
Abstract
Introduction
Acute pancreatitis is a sudden inflammatory condition of the pancreas (most commonly caused by biliary stone disease or ethanol consumption) that can present in a mild, self-limiting form or in a severe form that can lead to multiple-organ failure (122). The pathogenesis of the disease is unclear. However, sustained global calcium (Ca2+) signaling (126), pathologic activation of digestive enzymes (138), and the proinflammatory transcription factor nuclear factor-κB (NF-κB) (135) are thought to be common factors in the initiation of pancreatitis.
Nitric oxide (NO), in conjunction with reactive oxygen species (ROS) and reactive nitrogen species (RNS), contributes to pancreatic physiology and pathophysiology (26). Following a general overview of NO metabolism, signaling, and function, this review will specifically discuss the effects of NO in the exocrine pancreas. Further, we will focus on the role of NO in the pathogenesis of acute pancreatitis and pancreatic ischemia/reperfusion (I/R) injury.
Summary I
• NO and RNS regulate pancreatic function both under physiological and pathophysiological conditions.
The Importance of NO
NO was discovered as the endothelium-derived relaxing factor in 1987 by Palmer et al. (120), which mediates vascular relaxation in response to acetylcholine, bradykinin, and substance P. NO is a diatomic gas that readily diffuses through membranes (119). Therefore, it can diffuse from where it is synthesized into surrounding cells. NO is a ubiquitous (intra- and intercellular) messenger that has both physiological and pathophysiological functions (119). In fact, due to its diverse effects, NO was named “Molecule of the Year” in 1992 (32). In addition, The Nobel Prize in Physiology or Medicine in 1998 was awarded to Drs. Robert Furchgott, Louis Ignarro, and Ferid Murad for their discoveries concerning NO as a signaling molecule.
NO has important roles in the cardiovascular, immune, nervous, and gastrointestinal systems. In fact, NO is involved in the regulation of vascular homeostasis, nonspecific host defense (cytotoxic agents released by macrophages), and neurotransmission (119). The free radical gas has important functions in regulating vascular permeability and the relaxation of vascular smooth muscle cells, and is involved in angiogenesis (145). NO inhibits thrombocyte aggregation (132) and leukocyte activation, adhesion, and migration (93). NO-generating compounds (such as sodium nitroprusside [SNP] and S-nitroso-N-acetylpenicillamine) have been shown to activate NF-κB in peripheral blood mononuclear cells (95). Further, NO is also thought to play part in the regulation of cell proliferation and apoptosis.
In the gastrointestinal system, besides the above-mentioned roles, NO contributes to numerous physiological functions, including maintenance of mucosal integrity, secretion, and motility. NO appears to be the predominant nonadrenergic, noncholinergic (NANC) inhibitory neurotransmitter in the enteric nervous system. These topics are reviewed in detail by Shah et al. (146) and Stanek et al. (152).
Summary II
• NO is a gaseous signaling molecule that has diverse effects on the cardiovascular, immune, nervous, and gastrointestinal systems.
Metabolism and Signaling of NO
The main source of NO synthesis is NO synthase (NOS). However, NO can also be generated by reduction of nitrite and degradation of S-nitrosothiols. The genetrated NO will then be directly or indirectly (e.g., via peroxynitrite) involved in fundamental signaling processes or will be deactivated by oxidation. They can also give rise to RNS, especially when there is a simultaneous activation of superoxide synthesis.
Generation of NO
NO synthase
The de novo formation of NO is mainly catalyzed by NOS from the guanidino group of L-arginine and molecular oxygen (119). NOS has three isoforms: the constitutively expressed neuronal (nNOS, type I) and endothelial (eNOS, type III) and the inducible (iNOS, type II). nNOS and eNOS are also referred to as constitutive NOS (cNOS) (119). NOSs are unique homodimeric enzymes with each monomer consisting of two domains, an N-terminal oxygenase and a C-terminal reductase domain, that require five bound cofactors/prosthetic groups for NO production: flavin adenine dinucleotide, flavin mononucleotide, heme, 5,6,7,8-tetrahydrobiopterin (BH4), and Ca2+-calmodulin (CaM) (Fig. 1). Dimerization is an absolute requirement for NOS activity. If any of the cofactors are lacking, then in many cases NOS produces superoxide instead of NO. In oxidative stress conditions, cofactors become oxidized and NOS function is uncoupled. Further, when oxygen is limiting (e.g., in conditions of hypoxia), NO production by NOS is greatly reduced.

Recent data have demonstrated that mitochondria may contain their own NOS isoform (58, 94). Mitochondrial NOS activity decreases mitochondrial Ca2+ uptake, oxygen consumption, membrane potential, and, consequently, the formation of ATP (94). NO produced by mitochondrial NOS reacts readily with the superoxide anion to produce peroxynitrite. Consequently, peroxynitrite causes the release of cytochrome c, increases the peroxidation of mitochondrial membrane lipids, and oxidatively damages susceptible targets.
eNOS and mitochondrial NOS are membrane associated, whereas nNOS and iNOS are cytosolic. eNOS and nNOS are strongly regulated by changes in intracellular Ca2+ concentration, whereas iNOS is not (iNOS forms complex with CaM at very low concentrations of Ca2+). eNOS activity is altered by changes in phosphorylation and also by post-transcriptional processes (47). iNOS expression is upregulated in cells (e.g., macrophages) involved in inflammation by various pro-inflammatory signals (such as cytokines and lipopolysaccharide [LPS]) via NF-κB-dependent mechanisms (162). In general, the NO concentrations produced by cNOS in stimulated endothelial and neuronal cells are much lower (nM) than those generated by iNOS in macrophages (μM) (119). iNOS does not produce NO at a substantially greater rate than that measured for nNOS or eNOS, just more of the enzyme can transiently be induced and normal Ca2+ levels are sufficient to fully activate it. iNOS has been linked to numerous inflammatory diseases such as septic shock, rheumatoid arthritis, asthma, and acute pancreatitis.
Nitrite reduction
Apart from de novo synthesis of NO by NOS, recent studies have demonstrated that nitrite reduction can also serve as a possible source of biologically relevant NO (60). Thus, nitrate and nitrite can serve as storage pools of NO during metabolic stress, with their bioactivation involving both enzymatic and nonenzymatic reactions. Xanthine oxidase, hemoglobin, myoglobin, neuroglobin, respiratory chain enzymes, cytochrome P 450, aldehyde oxidase, carbonic anhydrase, and even NOS can reduce nitrite. Almost without exception, the latter enzymes generate NO at much greater efficacy under hypoxic conditions. This pathway of NO generation may be especially important in situations when oxygen-dependent NOSs become dysfunctional.
Degradation of S-nitrosothiols
An important biological reaction of NO is S-nitrosylation, the conversion of thiol groups (including cysteine residues in proteins) to form S-nitrosothiols (50). S-nitrosothiols can be degraded to NO and thiol by a number of nonenzymatic (such as metal ion catalysis—although the physiological relevance of these processes is small) and, more importantly, enzymatic reactions (e.g., xanthine/xanthine oxidase, thioredoxin/thioredoxin reductase, γ-glutamyl transpeptidase, glutathione peroxidase, copper/zinc superoxide dismutase, and glutathione-dependent formaldehyde dehydrogenase) (56).
NO signaling pathways
The best characterized downstream NO signaling pathway is mediated by soluble guanylyl cyclase (52) (Fig. 2). The enzyme contains the same heme protoporphyrin IX as hemoglobin with iron in the ferrous form that binds NO with great affinity. Consequently, soluble guanylyl cyclase generates cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP). NO can also act independently of guanylyl cyclase stimulation by directly or indirectly (e.g., through peroxynitrite) modifying cellular components, especially proteins. Target proteins of the NO cascade include protein kinases (such as protein kinase G) and phosphatases, phosphodiesterases, ion channels, and transcription factors that mediate effector functions.
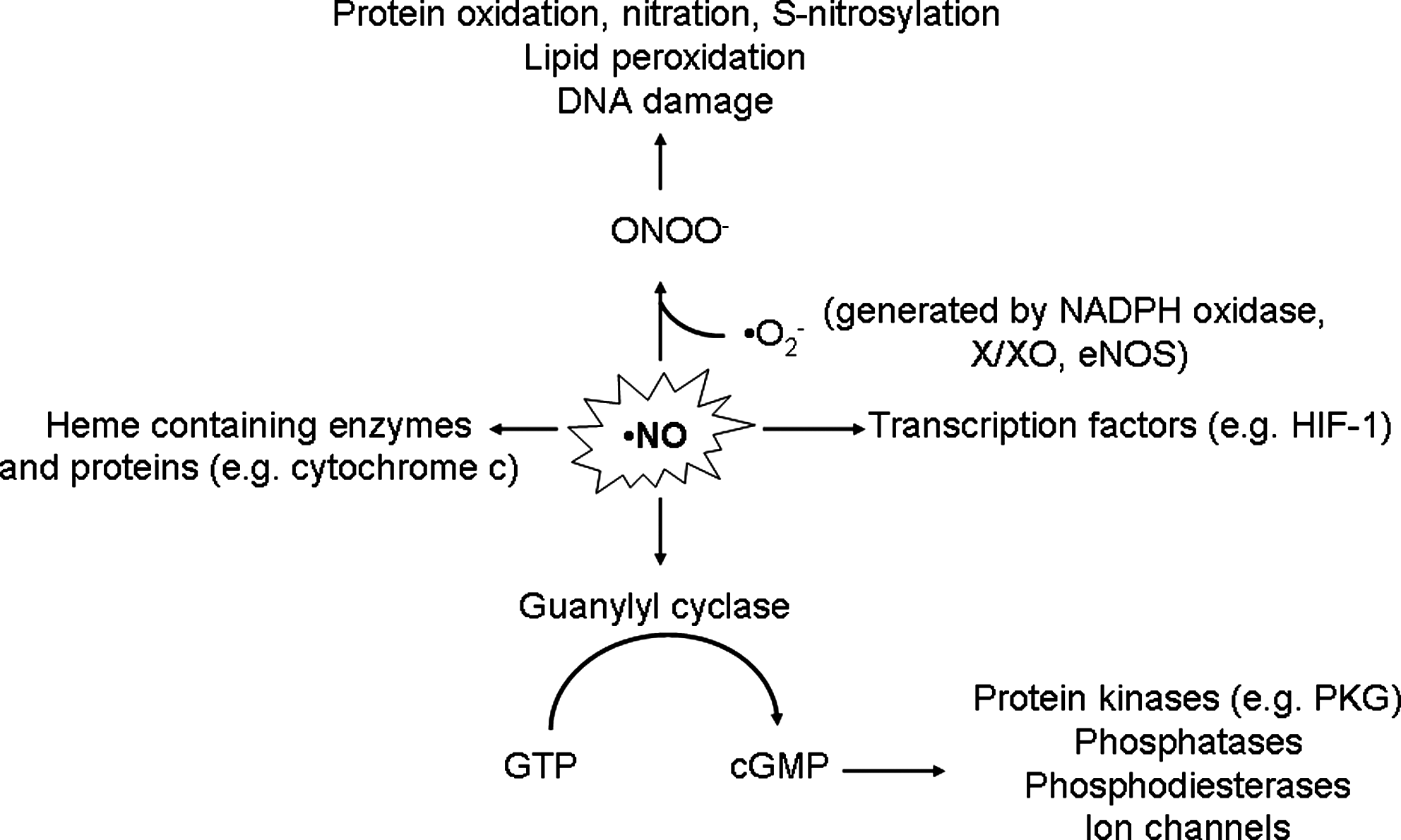
Recently, it has become clear that many effects of NO are mediated by S-nitrosylation. S-nitrosothiols are important in post-translational and transcriptional regulation of protein expression, as well as in the regulation of various intra- and extracellular protein functions (56). Moreover, molecules containing thiol groups, like N-acetylcysteine, albumin, and hemoglobin, can not only act as NO scavengers, but may also serve as depot and NO carriers that release active NO at a distance from the original site of NO production. Hypo- or hyper-S-nitrosylation of specific protein targets (which result in alterations in protein function) have been shown to be directly implicated in the etiology and symptomatology of numerous human diseases (50).
Deactivation of NO, formation of reactive nitrogen species
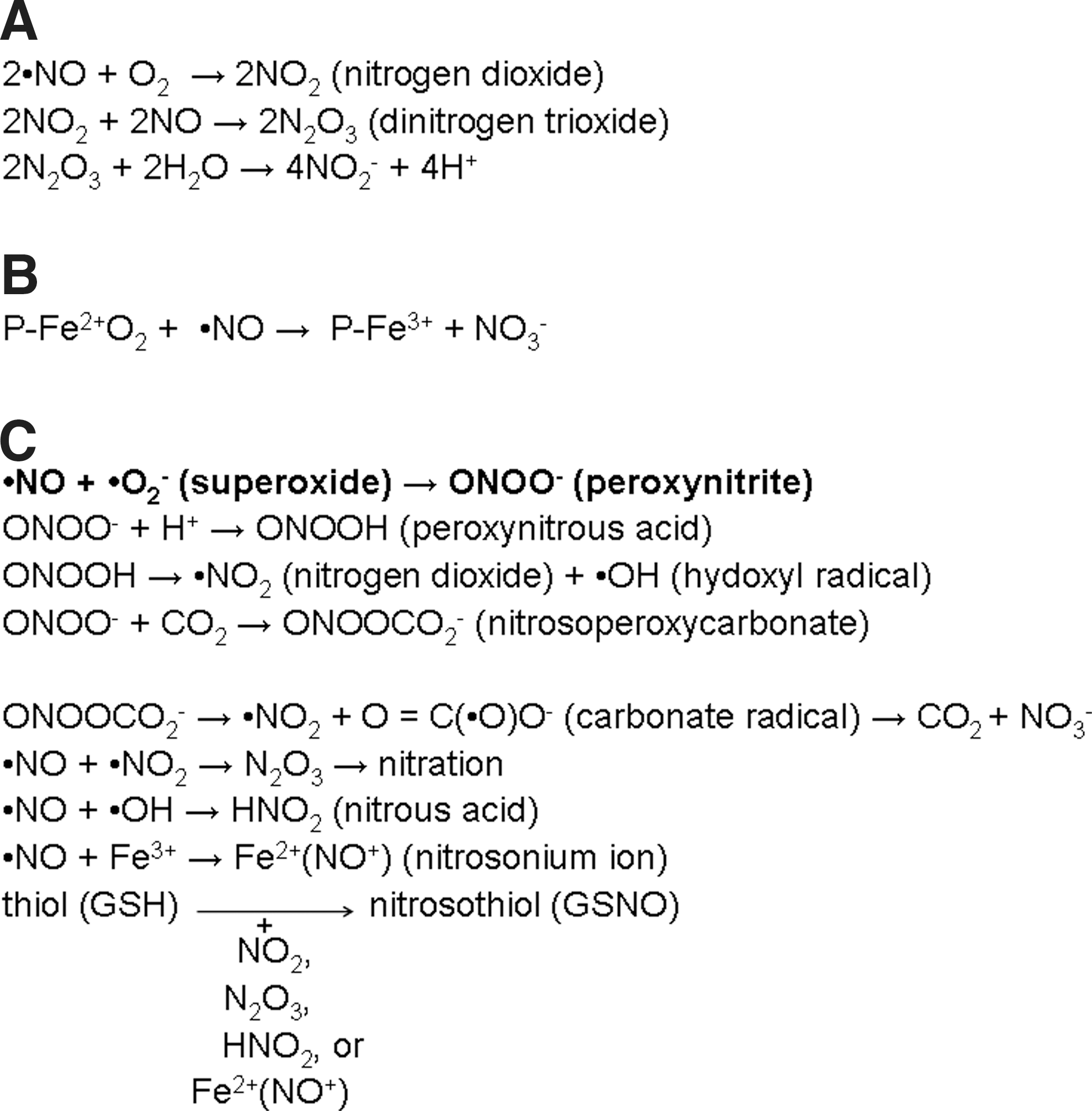
NO is very rapidly deactivated in biological fluids by oxidation to nitrite and nitrate (119) (Fig. 3A, B). Alternatively, NO can react with free radicals generating RNS such as nitroxyl, nitric dioxide, S-nitrosothiols, and dinitrosyl iron complexes (Fig. 3C). Consequently, these RNS can damage cellular proteins, lipids, or nucleic acids. One of the most important reactions of NO is combining with superoxide (•O2 −) to form the much more powerful oxidant peroxynitrite. Peroxynitrite can react with proteins through three possible pathways: directly with cysteine, methionine, tryptophan, and tyrosine residues, or transition metal centers and selenium-containing amino acids, or indirectly via secondary free radicals arising from peroxynitrite (50). A characteristic molecular footprint left by the reactions of reactive nitrogen species is the nitration (i.e., addition of nitro group, −NO2) of biomolecules like protein tyrosine residues to 3-nitrotyrosine (131).
Detection of NO
The half-life of NO is quite short (119); therefore, direct detection of NO is very difficult in vivo. Imaging techniques, such as optical (fluorescence and chemiluminescence), electron paramagnetic resonance, magnetic resonance imaging, and positron emission tomography can be used for direct detection of NO and RNS (72).
More often, indirect methods of NO detection are used. Most commonly, determination of serum/plasma nitrite and nitrate concentrations are performed (by the spectrophotometric assay based on the Griess reagent) as an index of NOS activity and NO production (88). Although, in fasting animals, a large proportion (70%–90%) of plasma nitrite is derived from endogenous NO production, an important consideration is that diet (consumption of meat, vegetables, and drinking water) also influences this parameter (88). Further, anaerobic commensal bacteria in the gastrointestinal tract reduce nitrate to nitrite (and NO) (151). However, besides nitrate reduction by bacteria, mammalian cells can also reduce nitrate, which can take place under normoxic conditions (77). In the plasma, NO is oxidized almost completely to nitrite, where it remains stable for several hours. However, in whole blood, nitrite is rapidly oxidized to nitrate. Oxyhemoglobins are thought to be involved in the latter process (see Fig. 3B).
Pharmacological compounds commonly used in NO/NOS research
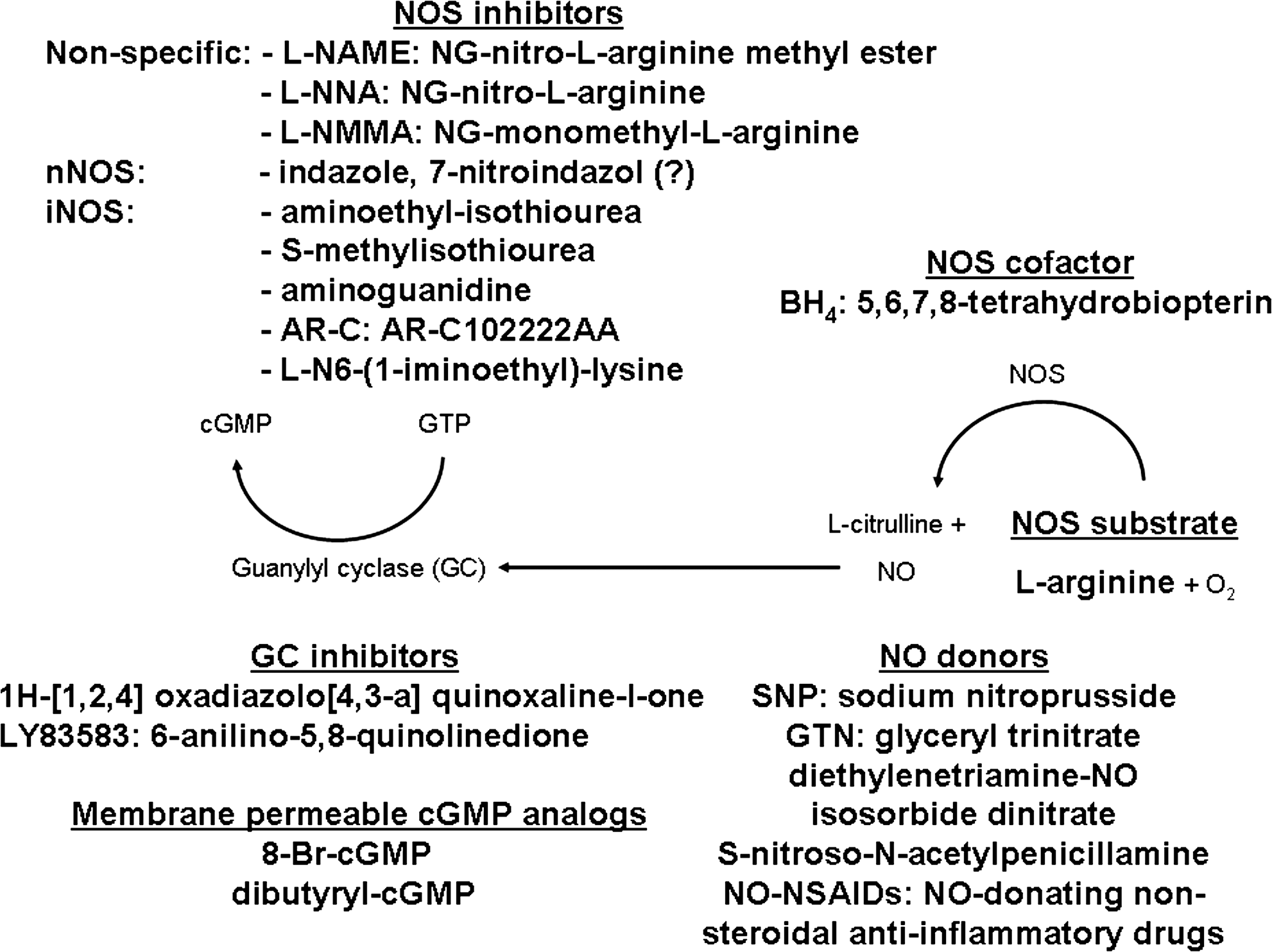
A brief summary of pharmaceutical compounds commonly used in NO/NOS research is provided in Figure 4. (i) The substrate of NOS (L-arginine) is administered parenterally in excess amounts to increase generation of NO. (ii) Molecular carriers of NO (NO donors) stabilize the radical until release of the messenger molecule. (iii) There is a wide variety of inhibitors (which are isoform selective or nonselective) available to block NOS activity. (iv) BH4 is a critical cofactor of NOS; therefore, it can be used effectively when its levels are decreased. (v) Guanylyl cyclase inhibitors can block the formation of cGMP from GTP, whereas (vi) membrane-permeable cGMP analogs can directly cross the cell membrane and mimic the effects of cGMP.
Summary III
• NO is mainly synthesized de novo by the constitutive NOS isoforms eNOS and nNOS or the inducible NOS isoform iNOS. In addition, mitochondria may have their own NOS isoform.
• Nitrite and S-nitrosothiols can also serve as substrates for NO synthesis.
• NO activates soluble guanylyl cyclase, which generates cGMP, a downsteam signaling molecule.
• In biological fluids, NO is rapidly oxidized to nitrite and nitrate (the concentrations of which are often measured to estimate NOS activity) or it can react with free radicals generating RNS.
• NO can combine with superoxide, giving rise to the highly reactive peroxynitrite.
• Nitric oxide donors and NOS inhibitors are useful tools in NO/NOS research.
Sources of Pancreatic NO Synthesis
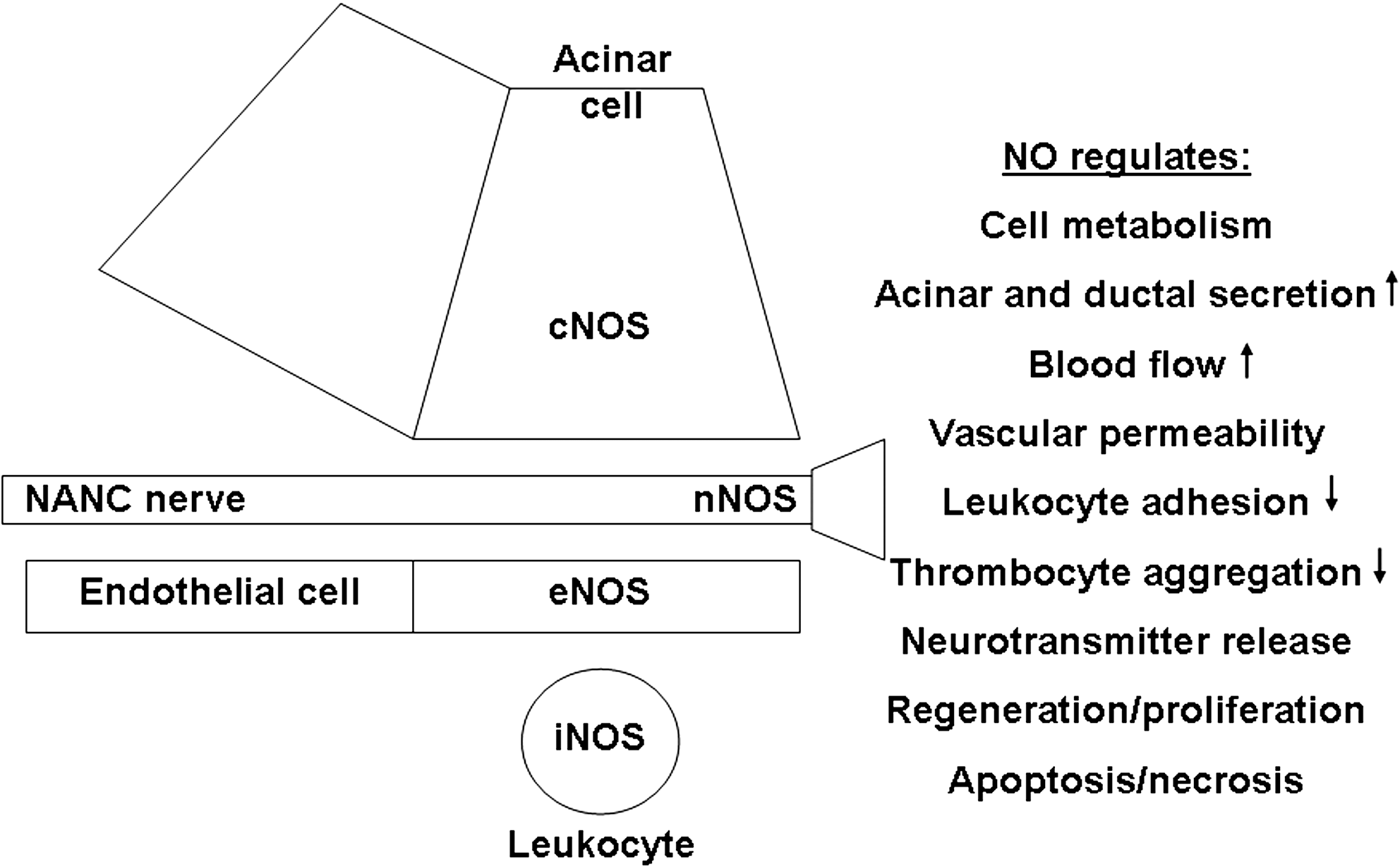
Pancreatic NO is synthesized by NOS or denitrosylation. The general agreement is that the main sources of pancreatic NO production by cNOS are neurons (87, 169) and vascular endothelial cells (182). Under basal conditions, iNOS protein expression is completely absent from the exocrine pancreas as determined by immunohistochemistry (5, 11, 12, 97, 101, 173) or Western blotting (42, 168). NOS expression in different cell types of the exocrine pancreas and the functions of NO are summarized in Figure 5.
Pancreatic NOS expression
In neurons, endothelial, and ductal cells
Physiologically, the rat and human pancreas contain eNOS in the vascular endothelium and nNOS in the intrapancreatic ganglia and neurons innervating acini, ducts, and blood vessels (179). NOS immunostaining in ganglion cells, nerve fibers, and endothelial cells colocalized with nicotinamide adenine dinucleotide hydrogen phosphate (NADPH) diaphorase staining, but not in islet and ductal epithelial cells (179). Since NADPH diaphorase histochemistry has often been used as a marker for NOS, the interpretation of these data needs caution. nNOS expression in the guinea pig pancreas was distributed in ganglia and nerves throughout the organ (98). The nitrergic nerves were most abundant along the blood vessels, especially in the head and body of the organ. Some of the nerves were associated with the main pancreatic duct, acini, and the islets of Langerhans. Interestingly, Umehara et al. (169) noted a species difference in distribution of pancreatic nNOS-positive nerve fibers. In the dog pancreas, nNOS-positive nerve fibers were abundant around ducts and moderate around the arteries and the acini but few in the islets. However, in the rat pancreas, nNOS-positive fibers were fewer around the pancreatic ducts and acini and more abundant in the islets. nNOS expression in dog islet cells was absent; weak immunoreactivity was seen in rat islet cells. NADPH-diaphorase staining in intrapancreatic ganglion cell bodies was higher in the rat versus dog.
The literature is relatively consistent regarding the lack of NOS expression in pancreatic ductal epithelia. Only Keklikoglu (85) has found iNOS expression in rat pancreatic duct cells. Wörl et al. (179) could not detect eNOS or nNOS protein by immunohistochemistry (using different fixation methods) in the pancreatic ducts of rats and humans. Similarly, iNOS messenger RNA (mRNA) and protein expression was absent in human pancreatic duct cells isolated from organ donors (124).
In acinar cells
Expression of cNOS in acinar cells has been demonstrated by a number of investigators (8, 42, 78, 109, 182). However, others could not confirm these results (5, 46, 149, 169). cNOS, but not iNOS, mRNA expression was detected in isolated rat pancreatic acini (78). Xu et al. demonstrated by both Western blot analysis and immunohistochemistry that rat pancreatic acinar cells do not express iNOS (182). nNOS was expressed at high levels in acinar cells next to the plasma membrane. Similarly, Nam et al. (109) found by immunohistochemical analysis that bovine pancreatic acinar cells show strong nNOS expression. Mouse pancreatic acinar cells were found to express all NOS isoforms (including iNOS) (8). In contrast, Dimagno et al. (41) found only weak eNOS and nNOS expression. According to the results of Western blot analysis of whole pancreas and acinar cell lysates, expression of both eNOS and nNOS is primarily extra-acinar. Acinar nNOS expression was actually attributed to contamination from other cell types. Overall, we believe that cNOS is expressed at low levels in acinar cells, which is further supported by functional assays (see below).
Several studies have confirmed detectable NOS activity using biochemical assays in isolated rat acinar cells (2, 63, 78, 107, 180, 182), although there are exceptions (186). Jaworek et al. (78) showed spontaneous NO release from acinar cells; however, it is unclear whether this was actually the result of NOS activity. Acinar cells are capable of converting L-arginine to L-citrulline with a parallel increase in cellular nitrite and cGMP levels (180). The changes in these parameters were significantly reduced by the NOS inhibitors NG-monomethyl-arginine (L-NMMA) and NG-nitro-L-arginine (L-NNA). Treatment of acinar cells with CCK-octapeptide (CCK-8) resulted in an increase in the L-arginine conversion to L-citrulline, the amount of nitrite/nitrate (NOx), and the level of cGMP (2). The Ca2+-mobilizing agonists carbachol and bombesin significantly increased cGMP levels via a NOS-dependent mechanism as indicated by the effects of maximal inhibitory concentrations of 7-nitroindazol, reduced hemoglobin (scavenger of NO), and 1H-[1,2,4] oxadiazolo[4,3-a] quinoxaline-l-one (inhibitor of the soluble guanylyl cyclase).
Generation of NO by denitrosylation
As mentioned before, generation of NO can occur independently of NOS activity. The fast initial increase in NO levels (as determined by NO-sensitive fluorescent dyes) induced by supramaximal acetylcholine stimulation in mouse pancreatic acinar cells had little sensitivity to inhibition of NOS; however, pretreatment with NO donors (increasing cellular S-nitrosothiol content) substantially enhanced this response (27). Tepikin's group went on to elegantly show that this Ca2+-dependent generation of NO in acinar cells was mediated through the denitrosylation of proteins (27). In fact, NO release was dependent on calpains, but inhibition of CaM and protein kinase C had no effect on NO responses.
Summary IV
• The synthesis of pancreatic NO is catalyzed by NOS or denitrosylation.
• The main cellular sources of pancreatic NO production are neurons and endothelial cells, but acinar cells can also produce NO.
The Physiological Role of NO in the Pancreas
cGMP/guanylyl cyclase signaling pathway
The major signal transduction pathway of NO is the production of cGMP by guanylyl cyclase. Both
Intracellular Ca2+ signaling and tyrosine phosphorylation
Ca2+ mediates numerous processes in pancreatic acinar cells. Most studies have shown a stimulatory effect of NO donors on Ca2+ oscillations and influx (13, 55, 63, 121, 181, 182), whereas others described minimal or no effects (59, 183, 186).
Xu et al. (181) pointed out that cGMP has a dual action on Ca2+ entry in pancreatic acinar cells: a 10-fold increase of cGMP concentration (elicited by secretagogues) activates Ca2+ entry, whereas a large increase of cGMP (up to 80-fold over basal, caused by high concentrations of SNP) inhibits Ca2+ entry.
Changes in intracellular Ca2+ concentration will also affect cNOS activity. NOS activity in guinea pig pancreatic acinar cells was stimulated by agents increasing cytosolic Ca2+ concentration (such as carbachol) and inhibited by intracellular Ca2+ chelators (64). In contrast, intracellular cGMP formation by guanylyl cyclase was inhibited by increase in cytosolic Ca2+ concentration.
García-Benito et al. (54) demonstrated that exogenous NO (1 mM SNP) administration increases tyrosine phosphorylation of the nonreceptor tyrosine kinase p125 focal adhesion kinase and the cytoskeleton-associated protein paxillin (which are important in control of mitogenic signal pathways, oncogenic transformation, cell adhesion, and migration) in rat pancreatic acini. Further, the same effect was observed after incubating acini with 8-Br-cGMP, indicating that soluble guanylyl cyclase activation and cGMP production play a role in these processes.
Pancreatic secretion
NO is thought to have a stimulatory effect on both basal acinar and ductal secretion. In the case of acinar secretion, we will discuss basal and stimulated secretion in separate sections.
Pancreatic acinar secretion
Basal secretion
Administration of the exogenous NO donors SNP (186), glyceryl trinitrate (GTN) (90), or
Further, inhibition of NOS in vivo by L-NNA significantly reduced basal protein secretion in the pancreatic juice of rats (91). This inhibitory effect was partially reversed when L-zarginine was coadministered with L-NNA. L-arginine given by itself did not affect basal pancreatic secretion. Long-term L-NNA administration (10 or 30 mg/kg for 4 weeks) in dogs produced dose-dependent elevations (1.3–10-fold above control) in serum activities of pancreatic enzymes with peak elevations occurring during the first week (89). In contrast, Trulsson et al. (163, 164) found significantly decreased plasma amylase activity in rats after administering L-NNA at much higher concentrations for 3–4 days (3×120 mg/kg i.p./day and 2×80 mg/kg i.p./day, respectively). In experiments performed on rats, total protein and amylase output showed a biphasic secretion pattern with an increase during intra-arterial infusion of L-NNA (0.48 mg/kg/h) (indicating an inhibitory role of NO on pancreatic secretion) followed by a decrease when the infusion ceased and further augmentation 1 h later (190). In animals with surgical pancreatic denervation, L-NNA caused a sustained decrease in pancreatic secretion (indicating that intra-pancreatic NO release is regulated by extrapancreatic nerves) followed by an increase 1 h later. Both nonspecific blockade of NOS by L-NAME (30 mg/kg i.v.) (107, 171) and blockade of nNOS by indazole (100 mg/kg i.p.) significantly reduced baseline amylase output in rats (171). Not surprisingly, aminoguanidine (40 mg/kg i.v.), an inhibitor of iNOS, had no detectable effect on basal amylase output. Taken together, these data suggest that acinar cNOS and nitrergic nerve fibers may play an important role in controlling basal pancreatic secretion.
Stimulated secretion
Pancreatic stimulants by themselves can induce NO generation in the pancreas (2, 171). For example, Molero et al. (107) have shown that physiological doses of cerulein induced a marked amylase secretion and a small release of NO (as determined by NO2/NO3 concentration) in rat. High doses of cerulein mildly increased amylase secretion and markedly increased NOx levels. L-NAME inhibited the effects of cerulein-induced NO formation and amylase secretion. The results of in vivo experiments were partly confirmed on isolated acini. Cerulein dose-dependently induced NO release (which was inhibited by L-NAME); however, the amylase dose–response curve was not modified by NOS inhibition. Similarly, carbachol was found to increase [3H]arginine conversion to [3H]citrulline, which indicates NO synthesis in guinea pig acini (63).
Overall, it seems that exogenous and endogenous NO have differential effects on stimulated pancreatic secretion. When exogenous NO was combined with carbachol, CCK-8, or cerulein, it had no effect on secretory responses from both pancreatic segments and acinar cells (46, 90, 107, 177, 183, 186). Similarly, combination of 8-Br-cGMP with acetylcholine had no significant effect on the amylase output compared with that of acetylcholine alone (46). However, nonselective pharmacological inhibition of NOS (the main source of endogenous NO synthesis) reduced stimulated pancreatic amylase/protein secretion by CCK analogs in rats (91, 107, 170), mice (42), cats (123), dogs (90), and humans (92) in vivo. L-NAME and L-NNA also significantly reduced nerve- and vasoactive intestinal polypeptide-stimulated exocrine secretion in pigs (71). Indazole treatment did not influence the effect of physiological concentrations of cerulein (170). However, the nNOS inhibitor partially reversed the suppression of secretory activity elicited by supramaximal doses of cerulein. Similar results were found when afferent nerve fibers were ablated by capsaicin. L-NNA dose-dependently inhibited the pancreatic secretion of protein stimulated by duodenal infusion of casein, exogenous CCK (0.06 μg/kg/h), and a meal (80). L-arginine significantly reversed the L-NNA-induced inhibition of pancreatic secretion in all experiments. The effect of endogenous NO on pancreatic secretion was also highlighted by Kawabata et al. (84), who demonstrated that NO participates in the in vivo protease-activated receptor 2 (PAR2)-mediated amylase release in the mouse. Pretreatment with capsaicin did not modify the PAR2-mediated secretion of amylase.
In in vivo studies, the administration of NOS inhibitors may have indirect effects on pancreatic secretion. L-NNA in fed dogs caused an initial increase in intestinal motility and about 60% inhibition of pancreatic secretion. The infusion of L-arginine in addition to L-NNA reversed, in part, both intestinal motility and pancreatic secretory effects (104). L-NNA administration (2.5 mg/kg/h) significantly decreased postprandial amylase output in conscious dogs (160). At least part of this effect is due to decreased gastric emptying or gastrointestinal blood flow, or contraction of the sphincter of Oddi (160). The effects of inhibiting NO synthesis on pancreatic secretion in vivo are thought to be associated with changes in pancreatic blood flow or neurotransmitter release in response to NO (90, 143).
Most studies reported no effect of NOS inhibition by L-NMMA or L-NNA on carbachol- or CCK-8-stimulated secretion in rats (87, 90, 91, 107, 186), mice (42), and dogs (90); however, in vitro NOS inhibition significantly reduced carbachol-stimulated amylase secretion as well as elevation of acinar cell cGMP concentration in pancreatic acini (180). Electrical field stimulation (to activate intrinsic secretomotor nerves in the isolated pancreas) and acetylcholine caused large increases in amylase output from isolated rat pancreatic segments (183), whereas both SNP (1 mM) and 8-Br-cGMP (0.1 mM) inhibited amylase secretion. Electrical field stimulation combined with either SNP or 8-Br-cGMP resulted in a marked decrease in amylase output compared to electrical field stimulation alone. However, when extracellular Ca2+ concentration was increased from 2.56 to 5.12 mM, SNP failed to inhibit the response to electrical field stimulation. In contrast, neither SNP nor 8-Br-cGMP had any significant effect on the amylase response to acetylcholine.
Nonselective NOS inhibition and eNOS gene deletion reduced CCK-8- and carbachol-stimulated in vivo pancreatic secretion in mice (41). This was likely to be due to modulation of nonacinar cell events as in vitro CCK-8-stimulated secretion of amylase from isolated acini was unaltered by NOS blockade and eNOS deletion. In contrast, nNOS gene deletion augmented CCK-8- but not carbachol-stimulated pancreatic secretion in vivo. Overall, the findings of DiMagno et al. (41) suggest that eNOS plays a dominant role and that nNOS plays a minor role in pancreatic secretion. It was speculated that eNOS acts on pancreatic microvasculature, whereas nNOS tonically inhibits acetylcholine release from pancreatic neurons.
Pancreatic ductal secretion
Experimental data suggest that NO affects not only the secretory function of acinar but also ductal cells. Patel et al. (123) have shown that intra-arterial infusion of SNP resulted in increased pancreatic secretion of fluid, bicarbonate, and protein without any change in pancreatic microvascular blood flow of anesthetized cats. Inhibition of NOS by L-NMMA did not affect basal secretion; however, it significantly reduced both secretin and CCK-stimulated pancreatic secretion. The study also suggests that NO-mediated stimulation of pancreatic secretion after secretin administration was not due to changes in pancreatic microvascular blood flow, whereas CCK-induced pancreatic secretion of protein was. Jyotheeswaran et al. (80) have shown that endogenous NO production may have a role in stimulation of pancreatic ductal secretion in rats. Intravenous infusion of L-NNA (5 mg/kg/h) significantly inhibited the pancreatic secretion of fluid and bicarbonate stimulated by either endogenous or exogenous secretin, and the inhibition was reversed by L-arginine. The administration of L-NNA did not influence the plasma concentrations of vasoactive intestinal polypeptide, secretin, or CCK. In contrast to the findings of Patel et al. (123) and Jyotheeswaran et al. (80), Konturek et al. (92) have shown that L-NMMA administration dose dependently reduced the secretin-cerulein-stimulated pancreatic enzyme secretion in humans without any alterations in the volume flow and bicarbonate output.
Proliferation and apoptosis
Endogenous NO may regulate the delicate balance between proliferative and apoptotic processes of the pancreas. Inhibition of NO synthesis by L-NNA reduced the urinary excretion of NOx and increased serum L-arginine concentration of rats (163). Further, L-NNA administration for 3 days (3×120 mg/kg i.p. daily) or 4 days (2×80 mg/kg i.p. daily) caused pancreatic hypotrophy (pancreatic weight, DNA, and protein contents were decreased) and increased the rate of pancreatic apoptosis during both basal and CCK-8 stimulated conditions (163, 164). L-NNA significantly reduced proliferation of both acinar and ductal cells under basal conditions (163). Interestingly, NOS blockade had an opposite effect on CCK-induced proliferation of acinar cells and ductal cells, which were stimulated and inhibited, respectively. Overall, it seems that NO has a tonic inhibitory effect on both mitogenesis and apoptosis of acinar cells.
Modulation of ion channel activity
In situ hybridization experiments showed high levels of TALK-1 and TALK-2 K+ channel expression in the acinar cells of human pancreatic tissue (45). These K+ channels may be involved in the control of pancreatic secretion. Interestingly, the activity of TALK-2 was markedly upregulated by the administration of SNP/dithiothreitol in Xenopus oocytes. This effect was not mediated by cGMP activation, as a membrane-permeable cGMP analog did not produce an activation of TALK-2.
Summary V
• NO donors increase cGMP and Ca2+ concentrations and basal secretion of pancreatic acinar cells.
• NO donors also increase the secretion of pancreatic ductal cells.
• Endogenous NO increases proliferation and decreases apoptosis of pancreatic acinar cells.
The Effect of Acute Pancreatitis on NOS Expression and Activity
There is mounting evidence that the NO signaling system is significantly altered in acute pancreatitis. Serum/plasma NOx levels are significantly increased indicating elevation of NOS activity. In parallel, iNOS activity is significantly increased, whereas eNOS activity is decreased during acute pancreatitis. The inhibition of eNOS activity may have detrimental effects on the circulation of the pancreas. Pancreatic RNS generation is evident as detected by markedly increased levels of 3-nitrotyrosine. In this section we will review changes in pancreatic and extrapancreatic NOS expression and activity.
Pancreatic NOS
Serum or plasma NOx levels were significantly increased in response to injections of supramaximal doses of cerulein (5, 8, 15, 57, 65, 101, 130), pancreatitis induced with ethanol and CCK administration (38), pancreatic duct obstruction combined with secretagogue stimulation (83), and in taurocholate-induced (101, 136, 144) pancreatitis. Supramaximal doses of cerulein significantly increased pancreatic contents of NOx in rats (9, 24) and mice (8). However, it must be noted that serum NOx levels may not necessarily reflect that of the pancreatic tissue. Injection of ethyl alcohol (48%, 1 ml) into the common bile duct of rats significantly increased NOx levels in the pancreas and lungs, but did not influence serum levels (6). On the other hand, Sugiyama et al. (155) found significantly reduced pancreatic NOx levels in rats repeatedly injected with cerulein, which were not influenced by L-NAME or aminoguanidine treatments. Correspondingly, cerulein-induced pancreatitis significantly reduced the release of NO from isolated rat pancreatic acini to about half the control value (78). It is likely that increased serum NOx levels originate from nonacinar cell types such as endothelia, neurons, and/or leukocytes.
Baseline pancreatic eNOS (42, 156) and nNOS (42) expression was not affected by the injections of cerulein in rats (156) and mice (42). Pancreatic eNOS dimer (which represents the catalytically active enzyme) immunoreactivity was markedly reduced following the injections of cerulein (156). The reduction in eNOS dimer amount was partially due to reduced endogenous BH4 levels. Ang et al. reported significantly increased nNOS and decreased eNOS protein expression in mouse pancreatic acinar cells treated with supramaximal concentrations of cerulein in vitro (8). eNOS Thr495 dephosphorylation was reported by DiMagno et al. (42) in the initiation phase of cerulein-induced acute pancreatitis in mice, which has been shown to substantially increase eNOS activity (47).
iNOS expression is upregulated by various inflammatory signals. Therefore, it is not surprising that pancreatic iNOS expression is significantly increased in various in vivo models of acute pancreatitis. Supramaximal doses of cerulein induced iNOS expression in mice (8, 167, 173) and rats (5, 65, 168). iNOS expression during cerulein-induced pancreatitis in rats was localized to the endothelium and smooth muscle cells (5, 101). Ueno et al. (167) have shown that interleukin (IL)-18 seems to play an important role in inducing pancreatic iNOS expression and releasing NO into the systemic circulation during cerulein-induced acute pancreatitis in mice. Also, pancreatic iNOS protein (116) and mRNA (78) expression were greatly increased after injection of rodents with the combination of cerulein and LPS. Similarly, a marked upregulation of iNOS protein (101, 103, 136) and mRNA (74, 136, 170) was detected in rats with taurocholate pancreatitis. Folch-Puy et al. (49) found increases in iNOS levels after infusion of contrast media (to mimic post-endoscopic retrograde cholangiopancreatography [ERCP] pancreatitis) into the pancreatic duct of rats.
In accordance with the above-mentioned results, the activities of pancreatic cNOS and iNOS changed oppositely in secretagogue- (5, 157) or L-arginine-induced (158) acute pancreatitis in rats. cNOS activity significantly decreased, whereas iNOS activity increased. Pancreatic iNOS activity was also significantly increased 6–24 h after the induction of taurocholate-pancreatitis combined with pancreatic ischemia (96). Similarly, taurodeoxycholate-induced acute pancreatitis resulted in marked iNOS expression in the beta cells of pancreatic islets, whereas cNOS expression and activity was decreased (128).
Increased NO synthesis induces the generation of RNS in the pancreas. Correspondingly, pancreatic levels of nitrotyrosine, a marker of nitrosative stress, were significantly increased in mice with cerulein- (15, 33 –35, 57) and in rats with L-arginine- (36) and taurocholate-induced (136) pancreatitis. Al-Mufti et al. (5) localized 3-nitrotyrosine staining in cerulein-induced pancreatitis to the perivascular tissue. Acinar nitrotyrosine content was significantly enhanced in common biliopancreatic duct ligation pancreatitis (22). In taurocholate-pancreatitis nitrotyrosine was localized to cells of the vascular endothelium, especially at the sites of intense inflammatory response, and was noted in the perivascular area and in the stroma (21). Nitrotyrosine staining was also observed focally in the cytoplasm of necrotic adipocytes.
Extrapancreatic NOS
Acute pancreatitis (especially its severe form) can affect other organs (like the lungs and liver) besides the pancreas. Therefore, a number of investigators have looked at changes of NOS expression in response to pancreatitis.
In the lungs, there was increased cNOS protein expression, whereas iNOS was significantly overexpressed at a later time point during cerulein-induced pancreatitis (8). Pulmonary iNOS mRNA expression was also significantly increased soon after the induction of taurocholate-induced acute pancreatitis (82). Alveolar macrophages isolated from rats with acute necrotizing (taurocholate-induced) pancreatitis showed increased levels of NO generation (29, 137). Further, Closa et al. (29) showed that the liver plays an active role in the activation of alveolar macrophages in this experimental model. Alveolar macrophages isolated from rats with selective pancreatic duct ligation expressed iNOS mRNA 6 h after the induction of pancreatitis and generated large amounts of NO and superoxide and demonstrated strong cytotoxicity against human umbilical vein endothelial cells (166). This cytotoxicity was reduced by the administration of L-NMMA (50 mg/kg s.c). NG-monomethyl-L-arginine administered to rats with pancreatitis apparently reduced lung edema and improved levels of partial pressure of oxygen in arterial blood. Taken together, these results suggest that activated alveolar macrophages may contribute to pancreatitis-induced lung injury. NO production and iNOS mRNA expression were significantly elevated in alveolar macrophages along with significant increases in lung histological abnormalities, myeloperoxidase (MPO) activity, tumor necrosis factor-α expression, and bronchoalveolar lavage proteins in taurocholate-induced pancreatitis (25). These parameters were further enhanced by pretreatment with L-arginine and attenuated by pretreatment with L-NAME.
The activation of NF-κB and iNOS mRNA expression were detected in peritoneal macrophages isolated from both rats with cerulein- and taurocholate-induced pancreatitis (144). Interestingly, iNOS protein expression was only observed in the peritoneal macrophages after the induction of taurocholate pancreatitis, but not cerulein-induced pancreatitis (144). Further, the supernatant of tauroholate pancreatitis ascites could induce iNOS in the peritoneal macrophages of normal rats in vitro, but the peritoneal lavage fluid of cerulein pancreatitis rats could not (144). Similarly, NO concentration was significantly increased in both the serum and culture medium of peritoneal macrophages of rats with taurocholate-induced pancreatitis, along with the upregulation of the expression of NF-κB and iNOS in peritoneal macrophages (102).
Tanjoh et al. (161) investigated the iNOS and cNOS mRNA expression of cultured monocytes isolated from patients with mild or severe acute pancreatitis. iNOS mRNA expression was detected in eight of nine patients with severe acute pancreatitis. However, no iNOS expression was found in patients with mild acute pancreatitis. cNOS mRNA was not found in either of the groups. There were no detectable levels of iNOS mRNA expression in the peripheral blood mononuclear cells of 18 patients with late-stage alcoholic chronic pancreatitis (66).
Besides the lungs and macrophages, NOS expression was also investigated in the liver and adipose tissue of rodents with severe acute necrotizing pancreatitis. Hepatic iNOS mRNA expression was significantly increased in mice suffering from cerulein/LPS-induced acute pancreatitis (184). In contrast, splenic and hepatic iNOS mRNA expression was unchanged (as determined with semiquantitative PCR) after the induction of acute pancreatitis with cerulein in mice (167). iNOS mRNA expression was significantly upregulated in necrotic versus non-necrotic peritoneal white adipose tissue of rats with taurocholate-induced pancreatitis (51).
Summary VI
• Pancreatic and extrapancreatic iNOS expression and activity are increased, whereas pancreatic eNOS activity is decreased during acute pancreatitis.
The Effect of NO Donors and NOS Inhibitors on Acute Pancreatitis
There is overwhelming evidence that NO generation exerts a beneficial effect in acute pancreatitis, although some researchers have found a detrimental or no effect. Variations in the effects of NO/NOS on acute pancreatitis may be attributed to differences in species, administered drugs (e.g., NOS inhibitors have unequal enzyme selectivity), and their dosing and timing (pre- and post-treatment), and acute pancreatitis models (mild edematous vs. severe necrotizing).
Protective effect of NO generation
Most studies have described a beneficial effect of NO donors and a detrimental effect of NOS inhibition on the severity of acute pancreatitis. L-arginine (125 or 250 mg/kg) dose dependently ameliorated the severity of hemorrhagic pancreatitis and improved the pancreatic blood flow (99). L-arginine and SNP significantly reduced severity of cerulein- and glycodeoxycholic acid-induced pancreatitis in rats (176, 177). The NO donors reduced pancreatic edema formation, trypsinogen activating peptide levels, and histological damage (176). L-NAME administration increased the inflammatory response in pancreatitis, while decreasing pancreatic tissue oxygenation. Intracellular trypsinogen activation in pancreatic acini stimulated with supramaximal concentrations of cerulein was not influenced by either NO donors or inhibitors (177), which suggests that extra-acinar factors are mediating the effect of NO on cerulein-induced pancreatitis. L-NNA administration exacerbated cerulein-induced pancreatitis (42, 43, 86, 99) and caused a decrease in pancreatic blood flow (99). L-NNA treatment augmented the effect of cerulein by triggering a greater increase in intrapancreatic trypsin and serum lipase activities compared with controls (42). In rat cerulein-induced acute pancreatitis, L-NAME increased amylasemia and pancreatic MPO activities, whereas NO donors (SNP and GTN) reduced amylasemia, lipasemia, and pancreatic histological damage (106). L-NNA significantly potentiated the inflammatory changes in the pancreas caused by cerulein (7, 75, 91). Addition of L-arginine enhanced the pancreatic blood flow and ameliorated the pancreatitis induced by cerulein alone or in combination with the nonselective NOS inhibitor L-NNA. Further, L-NNA treatment enhanced the ultrastructural degenerative alterations of cerulein-induced pancreatitis (mitochondrial damage, dilation of cisternae of Golgi apparatus, focal degranulation of rough endoplasmic reticulum, reduced number of zymogen granules and condensing vacuoles, and autophagosome formation in acinar cells) (7). Coadministration of L-arginine reversed the deleterious effect of L-NNA to some extent.
The administration of the NOS cofactor BH4 (30 mg/kg i.p.) ameliorated the severity of cerulein-induced pancreatitis, most likely by restoring the dimeric form of eNOS (156). Ueno et al. (167) have reported that the protective effect of iNOS induction in cerulein-pancreatitis may be mediated by IL-18. Pancreatitis severity was significantly higher in IL-18 knock-out versus wild-type mice (167). Laboratory and morphological signs of pancreatitis in both types of mice were improved by dose-dependent pretreatment with recombinant IL-18. Overall, IL-18 appears to protect the pancreas by inducing iNOS, as its protective effect was abolished by aminoguanidine. The influence of RNS on ROS during cerulein-induced acute pancreatitis was investigated by Sánchez-Bernal et al. (139). They found that endogenous NO synthesis appears to protect pancreatic subcellular fractions against oxidative stress. The complexity of the NO/NOS system is highlighted by the fact that laboratory and histological parameters of cerulein-induced acute pancreatitis in rats were decreased by pretreatment with either L-arginine or L-NAME (118).
Pretreatment of rats with low doses (1 mg/kg i.p.) of LPS protected the pancreas against cerulein-induced (5 μg/kg/h for 5 h s.c.) damage (78). This effect was attributed, at least in part, to the activation of L-arginine-NO system, since pretreatment with L-NNA (20 mg/kg i.p., which by itself aggravated cerulein-induced pancreatitis) partly reversed the LPS-induced protection. However, these results are in disagreement with Kikuchi et al. (86), who have shown that LPS (2 mg/kg) aggravates pancreatic inflammation in the course of cerulein (4×20 μg/kg i.p.) pancreatitis in mice. Pretreatment with L-NNA (10 mg/kg i.p.) significantly reduced the serum amylase activity of LPS + cerulein-injected mice (however, the histology of the pancreas was unaffected). The effects of L-NNA were reversed by the administration of L-arginine (5×200 mg/kg i.p.), but were not affected by D-arginine. The discrepancy between the results of Jaworek et al. (78) and Kikuchi et al. (86) could be due to differences in species and dosing of drugs.
Capsaicin-induced ablation of afferent neurons significantly increased pancreatic damage in cerulein-induced pancreatitis (40). Inhibition of NO synthesis enhanced pancreatic damage, and this was reversed by administration of L-arginine. Both GTN and L-arginine treatment resulted in attenuation of biochemical parameters in cerulein-induced pancreatitis, but not pancreatic morphology was unaltered (79). Treatment of rats with L-arginine resulted in augmented cell proliferation after induction of acute pancreatitis followed by more rapid recovery in comparison to untreated or the L-arginine and L-NNA-injected group.
Inhibition of endogenous NO production was detrimental, whereas exogenous NO donors were beneficial in several studies investigating necrotizing pancreatitis models. The administration of L-NNA significantly exacerbated the severity of tauorocholate-induced pancreatitis (43). The NOS inhibitors L-NAME and aminoethyl-isothiourea increased the mortality of rats suffering from acute necrotizing pancreatitis from about 50% to 80%. However, there was no effect on volume of ascites and degree of pancreatic damage (3). L-NAME significantly aggravated closed duodenal loop pancreatitis in rats, whereas the iNOS inhibitor aminoguanidine did not change the severity of the pancreatitis (111). Administration of S-nitroso-N-acetylpenicillamine (which provides a slow, sustained release of NO) from 15 min before the induction of taurocholate-pancreatitis significantly ameliorated disease severity (70). Cosen-Binker et al. (31) investigated the effects of NO-donating nonsteroidal anti-inflammatory drugs (NO-NSAIDs) on biliopancreatic duct outlet exclusion-closed duodenal loop pancreatitis model in rats. The results showed that NO-NSAIDs have a protective role when administered before or during the first hour after the induction of pancreatitis. The compounds were even more effective when the 1-h preadministration was combined with a 4-h postadministration. The most effective drug in reducing pancreatitis severity was NO-flurbiprofen. The combination of the NO donor diethylenetriamine-NO and a low-dose glucocorticoid injected 1 h before triggering biliopancreatic duct outlet exclusion-closed duodenal loop acute pancreatitis ameliorated morphological damage (30).
NO is involved in the maintenance of pancreatic vascular perfusion. Satoh et al. (143) and Konturek et al. (91) were one of the first to provide evidence that endogenous NO synthesis may maintain pancreatic perfusion during the administration of large doses of CCK/cerulein. Satoh et al. (143) found that although administration of L-NNA (0.5–30 mg/kg) did not affect the basal pancreatic blood flow, 5 mg/kg L-NNA completely inhibited the cerulein-induced increase in pancreatic perfusion. These results are supported by the observations of Dimagno et al. (42), who found that eNOS gene deletion had no effect on basal pancreatic blood flow, but nearly abolished the increase in blood flow 30 min after cerulein treatment. In contrast, Konturek et al. (91) reported that both L-NNA and cerulein reduce pancreatic blood flow. Dobosz et al. (44) tested the effects of L-arginine (2×100 mg/kg) and L-NNA (2×25 mg/kg) on organ microcirculation (pancreas, liver, kidney, colon, and skeletal muscle) by laser Doppler flowmetry in experimental acute pancreatitis induced by intraperitoneal injections of cerulein (4×15 μg/kg). Acute pancreatitis resulted in a significant decrease of microperfusion in all examined organs. L-arginine administration improved the microcirculation and lowered hematocrit levels. L-NNA treatment caused aggravation of edematous to necrotizing acute pancreatitis. Central (intracisternal) injection of a stable thyrotropin-releasing hormone analog has been shown to increase pancreatic blood flow through vagal and NO-dependent pathways in rats (61). Further, central, but not peripheral (i.v.), administration of the hormone protected against cerulein-induced acute pancreatitis (185).
The results of Lomis et al. (100) suggest that the progressive and severe hypotension associated with a rat model of pancreatitis (intraductal infusion of very low concentrations of glycodeoxycholic acid with intravenous cerulein) may actually be mediated by NOS activity, since administration of aminoguanidine or L-NMMA prevented this effect on blood pressure. Rats with taurocholate-induced pancreatitis develop hypotension around 3 h after the induction of the disease (53). Administration of L-NAME (25 mg/kg) caused a similar increase in mean arterial pressure in control rats and rats with severe necrotizing pancreatitis (53). This indicates the importance of the vasodilatory effect of NO in both conditions. iNOS inhibition by aminoguanidine (40 mg/kg) did influence the mean arterial blood pressure in normal and pancreatitic animals (53, 70). The latter findings indicate that cNOS (but not iNOS) is likely to be involved in the development of the early hypotension of animals with pancreatitis. PAR2 can be activated by trypsin released during acute pancreatitis, which can lead to hypotension. Activation of PAR2 in human umbilical vein endothelial cells caused stimulation of NO (110). PAR2 activation may play a role in pancreatitis-induced hypotension as infusion of trypsin (at concentrations found in rat serum after the induction of acute pancreatitis) or PAR2 activating peptide caused an immediate decrease in mean arterial pressure of rats.
NO may not only affect pancreatic blood flow, but possibly neutrophil recruitment. Administration of L-NNA to rats treated with cerulein significantly enhanced pancreatic inflammatory cell infiltration (by decreasing NO synthesis, leukocyte adhesion will increase) and also increased the extent of acinar cell injury (7, 75). Also, SNP administration inhibited and L-NAME treatment exacerbated cerulein pancreatitis-induced lung injury (115). Taken together these results indicate that both endogenous and exogenous NO may be beneficial in edematous acute pancreatitis.
Small doses of L-arginine (up to several hundred mg/kg) have usually been shown to be protective against acute pancreatitis. However, large i.p. doses (2.5–5 g/kg) of L-arginine can induce severe acute necrotizing pancreatitis in rats and mice (39, 69). For many years researchers have thought that excessive NO formation may play an essential (detrimental) role in the pathogenesis of L-arginine-induced pancreatitis. Certainly, this was a tempting speculation as L-arginine is the direct precursor of NO. However, we have recently found, by measuring serum concentrations of L-ornithine and L-citrulline, that metabolism of a large L-arginine dose (3.5 g/kg i.p.) in rats is mainly catalyzed by arginase rather than cNOS activity (134). Similarly, after a load of L-arginine (2×2.5 g/kg i.p.), Trulsson et al. (165) observed increased serum levels of L-arginine and L-citrulline at 8 h after injection, but these fell below control levels after 24 h as well as amino acids in the glutamate family (ornithine, proline, histidine, and glutamine). Inhibition of arginase activity significantly decreased the severity of L-arginine-induced pancreatitis (20). Further, i.p. injection of L-ornithine, compared to L-arginine, results in a more severe pancreatitis (134). The reason for the latter finding is unknown. However, L-arginine in itself may be less toxic than L-ornithine and/or NO formation from L-arginine via cNOS may have a protective effect against basic amino acid-induced pancreatic damage.
Detrimental effect of NO generation
There are numerous reports arguing against the beneficial effect of NO in the pathogenesis of acute pancreatitis. L-arginine treatment has been shown to improve pancreatic perfusion; however, it potentiated morphological alterations induced by cerulein-pancreatitis (43). The nonspecific inhibition of NOS activity by oral administration of L-NAME (10 and 30 mg/kg) dose-dependently prevented the increase in serum amylase activity, pancreatic weight, and MPO activity in cerulein-induced acute pancreatitis in rats (156). The effect of L-NAME (30 mg/kg) pretreatment, as compared with dexamethasone, was more potent against mild pancreatitis and was less potent against severe pancreatitis (155). Coadministration of L-arginine (500 mg/kg s.c.) with L-NAME antagonized the effects of the NOS inhibitor. Similarly, L-NNA significantly lowered the edema index, the wet/dry weight ratio of the pancreas, and Evans blue extravasation in rats with cerulein-induced acute pancreatitis (1). Since aminoguanidine (30 mg/kg per os), a relatively selective iNOS inhibitor, had no effect on pancreatitis severity (156), these data indicate that inhibition of cNOS isoforms is responsible for the observed effects. In contrast, administration of aminoguanidine (2×100 mg/kg i.p.) tended to decrease the serum amylase and lipase activities and acinar vacuolization in cerulein-induced pancreatitis, suggesting a detrimental effect of NO produced by iNOS in mice (167).
Pancreatic NO level was found to be significantly elevated (2.5-fold) 1–3 h after the induction of cerulein-induced pancreatitis (24). The velocity of pancreatic microcirculation was significantly reduced by about 30%. Both microcirculatory changes and NO elevation were significantly alleviated in cerulein-induced rats pretreated with the NOS inhibitor L-NNA. Moreover, pancreatic NO level correlated well with the number of adherent leukocytes in the pancreas, suggesting that during the initial phases of acute pancreatitis, NO could play an adverse role.
Inhibition of iNOS by S-methylisothiourea significantly reduced the serum amylase activities, and bacterial translocation to mesenteric lymph nodes in taurocholate-induced pancreatitis (117, 150). Also, the administration of selective iNOS inhibitors AR-C102222AA and L-N6-(1-iminoethyl)-lysine had beneficial effects in experimental acute pancreatitis in Australian possums (141). Pancreatic and pulmonary damage was markedly attenuated in L-arginine-induced acute pancreatitis by the selective iNOS inhibitor aminoguanidine (although inhibition of NO synthesis was not confirmed in this study) (148). Aminoguanidine pretreatment also significantly decreased endothelial permeability, neutrophil sequestration, and pro-inflammatory cytokine concentrations.
Administration of SNP in rats with taurocholate-induced acute pancreatitis significantly increased parameters of oxidative stress, and intensity of the oxidative stress was significantly reduced in rats treated with L-NAME (37). In contrast, Closa et al. (28) found that inhibition of endogenous NO synthesis by L-NAME did not influence oxygen free radical damage in taurocholate-induced acute pancreatitis.
No effect of NO generation
One of the earliest studies on NO reported that neither administration nor inhibition of NO (by GTN and L-NAME, respectively) had any significant effect on the severity of pancreatitis induced by cerulein (175). Similarly, Kikuchi et al. (86) showed that pancreatic edema and histology were not affected by L-NNA pretreatment in mice with cerulein-pancreatitis. The results of Jurkowska et al. (79) also suggested that the inhibition of NO synthesis by L-NNA during cerulein-induced pancreatitis had no effect on pancreatic injury and recovery in rats. Inhibition of nitrergic transmission by indazole did not influence hyperamylasemia and pancreatic water content, but paradoxically reduced MPO activity in cerulein-induced pancreatitis (170). Unfortunately, pancreatic histology was not assessed in this study, so it can not be conclusively decided whether the severity of pancreatitis was altered.
Lessons learned from NOS KO mice
Besides pharmacological inhibitors of NOS (which often are not completely selective for NOS or a specific NOS isoform), transgenic mice have also been used to determine the role of NO in pancreatitis. Deletion of iNOS was found to be beneficial (33), inconsequential (42) or deleterious (130) in various studies using the cerulein-induced pancreatitis model. Mortality, the degree of pancreatic histological damage, upregulation/expression of P-selectin and inter-cellular adhesion molecule-1, the staining for nitrotyrosine and poly (ADP-ribose) synthetase, trypsinogen activation, and lipid peroxidation were markedly reduced in cerulein-treated (5×50 μg/kg i.p.) iNOS-deficient versus wild-type mice (33). Further, a significant amelioration of IκB-α degradation (and consequently NF-κB activation) was observed in acinar cells collected from iNOS-deficient mice subjected to cerulein treatment. Taken together, the findings of Cuzzocrea et al. support a pro-inflammatory role of iNOS in the acute pancreatitis caused by cerulein in mice (33). Qui et al. (130) could not confirm these results. Pancreatic edema, serum amylase, and pancreatic MPO activities were significantly higher in iNOS knock-out versus wild-type mice injected with 7×60 μg/kg (i.p.) cerulein. The administration of the NO donor isosorbide dinitrate (4×10 mg/kg by oral gavage) during cerulein-pancreatitis in iNOS knock-out animals significantly reduced the latter parameters to levels observed in cerulein-treated wild-type mice. Although pancreatic morphologic damage was not assessed by Qui et al. (130), their results suggest a protective role of iNOS during pancreatitis. Further adding to this puzzling picture, DiMagno et al. (42) have found that only eNOS deletion, and not nNOS or iNOS deletion, affected the initiation of cerulein-induced acute pancreatitis. Compared to wild-type mice, eNOS knock-out mice exhibited greater increase in intrapancreatic trypsin and serum lipase activities 30 min after the injection of cerulein (50 μg/kg i.p.). These results suggest that eNOS-derived NO may be beneficial in the initial phases of the disease. We do not have a clear explanation to the marked differences between the above-mentioned studies.
Coxsackie virus B4 infection causes severe pancreatitis in mice. The virus replicates in acinar cells causing their destruction and inflammatory infiltration. Coxsackie virus-infected young (3 weeks of age) mice lacking iNOS develop much more severe acute pancreatitis and die more rapidly compared to wild-type mice (189). It is not proven whether the protective effect of iNOS expression is due to a systemic (e.g., immune) or local (pancreatic) response. However, a systemic effect is more likely, considering that iNOS is involved in the elimination of pathogens. Also, it must be noted that using older mice (8–12 weeks of age), Flodström et al. (48) showed that Coxsackie virus caused chronic pancreatitis with extensive damage to the exocrine pancreas in both the iNOS and the wild-type strains.
Is NO important in human acute pancreatitis?
NO may play a role in the pathogenesis of human acute pancreatitis, although evidence is not strong. Severe acute nectrotizing pancreatitis in humans was associated with elevated serum NOx levels in the early stage of the disease (106), which may reflect increased NOS activity. Patients with higher serum NOx levels were at a significantly higher risk of sepsis and mortality. Interestingly, NOx levels were not affected by the occurrence of local complications or distant-organ failure. According to Que et al. (129) there is a correlation between plasma NO levels and the severity of acute pancreatitis (as determined by APACHE-II scores). In contrast with these findings, total urine nitrite excretion over a 24-h period (which has been shown to reflect NO synthesis) correlated strongly with both intestinal permeability and markers of systemic endotoxin exposure but not with serum C-reactive protein concentrations or APACHE-II scores in acute pancreatitis (133).
Sandstrom et al. (140) have found that patients with acute pancreatitis had lower serum L-arginine and L-citrulline concentrations than controls. Patients with gallstone and idiopathic pancreatitis also had reduced urinary concentrations of nitrite and nitrate (indicating a defect in NO formation), but this was not seen in patients with alcoholic pancreatitis. In a self-control study, serum phenylalanine and glutamate were increased, whereas arginine, citrulline, ornithine, and glutamine were decreased compared with levels after recovery (142). Urine NOx concentration was significantly increased on day 1 compared to day 5 after admission.
Most animal studies have found a beneficial effect of NO donors on acute pancreatitis. Whether the results of these animal experiments can be extrapolated to humans is hard to tell. Also, it must be noted that in many studies investigating the effects of exogenous NO on animal models of acute pancreatitis, drugs are administered prophylactically. Usually, this experimental design does not mimic the clinical situation. However, one exception in which prevention of acute pancreatitis could be achieved is post-ERCP pancreatitis. Numerous studies have investigated the effect of prophylactic GTN administration on post-ERCP acute pancreatitis. However, the results obtained from these are controversial. Some randomized double-blind controlled trials have found a protective (67, 108), whereas others found no effect (17, 81, 112, 154) of GTN. Similarly, in meta-analyses of randomized double-blind controlled trials, the usefulness of GTN was not clearly proven (14, 16, 147). The optimal dosage, route, and timing of GTN administration need further investigation.
Summary VII
• Most studies have shown a beneficial effect of endogenous and exogenous NO in experimental acute pancreatitis.
The Role of NO Generation in Ischemia/Reperfusion and Transplantation
As cNOS activity is essential in regulating vascular tone and integrity, it is not surprising that NO may be especially important in the pathomechanism of I/R injury. I/R injury can be a major clinical problem during shock, pancreatic surgery, and transplantation (all of which affect the blood supply of the pancreas).
I/R injury of the rat pancreas resulted in significantly increased blood levels of NOx (23, 97, 127, 159, 172). Increased generation of NO was further demonstrated by nitrosylhemoglobin detection by electron spin resonance in the blood after reperfusion (159). Total NOS activity in the pancreas and lung was significantly elevated in a rat model of I/R-induced acute pancreatitis (172), which was mainly due to activation of the inducible isoform of the enzyme. iNOS and nitrotyrosine expression was confirmed by immunohistochemistry in both the pancreas and lung. Four hours after reperfusion following pancreaticoduodenal transplantation, iNOS activity in pancreatic tissue was significantly increased, but cNOS activity was not altered (97). Pancreatic iNOS staining was mainly localized to the vascular endothelium and smooth muscle, and islet cells.
Interestingly, in most of the studies, both NO supplementation (by L-arginine or SNP administration) and NOS inhibition appears to reduce pancreatic I/R injury (18, 19, 73, 97, 113, 114, 127, 187, 188). In a rat model of pancreatic transplantation, Vollmar et al. (174) have shown that the i.v. injection L-arginine (50 mg/kg immediately before and 100 mg/kg during the first 30 min of reperfusion) improves microvascular perfusion and attenuates pancreatic edema formation and neutrophil infiltration. A protective effect of SNP and L-arginine was shown on pancreatic morphology, tissue oxygenation, blood flow, and lipase release in pigs with I/R injury (18, 19). A mild protective effect was also seen in some parameters with L-NAME administration (18). The administration of L-arginine and/or SNP is protective in I/R injury of the rat pancreas as indicated by an improvement in histological damage, postischemic tissue oxygenation, functional-capillary density, and extent of leukocyte adherence (113, 114). I/R-induced (1 h ischemia, followed by a 6-h reperfusion period) pancreatic injury was also significantly reduced by treatment with the relatively selective iNOS inhibitor L-N6-(1-iminoethyl)-lysine (6 mg/kg/h) (12). The administration of aminoguanidine (80 mg/kg i.v.) or L-NAME (10 mg/kg i.v.) significantly reduced pancreatic injury induced by pancreatic transplantation (73, 97, 127). In contrast, Yuan et al. (187) showed that L-arginine (2×200 mg/kg i.v.) ameliorated and L-NAME (2×5 mg/kg i.v.) aggravated pancreatic damage after pancreaticoduodenal transplantation in rats. In accord with the latter findings, L-NAME administration significantly reduced plasma NOx concentrations and increased pancreatic injury in response to incomplete I/R, indicating a beneficial effect of NO generation (159).
Maglione et al. (105) found that the pancreatic concentrations of the NOS cofactor BH4 were significantly decreased following prolonged cold ischemia in murine pancreatic grafts. Treatment of mice with the NOS cofactor significantly reduced pancreatic postischemic deterioration of microcirculation as well as histologic damage and nitrotyrosine formation after pancreas transplantation.
Summary VIII
• The administration of NO donors has been shown to reduce the severity of pancreatic I/R injury.
Conclusions
NO is one of several key messenger molecules in the exocrine pancreas. Physiologically, the main sources of pancreatic constitutive NO synthesis are neurons and vascular endothelial cells. Acinar cells, but not ductal cells, are also likely to generate NO via NOS. There is ample evidence that the synthesized NO influences pancreatic circulation and stimulates pancreatic secretion.
Acute pancreatitis and I/R injury induce pancreatic iNOS expression and activity, but eNOS activity is significantly reduced. The beneficial effect of NO donors in pancreatic I/R injury and in the initial phases of experimental acute pancreatitis is more or less proven. However, the extrapolation of the results of these animal experiments into a clinical response in humans is difficult since patients usually present at a late stage of the disease. Besides, the beneficial effect of GTN in preventing post-ERCP pancreatitis is not proven.
The role of NO in pancreatic physiology and pathophysiology remains controversial in numerous areas. Many questions regarding the messenger molecule still remain unanswered. Numerous obstacles prevent us from investigating the relevance of NO/RNS signaling. One major difficulty is measuring their production. There also seems to be great variations in species- and cell-specific expression of NOS isoforms. Further, most of the available NOS inhibitors are not isoform specific and, if applied in vivo, not only act on the organ of interest (i.e., the pancreas). Unfortunately, the use of transgenic animals (especially iNOS knock-out mice) has not been of much help to understand the effect of NOS in pancreatitis and often lead to completely opposite results. All these things make the interpretation of results very difficult.
Taken together, there is still much to investigate in the area of NO signaling in the exocrine pancreas. Probably, the least is known about the downstream targets of NO/RNS, which need to be identified, especially at the molecular level.
Footnotes
Acknowledgments
Our research is supported by National Development Agency grants (TÁMOP-4.2.1.B-09/1/KONV-2010-0005, TÁMOP-4.2.2-08/1/2008-0002 and 0013), the Hungarian Scientific Research Fund (K78311 to Z.R. and NNF 78851 to P.H.), and the Hungarian Academy of Sciences (BO 00334/08/5 to P.H. and BO 00174/10/5 to Z.R.).