yet clear, it has been assigned that of a villain in cardiomyopathy and ischemic injury (39), aging (205), and Alzheimer's disease (AD) (189).
Mitochondrial ROS are thought to be relevant for a host of cellular processes. They, in particular H2O2, act as intracellular messengers, whose effects depend on the cellular context and on the quantitative and temporal aspects of their production (167). For example, they are a signal for autophagy and play a major role in apoptosis (see below).
Elevated ROS production can be a cause as well as a consequence of genetic damage. When not repaired, these lesions lead to genome instability, cellular senescence, mitochondrial dysfunction, and pathologies such as neurodegeneration, cancer, and aging. Proteins can undergo various types of oxidative stress-induced modification imparing their functionality (32, 141). In particular, carbonylation is an irreversible process that may lead to the accumulation of defective protein aggregates, often observed in senescence-linked disorders. Oxidative stress also has a negative impact on the ubiquitin-dependent protein degradation system (proteasome), which itself is charged with eliminating damaged proteins. This results in an amplification of damage, since it leads to the accumulation of misfolded and/or damaged proteins (61).
ROS and aging
A working model of aging identifies mitochondrial ROS as a key culprit: ROS-inflicted damage to macromolecules, including mtDNA, leads to a progressive and self-reinforcing loss of cellular function, which impacts on systemic functionality (69). This scheme is now under attack (102, 105, 149).
The debate has been fueled largely by the creation of “mitochondrial DNA mutator” mice, expressing a mtDNA polymerase γ lacking its proofreading 3′-exonuclease activity: a mouse expressing D181A polγ in heart mitochondria (241), and two animals ubiquitously expressing D257A polγ (94, 204). These mice accumulate random mtDNA mutations as well as large deletions at a several-fold higher rate than WT counterparts, with consequent problems in the oxphosph system. Note that the mutated mitochondrial genotype is very likely to differ from cell to cell. The homozygous D257A polγ mice display a severe premature aging-like phenotype and have a shortened lifespan, thus confirming the relevance of mitochondria for these processes (201, 204). No evidence for increased oxidative stress or ROS production was found in either model (71, 202). The conclusion was therefore reached that while mtDNA mutations and/or deletions indeed induce age-associated organism deterioration, this effect is not mediated by ROS, but is rather the direct consequence of the decrease in mitochondrial functionality. Yet, other studies confirm an increased ROS production by mitochondria with defective respiratory chain components (e.g., 1).
A way out of the conundrum may be provided by a tendency toward excess apoptosis both in the D257A polγ mutant (94, 203) and in normal aging (94). What may the apoptotic stimulus be? An excessive generation of ROS would be fully expected to prompt cell death (see the section on ROS and apoptosis). Dead cells would be disposed of, escaping observation. The surviving cells, in the mutator mice as well as in normal aging, may be those in which the random mutations have not yet resulted in an above-threshold oxidative stress (in this hypothesis mitochondria are allowed to be dysfunctional without necessarily producing ROS). Oxidative stress marker levels would be close to normal because the cells still in place would have been exposed to not-too-high stress levels. Autophagy, selectively promoted by mitochondrial depolarization and dysfunction and by ROS (see section on mitophagy), would be expected to eliminate the more heavily damaged mitochondria and other intracellular components, thus keeping stress markers at approximately normal levels until overwhelmed, and/or intervening as autophagic death when the situation becomes unmanageable. An analysis of autophagy in polγ mutants may well be worth the effort. Maintenance-by-elimination would be expected to result in a higher rate of cell loss, and thus in a more rapid organism senescence (94), in the mtDNA mutators. Surviving cells may still suffer, and systemic responses such as cardiac hypertrophy or mitochondrial mass increase are not surprising. Cardiac mtDNA deletions, protein oxidative damage, apoptotic markers (activated casp-3), senescence, and hypertrophy were all decreased in the heart of D257A mice overexpressing mitochondria-targeted catalase in the heart (31). mtCAT also reduced the activation of the mitochondrial biogenesis system (PGC1α, TFAM, and NRFs) caused by the expression of the defective polγ.
In summary, a role of mitochondrial ROS in causing DNA mutations, oxidative alterations of proteins, apoptosis, and somatic senescence due to cell loss may still be compatible with the observations obtained with mtDNA mutators, as it is with the effects of a variety of genetic manipulations of redox enzymes in the mouse (149, 150, 215).
Oxidative stress, attendant biochemical alterations, and iron dyshomeostasis on one side, housekeeping autophagy and rejuvenating mitogenesis on the other thus appear to be key factors in the process of aging-related deterioration of mitochondrial and cellular functionality. Contrasting oxidative stress, potentiating the autophagic system, and increasing biogenesis to generate new, clean mitochondria may help to slow down or prevent aging, and age-associated neurodegeneration, sarcopenia, and diabetes. The possibilities offered by phytocompounds in these respects are mentioned in the appropriate sections.
ROS and cancer
In oncology mitochondria are emerging as a target of chemopreventive or chemotherapeutic intervention (e.g., 116, 160, 200, 211), whereas natural compounds are seen as multi-task agents capable of exerting pleiotropic effects (3). Over 200 active compounds have been identified using biological response as a criterion (130, 185). Most of the drugs approved for cancer treatment over the past quarter-century were natural products or were inspired by natural products (e.g., vinblastine and paclitaxel) (3). Since we have coexisted with them for ages, natural products are less likely to have deleterious side effects (although this does not necessarily apply to derivatives).
The most important connection between mitochondria, natural compounds, and cancer is provided by ROS (Fig. 4). For example, in mice one of the consequences of partial deficiency of SOD2 (the mitochondrial form) is a higher incidence of cancer (210). The effects of ROS may vary depending on the stage of carcinogenesis. In the tumor initiation phase, ROS may cause DNA damage and mutagenesis, whereas in established cancer ROS acting as signal mediators may stimulate proliferation. H2O2 is a recognized mitogen, acting via transactivation of growth factor receptors and/or of downstream effectors such as the Akt pathway. These effects are largely ascribed to the inhibition, via sensitive cysteine residues, of phosphatases (e.g., phosphatase and tensin homolog [PTEN] and phosphatases of the mitogen-activated protein kinase [MAPK] cascade). Redox-sensitive transcription factors, notably AP-1, nuclear factor κB (NFκB), and p53, are also activated by ROS (211). ROS can induce activation of hypoxia inducible factor, which promotes cancer stem cell proliferation and self-renewal, metabolic switch to glycolysis, adaptation to acidosis, modulation of histone methylation, expression of genes contributing to aggressiveness and metastasis, and induction of microRNAs associated with poor prognosis (78). An increased production of ROS has been reported for several different cancer types (116). Alterations in antioxidant enzyme activities have also been reported in many tumor cell lines, resulting in increased oxidative stress.

On the other hand, it is clear (see sections on apoptosis and necrosis) that ROS can induce cell death and thus act as anti-cancer agents. Since cancerous cells in many cases have a higher-than-normal basal rate of ROS production, despite their enhanced redox defenses an additional oxidative stress may push them over the brink of death more easily than normal cells (e.g., 116, 159, 227). Thus, both antioxidant and pro-oxidant anti-cancer approaches have been proposed (54, 158 –160, 200, 221, 227). Whether one or the other might be more useful is likely to depend on each individual case. These considerations explain the booming development of pharmacological tools allowing the modulation of the mitochondrial redox state, which in most studies take the form of mitochondria-targeted redox-active compounds (Smith et al., this issue; 13, 188). At least in the case of mitoVES (40) and of polyphenol-triphenylphosphonium conjugates synthesized by our group (unpublished results), these compounds can indeed act as pro-oxidant cell killers (see the contributions by Smith et al., Neuzil et al., and Huang in this issue).
Antioxidant phytocompounds and hormesis
ROS have long been the target of interventions deploying a variety of antioxidant natural compounds. The results have been mixed (29, 168, 226). The point has repeatedly been made that in vivo a direct ROS-scavenging action can hardly be relevant, given that exogenous antioxidants are not expected to reach significant levels in comparison with the cellular detoxifying antioxidants already present. Further, antioxidants can be turncoats, switching to the pro-oxidant side (e.g., 66). Both behaviors descend from a relatively low oxidation potential. Whether one or the other mode prevails depends on circumstances such as the presence of metal ions that can induce redox cycling, the concentration of the redox-active species itself, and the status of the cellular glutathione pool. The view is now widespread that even the antioxidant effects are to be largely attributed to a pro-oxidant behavior, according to the paradigm of hormetic action.
Hormesis refers to a nonlinear (biphasic) dose response characterized by opposite effects in the low and high ranges. Another definition is that of a response to a low level of stress or toxicant, affording protection against higher doses. The concept is useful to describe/rationalize at least in part the positive effects of exercise, dietary restriction, and the protection by dietary phytocompounds against disorders such as cancer, inflammatory diseases, and cardiovascular and neurodegenerative problems (129).
This effect is believed to be largely due to an induced low-level production of ROS, that is, to the action of plant-derived compounds as mild pro-oxidants (or indirect antioxidants) in a physiopathological context in which ROS have major roles (Fig. 5). Hormesis has been proposed as an explanation of the protective effects of isoflavones (187), flavonoids (106), resveratrol (19), α-tocopherol (45), hydroxytyrosol (243), sulphoraphane and other isothiocyanates from cruciferae (207) and other natural as well as some synthetic compounds. This pro-oxidant action has been suggested to take place mostly at the level of mitochondria (168). However, it is difficult to be sure of what is happening in vivo. Since ROS production by redox compounds is very dependent on the local environment, a pro-oxidant effect by a compound in cell culture is not necessarily and indication that it produces ROS in vivo.
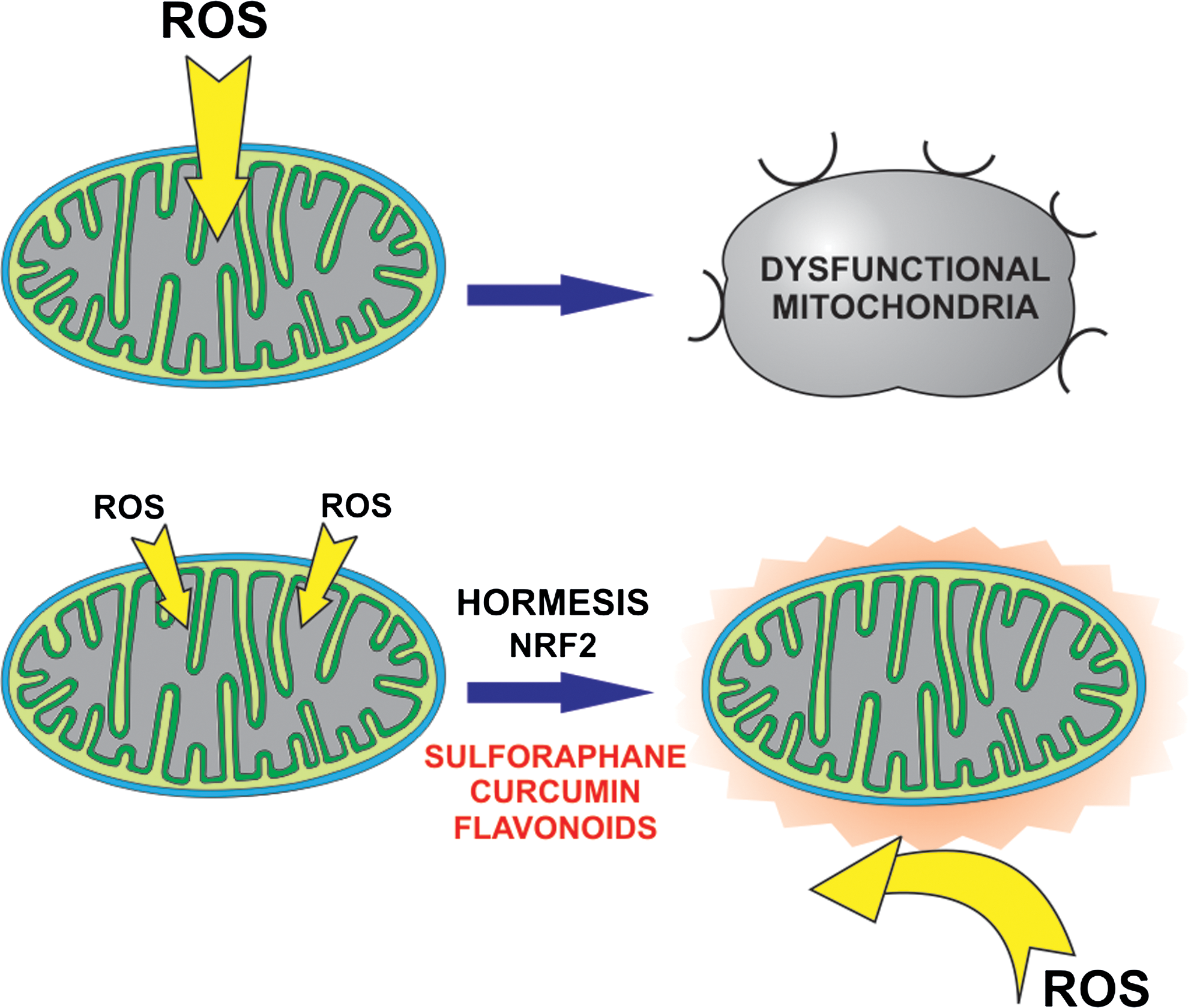
Mechanistically, most of the attention in this context is focused on the redox-sensitive transcription pathway of Kelch-like ECH-associated protein 1 (Keap1)/nuclear factor (erythroid-derived 2)-like factor (Nrf2) (56). This pathway potentiates the expression of many genes downstream of promoters containing an antioxidant response element sequence, including detoxifying Phase II enzymes, heat shock proteins, ROS-neutralizing SOD and catalase, enzymes involved in glutathione and NADPH homeostasis. Keap1 is a scaffolding protein that promotes Nrf2 degradation by the proteasome. Oxidation of some of the several cysteines of Keap1—and/or serine phosphorylation of Nrf2 by redox-sensitive kinases PKC or phosphoinositide 3-kinase (PI3K)—allows Nrf2 to migrate to the nucleus. Three critical cysteines of Keap1 react readily with the isothiocyanate group of sulphoraphane and analogous compounds or with conjugated systems such as that of curcumin, which are therefore particularly efficient inducers. This pathway has been shown to be relevant for protection against cancer, neurodegeneration, and cardiopathy.
Mitogenesis
The cellular mitochondrial pool is maintained at a steady state by the concomitant generation of new mitochondria and elimination of weakened mitochondria via mitophagy. Mitochondrial biogenesis decreases with age, and this is an important component of the functional decline in aging (212). Vice versa, mitochondrial proliferation is an essential component of exercise-induced (212) and calorie restriction-induced (127) myogenesis. Interestingly, the premature aging phenotype of mtDNA mutator mice (see above) can be rescued by exercise-induced mitochondrial rejuvenation (174). Mitochondrial biogenesis is a complex affair, with more than 150 proteins potentially involved in the process (236). We focus here on three major cooperating players: PGC1α (Peroxisome proliferator-activated receptor-γ coactivator-1α), Sirtuin (Sir2 protein; Silent mating-type Information Regulation 2 protein) (SIRT1), and AMP-activated kinase (AMPK) (Fig. 6).
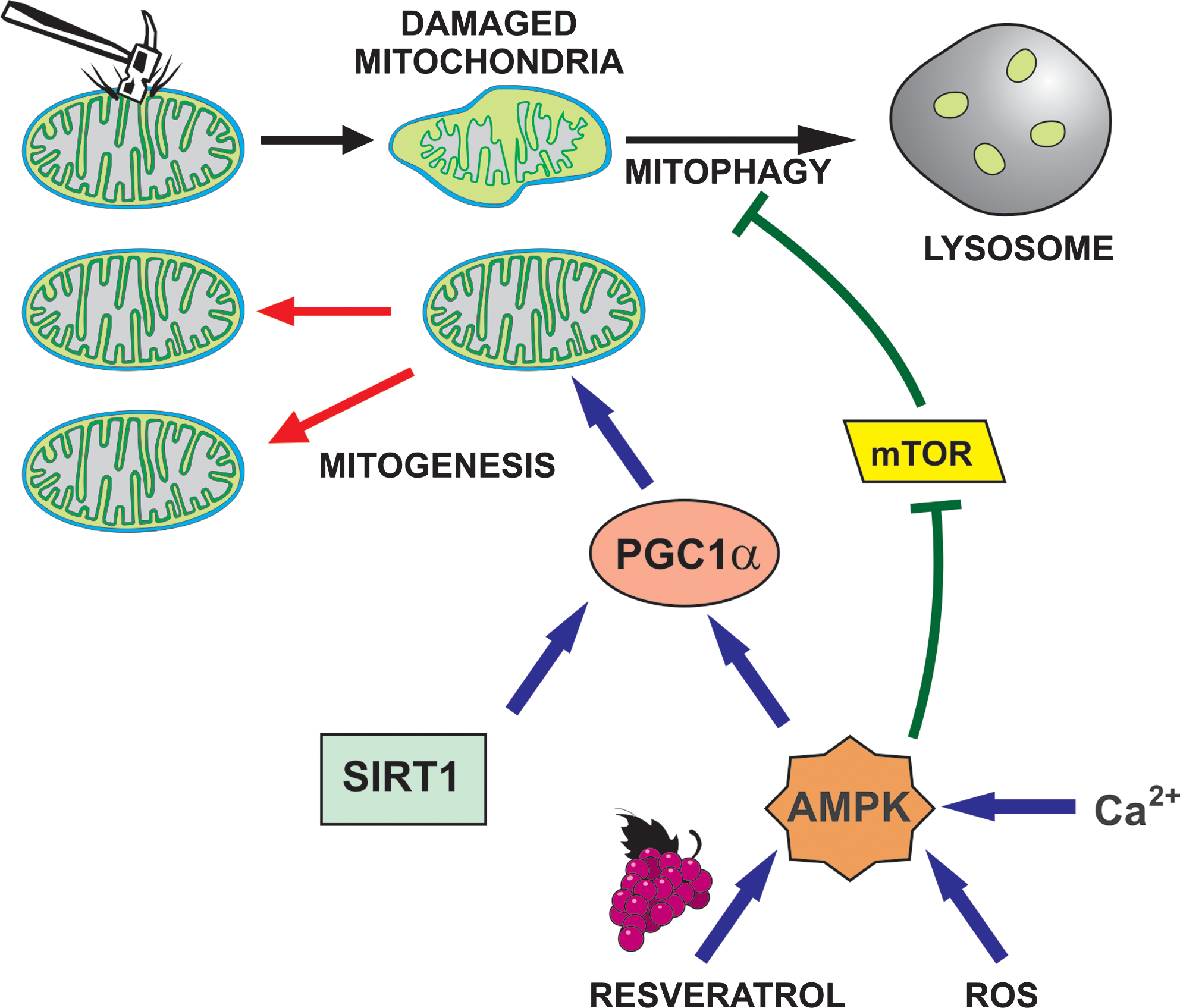
PGC1α is a transcriptional co-activator with multiple functions, including a major role in mitochondrial biogenesis and functionality (175, 212). Besides peroxisome proliferator-activated receptor γ and cAMP response element-binding family members, among its functional partners are the estrogen-related receptor α and Nrf1/2, which control components of the respiratory chain and many other nucleus-encoded mitochondrial genes. Among the proteins downstream of PGC1α are thioredoxin, SOD2 (MnSOD; mtSOD), and mitochondrial transcription factors TFAM and TFB2M.
SIRT1 has various enzymatic activities, including that of an NAD+-dependent deacetylase acting on histones, p53, and PGC1α (and others). Deacetylation (along with phosphorylation, see below) activates PGC1α.
AMPK (20) is a Ser/Thr kinase activated by AMP and inhibited by ATP. Nucleotide binding also affects phosphorylation and consequent activation by upstream kinases Serine/threonine kinase 11 (STK11) (LKB1) and Calcium-calmodulin dependent protein kinase kinase (CaMKK). AMPK thus responds to the AMP/ATP ratio, a sensitive measure of the metabolic status of the cell. Importantly, the enzyme can also be activated by mitochondrial ROS (44). PGC1α seems to be the fundamental downstream effector of AMPK for mitochondrial biogenesis. AMPK can directly phosphorylate PGC1α, and this is thought to allow deacetylation by SIRT1. SIRT1 activity is upregulated by an AMPK-mediated increase in NAD+ (for details see Ref. 20).
Mitochondrial biogenesis is also influenced through Ca2+ signaling: activation of CaMK initiates a cascade that leads to activation of p38 MAPK. P38 phosphorylates PGC1α and transcriptional cofactors. This pathway has been reported to be important both in exercise-induced muscle mass increase (114) and in neurons subjected to chronic depolarization (111).
Resveratrol and mitogenesis
Resveratrol has a stimulating effect on mitochondrial biogenesis (99). The mechanism involves the three actors mentioned above: PGC1α, SIRT1, and AMPK. A connection between resveratrol and SIRT1 was proposed when the resveratrol-induced life extension of yeast was attributed to a direct activating effect on Sir2 (the yeast orthologue of SIRT1) (74). Resveratrol (along with several other less effective polyphenols) was observed to increase the response in fluorescence assays measuring SIRT activity on the acetylated tetrapeptide “Fluor de Lys” (74). Subsequent reports (e.g., 143) indicated that the SIRT-activating effect is substrate dependent; that is, it is only observed when the Fluor de Lys peptide is used as a substrate for the enzyme, in in vitro assays. While a debate is still going on, a majority of investigators now believe that resveratrol does not act directly on SIRT1. However, it remains likely that SIRT1 is activated in cells exposed to resveratrol and other polyphenols [including flavonoids, butein, catechins, and curcumin; (26)] by an indirect mechanism, and that the partnership of resveratrol and SIRT1 accounts at least in part for resveratrol's effects.
Activation of AMPK by resveratrol and other phytochemicals is clearly supported by data from several labs (9, 34, 213). Thus, the resveratrol-induced signaling cascade impinging on mitochondrial biogenesis is now considered to involve AMPK as one of its top-level mediators (Fig. 6). How resveratrol brings about AMPK activation remains uncertain. The effect has been reported to depend on LKB1 (34), and/or on an induced increase of cytosolic Ca2+ levels and activation of CaMKKβ (213). A resveratrol-induced increase of PGC1α transcription/activity has been observed directly (9, 99).
Given that ROS (and NOS) have a stimulating effect on mitochondrial biogenesis, one would expect antioxidants to antagonize mitogenesis, and pro-oxidants to increase it. However, it is not straightforward to predict which way a given compound will act. The literature contains apparently contradictory reports, but the general trend seems to indicate a positive effect by plant compounds such as quercetin (36), hydroxytyrosol (67), and isoflavones (161), possibly because of an hormetic action. They may therefore join resveratrol and other supplements, such as lipoic acid or acetyl-L-carnitine, aiming to improve bioenergetic parameters.
Mitophagy
Autophagy, or more precisely macroautophagy (165, 176, 234), can be defined as a catabolic function in which portions of a cell are degraded in lysosomes to allow their recycling. Autophagy constitutes a cellular mechanism for housekeeping and renovation, and a defensive response against stressful conditions such as nutrient deprivation, ischemia/reperfusion (I/R), infection, or organelle malfunction. Depending on environmental clues and active signaling pathways, the autophagic response may serve purposes ranging from the elimination of defective or damaged proteins to autophagic cell death. It has important roles in development, in caloric limitation-induced lifespan extension, and in opposing aging, neurodegeneration, diabetes, and other major pathologies.
Regulation is complex. An apical position in the signaling cascade is occupied by mammalian Target of Rapamycin (mTOR) kinase Complex 1, a key sensor of the cellular nutrient status, which acts as an autophagy repressor. In turn mTOR is regulated downstream of PI3Ks. Activation of AMPK promotes autophagy by downregulating mTOR. Since AMPK can be activated by CamKKβ, cytosolic Ca2+ can induce autophagy. SIRTs also play a prominent role (157).
Upregulation of autophagy is associated with cancer (223) and neurodegeneration (228). Its function in these major pathologies is still debated, but it may mainly represent a pro-survival/proliferation strategy. Conversely, defects in autophagy are associated with inflammation (109, 223), an oncogenic factor. Thus, cancer prevention may benefit from autophagy-enhancing interventions, while autophagy repression may be a therapeutic approach (223).
Autophagy can be selective, targeting one or the other cellular component: mitochondria, peroxisomes, the ER, the Golgi apparatus, endosomes, invading bacteria and viruses, and protein aggregates. Selectivity is achieved through the use of markers that can link the specific target and autophagosomal membranes.
Mitophagy (238) is of particular interest here. The same signals, AMPK activation and ROS (176), promote both mitochondrial biogenesis and mitochondrial elimination (Fig. 6), ensuring coordination. Mitophagy is prompted by mitochondrial damage or by generalized stress. Among the genes/proteins necessary for the process is Nix/Bnip3L, a BH3-only member of the Bcl-2 family that constitutes a link between autophagy and cell death. The MPT serves as a strong inductive stimulus for the selective autophagic elimination of mitochondria (89). Depolarization results in the recruitment to mitochondria of Parkin, an ubiquitin ligase instrumental for their lysosomal degradation (238). Loss-of-function mutation of Parkin causes Parkinson's disease (PD). PTEN-dependent protein kinase-1 (PINK1) is a mitochondrial as well as cytosolic Ser/Thr kinase involved in mitochondrial dynamics that acts also in the same signaling pathway as Parkin and thus constitutes a link between the two processes. In fact, increased fission correlates with increased autophagy.
ROS have been shown to be instrumental in inducing autophagy in a variety of systems (176). Whether the relevant species is H2O2 or superoxide (or both depending on circumstances), what the targets are and how exactly the effect takes place are, however, still open questions.
Phytocompounds and mitophagy
Since resveratrol activates AMPK, it generally increases autophagy (213). This effect has been linked to its life-prolonging action on model organisms (119). Avicin D, a plant triterpenoid, is also reported to induce autophagy (and autophagic death, along with apoptosis) via AMPK activation (231).
As was the case for mitogenesis, the involvement of ROS in the induction of autophagy points to the possibility that antioxidants might antagonize it. Again, the literature contains reports in apparent contrast with this notion. Curcumin (79), the flavonoids quercetin (155) and fisetin (191), the chalcone flavokawain B (97), and ascorbate (125) have been reported to increase autophagy. In this case too, and depending on concentration and experimental circumstances, antioxidants may actually act as pro-oxidants.
Apoptosis
The fundamental contribution of mitochondria to apoptosis is described in a number of reviews (60, 145, 220). We assume that the reader is familiar with the main features of the apoptotic process, and discuss here only two aspects of particular relevance for our topic and recent results from our group: the roles of ROS and of ion channels.
ROS and apoptosis
A vast consensus exists that ROS can induce cell death (27). Relatively low-intensity oxidative stress leads preferentially to apoptosis, with higher levels leading instead to necrosis (Fig. 4). Oxidative activation of kinase-based pro-apoptotic signaling is of fundamental importance, and it has been recently reviewed in depth (198). Apoptosis Signal-Regulating Kinase 1, a PKCδ isoform (which migrates to mitochondria in ischemic preconditioning and upon reperfusion of ischemized organs), and Ca2+-activated Death-Associated Protein Kinases are some of the kinases involved. A major pathway is the stress response of the JNK and p38 MAPK (Stress Activated Protein Kinases) cascades. Transcription factors such as c-Jun, p53, and p73, other kinases and members of the Bcl-2 family act downstream of JNK and p38 to induce apoptosis (198).
Bax, one of the key players in mitochondrial apoptosis, can be activated by a number of signals, including oxidative stress (52). Homodimerization via formation of an S-S bridge between cysteines and phosphorylation at Thr167 by redox-sensitive JNK/p38, required for mitochondrial translocation, account for this. Pro-caspase 9 belonging to the mitochondrial pool can form dimers via disulfide bridges and perform auto-processing to generate the active enzyme (85). Oxidation of cardiolipin allows the release of cyt c (179) and is also thought to be important for the formation of the permeability transition pore.
On the other hand, ROS are well known to also be capable of anti-apoptotic, proliferative effects, a matter of great importance in carcinogenesis (Fig. 4). Again, quantitative aspects and concurrent cellular signaling are very relevant for the outcome.
Mitochondrial channels and apoptosis
Mitochondrial ion channels have been proposed to participate in apoptotic signaling contributing prevalently to three main interconnected aspects: release of pro-apoptotic factors, ROS production, and calcium accumulation. The mitochondrial permeability transition (pore) (MPTP) is discussed in the section on the mitochondrial permeability transition.
Outer membrane permeabilization is necessary for the release of pro-apoptotic proteins from the mitochondrial inter-membrane space. The mechanisms of this process are still debated. Pro-apoptotic Bax oligomers form pores in artificial bilayers (black lipid membranes [BLM]), and oligomerization has been proposed to underlie OMM permeabilization. However, channels comprising only Bax do not wholly account for cyt c release, given that a single point mutant of Bax did not mediate cell death when expressed in Bax/Bak-less mouse embryonic fibroblasts despite forming pores with properties similar to WT Bax in BLM (195). A channel proposed to include Bax and with an estimated pore diameter sufficient to allow the passage of cyt c has been observed by patch clamp (126).
Bak and Bax as well as anti-apoptotic Bcl-xL interact with mitochondrial voltage-dependent anion channels (VDACs). VDAC2 has been proposed to allow the mitochondrial recruitment of Bak, thereby controlling tBID-induced OMM permeabilization and cell death (173). Shoshan-Barmatz's group reported that apoptosis induction by various stimuli is accompanied by VDAC oligomerization, and proposed that cyt c efflux may take place via VDAC oligomers (184).
Inner mitochondrial membrane (IMM) channels with a role in apoptosis include the calcium uniport, three K+ channels (Shaker-type mtKV1.3, ATP-inhibited mtKATP, and calcium-activated mtKCa1.1), possibly the inner membrane anion channel (IMAC), and uncoupling protein 2 (UCP2) (196).
Mitochondrial Ca2+ uptake via the calcium uniporter, corresponding to voltage-gated calcium channel(s) of unknown molecular identity, has been shown to perform multiple roles in the regulation of the respiratory rate and ATP production. Prolonged increases in [Ca2+]mit can induce the opening of the MPTP (see section on the permeability transition).
We have shown that OMM-integrated Bax directly interacts with and inhibits an IMM-located mitochondrial potassium channel, KV1.3, in lymphocytes (194, 195). According to the current model, block of KV1.3 by Bax results in hyperpolarization, due to reduction of depolarizing inward potassium flux, and this in turn causes an increase in ROS production and apoptosis.
Pharmacological activation of mtKATP and mtKCa1.1 has been reported to have a cytoprotective effect in I/R damage, and ROS-induced opening of the superoxide-permeable IMAC has been proposed to contribute to apoptosis, given that its blockage inhibited cell death (18). UCP2, which mediates proton leak, has also been proposed to regulate cell survival by lowering the mitochondrial membrane potential, thus decreasing formation of mitochondrial ROS (6). A role for UCP2 has been suggested in pathological states associated with oxidative stress, including degenerative diseases, atherosclerosis, stroke, aging, diabetes, and cancer. Please note, however, that in many cases the molecular identity of the mitochondrial channels is unknown (e.g., MPTP, mtKATP, and IMAC), thus preventing elucidation of their exact physiological and pathological roles. In the absence of the possibility to obtain gene deletions and/or dowregulation of expression for these channels, their involvement in the above-described processes cannot be considered as definitively proven and indeed is debated.
Plant-made compounds and apoptosis
The vast literature dealing with this topic, closely connected to cancer antagonism, has been the subject of some specific recent reviews (50, 87, 112). Matters are complicated by the possibility of a biphasic dose dependence of the effects.
In vitro, and in the high-μM range, resveratrol can induce apoptosis by multiple effects on cellular signaling pathways (68). Its impact on apoptosis-related gene expression patterns has been described in several reports. For example, it induced a decrease of the expression of Bcl-2, and an increase of Bax and of Growth arrest- and DNA damage-induced gene 45α (110). High concentrations of resveratrol cause an increase of mitochondrial ROS production, depolarization, and concomitant decrease in cell viability (62). The mechanism remains to be clarified. The compound has been reported to be a (rather poor) inhibitor of the respiratory chain (244) and of the ATP synthase (57).
Green tea catechins, in particular EpiGalloCatechin-3-Gallate (EGCG), are renowned antioxidants, displaying positive effects in variety of pathophysiological contexts. Given what has been said above, an antioxidant effect can be considered, as a first approximation, to be equivalent to an anti-apoptotic effect. Here too it is a matter of concentrations and context. On the antioxidant side, EGCG has been reported to concentrate in the mitochondria of cultured neurons, and to exert a protective effect against apoptosis mediated by oxidative stress (178). At high concentrations EGCG can also induce apoptosis or necrosis via ROS generation and mitochondrial depolarization (75, 136) as well as through promotion of p53 phosphorylation and proteasome inhibition (87, 100). Green tea polyphenols have been tested in several in vivo models and in clinical trials.
Similar considerations apply to quercetin and a variety of other flavone and isoflavone derivatives, which, again, can have an antioxidant effect or induce apoptosis via redox processes that impact on signaling cascades and protein expression and activity. Relevant effects may also descend from the inhibition of the proteasome and/or of kinases (87). The effects of curcumin, much touted as anticancer agent, again depend in part on its pro-oxidant character and its hormetic promotion of Nrf2 action. At least in vitro, high doses can again induce apoptosis via the redox pathway impacting on mitochondrial potential and Ca2+ homeostasis as well as by induction of ER stress via inhibition of SERCA pumps, inhibition of the Akt/NFκB signaling axis, modulation of the expression a variety of proteins relevant for apoptosis, or combinations of these effects (230). Gossypol, a polyphenolic compound from cotton, induces both apoptosis and autophagy by interacting with anti-apoptotic members of the Bcl-2 family (50) and upregulates, via ROS, Death Receptor 5 (TNF-related apoptosis-inducing ligand [TRAILR]).
Synthetic analogs of the various compounds are coming onto the scene. Thus, for example, a water-soluble derivative of gossypol has been recently produced and found to have improved properties against colon cancer (233). α-Tocopherylsuccinate (41, 42) targets mitochondrial succinate dehydrogenase, thereby causing redox stress and cell death. α-Tocopherol ether-linked acetic acid acts by binding to death receptors (240). Vitamin E metabolites containing a long aliphatic chain also caused oxidative stress and apoptosis (16). Digalloylresveratrol is intended to combine the properties of gallic acid and resveratrol (120).
Mitochondrial channels can also be affected by phytomolecules. Genistein, flavonoids, and curcumin have been reported to directly activate the calcium uniporter at concentrations in the μM range (5, 133). Several polyphenols, including cardamonin, resveratrol, kaempferol, purearin, and naringenin, activate KCa1.1 (137), mostly indirectly via kinases, resulting in some cases in a cardioprotective effect. Recently, morphine has been shown to induce preconditioning via activation of the mitochondrial calcium-activated K+ channels (48). EGCG seemingly reduces infarct size via activation of mtKATP (190). Concerning mtKV1.3, one of its potent inhibitors, Psora-4, a derivative of 5-methoxy-psoralen from Ruta graveolens, has been shown to induce cytochrome c release from isolated mitochondria (194). While most of these compounds have pleiotropic effects, the possibility to alter cell death and survival via modulation of mitochondrial ion channels by them needs to be considered.
Necrosis
Necrosis occurs under many circumstances of clinical relevance, from I/R injury to neurodegeneration. Long considered as a sort of uncontrolled, haphazard form of cell death, characteristically determined by elevated redox stress, it is now understood to comprise a set of regulated, interacting signaling cascades and biochemical phenomena forming a continuum with other forms of cell death (142). Depletion of ATP is often cited as a key factor shifting cell death toward necrosis rather than apoptosis. For example, DNA damage can induce either apoptosis or necrosis: a classical view is that moderate damage can be repaired, or it may lead to apoptosis if unrepearable, whereas extensive damage may lead to hyperactivation of poly-ADP-ribose polymerase (PARP), with consequent depletion of NAD+ and ATP, and necrotic cell death.
Necrosis can be classified into subtypes, still in the process of consolidation in usage. Accidental necrosis is induced by severe damage with profound ionic imbalance and cellular swelling. It is distinct from necroptosis (involving receptor-interacting serine/threonine-protein kinase 1 [RIP1] and also the MPT), programmed necrosis (involving the MPT and apoptosis-inducing factor [AIF] and endonuclease G release from the mitochondria), and secondary necrosis (following on the heels of apoptosis), all leading to morphologically similar end results (209).
Necroptosis (208) appears to be involved in excitotoxicity, neurodegeneration, and stroke. It can be launched by engagement of the same death receptors transducing extrinsic apoptosis signals carried by tumor necrosis factor, FasL, and TRAIL. It involves RIP1, a kinase and adapter that has roles also in apoptosis and in NFκB activation. Protein–protein interactions in alternative molecular complexes determine at least in part the fate of the cell. The events in the necroptosis program have not been fully defined. ROS, produced downstream of RIP1-mediated activation of JNK, p38, NADPH oxidases, and Glutamate dehydrogenase 1 (in mitochondria), have an important role.
Loss of Ca2+ homeostasis, for example, via excessive N-methyl-D-aspartate receptor activation in neurons, may lead to the activation of calpains (232). These Ca2+-dependent cysteine proteases destabilize the lysosomal membrane releasing lysosomal enzymes, in particular cathepsins, a group of proteases that proceed to act on a vast number of substrates, including mitochondria, determining generalized failure of cell bioenergetics, and death. Calpain-involving and caspase-based death pathways interact. For example, cathepsin B has been reported to be able to induce cyt c release from mitochondria. ROS are thought to necessarily cooperate with calpains in determining lysosomal rupture, which may well have to do with the generation of very reactive species in Fenton reactions catalyzed by the iron-rich, H2O2-permeable lysosomes (98).
Other variations on the necrosis theme exist. The necrotic impact of PARP hyperactivation is not due simply to ATP and NAD+ depletion: polyADP-ribose induces release of mitochondrial AIF, which provokes a caspase-independent type of death christened parthanatos (35). Activation of a mitochondrial calpain by a sustained Ca2+ increase also appears to be involved in AIF release (140). This mechanism contributes to cell death in models of stroke, diabetes, and neurodegeneration. Autoschizis is used by some researchers to indicate heavy redox stress-induced death (77), considered to involve widespread damage.
The impact of natural compounds on the various types of necrotic processes and on the apoptosis-necrosis cross-talk largely remains to be explored. Redox-active molecules are obviously expected to be relevant.
The MPT
A key executioner of necrosis is now considered to be the MPT (163, 247). Basically, the MPT consists in the activation/opening of a large permeation pathway in the IMM, with consequent depolarization, loss of solutes such as Ca2+, ATP, and NAD(P)(H) from the mitochondrial matrix, generation of ROS, and functional impairment. Onset of the MPT can be determined by several factors that may act synergically: accumulation of Ca2+ in the mitochondrial matrix, oxidative stress, partial depolarization, and basification of the mitochondrial matrix. A large number of pharmacological agents have been identified that can induce the MPT, whereas a more restricted number of molecules (including ATP) can inhibit it. The molecular composition of the MPT pore (MPTP) is still a matter of debate, but it includes Cyclophilin D (CypD, a prolyl isomerase) and probably a mitochondrial transporter, normally the adenine nucleotide translocator.
While Ca2+ is a key effector at least in vitro, activation by oxidizing conditions is also considered to be an essential feature of the MPT (93). Possibly caused by Ca2+-induced alterations of cardiolipin–protein interactions, ROS production is one mechanism for the transduction of Ca2+ binding into MPTP opening via oxidation of thiol groups. Thus, antioxidants may be expected to inhibit the phenomenon, and pro-oxidants to favor it, as indeed has been observed in several studies.
While currently MPT occurrence tends to be identified with necrosis, its participation in apoptosis cannot be dismissed. For one thing, activation of the MPTP may be involved in regulating cyt c release through remodeling of cristae (180). The MPTP is clearly a pharmacological target in pathologies involving necrosis. Unexpectedly, it has turned up in collagen VI deficiency syndromes (12). Its induction by redox stress is one of the strategies considered in the emerging field of anti-cancer mitochondrial medicine (51).
The MPTP can be modulated by cellular signaling. These aspects have been investigated mainly in studies on the molecular events taking place during I/R and the protection afforded by pre- and postconditioning, and on the repression of the MPT in tumor cells. In preconditioning, ROS produced under hypoxic conditions are thought to be a major factor determining a feedback response by the cell, which results in downregulation of the MPTP and thus protection from subsequent insults. This effect is proposed [the model is still controversial (138)] to be mediated by the activation of reperfusion injury salvage kinases, a group including Akt, ERK1/2, PKC-ɛ, PKG, and P70s6K (70). These kinases in turn determine inactivating serine phosphorylation of constitutively active glycogen synthase kinase-3β (GSK3β) with consequent desensitization of mitochondria to MPT induction (70). The MPTP component/regulator CypD is phosphorylated by GSK3β (162). During severe hypoxia oxidative phosphorylation is reduced, and cellular ATP is depleted. This is expected to stimulate AMPK activity. AMPK activation has an inhibitory effect on GSK3β and thus is expected to inhibit the MPTP as well. Further kinases such as PKC-ɛ, Akt, and PKA may act directly on mitochondria to achieve protection from the MPT. Mitochondrial K+ channels and NO are also believed to be involved (246).
The pathological context in which most attention has been devoted to the MPT is probably I/R damage (65, 163). Oxygen deprivation causes a decrease of ATP and reduction of the respiratory chain. Glycolysis leads to acidosis. Na+ enters the cytoplasm via the Na+/H+ exchanger operating in reverse and cannot be pumped out efficiently by the Na+/K+ ATPase. When reperfusion allows the pH to normalize, a massive Ca2+ influx ensues due to reactivation of the Na+/Ca2+ exchanger, previously inhibited by acidosis. Since the mitochondria have resumed respiration, much of this Ca2+ is taken up into the organelles. As oxygen is readmitted a burst of ROS is produced because single electrons leak from the reduced respiratory chain onto oxygen (135). Ca2+ accumulation into the mitochondrial matrix, re-adjustement of matrix pH, and oxidative stress are a recipe for the MPT. Occurrence of the MPT leads to cell death, and in fact tissue damage can be reduced by pharmacological inhibitors of the MPTP or by deletion of CypD.
Phytochemicals and the permeability transition
Since resveratrol activates AMPK, and AMPK inactivates GSK3β, resveratrol may have an AMPK- and GSK3 β-mediated protective effect against the MPT and ischemic damage, as reported (43, 164, 183). Isoliquiritigenin (25), a chalcone of liquorice, and sauchinone (91), a lignan of traditional Chinese medicine, behaved analogously, with AMPK activation downstream of LKB1. Another mechanism involved in protection by resveratrol is AMPK-mediated stimulation of autophagy (64). Autophagy is upregulated in ischemia and can help limit I/R damage.
One would also expect pro-oxidative hormesis to limit the occurrence of the MPT and infarct size, and prolonged previous administration of quercetin and α-tocopherol (156) or EGCG (38) had the expected effect in rat models. Observations such as these are promoting the idea of chemical preconditioning, the reinforcement of defenses against I/R damage by the chronic administration of protective agents (nutraceuticals).
Dynamics of the mitochondrial network
Mitochondria form a dynamic network, displaying fusion/fission and movement along cytoskeletal elements (37). Fusion of the outer and inner membranes occurs in separate steps, involving proteins with GTPase activity: mitofusin-1 and −2 (MFN1/2) for the outer membrane, and optic atrophy protein 1 (OPA1) for the inner one. MFN act as tethers, forming homo- or heterocomplexes. MFN2 is also directly involved in mitochondria-ER interaction (37) and in axonal mitochondrial transport (132). PINK1 and Parkin-mediated ubiquitination of MFN1/2 form a link between mitochondrial fusion/fission and mitophagy (245) (Fig. 7).
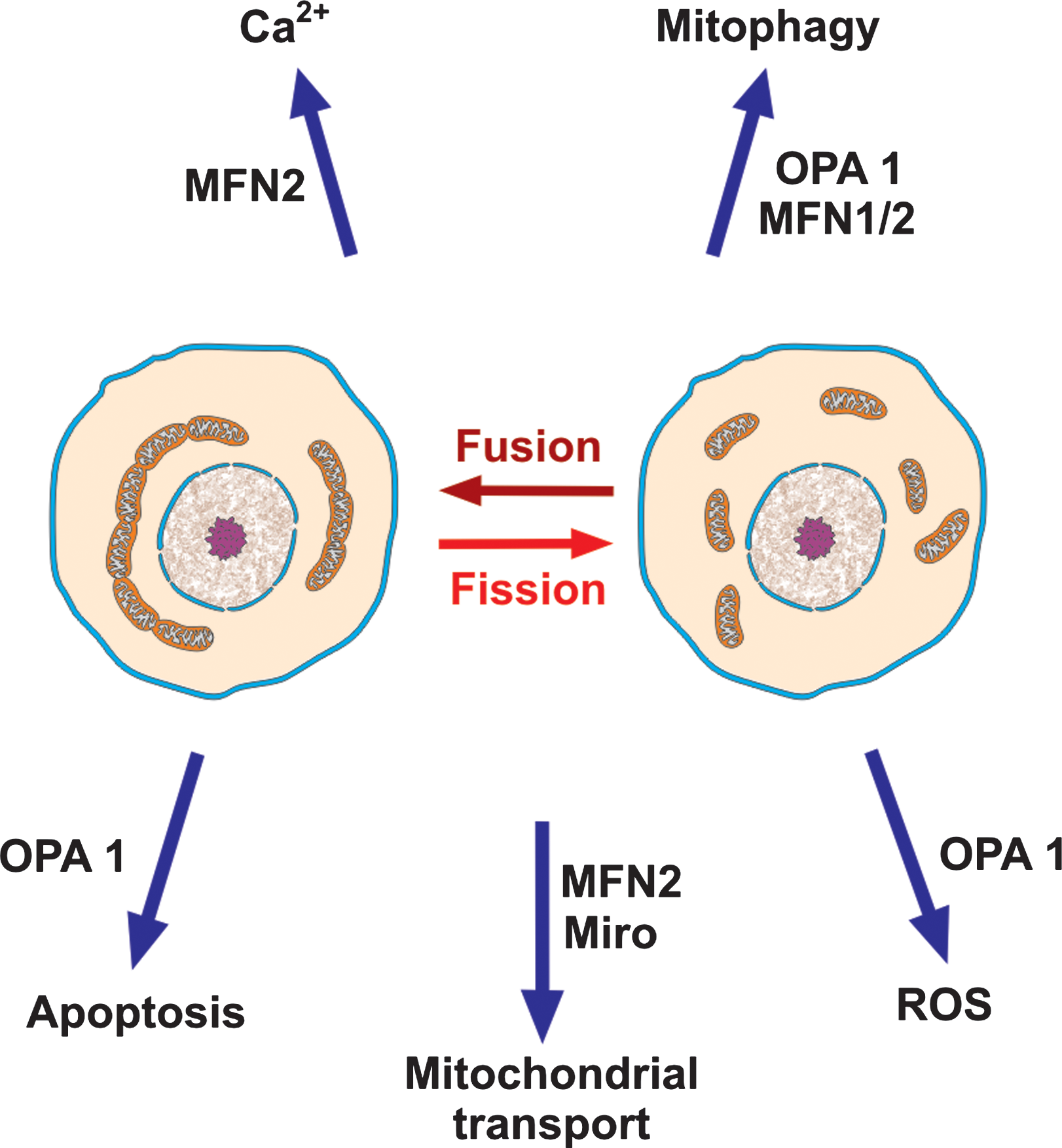
OPA1 cooperates with Bcl-2 family members and prohibitins to control IMM fusion, the shape of cristae and cytochrome c release during apoptosis (148). Defects in OPA1 result in enhanced ROS production (199), which might account in part for the mtDNA instability observed in patients carrying OPA1 mutations. OPA1 also links mitochondrial dynamics and mitophagy: a prolonged decrease in mitochondrial potential, a sign of mitochondrial damage, induces degradation of OPA1, which prevents the fusion of these mitochondria to the network and destines them to elimination (59). Mutations in the OPA1 gene have been associated with various syndromes, prominently hereditary optic neuropathies.
The cytosolic GTPase dynamin-related protein 1 (Drp1) and its receptor on the OMM Fis1 are required for fission. Drp1 autoassembles in rings identifying the division plane around mitochondria, and this process then leads to constriction and eventually fission (22).
Important links exist between mitochondrial network dynamics and apoptosis (84): enhanced fission is associated with apoptosis, whereas enhanced fusion acts the opposite way. The molecular apparatuses involved in the two processes share some interacting elements: members of the Bcl2 family (Bcl2, BclxL, Bax, Bak, and Bnip3) interact with members of the fusion/fission machinery (MNF1/2, Opa1, and Drp1) and some of these proteins have been observed to co-localize in distinct foci on the OMM.
Fission can be induced by Ca2+ (73) and can reciprocally influence signaling by this ion, in particular the propagation of Ca2+ waves (193). Excess ROS production has been observed to accompany Ca2+-induced mitochondrial fragmentation, again demonstrating the pervasive interconnections of cellular processes. In turn, the fusion/fission system appears to be redox-sensitive, with oxidative and nitrosative stress leading to enhanced fission (229).
Under normal circumstances the fusion/fission process helps the cell get rid of dysfunctional mitochondria, which are separated from the healthy pool and undergo mitophagy. Another fundamental task is the maintenance and distribution of the mtDNA pool. Problems with the fusion/fission apparatus cause mitochondrial heterogeneity and increase mtDNA mutations and deletions, as well as mtDNA depletion, and contribute to a number of pathologies, including neuropathies such as autosomal dominant optic atrophy (24), neurodegeneration, and muscle atrophy (172).
A degree of control of mitochondrial function is also associated with the position of the organelles, and their transport along microtubules (49). This is particularly important in neurons, where mitochondrial transport is associated with the formation of new structures (plasticity). The mitochondrial fusion/fission machinery has a role in this type of process as well. Anterograde transport uses specific kinesins, linked to a mitochondrial complex containing Miro (an OMM GTPase), PINK1 (the kinase also involved in mitophagy), mitofilin (a protein of the IMM), and MFN2. This explains why mutations in MFN2 lead to selective degeneration of long peripheral axons (Charcot-Marie-Tooth type 2A disease).
Iron and copper handling
Iron is used to synthesize heme and Fe/S clusters in mitochondria, and mitochondria play an important part in the control of this ion, potentially dangerous because it can catalyze the Fenton reaction. Iron handling is a complex and incompletely understood process, the complexity being due largely to the need to keep the ion(s) constantly sequestered (235). Schematically, cells acquire iron either by importing heme via heme carriers or by binding iron-loaded transferrin, which is then endocytosed. Fe2+ is eventually released to form the cytoplasmic chelatable iron pool, whose exact nature is uncertain, and is then bound by cytoplasmic ferritin. Heme precursor is synthesized in the cytoplasm in a series of steps leading to coproporphyrinogen III, which must then reach the mitochondrial intermembrane space to be converted into protoporphyrinogen IX and then protoporphyrin IX (PPIX). The final step of heme synthesis, Fe2+ insertion, is accomplished by ferrochelatase on the matrix face of the IMM, and must therefore be preceded by transmembrane transport of PPIX. Heme must then be delivered to the utilizers, which include apocytochrome c and extramitochondrial proteins. How this occurs is only poorly known. An important role is attributed to the ER, where transiting apoproteins appear to acquire heme.
How free iron is transferred into mitochondria is uncertain, and more than one mechanism may apply. Transient docking and fusion of the endosome to the mitochondrion has been proposed to provide an efficient delivery system in developing erythrocytes, which need large amounts of iron. Mitochondria also possess at least one iron transporter, Mfrn1/2 (mitoferrin). Fe/S clusters are assembled in a multi-step process on a scaffold protein and then transferred to apoproteins. Mutations in some of the proteins involved in this process give rise to human pathologies (235). A trinucleotide repeat expansion in the gene encoding one of these proteins, frataxin, resulting in a decrease of its expression, causes Friedreich's ataxia.
Copper is needed by mitochondria for cytochrome c oxidase biogenesis. The trafficking system of this ion also makes intensive use of metallochaperones (171) to keep the redox-active ions under control. The mechanisms of transport and incorporation into apoproteins, analogously complex, have been studied largely in yeast. Redox-sensitive Cu-coordinating cysteines in transport ATPases are important.
Several plant polyphenols can chelate with high affinity iron or copper, especially the most highly oxidized ions, inhibiting Fenton chemistry (151). The possibility of a protective biological role as metal chelators has been proposed at least for EGCG (122), quercetin and other flavonoids (214), and sauchionone (91). The possibility that a pro-oxidant effect may arise via cyclic reduction of the metal ion (Fe3+→Fe2+, Cu2+→Cu+) by the polyphenol is also there, especially in the case of copper, but given the generally reduced state of these ions in biological systems, this mechanism of ROS generation is not likely to be a major one (151).
Diabetes, neurodegeneration, and phytochemicals
Cancer, ischemias, diabetes, and neurodegeneration are the major modern diseases of the Western world. All four involve mitochondria and can in principle be antagonized by plant-made molecules. Ischemia and cancer have already been touched upon in preceding sections.
Mitochondria are fundamental for insulin secretion (81, 224). Diabetes can arise as a disease of mitochondrial aging, that is, downstream of the accumulation of mtDNA mutations. High levels of glucose metabolism are associated with increased superoxide production, whereas fatty acids lead to H2O2 production in peroxysomes. Hyperglycemia may thus produce a positive (pathology-enhancing) feedback. Part of these ROS are believed to be generated by the respiratory chain of mitochondria. Clearance of defective mitochondria would be expected to antagonize diabetogenic dysfunction. In fact, autophagy has been recognized as relevant for the onset (or prevention) of diabetes (82).
Mitochondrial malfunction and excess ROS are also involved in the etiology of insulin resistance and its complications (83, 107). AMPK has a large role in glucose-stimulated insulin secretion, so much so that a major approach to treatment for type 2 diabetes currently relies on AMPK activators such as metformin, but how it operates is still unclear. SIRT1 seems to be involved (26).
ROS production, mitochondrial biogenesis, mitophagy, and AMPK activity can therefore be identified, again, as target processes for an intervention with phytochemicals. Indeed, the results of studies with resveratrol are promising (197). Flavonoids have been shown to exhibit hypoglycemic activity in vivo (7, 23, 118) and in vitro (e.g., 248), an effect linked to the ERK1/2 pathway (237). Together, these compounds probably account for the positive results reported in many studies with preparations and extracts of plant material.
Neurodegenerative diseases are an heterogeneous group of disorders, generally characterized by the progressive loss of specific anatomically or physiologically related neurons. In all these pathologies mitochondria have been shown to be involved in etiology and/or in disease progression (e.g., 53, 113). Among the mitochondrial dysfunctions associated with neurodegeneration are mtDNA mutations/deletions, defective respiratory chain, mutations in enzymes of Krebs' cycle, increased ROS production, alterations of Ca2+ homeostasis, abnormal mitochondrial dynamics, impaired mitochondrial quality control system, enhanced permeability transition, apoptosis, and mismanagement of brain iron. Some of these features are common to normal brain aging and neurodegeneration.
For example, oxidative damage occurs early in AD, before plaques appear; abnormal mitochondrial dynamics and mitophagy contribute to disease onset and progression (100). Mitochondria from AD brains have an abnormal distribution inside the cell and morphological alterations: reduced number, increased size, and broken internal membrane cristae. Oxidative stress and impaired mitochondrial respiration have a key role in the development of PD, as underscored by studies on familial PD forms (242). Selective vulnerability of dopaminergic neurons in PD may be due to their autonomous pacemaking activity, which relies on Ca2+ influx through CaV1.3 channels. This implies that the magnitude, continuity, and spatial extent of Ca2+ influx is much larger than in other neurons, and that the functionality of the intracellular organelles charged with keeping Ca2+ under control, the ER and mitochondria, is of even greater than usual importance. Mitochondrial impairment may thus affect these neurons before others, or, hypothetically, might be eventually caused by the continuous exposure to elevated Ca2+ transients (21).
Production of ROS, propensity to undergo the permeability transition, altered mitochondrial dynamics, embattled autophagy, insufficient mitogenesis, and iron dysregulation are all features having to do with the mitochondrial system in neurodegeneration and which may represent profitable targets for phytochemicals (33, 90, 166, 192).
One would expect resveratrol to oppose neurodegeneration, provided it can reach the relevant brain district at sufficient concentrations, as it may since it was found to induce an increase of mitochondrial mass and MnSOD in the brain of healthy mice (170). A positive impact of resveratrol on the systemic and cellular manifestations of Alzheimer's-like disease in animal models has been reported (e.g., 88, 166). On the basis of in vitro studies, these effects have been attributed to the upregulation of AMPK and SIRT1 (4, 88, 213), and/or to a modulation of the proteasome to favor elimination of the toxic peptides (123).
Resveratrol also showed a protective action in pharmacological rodent models of PD (80, 117) and Hungtington's disease (95) and was among a few small molecules emerging from a screen for antagonists of amyotrophic lateral sclerosis (8). Positive effects of resveratrol have also been reported in models of excitotoxic damage and glial inflammation.
Data on the effects of resveratrol or other phytocompounds on the course or development of neurodegenerative disease in humans are limited. Of note, a statistical study on a large cohort concluded that moderate wine drinkers may have a considerably decreased incidence of dementia and AD in old age compared with nondrinkers (103).
Anti-amyloidogenic and symptoms-reducing effects have been observed also for curcumin in rodent models of Alzheimer's (90) and Parkinson's (239) diseases. Green tea catechins may also help against AD (90). Antioxidant action, including hormetic effects, are credited.
Perspectives
The benefits that can potentially be derived from phytochemicals are evident. As is often the case, there is a but, namely, that difficulties may arise because of the limited absorption and bioavailability of some of these compounds. This is especially true of dietary polyphenols, which are ready-made substrates for Phase II metabolism because of their hydroxyls. They are rapidly converted by sulfo- and glucuronosyl-transferases in enterocytes into metabolites that are, to a large extent, re-exported to the intestinal lumen. Liver enzymes then intervene on the molecules that have entered the circulation, administering other rounds of Phase II metabolism (101, 121, 218, 219).
Because of the action of these enzymes, only low (nM–μM) concentrations of any given polyphenol are found in plasma and lymph even after a polyphenol-rich meal, and mostly in the form of conjugates. Sulforaphane and tocotrienols experience analogous difficulties. The biological activities of metabolites are now being investigated, with dozens of articles (but few reviews: 46, 101, 225) already published. In general, metabolic modification seems to entail a decreased effectiveness, for example in the case of resveratrol as an inhibitor of cancerous cell growth (86, 134).
It follows from these considerations that hormesis may be the most relevant mode of action of dietary phytochemicals. More acute effects are probably limited to modes of administration and body districts (gastrointestinal tract, derma, and blood circulatory system) that allow direct access of the phytocompounds to targets. To potentiate hormesis, and even more so to promote a pharmacology in acuto, efforts are beginning to be made to apply pharmaceutical delivery systems (108, 186) and to develop prodrugs (e.g., 14) of phytocompounds. Increasing attention is also being devoted to the elaboration of dietary compounds into a vast—and to some degree individually specific—range of potentially active derivatives by the colonic flora (153). Another important line of evolution is the creation of molecules inspired by natural compounds and by knowledge of their mechanisms of action. Derivatives of natural molecules addressed to mitochondria (Smith et al., this issue) are one direct result of this line of reasoning. The combination of improved systemic delivery and mitochondrial targeting may produce remarkable results. As summarized above, phytochemicals clearly can also affect mitochondria indirectly, by influencing their biogenesis, turnover, participation in cell death. These mitochondrial modes of action may contribute in a major way to the overall effects, and ought to be paid due attention by researchers, nutritionists, and health professionals.
Footnotes
Acknowledgments
We thank Mr. Renzo Mazzaro for the artwork. Research in the authors' laboratories is supported by grants from the Fondazione Cassa di Risparmio di Padova e Rovigo (CARIPARO) (to M.Z.) and the Italian Association for Cancer Research (AIRC) (to I.S.; n. 5118).