
Abstract
Introduction
Selenoproteins are proteins containing the 21st proteinogenic amino acid selenocysteine (Sec). Sec is incorporated cotranslationally into at least 25 and 24 selenoproteins encoded by the human and mouse genomes, respectively (19, 23). Most of the characterized selenoenzymes participate in redox reactions and accordingly, many are predicted to adopt a thioredoxin-like folded structure (23). The best-known redox enzymes among selenoproteins are the GPx, which represent the prototypic selenoenzymes (16, 28) in mammals. Thioredoxin reductases are also Se dependent in mammals, as well as methionine-R-sulfoxide reductase. Three enzymes of the iodothyronine deiodinase (Dio) family reductively remove iodine atoms from iodothyronines, and thus locally activate thyroid hormone (from L-3,3′,5,5′-tetraiodothyronine [thyroxine, T4] to L-3,3′,5-triiodothyronine [T3]) or inactivate T4 to L-3,3′,5′-triiodothyronine (rT3) or T3 to L-3,3′-diiodothyrione (T2) (35). Dio activities in tissues can be affected by Se deficiency or mutations in selenoprotein P (32) or tRNA[Ser]Sec (31, 34). Patients with inherited defects in SECIS-binding protein 2, a biosynthetic factor for selenoproteins, display reduced Dio activities and abnormal thyroid hormone serum concentrations (14). The thyroid gland is among the organs with the highest Se content in humans (12). Se status can influence thyroid function and the course of autoimmune thyroid disease (30). Thyroid hormone levels and metabolism are directly altered in Se-deficient rats (1, 5), although the thyroid itself is preferentially supplied with Se in Se-deficient transgenic mice (31). Accordingly, the effects of Se supplementation on circulating thyroid hormones were only marginal in nondeficient healthy adults (9, 27, 38).
Innovation
The thyroid is among the tissues with the highest selenium (Se) content and expresses many selenoproteins. Low Se levels have been associated with thyroid diseases and Se supplementation is reportedly beneficial for the clinical course of thyroiditis. Thyroid hormone biosynthesis requires hydrogen peroxide (H2O2) production and thus the gland must be protected from excess oxidative damage from (Se-dependent) peroxidases. Moreover, key metabolic enzymes for thyroid hormones, deiodinases (Dios), are selenoenzymes. It is therefore widely assumed that selenoproteins play vital roles in thyroid biology, cellular survival, and hormone production. Our genetic loss-of-function approach targeting selenoprotein biosynthesis provides strong evidence that selenoproteins are not essential for thyroid gland integrity. However, the finding of elevated thyrotropin (TSH), but normal thyroid hormone levels, reveals a novel role of an unidentified selenoprotein in regulation of thyroid hormone production. This work, therefore, provides the framework for future studies that should (i) identify one selenoprotein required for euthyroidism out of 24 selenoproteins and (ii) dissect which biochemical or cell biological process during thyroid hormone biosynthesis, storage, release, transport, or activation is affected by this selenoprotein. From a clinical point of view, the mechanistic basis for the potential benefit of Se supplementation is needed. This report will thus represent a key reference for thyroid hormone economy in both basic and clinical contexts.
Conditional inactivation of tRNA[Ser]Sec provides an elegant way to probe whether selenoproteins are essential in a given tissue or cell type, because this method allows for complete abrogation of all selenoprotein translation (4, 10). We report here conditional inactivation of tRNA[Ser]Sec in thyroid epithelial cells. With this model, we set out to test two hypotheses: (i) that selenoenzymes are essential for thyroid integrity, that is, cell survival, and (ii) that type I-deiodinase (Dio1) activity in thyrocytes contributes to thyroid hormone homeostasis.
Results
Targeted inactivation of tRNA[Ser]Sec during embryonic development
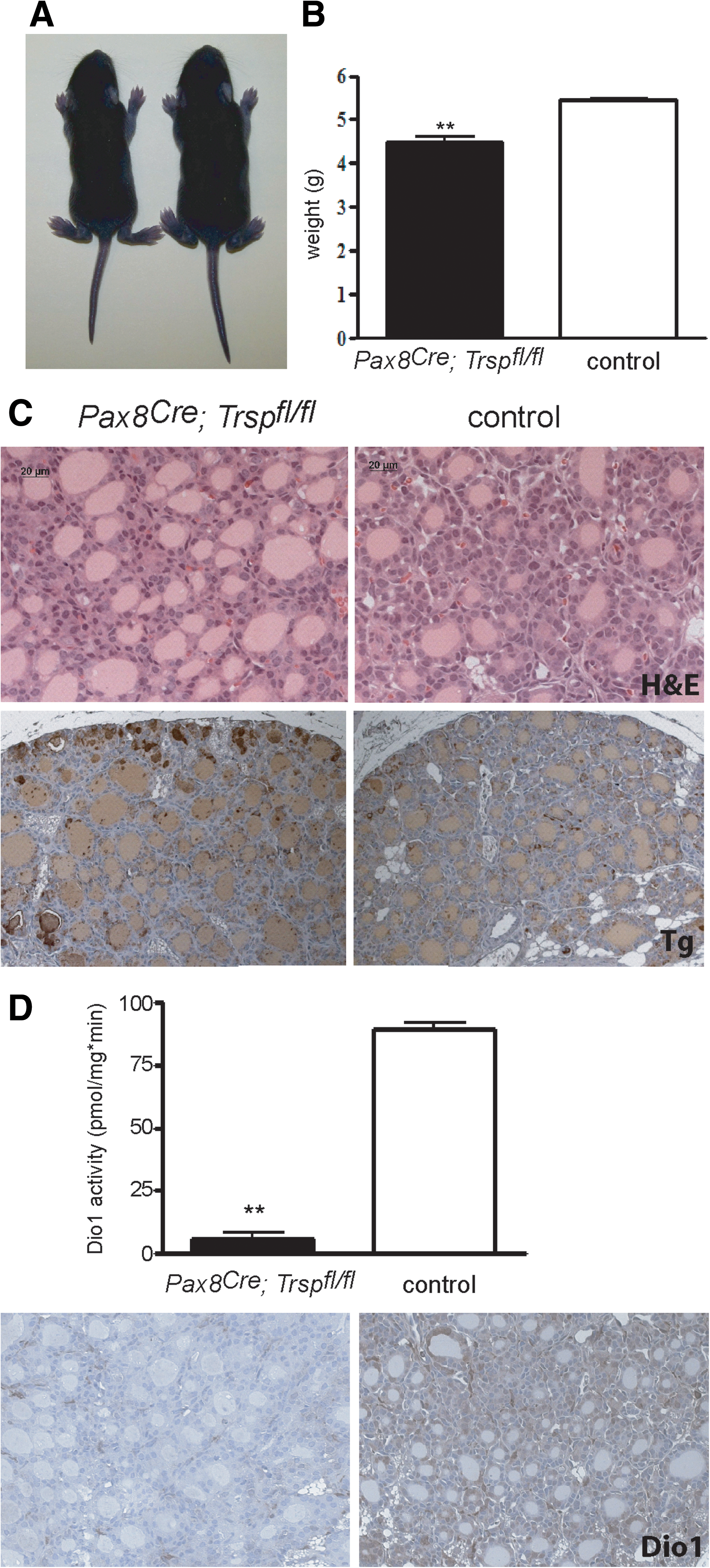
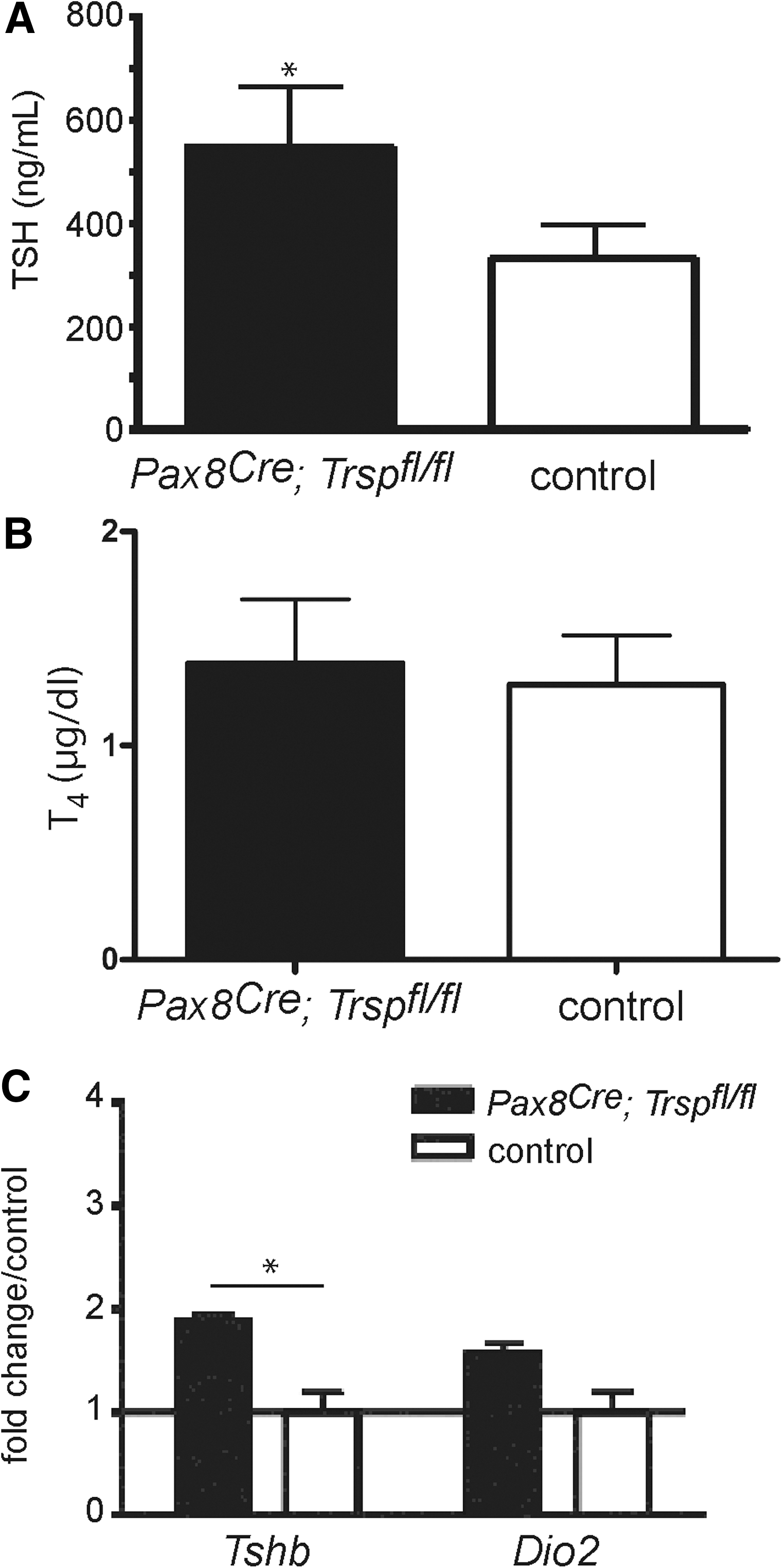
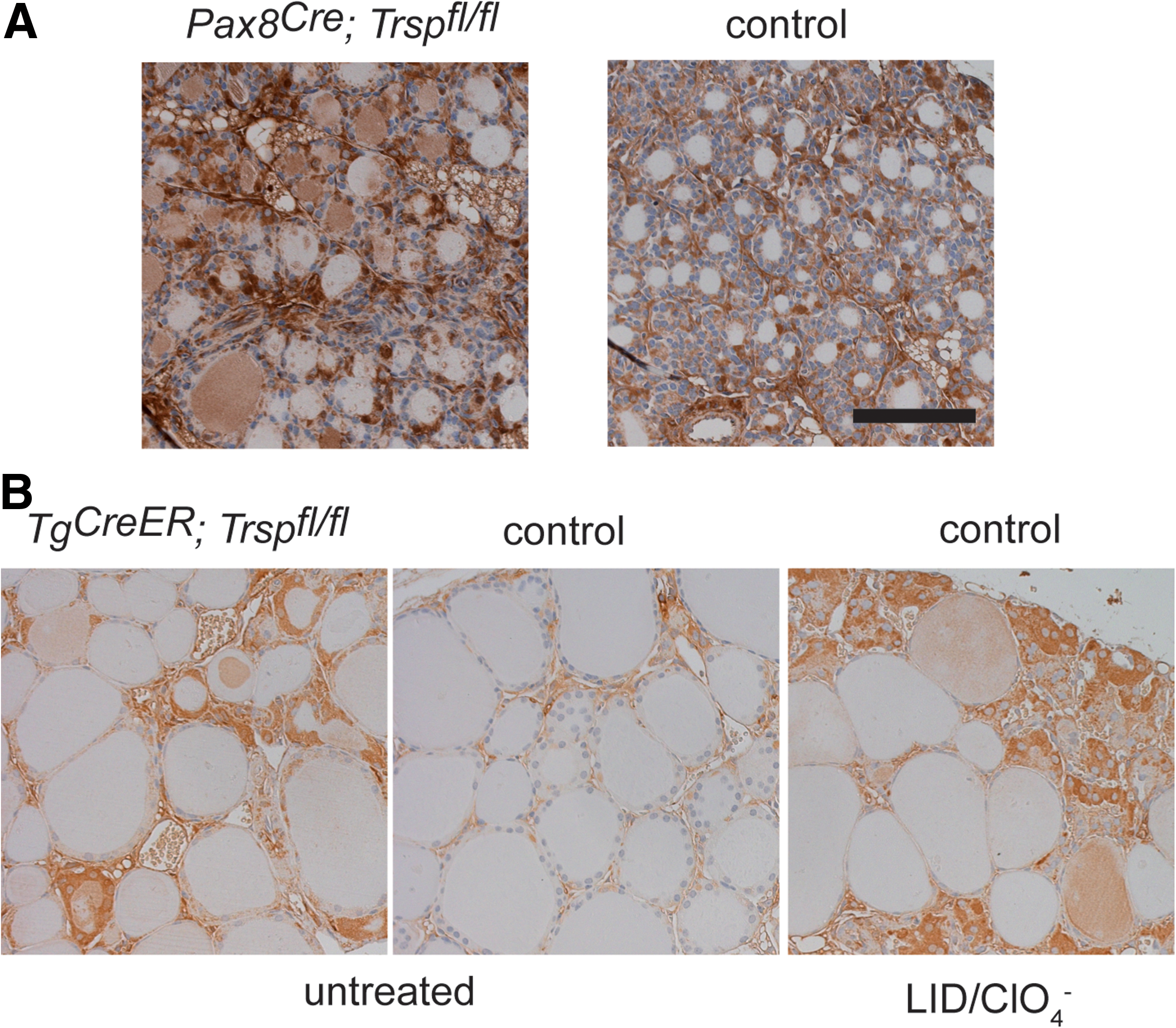
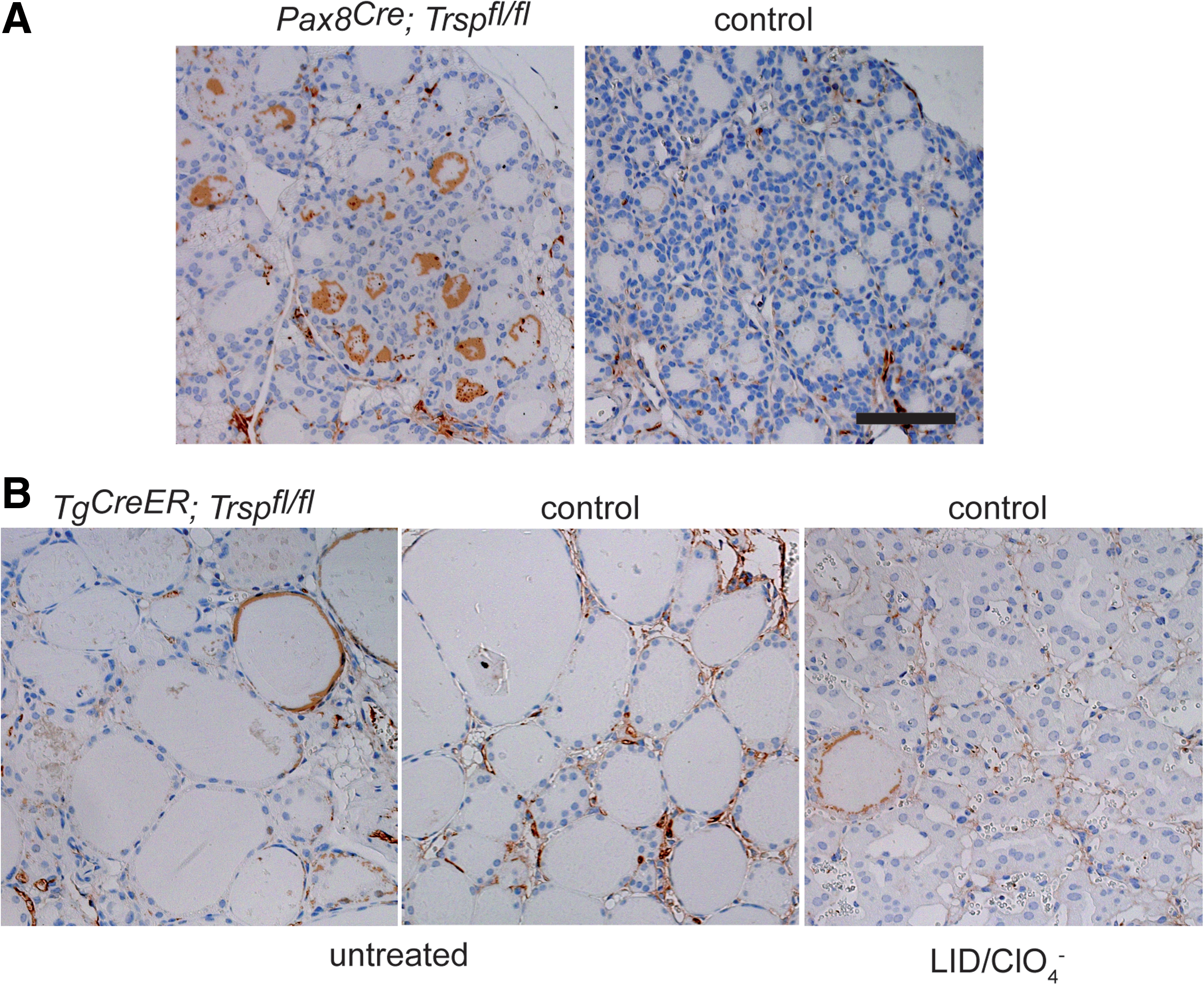
Thyroid epithelial progenitor cells are known to express the transcription factor Pax8 from embryonic day 9 (11). Targeted inactivation of both Pax8 alleles in mice results in athyreosis (25), while heterozygous deletion of Pax8 alone leaves the thyroid intact. We employed mice with a heterozygous Cre insertion into the Pax8 locus (Pax8Cre ) to conditionally remove tRNA[Ser]Sec (Trsp) early during thyroid development. Pax8+/Cre ; Trspfl/fl (Pax8-selenoprotein knockout) mice were born at the expected Mendelian ratio, but did not survive until weaning. Since mutant animals showed retarded growth (Fig. 1A, B), we suspected congenital hypothyroidism due to Trsp gene targeting in thyroid progenitors, and analyzed Pax8-selenoprotein knockout mice along with controls at postnatal day 8 (P8). Histological analysis revealed thyroid morphology with a mixture of smaller and larger follicles as appropriate for early postnatal development in mice (Fig. 1C). Normal differentiation of mutant thyroids was observed as judged by normal biosynthesis of Tg (Fig. 1C). We then tested the activity of Dio1 in thyroid extracts of Pax8-selenoprotein knockout mice and controls, demonstrating quantitative loss of activity in the mutants (Fig. 1D). Similarly, immunohistochemical staining for Dio1 supported the absence of selenoprotein biosynthesis in the vast majority of thyroid epithelial cells in Pax8-selenoprotein knockout mice (Fig. 1D). To probe the thyroid hormone axis, we determined serum thyrotropin (TSH) values, which showed a moderate, but significant, elevation in Pax8-selenoprotein knockout mice (Fig. 2A). Total T4 was normal in mutant mice at P8 (Fig. 2B). Pituitary expression of TRH-responsive genes was probed by qPCR, indicating a significant increase of Tshb while Dio2 transcripts were unchanged in Pax8-selenoprotein knockout mice (Fig. 2C). However, while the moderate elevation of TSH serum levels and pituitary transcripts is consistent, normal T4 levels rule out congenital hypothyroidism in Pax8-selenoprotein knockout mice and do not explain the premature death of mutant animals. We thus speculated that Pax8Cre expression in kidney may confound our analysis (3). In fact, GPx1 expression is reduced in Pax8-selenoprotein knockout kidneys at P8 (not shown). Our results with Pax8Cre mice suggest that selenoproteins are not essential for thyroid development, but may participate in thyroid function. We therefore chose to investigate a more specific mouse model with Cre expression restricted to thyroid epithelial cells.
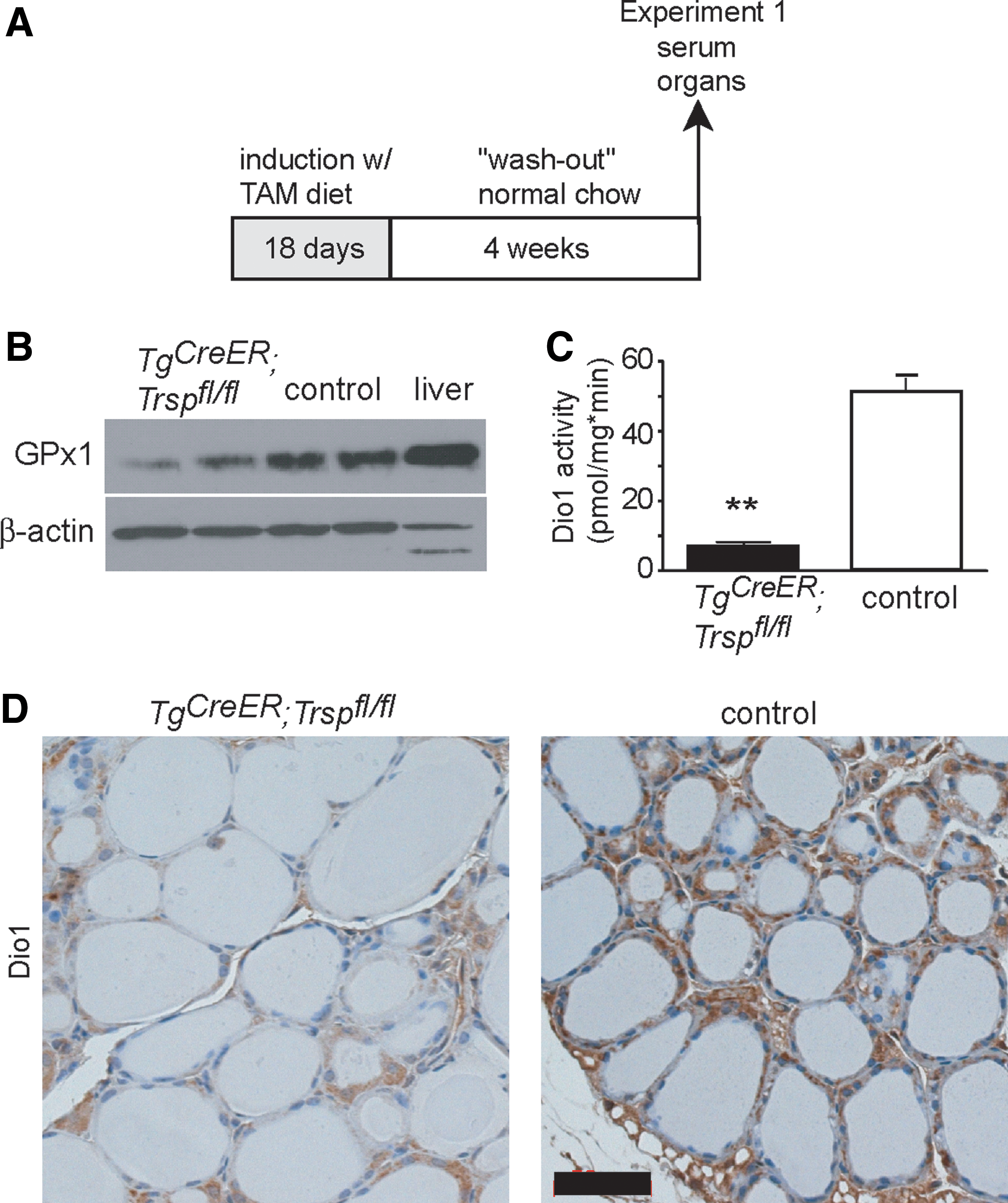
Inducible inactivation of tRNA[Ser]Sec in the thyroid reduces selenoprotein expression
To generate an improved model, we crossed Trspfl/fl mice with Tg-CreER mice. In this construct, the Cre coding region is fused to a mutant estrogen receptor ligand-binding domain (TgCreER ), which does not bind estrogen but the synthetic antiestrogen tamoxifen (TAM). Hence, Cre-mediated recombination can be induced specifically in thyrocytes by feeding TAM to adult animals. We refer to TgCreER ; Trspfl/fl mice after TAM induction as Tg-selenoprotein knockout mice. Of note, without TAM treatment, TgCreER ; Trspfl/fl mice behaved like controls. In preliminary experiments, we determined that feeding Tg-selenoprotein knockout mice a diet containing 0.36 g/kg TAM citrate for 18 days leads to maximal and almost complete inactivation of Trsp in the thyroid (Fig. 3A). After a TAM washout period of 4 more weeks, thyroidal GPx1 expression was reduced (Fig. 3B). Thyrocyte-specific Dio1 activity was decreased by 90% in Tg-selenoprotein knockout mice (Fig. 3C). Almost quantitative loss of Dio1 in thyrocytes is supported by immunohistochemistry (Fig. 3D).
Thyroid phenotype and thyroid hormone levels
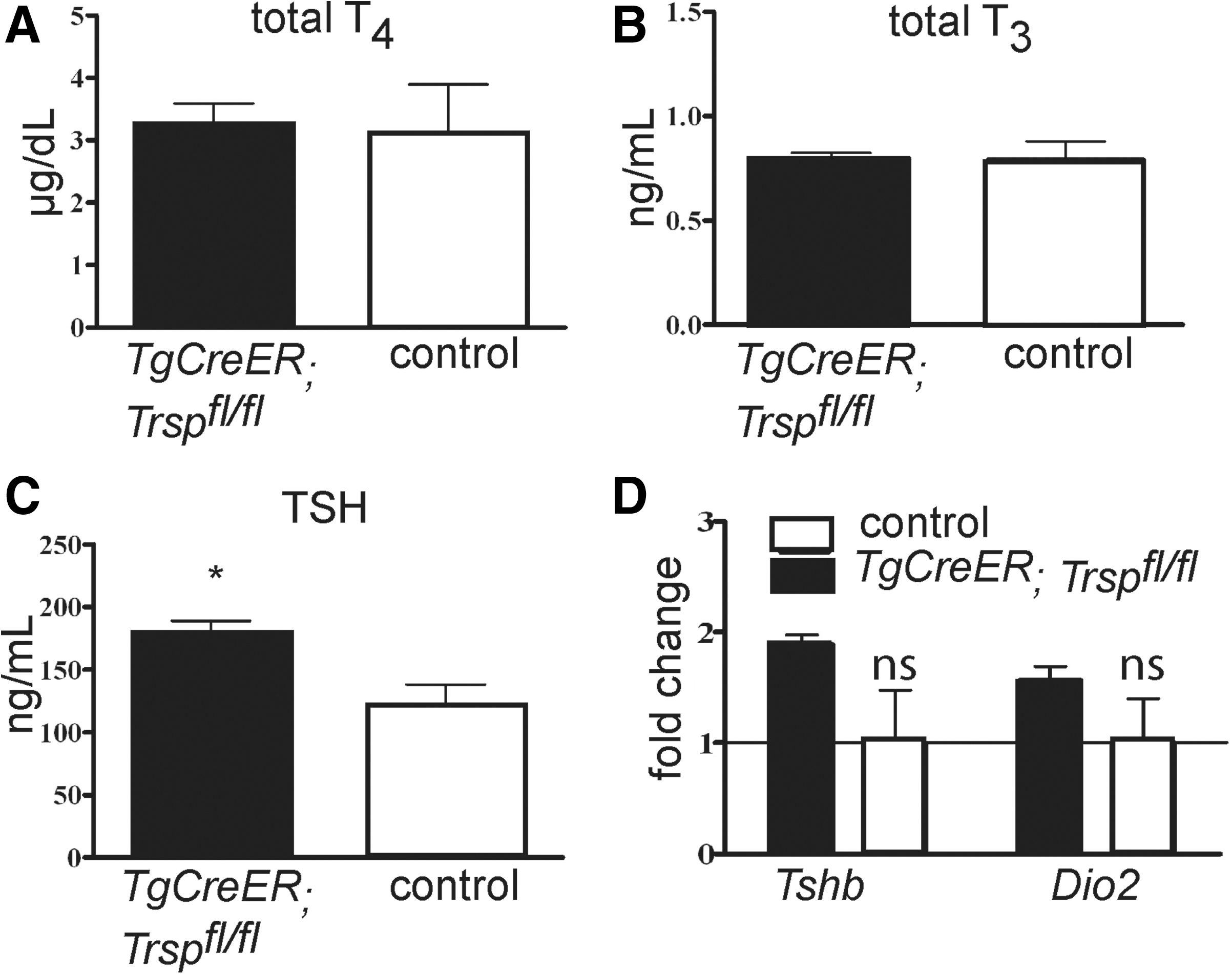
Thyroid hormones, total T4 (Fig. 4A) and total T3 (Fig. 4B), were not changed in Tg-selenoprotein knockout mice, while TSH was moderately increased (Fig. 4C). The increase of plasma TSH was reflected by a moderate elevation of Tshb and Dio2 transcripts in the pituitaries of Tg-selenoprotein knockout mice, which however failed to reach statistical significance (Fig. 4D).
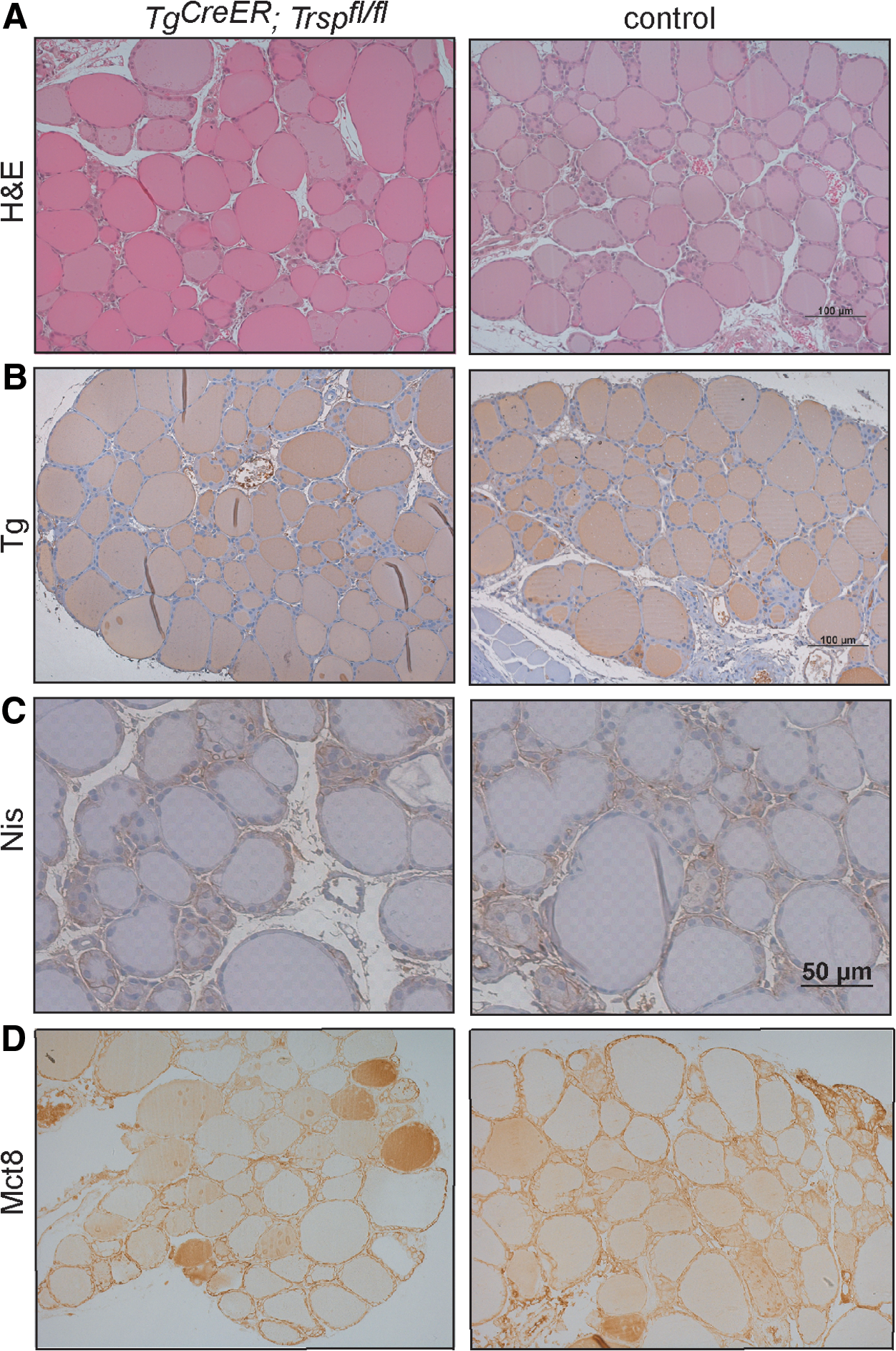
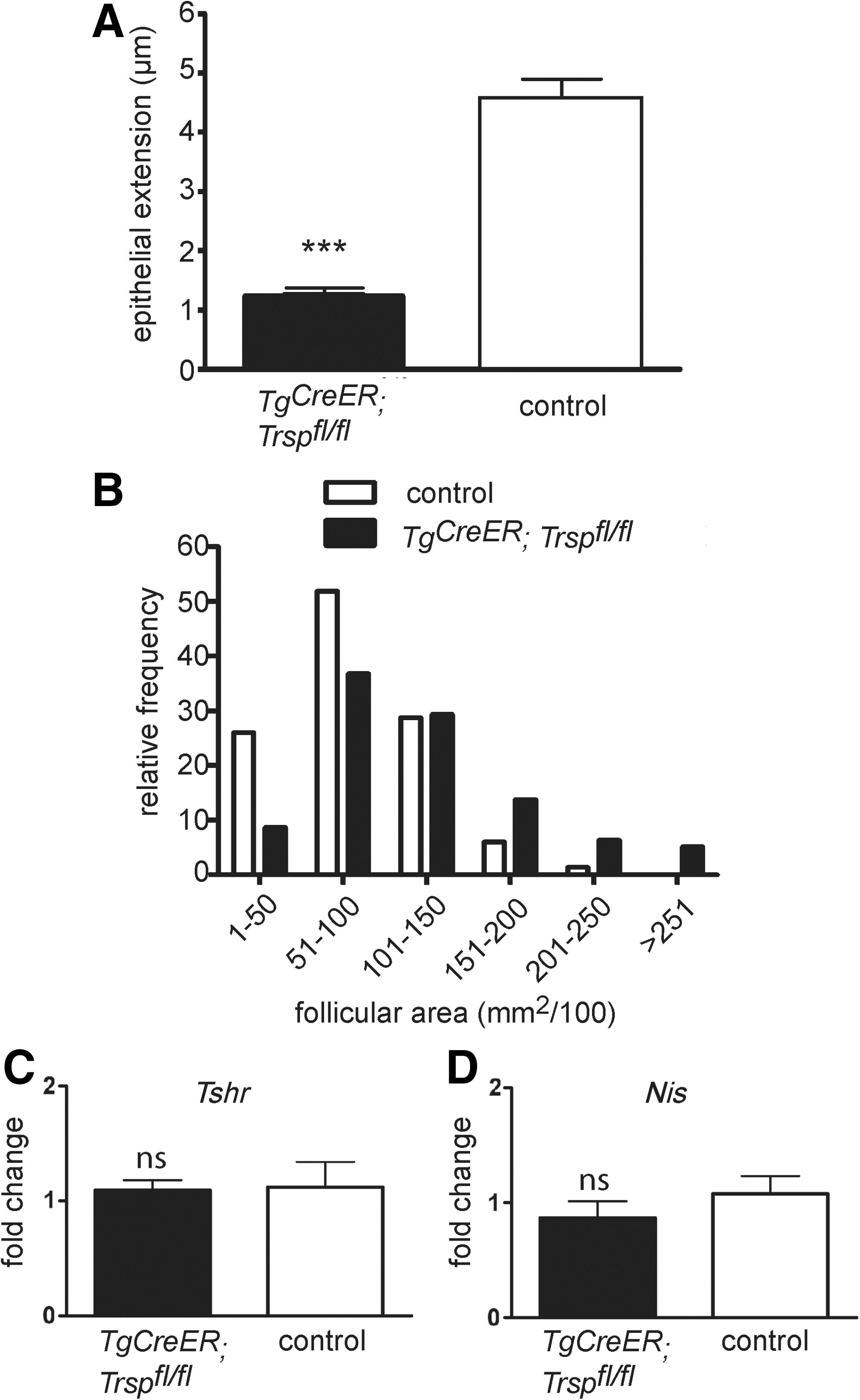
Despite the drastic reduction of selenoproteins in the thyroid, hematoxylin and eosin (H&E) staining did not reveal major differences between genotypes (Fig. 5A). Immunohistochemical staining for functional markers of production and storage of thyroid hormones (Tg), iodide uptake (sodium-iodide-symporter [Nis]), and export of thyroid hormones from the thyroid gland (monocarboxylate transporter 8 [Mct8]) showed no differences between mutants and controls (Fig. 5B–D). However, we noted morphometric differences. Thyroid epithelia from Tg-selenoprotein knockout mice were significantly flattened (Fig. 6A), and the size distribution shifted towards larger follicles (Fig. 6B). The paradoxical flattening of epithelial cells in the presence of elevated plasma TSH could reflect a cellular resistance to TSH in Tg-selenoprotein knockout thyroids. We, therefore, first tested the expression of the TSH-receptor (Tshr) in the thyroids. We could not detect a reduction of Tshr by qPCR (Fig. 6C). To test TSH-receptor downstream signaling, we quantified Nis mRNA in thyroids, but did again not find evidence of decreased target gene expression (Fig. 6D).
Low-iodine challenge of the thyroid
We reasoned that an increase of TSH will activate thyrocyte metabolism, elevate H2O2 production, and consequently unmask the limited antioxidative defense system in Tg-selenoprotein knockouts. Instead of injecting TSH directly, we chose a more physiological approach and fed Tg-selenoprotein knockout animals a low-iodine diet combined with perchlorate in drinking water (LID/ClO4 −), a regimen known to rapidly induce goiter formation in C57Bl/6 mice (Experiment 2; Fig. 7A). As expected, thyroid weights doubled within 21 days (Table 1) in LID-treated animals. Accordingly, total serum T4 was decreased (Fig. 7B), while total T3 levels were maintained (Fig. 7C). As expected, TSH was induced upon LID treatment (Fig. 7D). With respect to hormone levels, Tg-selenoprotein knockout mice did not respond differently than controls. Goiter formation was similar in mutants and controls. Interestingly, we did not observe any signs of increased tissue degeneration, fibrosis, or malignancy in the mutants (Fig. 7E).
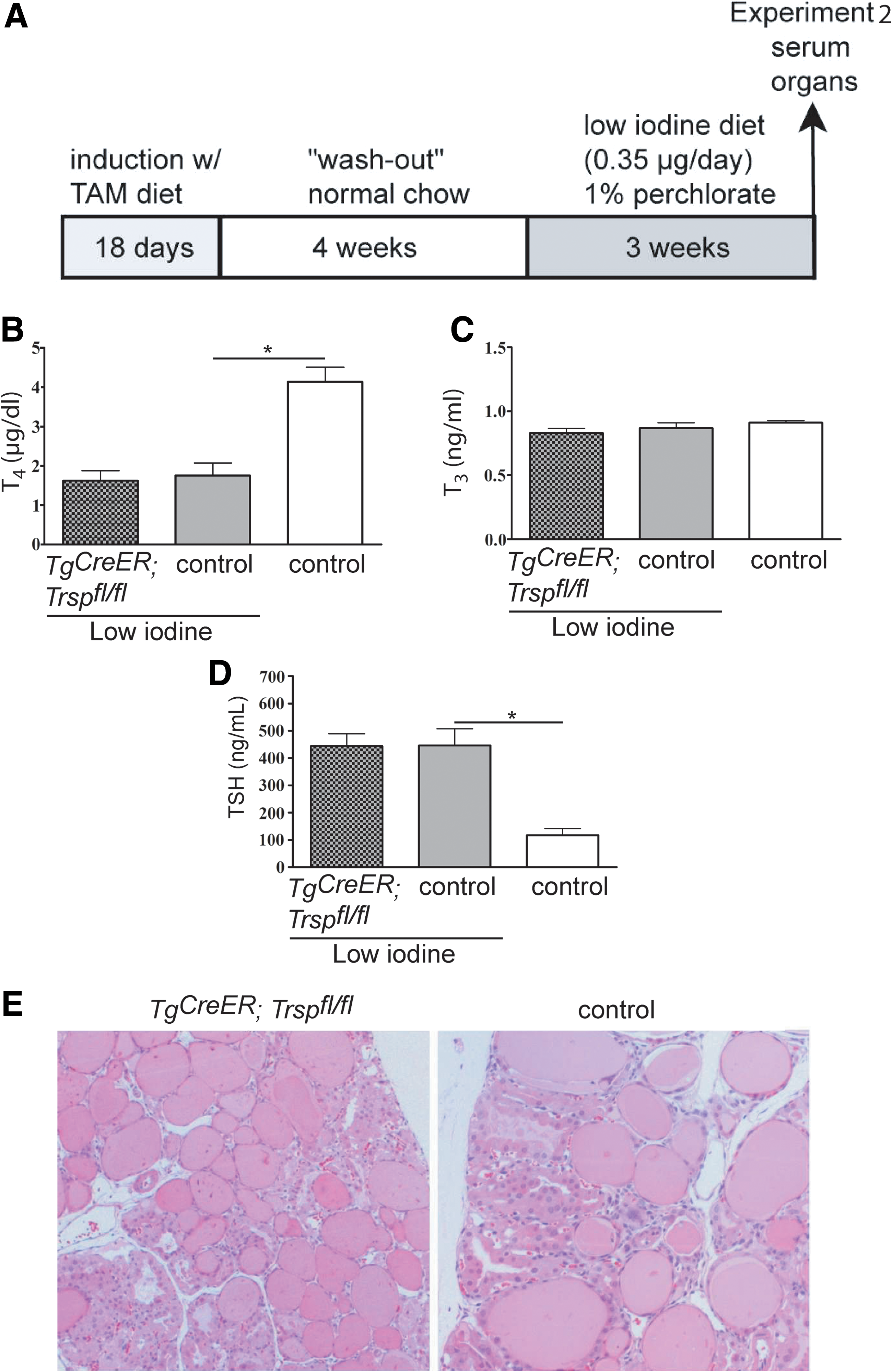
TAM treatment refers to the initial 18-day exposure as described in Experiment 2 (Fig. 7).
TAM, tamoxifen; LID/ClO4 −, low-iodine diet/perchlorate; Tg, thyroglobulin; Trsp, tRNA[Ser]Sec gene.
Increased lipid peroxidation in thyrocytes
Inactivation of selenoprotein biosynthesis in the thyroid is expected to abrogate expression of several antioxidative selenoenzymes, including GPx4, a lipid hydroperoxide-specific GPx, which is essential for the development of embryos and survival of fibroblasts (36) and neurons (39). Therefore, preservation of thyroid tissue integrity in Tg-selenoprotein knockout mice appeared, at first, a surprising finding. To test for potential oxidative stress in response to lack of selenoproteins, we stained thyroids from mutant mice and littermate controls for 4-hydroxynonenal (4-HNE), a stable marker of lipid peroxidation. 4-HNE immunoreactivity was present in a minor fraction of thyroid epithelial cells and in blood vessels in control animals at P8. In Pax8-selenoprotein knockout thyroids, the 4-HNE signal was much more intense occurring in a major fraction of thyrocytes within a given follicle and extending sometimes into the colloid (Fig. 8A). The increased 4-HNE staining supports successful inactivation of selenoenzymes, including GPx4. In adult control thyroids, 4-HNE was mainly confined to blood vessels and only weak immunoreactivity was sometimes seen in active (high) epithelial cells (Fig. 8B). In Tg-selenoprotein knockout animals, 4-HNE staining was much stronger in thyrocytes, in particular in active cells. Induction of oxidative stress during LID treatment was supported by massive 4-HNE staining in goitrous tissue. Inactivation of selenoprotein biosynthesis thus massively induces lipid peroxidation in thyrocytes.
Increased nitrosative stress in thyrocytes lacking selenoproteins
Formation of 3-nitro-tyrosine in proteins occurs in response to NO and peroxide exposure. While 3-nitro-tyrosine in proteins is emerging as a signaling device, its role as marker for oxidative stress is well established (26). We have stained Pax8-selenoprotein knockout thyroid sections with an antibody against 3-nitro-tyrosine and found massively increased immunoreactivity in the colloid and along the apical surface of the thyrocytes, while respective wild-type thyroids displayed weak immunostaining in some follicles and mostly along blood vessels (Fig. 9A). In adult thyroids, 3-nitro-tyrosine was still mostly confined to blood vessels and almost absent from colloid and thyrocytes. In contrast, some follicles in Tg-selenoprotein knockout thyroids displayed strong staining along the apical membrane of thyrocytes and frequently staining was found in thyrocytes (Fig. 9B).
Expression of antioxidative enzymes
We then tested whether lack of selenoprotein biosynthesis induced compensatory upregulation of Se-independent antioxidant enzymes. Western blots with tissue lysates obtained from three individual mice per genotype revealed that Sod1 and Sod2 protein levels were similar in mutants and controls, while catalase protein appeared moderately (1.8-fold, p=0.06) elevated in Tg-selenoprotein knockout thyroids (Fig. 10).

Discussion
Conditional inactivation of selenoprotein biosynthesis in thyrocytes was achieved using two different Cre-transgenic mouse lines. Selenoprotein expression was almost completely abrogated in both models as demonstrated by a reduction of both Dio1 activity and immunostaining. In contrast to our expectation that selenoproteins are essential for thyroid integrity and thyrocyte survival, we did not observe developmental (Pax8-selenoprotein knockout) or degenerative (Tg-selenoprotein knockout) alterations in thyroid structure. Expression of other antioxidative enzymes appears sufficient to protect the thyrocyte from lack of selenoprotein translation. Interestingly, both models exhibit a mild endogenous elevation of TSH without manifest reductions of thyroid hormones. These data suggest that lack of selenoproteins mildly impinges on thyroid hormone biosynthesis, but selenoproteins are not essential for tissue integrity. Massive increases in 4-HNE and 3-nitro-tyrosine staining in selenoprotein-deficient thyroids indicate, however, the occurrence of oxidative stress.
Pax8Cre-selenoprotein knockout mice
Pax8Cre expression affords quantitative inactivation of Trsp and thus of selenoprotein biosynthesis at least by P8. We have not re-investigated Pax8Cre expression ourselves, but it has been demonstrated that Pax8Cre leads to quantitative gene recombination in developing thyrocytes by embryonic day 15 (3). On P8, thyroid development and expression of functional markers appeared normal in Pax8-selenoprotein knockout mice. Plasma TSH was elevated as well as expression of Tshb mRNA in pituitary. However, serum T4 remained normal in these mice, indicating preserved thyroid hormone secretion by the gland. The mild elevation of TSH does not indicate congenital hypothyroidism that is expected to lead to a more than 10-fold induction of TSH as seen in Pax8 −/− mice (25). Genetic inactivation of Dio1 alone does not lead to hypothyroidism (29).
Pax8Cre also reduces selenoprotein expression in kidney and we suppose that renal lack of selenoprotein expression may have caused the death of Pax8-selenoprotein knockouts before weaning, although we did not explore further the possible kidney phenotype. Tissue responses to inactivation of Trsp suggest that selenoproteins exert very specific functions in different tissues and may be essential in one cell type, while not in another. For example, Trsp inactivation in mammary tissue or in hepatocytes does not cause a major phenotype (24, 34), while Trsp deletion in neurons or chondrocytes leads to tissue degeneration (13, 39). An association of Se status and kidney disease has been proposed (6, 41), and therefore podocyte-specific inactivation of Trsp was performed (2). Interestingly, these mice did not show a severe phenotype. Therefore, a dedicated study on kidney tubular function is warranted in the future.
TgCreER-selenoprotein knockout mice
TgCreER expression is specific for thyrocytes [our unpublished observations and (20)]. Reduction of Dio1 activity and Dio1 immunostaining suggests a more than 80% penetrance of Trsp deletion after TAM induction. Accordingly, we found increased plasma TSH in these mice, similar to Pax8-selenoprotein knockouts, as well as normal total T4 and T3 levels. It thus seems as if some thyroid insufficiency occurs upon selenoprotein inactivation, but this is readily compensated for by a moderate increase in TSH secretion and normal plasma T4 and T3 levels are ultimately reached. Biochemical and histological evidence suggest that selenoproteins are inactivated in the vast majority of thyrocytes.
Nevertheless, morphometric changes reveal that the thyroids responded to Trsp inactivation. Epithelial height is reduced and follicle diameter is increased in Tg-selenoprotein knockout mice. Flat epithelia are usually considered “inactive.” At the same time, however, colloid is not consumed in selenoprotein knockout thyroids (as in iodine deficiency). One could speculate that mobilization of thyroid hormones from colloid is compromised in Tg-selenoprotein knockout mice and at the same time hormone synthesis is reduced. However, this interpretation is in contrast to the normal plasma T4 levels observed. Morphological changes of the thyroids may suggest a failure to efficiently transduce the TSH signal. Nevertheless, Tshr expression was normal in mutant thyroids and Nis mRNA, a TSH-receptor target gene, was also not significantly affected. Another possibility is that selenoprotein-deficient thyrocytes are slightly impaired in their thyroid hormone production or release. A third possibility is that selenoprotein-deficient thyrocytes are completely inactive and the remaining functional thyrocytes are stimulated more strongly by increased TSH. Based on our present data we cannot decide amongst these possibilities. However, almost quantitative inactivation of selenoproteins in the Pax8Cre model with preserved T4 levels argues against a complete failure of knockout thyrocytes. Increased TSH is likely not caused by inactivation of Dio1, since genetic Dio1 deletion yields a different phenotype (29). Hepatic Dio1 activity is not increased in Tg-selenoprotein knockouts, arguing against the possibility that Dio1 in liver compensates for loss of Dio1 in thyroid gland (not shown). We have also performed the reverse experiment and determined Dio1 activity in thyroids of hepatocyte-specific Trsp knockouts (37). Again, no compensatory regulation in the thyroid was apparent (not shown). We therefore cannot support nor fully refute our other hypothesis, that is, that thyroidal Dio activity contributes to thyroid hormone homeostasis. Observed effects on thyroid morphometry may well be caused by the loss of any other selenoprotein.
Consistent with a role of selenoproteins in redox regulation and peroxide degradation, we found significantly increased 4-HNE and 3-nitro-tyrosine immunoreactivity, both established markers of oxidative stress. It is thus interesting that we did not find any indication of tissue degeneration in the mutants 6 months after TAM-induced Trsp recombination or after an LID challenge, which induced goiter in both wild-type and Tg-selenoprotein knockout animals. This finding is in sharp contrast to our other working hypothesis, that is, that selenoproteins are essential for thyroid gland integrity. It is thus possible that remaining, non-Se-dependent antioxidative enzymes are sufficient to protect thyrocytes from H2O2 produced during hormone synthesis.
While our results are clear in terms that selenoproteins are not essential for thyroid gland integrity or hormone biosynthesis, it remains unclear why Se supplementation reportedly exerts beneficial effects on thyroid disease, namely, autoimmune diseases like Hashimoto thyroiditis (15, 18). On the basis of our findings presented here, we are tempted to speculate that in these instances Se has influenced the immune system rather than thyrocytes directly.
Materials and Methods
Mouse models
Conditional gene targeting of Trsp (tRNA[Ser]Sec gene) abrogates selenoprotein biosynthesis in cells expressing transgenic Cre recombinase (24). We have bred conditional Trsp (Trspfl/fl ) mice with two Cre-transgenic mouse lines: Pax8Cre harbors a Cre minigene knocked into the Pax8 locus (3). The inducible thyrocyte-specific Cre mouse line (TgCreER ) was generated as described (20). Instead of the Cre cDNA, a cDNA encoding a fusion protein consisting of Cre and the mutated estrogen receptor ligand-binding domain (ERT2) was inserted into the Tg locus. All mice were on a C57Bl/6 genetic background. Mice were fed standard breeding diets (Ssniff, Hannover, Germany). For Cre activation and recombination, diets containing 0.36 g/kg tamoxifen (TAM) citrate (a gift from Dr. M. Brielmeier, Helmholtz-Zentrum München) were fed to both TgCreER; Trspfl/fl mice and controls (Trspfl/fl , Trspfl/+, with or without TgCreER transgene) for the indicated periods of time starting at an age of 2–4 months (21). The presence of the TgCreER transgene had no effect on any of the measured parameters. For genotyping, ear clippings were dissolved in alkaline PEG lysis solution (8) and 1 μl of DNA solution was directly applied to 20 μl PCR using the following conditions: Trsp [as described in Ref. (34)], Pax8Cre (using primers Pax8 intron2 TCTCCACTCCAACATGTCTGC, Pax8 exon3 CCCTCCTAGTTGATTCAGCCC, and Pax8 Cre AGCTGGCCCAAATGTTGCTGG; Pax8+ 389 bp; Pax8Cre 673 bp), TgCreER (using primers JkphCreAS1 AGTCCCTCACATCCTCAGGTT and JKGpromS1 ATGCCAACCTCACATTTCTTG; ∼500 bp). Animal protocols were approved by the local government authority LaGeSo Berlin.
Histology
Thyroid glands were excised together with the trachea, fixed overnight in 0.1 M Na-phosphate buffer (pH 7.4) containing 4% paraformaldehyde, dehydrated, and embedded in paraffin. Sections of 2 μm were cut on a rotating microtome (Leica) and stained with H&E by standard procedures. Tissue sections were examined under a light microscope (Axioskop; Zeiss), and pictures were taken using a digital camera (ActionCam; Agfa). Immunostaining against Tg (Thermo Fisher), NIS (Acris), Dio1 (a gift from Dr. T. Visser), 4-HNE (Calbiochem, Merck KGaA), nitrotyrosine (Acris), and GPx1 [Abcam, rabbit polyclonal, as in Ref. (6)] was revealed using Vector reagents (Vector Laboratories) and diaminobenzidine substrate according to the manufacturer's recommendations as described (31). Morphometry was performed based on published procedures (17). Briefly, H&E-stained sections were photographed with a 20× objective, and follicular diameters, circumferences, and areas were calculated by AxioVision software (Zeiss). Results were used to calculate epithelial extension as given by (17), and 280–300 follicles per genotype were evaluated for area.
Hormones
Determinations of TSH, T3, and T4 from mouse serum were performed as described previously (37).
Enzyme activity assays
GPx and Dio1 activity assays were performed with minor modifications according to the methods described (31). Because of limited sample size of single thyroid lobes from individual mice, tissue lysates were prepared and subjected to the assays without preparation of microsomal fractions.
qPCR
RNA from a single thyroid lobe was extracted using the RNeasy Mini Kit from Quiagen. cDNA synthesis was performed using ThermoScript Reverse transcriptase from Invitrogen. qPCR was done using SYBR Green (Abgene) on iCycler. Primer sequences were as follows: Tshb: fwd GTGGGTGGAGAAGAGTGAGC, rev AAGAGCAAAAAGCACGGAGA; Dio2: fwd CTCCAACTGCCTCTTCCTGG, rev GACGTGCACCACACTGGAAT; Tshr: fwd CCTTGACAGAGCTCCCCTTG, rev ATTGCATAGGCCCTGGAATG; Nis: fwd TACATGCCATTGCTCGTGTT, rev CAGCTGCCATAGCGTTGATA.
Western blot
Protein extraction was performed using single thyroid lobes. Thyroids were homogenized in homogenization buffer using a rotor-stator, followed by ultrasound. Determination of protein content and western blotting were performed as described before (40). Western blot signals were detected using ECL reagents (GE Healthcare) and exposure to X-ray films. Antibodies used in this study were the following: rabbit α-mouse catalase (1:4000; Abcam), rabbit α-mouse superoxide dismutase 1 and 2 (1:2000; Abcam), and rabbit α-mouse GPx1 (1:2000; Abcam); goat α-rabbit (1:2000; Dako). β-Actin (1:2000; Rockland) was used as loading control.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software as indicated in the figure legends. Data are depicted as means±SEM.
Footnotes
Acknowledgments
The authors thank Dr. Theo Visser, Rotterdam, for Dio1 antibody; Dr. Stefan Offermanns, Heidelberg, for TgCreER mice; Dr. Meinrad Busslinger, Vienna, for Pax8Cre mice; Dr. Dolph L. Hatfield, Bethesda, for Trspfl/fl mice; and Dr. Markus Brielmeier, Munich, for TAM-supplemented mouse chow. We thank Vartitér Seher for technical assistance with histology. J.C-.U. was supported by Deutscher Akademischer Ausstauschdienst (DAAD). N.F.-V. was supported by JCCM. Financial support was provided by Charité-Universitätsmedizin Berlin, Deutsche Krebshilfe, and Deutsche Forschungsgemeinschaft, SFB665.
Author Disclosure Statement
No competing financial interests exist.