
Abstract
Introduction
Lipid rafts are a microdomain of the plasma membrane, serving as a platform where receptor-mediated signal transduction is initiated (32, 38, 45). It has been shown that activated TLR4 is recruited to the lipid rafts to initiate signaling, which is controlled by ROS generated through NADPH oxidase (32, 45). TLR signaling pathways rapidly activate the NFκB transcriptional factor, which plays a key role in inflammation and the immune response by regulating the expression of numerous inflammatory cytokine genes, such as tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), transforming growth factor β (TGF-β), and interferon γ (IFN-γ) (1, 14, 20, 21). In this respect, controlling TLR signaling may prevent skin inflammation.
Innovation
Hyaluronic acid (HA) is extensively applied for cosmetic, therapeutic, and antiaging purposes on the skin. However, HA causes adverse effects such as inflammation when it is fragmented. Considering that UV-exposure produces fragmented HA and the skin is the first barrier to the environment, the precise mechanism of HA fragments (HAFs)-mediated skin inflammation needs to be clarified, and strategies for the prevention of skin inflammation are necessary. In this study, we showed that superoxide dismutase 3 (SOD3) suppresses HAF-mediated skin inflammation. Interestingly, we found that keratinocytes on skin are not directly responded to HAF- or SOD3-mediated pro- and anti-inflammatory response. Instead, macrophages and dentritic cells (DCs) are the primary role for HAF-mediated skin inflammation, producing inflammatory cytokines, which, in turn, stimulates keratinocytes to induce inflammatory molecules, and skin inflammation was progressed. SOD3 blocked production of inflammatory cytokines from macrophages and DC. Toll-like receptor 4 (TLR4) signaling pathways are involved in this inflammatory response including DC maturation. We report for the first time that HAF-promoted TLR4 trafficking to the lipid rafts, which is controlled by ROS generated through NADPH oxidase. Subsequently, HAF promoted recruitment of NFκB subunit p65 on the promoter of gene encoding inflammatory molecules. This inflammatory machinery was blocked by SOD3. Therefore, SOD3 may be useful for the treatment of skin inflammation mediated by HAF, UV, and pathogens.
Extracellular SOD, SOD3, is one of isoforms of SOD that scavenge superoxide radicals (28). Unlike the other isoforms, SOD3 is a glycoprotein that is primarily located in the interstitial matrix of tissues and the glycocalyxes of other cell surfaces where it is anchored to heparin sulfate proteoglycans (18, 27, 28). Since SOD3 is highly expressed in normal lung tissue and protects from oxidative stress, SOD3-mediated anti-inflammatory effects in the lung are well established (6, 9, 46), and an ischemic injury model has been reported (26). However, the role of SOD3 in skin inflammation has not yet been determined. Compared with cytosolic SOD1 or nuclear SOD2, SOD3 mainly acts in the ECM and cytosol. Furthermore, even though both HA and SOD3 exist in the ECM, their interplay is undefined. Since the ECM is essential for tissue homeostasis, any changes in the ECM microenvironment can be detrimental to cell function during inflammation. Therefore, SOD3 may offer protection from skin inflammation.
Considering that the skin is the first barrier for defense against the external environments, including UV exposure, and contains large amounts of HA, a strategy for the prevention of skin inflammation due to HAF is required. Furthermore, although HAF is involved in innate immune function via TLR, the precise molecular mechanism has not been clarified.
In this study, we demonstrated the role of HAF and SOD3 by using an in vivo acute inflammatory model adopting epicutaneously treated HAF on the skin of wild-type (wt) and SOD3 transgenic (tg) mice. HAF-induced skin inflammation was primarily mediated by macrophages and DC. However, SOD3 suppressed HAF-mediated skin inflammation. Moreover, SOD3 inhibits DC maturation. TLR4 signaling pathways are involved in these responses. Like LPS, HAF promotes the recruitment of TLR4 to lipid rafts, but SOD3 inhibits this translocation. NADPH oxidase-dependent ROS generation was, at least in part, involved in this trafficking. Consequently, the recruitment of NFkB subunit p65 to the promoters of inflammatory molecules was altered. Therefore, we suggest that SOD3 may be useful for preventing or alleviating skin inflammation mediated by HAF, UV, and pathogens.
Results
SOD3 alleviates HAF-mediated skin inflammation
To understand the role of HAF and SOD3 on the skin, HAF (200-kDa) were epicutaneously treated to the skin of wt or SOD3 tg mice. As shown in Figure 1A, HAF induced skin inflammation in the wt mice, but the level of inflammation was reduced in the SOD3 tg mice (Fig. 1A, left panel). For the control, we applied higher-molecular HA (2000 kDa) to the skin of wt mice. As expected, HA did not induce skin inflammation (Fig. 1A, right panel). Consistently, HAF up-regulates the expression of inflammatory markers, TNF-α, cyclooxygenase 2 (Cox-2), metalloproteinase 1 (MMP-1), and MMP-2 in the skin of wt mice, whereas the expression was diminished in SOD3 tg mice (Fig. 1B). Based on the results, we identified the infiltrating cells. As shown in Figure 1C, DC, macrophages, and neutrophils, which expressed CD11c, CD11b, and neutrophil marker (NIMP-R14), respectively, were identified as the infiltrating cells. However, their levels were diminished in SOD3 tg mice. Macrophages and DC appeared at day 2 and the level gradually increased until day 5, whereas infiltrated neutrophils increased, and the increased level remained until day 5 (Supplementary Fig. S1; Supplementary Data are available online at
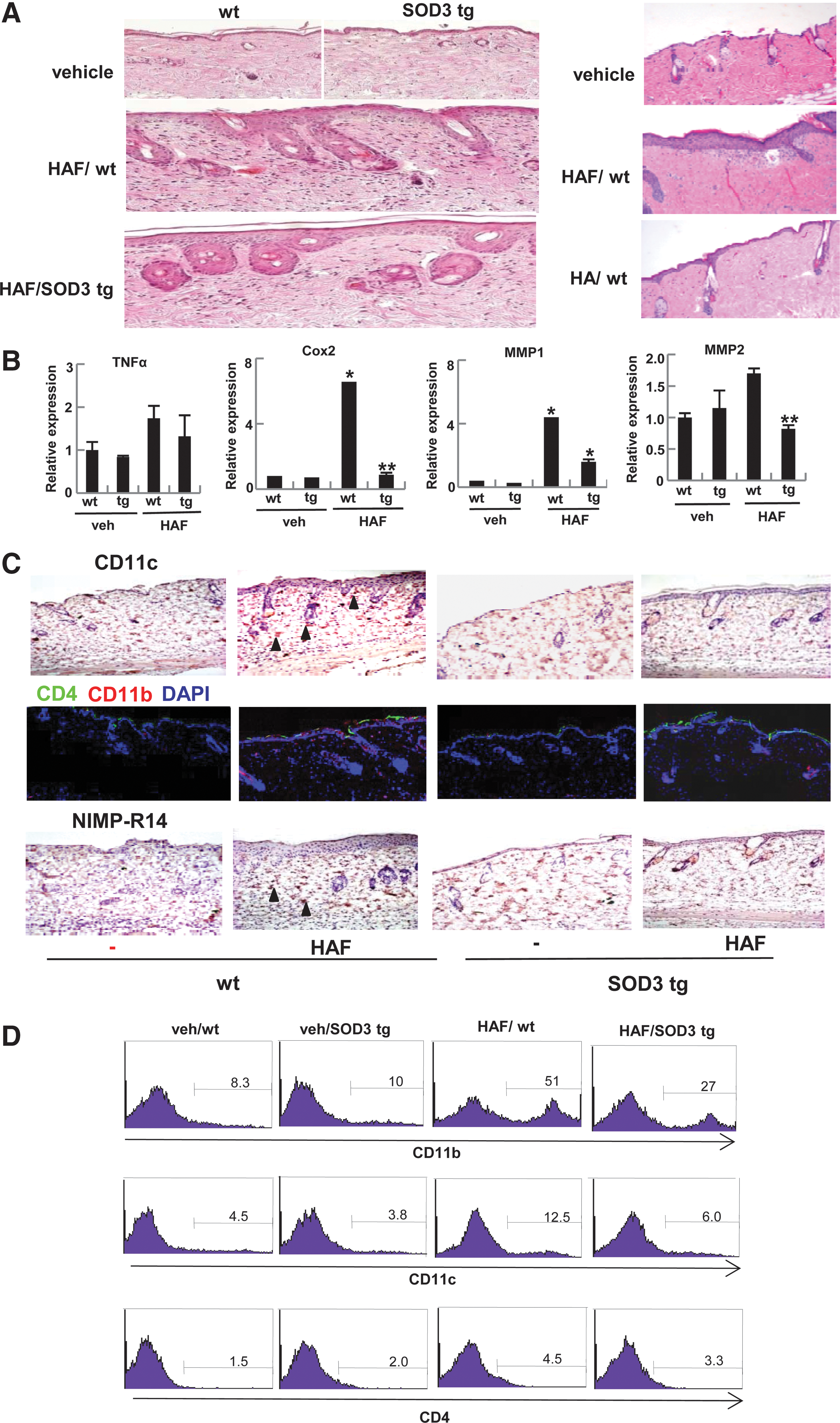
SOD3 inhibits the expression of HAF-induced chemokines and their receptors
Chemokines attract mononuclear cells to inflammatory sites for progression of inflammation (30). Since we observed infiltrated macrophages and DC in the skin, we investigated whether HAF or SOD3 controls the trafficking of immune cells to the skin. To do this, we assessed the expression of inflammatory chemokines in skin, keratinocytes, and fibroblasts, and chemokine receptors in macrophages and DC.
As shown in Figure 2A, the inflammatory chemokines cheomkine (c-x-c motif) ligand 1 (CXCL-1) and monocyte chemotactic protein 1 (MCP-1) were up-regulated in HAF-treated skin in the wt mice, whereas the expression level was reduced in the SOD3 tg mice. For recruitment of T cell, interferon-gamma-inducible 10-kDa protein (IP-10) expression in the skin was not altered by treatment with HAF in either wt or SOD3 tg mice (Fig. 2A, right panel). However, interestingly, CXCL-1 and MCP-1 level in keratinocytes or fibroblasts were not changed by treatment of HAF or SOD3 (Fig. 2B). Instead, corresponding inflammatory chemokine receptors, chemokine receptor 5 (CCR5), CCR1, and CCR2, were up-regulated in RAW 264.7 cells, bone marrow (BM)-derived macrophage (BMDM), and BM-derived dendritic cell (BMDC) by treatment with HAF (Fig. 2C–E), but the expression levels were greatly diminished in the presence of SOD3, indicating that HAF-induced and SOD3 suppressed the inflammation, are partly due to control of recruitment and trafficking of monocytes to the skin.

Macrophages and DC are primarily responsible for the HAF and SOD3 mediated pro- and anti-inflammatory effects
Since keratinocytes and fibroblasts are the dominant cells for skin inflammation (5, 33), we speculated that HAF or SOD3 may directly stimulate these cells. To test this, a human keratinocytes cells line, human immortalized keratinocyte (HaCaT), and primary cultured human dermal fibroblasts (HDFs) were treated with HAF and SOD3, and the expression of inflammatory genes was measured by quantitative real-time polymerase chain reaction (qRT-PCR). However, interestingly, the expression level of inflammatory cytokines, Cox-2, TNF-α, IL-6, and procollagen was not altered in either HAF or SOD3-treated HaCaT and fibroblasts, indicating that keratinocytes and fibroblasts do not directly respond to HAF and SOD3-mediated pro- and anti-inflammation of the skin (Fig. 3A, B).
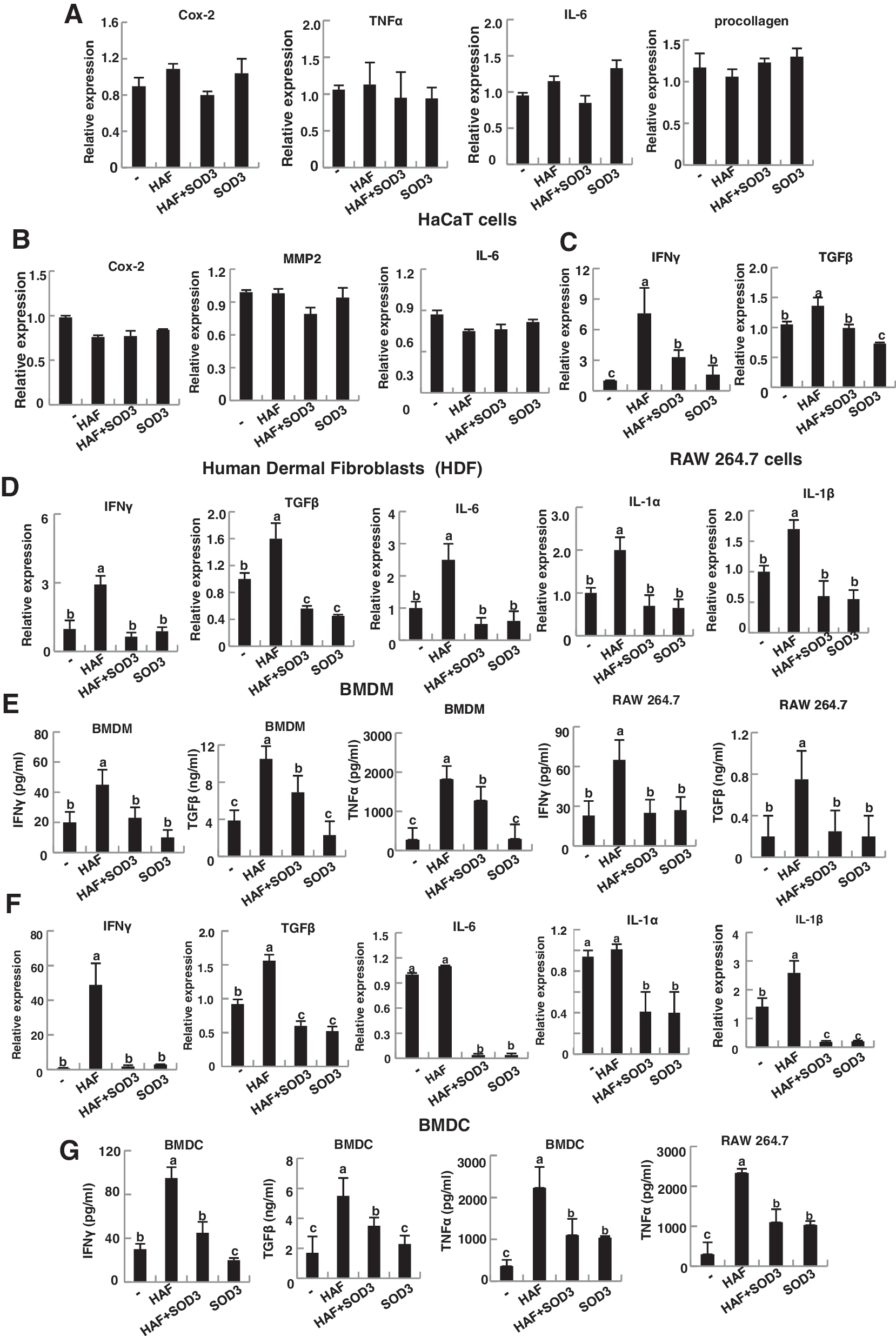
Instead, macrophages were directly responsible for this inflammation. As shown in Figure 3C and G, HAF-treated RAW 264.7 cells up-regulate inflammatory cytokines. Specifically IFN-γ, TGF-β, and TNF-α were greatly induced. However, the expression of these inflammatory molecules was drastically reduced in the presence of SOD3. Consistent results were observed in primary cultured BMDM (Fig. 3D, E). In addition, BMDM treated with HAF up-regulated other pro-inflammatory cytokines, IL-6, IL-1α, and IL-1β, while SOD3 reduced expression of these genes (Fig. 3D).
DC is the primary immune cell responsible for the inflammation, and we also observed increased DC infiltration during HAF-mediated skin inflammation. Thus, we investigated the role of HAF and SOD3 in DC. Consistent with the results for BMDM, BMDC directly responded to HAF and SOD3 (Fig. 3F, G).
Collectively, these results indicate that HAF- or SOD3-mediated pro- and anti-inflammation on the skin is primarily due to macrophages and DC.
Inflammatory molecules produced by macrophages and DC stimulate the epidermis, leading to skin inflammation
Since keratinocytes are the major cells responsible for the progression of skin inflammation, we speculated that HAF or SOD3 may indirectly stimulate these cells. Thus, it may be possible that secreted inflammatory molecules from macrophages and DC stimulate keratiocytes.
To verify this hypothesis, epidermal cells, mainly composed with keratinocytes, were isolated from mouse skin and treated with supernatants collected from macrophages and DC-treated HAF, HAF with SOD3, or SOD3. Since the supernatants contained released inflammatory molecules including cytokines, we tested the response of keratinocytes after treatment with these supernatants. As shown in Figure 4A, the epidermis responded to the supernatants. Inflammatory cytokines, TNF-α, IL-1β, and IL-6, intercellular adhesion molecule-1 (ICAM-1), defensin-β1, and chemokine, CXCL-1, and MCP-1 were greatly increased in epidermal cells treated with the supernatant collected from HAF-treated macrophages, but their levels were greatly diminished in the presence of the supernatant from SOD3-treated macrophages (Fig. 4A). Similar results were observed by treatment with the supernatant of DC (Fig. 4B). However, the effect was milder than that of macrophages (Fig. 4B).
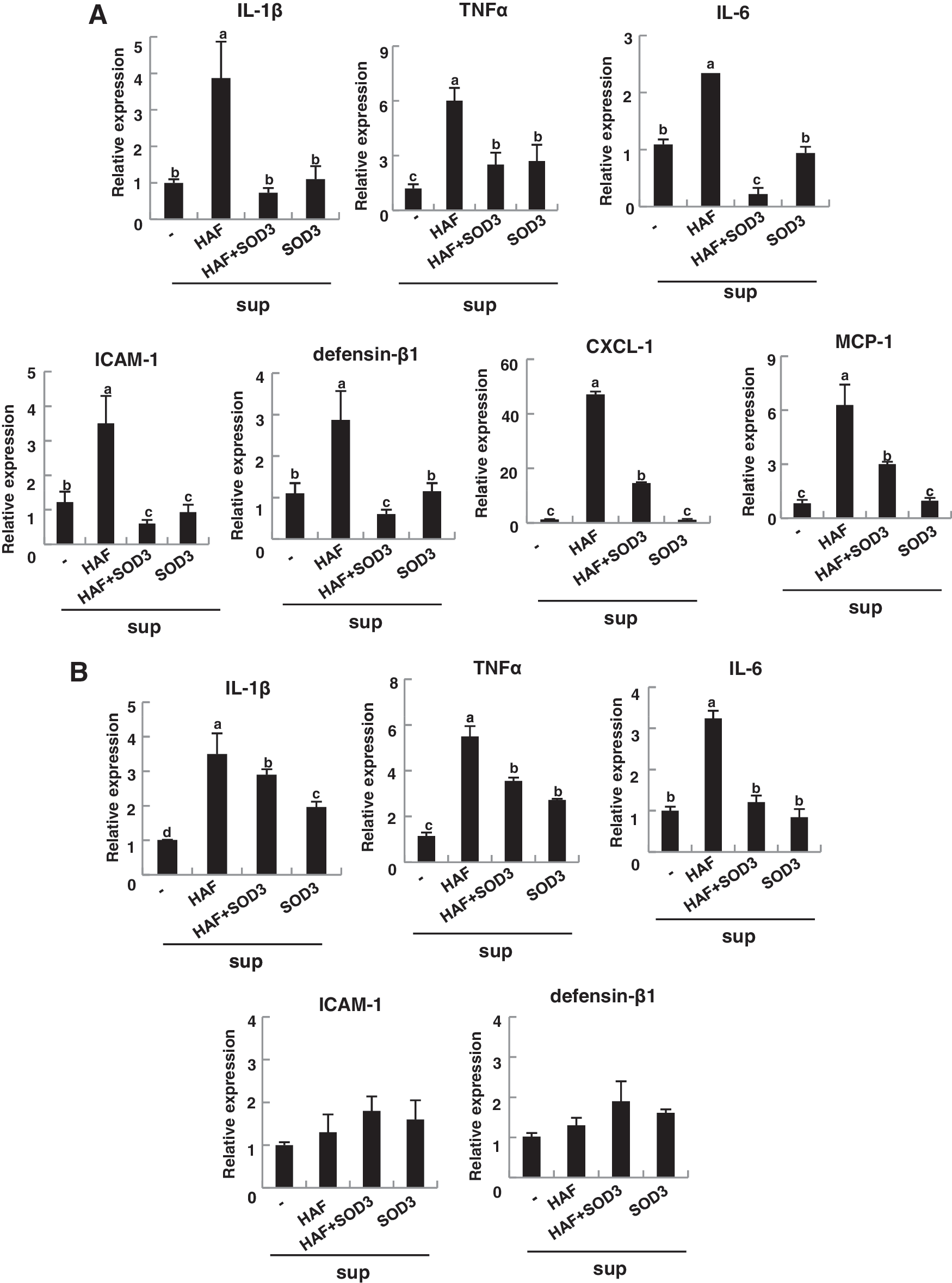
Taken together, these results suggest that HAF-mediated skin inflammation is primarily due to the response of infiltrated macrophages and DC to the skin and produced inflammatory cytokines, which, in turn, stimulate the keratinocytes to produce inflammatory molecules. Consequently, skin inflammation progressed. Treatment with SOD3 blocked these sequential events.
SOD3 attenuates DC maturation
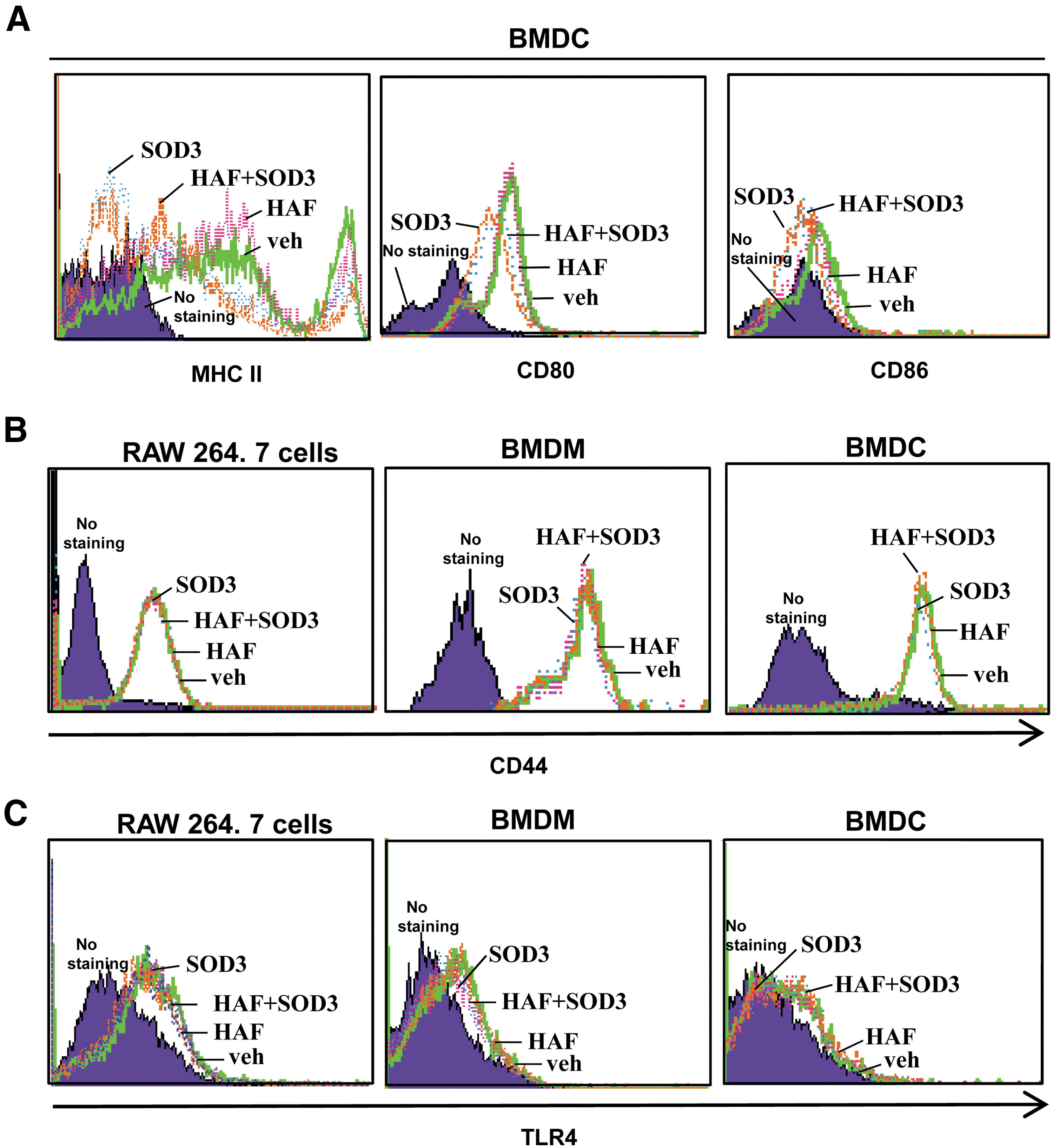
DC maturation is another function of DC in the inflammatory response. During DC maturation, major histocompatibility complex (MHC) II and its co-stimulatory molecules CD80 and CD86 are expressed on the cell surface, where they play a key role in the adaptive immunity by presenting antigens to the T cell. Consequently, T cell proliferation and differentiation are controlled. Thus, improper antigen presentation can cause failed T cell activation and poor adaptive immunity. Furthermore, T cells are infiltrated into the inflammatory site during inflammation. For these reasons, we investigated role of HAF and SOD3 in DC maturation. As shown in Figure 5A, HAF slightly up-regulated MHC II expression, but the expression level declined in the presence of SOD3. The expression of CD80 and CD86 was clearly reduced by SOD3 treatment (Fig. 5A). Taken together, these data suggest that SOD3 suppresses DC maturation.
SOD3 does not affect the expression of CD44 and TLR4
Next, we investigated how HAF or SOD3 regulates the macrophages or DC-mediated pro- or anti-inflammatory response. Since CD44 is a known receptor for HA, we tested whether the expression of CD44 is affected by HAF or SOD3. As shown in Figure 5B, CD44 expression in RAW 264.7 cells, BMDM, and BMDC was not affected by either HAF or SOD3. Since HAF acts as a ligand for TLR2 and TLR4 (37, 43), we also tested whether TLR4 surface expression was affected by HAF or SOD3. As shown in Figure 5C, surface expression of TLR4 in RAW 264.7 cells, BMDM, and BMDC was not affected by treatment with either HAF or SOD3.
TLR4 signaling pathways are involved in HAF- and SOD3-mediated pro- and anti-inflammatory effect
It has been shown that HAF triggers the immune response (29) and activates macrophages and DC via the TLRs signaling pathways (37, 43). Thus, we investigated whether SOD3-mediated anti-inflammatory effects are due to the control of TLRs signaling pathway. For this study, we used TLR4-deficient mice. As shown in Figure 6A, left panel, skin inflammation, and infiltrating DC and macrophages did not occur in HAF-treated TLR 4 dificient-C3H/HeJ mice. Furthermore, neither HAF nor SOD3 affected the expression or induction of inflammatory cytokines, IFN-γ, TGF-β, IL-1β, and TNF-α in BMDM of TLR4-deficient mice (Fig. 6B). In contrast, corresponding wt, C3H/HeN mice, had similar effects as in C57BL/6 mice (Fig. 6A, right panel, C).
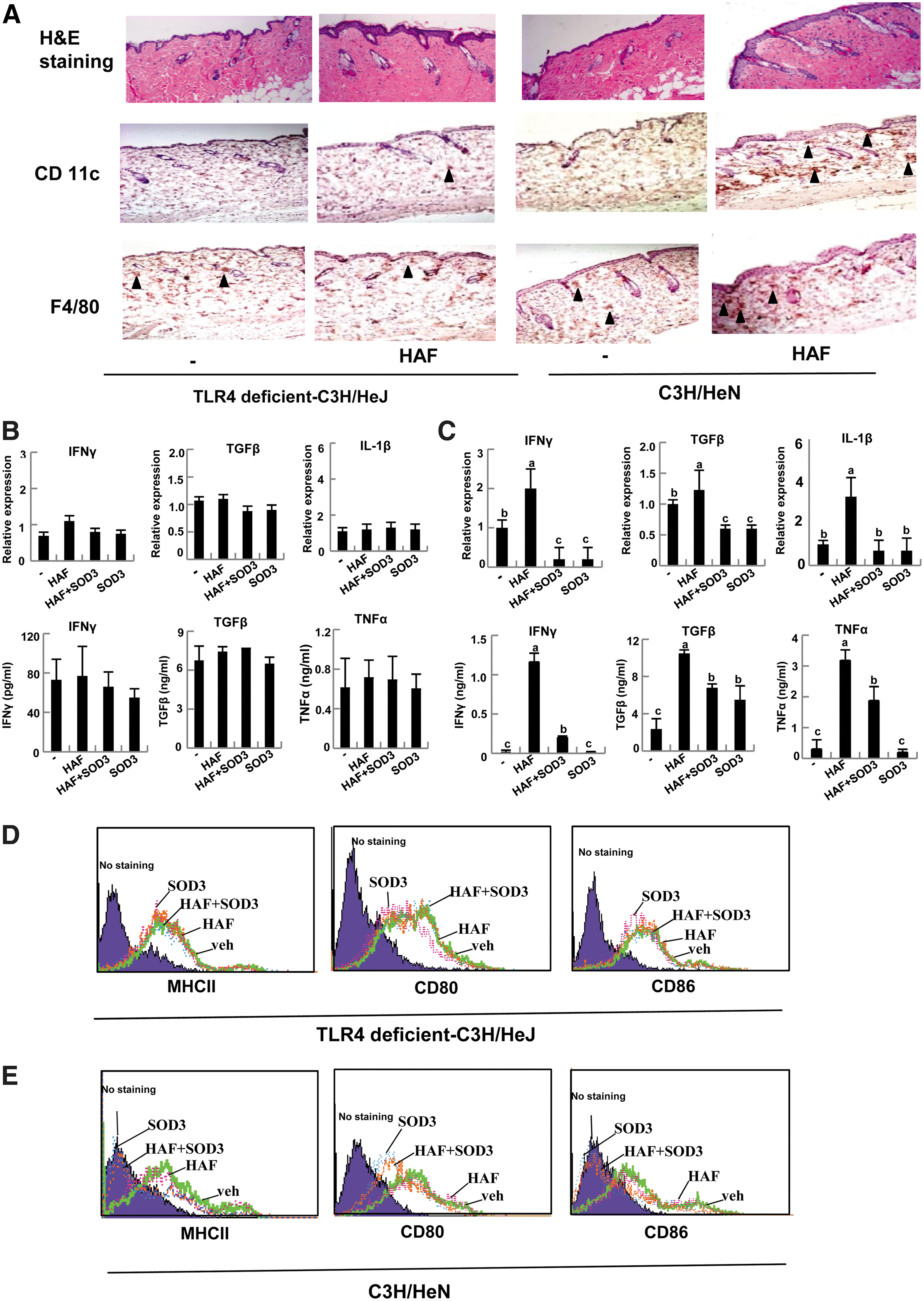
Interestingly, as shown in Figure 6D, HAF and SOD3 did not affect the surface expression of MHC II, CD80, and CD86 in BMDC of TLR4-deficient mice, but the expression of these molecules was increased in HAF-treated BMDC of wt mice, and drastically reduced in SOD3-treated BMDC of wt mice (Fig. 6E). Taken together, these results suggest that HAF-mediated inflammation, and its suppression by SOD3 operates through the TLR4 signaling pathway.
HAF promotes and SOD3 diminishes recruitment of TLR4 to lipid rafts
We next asked how HAF triggers TLR4 signaling and is inhibited by SOD3. Lipid rafts serve as a platform where receptor-mediated signal transduction is initiated (32, 38, 45). TLR4 is recruited into the lipid rafts after response to stimuli, and its signals are subsequently propagated downstream leading to up-regulation of NFkB transactivation (45). Furthermore, methyl-β-dextrin, a lipid raft inhibitor, significantly inhibits the LPS-induced expression of cytokines (45), suggesting that lipid rafts are essential for TLR4-mediated signal transduction and target gene expression. In addition, HA exist in ECM, and SOD3 mainly functions in the ECM (8). Thus, we tested whether HAF induced and SOD3 diminished inflammation through controlling the recruitment of TLR4 to the lipid rafts. To test this, we used fluorescein isothiocyanate (FITC)-conjugated cholera toxin B, which serves as a marker for lipid rafts by binding to Ganglionmonosialoganlioside 1 (GM1), a glycosphingolipid enriched in lipid rafts.
We performed confocal microscopy experiments in which lipid rafts were stained green, TLR4 was stained red by phycoerythrin (PE)-conjugated TLR4, and merging these two colors resulted in yellow staining when the lipid rafts and TLR4 co-localized. In resting BMDM cells, TLR4 did not co-localize into the lipid rafts, but instead localized in a condensed formation that was distributed throughout the cytoplasm (Fig. 7A, first panel). When the BMDM cells were treated with LPS, TLR4 was clearly trans-located into the lipid rafts (Fig. 7A, second panel). Like LPS, HAF leads to TLR4 translocation into the lipid rafts (Fig. 7A, fourth panel). However, LPS or HAF with SOD3, or SOD3 alone, failed to trigger the localization of TLR4 to the lipid rafts (Fig. 7A, third, fifth, and sixth panels).
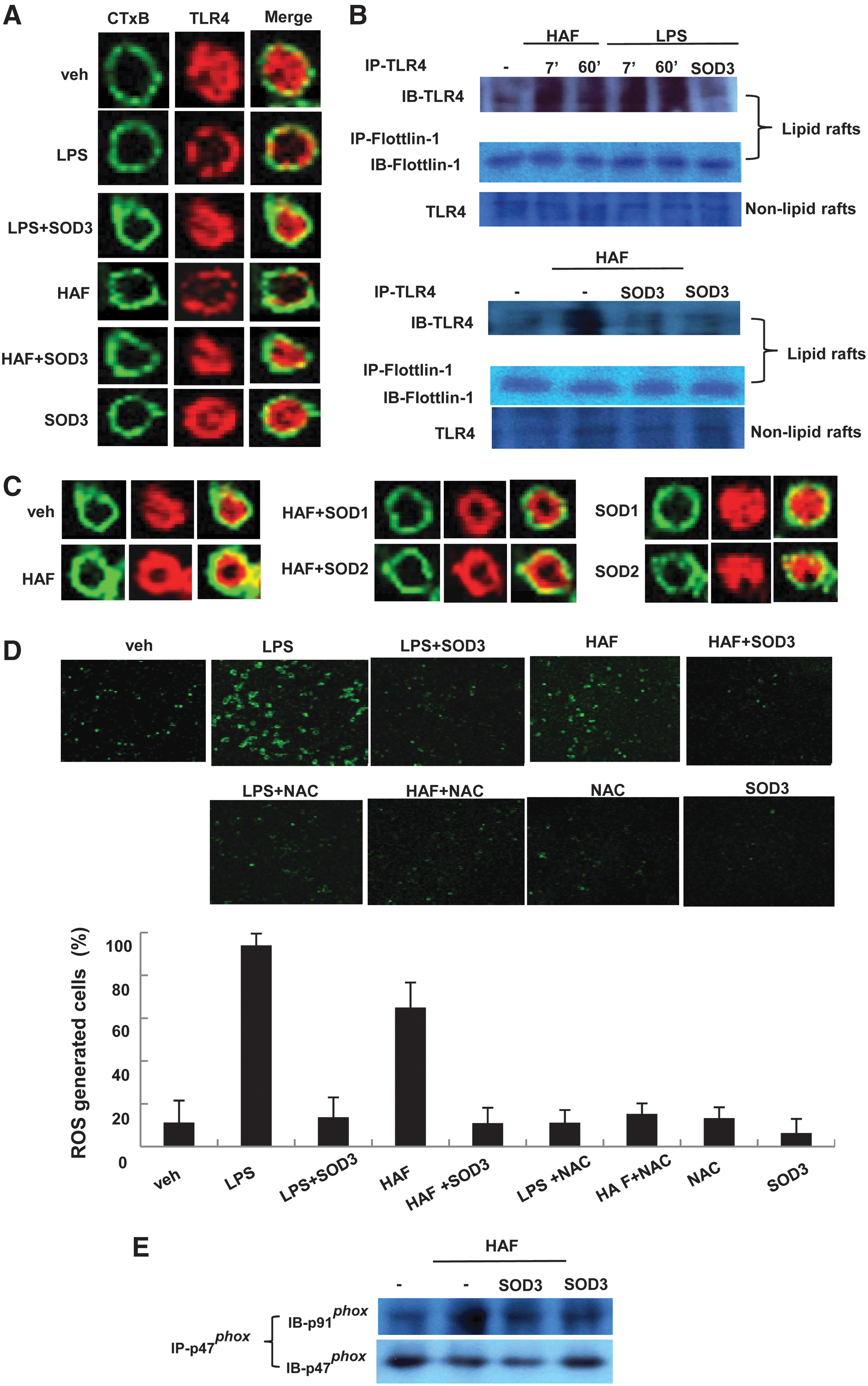
We confirmed the effects of HAF and SOD3 on TLR4 translocation by performing biochemical analyses using sucrose gradient ultracentrifugation. Consistent with results of confocal microscopy, both LPS and HAF caused TLR4 translocation into lipid rafts (Fig. 7B, first and fourth panels), but the levels of translocated TLR4 were greatly diminished in the presence of SOD3 (Fig. 7B, first and fourth panels). Both the hue and tone of TLR4 detected are due to glycated TLR4. However, the amount of TLR4 in regions other than lipid rafts (i.e., nonlipid rafts) was not altered by LPS, HAF, or SOD3 (Fig. 7B, third and sixth panels). Furthermore, neither SOD1 nor SOD2 inhibited HAF-mediated translocation of TLR4 to the lipid rafts (Fig. 7C).
Taken together, these results suggest that HAF or SOD3 regulates TLR4 translocation to the lipid rafts, which, in turn, controls TLR4 signaling.
HAF promotes and SOD3 inhibits ROS generation via NADPH oxidase, which controls TLR4 translocation to the lipid rafts
It has been shown that LPS produces ROS, which controls TLR4 translocation to the lipid rafts (32, 45). Considering that SOD3 quenches ROS generation, the modulation of TLR4 translocation by HAF or SOD3 may be regulated by ROS generation. To test this possibility, we assessed ROS generation of cells treated with HAF or SOD3 through confocal microscopy using 2′,7′-dichlorofluorescein-diacetate (H2DCFH-DA) dye. As shown in Figure 7D, similar to LPS, HAF clearly promotes ROS production in the cells, but this increase was diminished in the presence of SOD3. Furthermore, in the presence of N-acetyl-L-cysteine (NAC), an antioxidant, the HAF-induced generation of ROS was greatly diminished.
Next, we asked how HAF or SOD3 controls ROS generation. Activation of NADPH oxidase leads to the generation of ROS (3). It has been shown that LPS promotes TLR4 translocation via ROS generated by activation of NADPH oxidase (32). It is shown that diphenyliodonium, NADPH oxidase inhibitor, blocked TLR4 translocation to the lipid rafts (32). NADPH oxidase is composed of several membrane subunits such as gp91, p22, FAD, and rap1A, and cytosolic subunits such as p47, p67, and p40. NADPH oxidase is activated as the cytosol subunits are recruited and bound to one of the plasma membrane subunits (3). Thus, we further tested whether HAF or SOD3 controls the activation of NADPH oxidase. We treated RAW 264.7 cells with HAF, HAF with SOD3, or SOD3 alone and observed the interaction between the membrane and cytosolic subunits of NADPH oxidase. As shown in Figure 7D, the interaction of p91 phox and p47 phox was promoted by HAF (Fig. 7E), but the level was greatly diminished in the presence of SOD3.
Taken together, these results suggest that HAF-promoted and SOD3-suppressed skin inflammation is mediated by TLR4 signaling, at least in part, governed by ROS generation through NADPH oxidase, which controls TLR4 translocation into the lipid rafts to initiate signaling.
HAF promotes and SOD3 suppresses NFkB recruitment to the promoters of target genes: TNF-α, IFN-γ, TGF-β, IL-1β, and Class II transactivator
The TLR4 signaling pathway proceeds via NFkB transactivation. So far, we have observed that IFN-γ, TGF-β, TNF-α, IL-1β, and MHC II are dominantly affected by HAF or SOD3. Next, we confirmed whether HAF or SOD3 regulated these genes by controlling NFkB transactivation. Since NFkB binding affinity for the promoter reflects gene expression, we tested whether NFkB binding was affected by treatment with HAF or SOD3. We first examined the NFkB binding sites of each promoter. As shown in Figure 8A, we observed that the promoter of TNF-α, IFN-γ, TGF-β, IL-1β, and Class II transactivator (CIITA), a key regulator of MHC II, contain NFkB binding sites. Based on that information, we designed the promoter primers for a chromatin immunoprecipitation (ChIP) assay. RAW 264.7 cells were treated with HAF, HAF with SOD3, or SOD3 alone, and chromatin was prepared for a ChIP assay with anti-p65. As shown in Figure 8B, consistent with the gene expression patterns, HAF increased the recruitment of p65 to the promoters of TNF-α, IFN-γ, TGF-β, IL-1β, and CIITA (Fig. 8B). However, the recruitment was decreased in the presence of SOD3 (Fig. 8B).

Taken together, these results suggest that SOD3 inhibits the induction of HAF-mediated inflammatory cytokines through arrest of NFkB transactivation by inhibition of the TLR4 signaling pathway.
Discussion
Our study showed that SOD3 suppressed HAF-mediated skin inflammation by controlling the TLR4 signaling pathway. It has been reported that HAF leads to acute lung inflammation (15, 16, 42) that is mediated through either TLR2, or TLR4, or both (15, 39, 43). Macrophages are involved in this inflammation by producing a variety of chemokines. Thus, although HAF are not pathogen derived, they act like a pathogen associate molecular pattern (PAMP) by activation of the TLRs signaling pathway leading to inflammation. Considering that nonpathogen originated molecules such as Taxol (7), lauric acid (45), and heat shock protein (4) can induce sites of inflammation via activation of the TLR4 signaling pathway, the ligands of TLR4 are probably not limited to PAMPs. Noninfectious associated inflammation may be due to these nonpathogen originated molecular patterns.
The HAF-mediated inflammatory response might depend on the size of the fragments. Indeed, we observed that MHC II expression was slightly enhanced, with no significant difference in the expression of the co-stimulatory molecules CD80 and CD86 by treatment with 200-kDa HAF, whereas other groups showed that HAF greatly enhanced expression of MHC II, CD80, and CD86 (43). This discrepancy is probably due to different sizes of the HAF. Thus, it seems that the size of HAF may affect the strength of TLR4 signaling. A supportive study showed that 135-kDa HAF induced inflammatory molecular changes much more strongly than 50-kDa fragments (15). Further, although HA of 12–16 disaccharides induced the cytokine TNF-α in DC, the expression level was greater when the HA was polymers (29). Thus, it is likely that the size of the HAF and a subset of HA polymers contributed to inflammation with a greater magnitude.
Interestingly, we observed that the HAF-mediated skin inflammation is primarily due to macrophages and inflammatory DC, which produced inflammatory molecules including cytokines. This stimulated the epidermis to produce inflammatory molecules including chemokines that recruited monocytes to the skin leading to progression of inflammation. Another function of DC, DC maturation, and expression of MHC II whose function is to present antigen for T cell activation, leads to proper T-cell differentiation and proliferation. Thus, DC can also play a role in T-cell-mediated inflammation. However, current studies show that HAF-mediated skin inflammation is not mainly due to T-cell-mediated inflammation, as neither CD4 T-cell infiltration nor the expression of the chemokine IP-10, which is important for trafficking of T cells, was altered by HAF or SOD3. Furthermore, the expression of Th1 and Th17 cells and their transcriptional factors, T-bet and RORγt, in the skin were not comparable between wt and SOD3 tg mice by treatment with HAF (data not shown).
Considering all these data, our study suggests that HAF induces noninfectious skin inflammation by activating macrophages and DC to produce inflammatory cytokines, and by enhancing expression of chemokine receptors. This stimulates the epidermis to produce inflammatory molecules and chemokines for progression of skin inflammation. This noninfectious inflammatory machinery was blocked by SOD3.
In this study, we demonstrated that the SOD3-mediated anti-inflammatory effect was due to a blockade of the TLR4 signaling pathway. SOD3 inhibits recruitment of NFkB to promoters of inflammatory cytokines and CIITA, a key regulator of MHC II. Correspondingly, the expression of the NFkB-mediated inflammatory cytokines, IFN-γ, TNF-α, TGF-β and IL-1β, and MHC II, was attenuated. However, SOD3 does not affect the expression of TLR4 or CD44, a receptor for HA. Since TLR4 signaling initiates after its translocation to the lipid rafts as a complex with its ligand and SOD3 is in the ECM, the anti-inflammatory effect of SOD3 might be exerted through controlling TLR4 translocation to the lipid rafts. Since TLR4 translocation to the lipid rafts is controlled by ROS generation by NADPH oxidase, the anti-inflammatory effects of SOD3 are likely due to inhibition of ROS generation.
Given the importance of controlling ROS generation for the prevention of inflammation, it remains unclear how SOD3, rather than other isoforms of SOD, specifically affects HAF-mediated inflammation. It is possible that SOD isoforms perform different roles. Indeed, the level of SOD3 was significantly decreased with no change in the levels of SOD1 (Cu, Zn-SOD) or SOD2 (Mn-SOD) after the administration of LPS in the lung (44). In addition, on administration of TNF-α, the SOD3 level was drastically reduced, but SOD1 or SOD2 levels were not altered (44). Moreover, the antioxidant NAC did not exhibit any beneficial effects in elastase-induced lung inflammation in SOD3 KO mice (46). Thus, it seems that the SOD3-mediated anti-inflammatory effect was not simply due to radical scavenging.
Since SOD3 is primarily located in the ECM, we are tempted to speculate that the distinctive role of SOD3 is due to the blockade of signaling events at the plasma membrane, such as control of receptor-ligand complexes, interaction between signaling molecules, and translocation into the lipid rafts via control of ROS generation by NADPH oxidase. Although we observed that SOD3 inhibits TLR4 translocation to the lipid rafts, TLR4 needs to be dimerized and combined with other moleculars such as CD14 or MD2 on the plasma membrane to initiate signaling (11, 36, 47). SOD3 may inhibit the interaction of these signaling molecules on the plasma membrane. SOD3 may also control the strength of its signal by accelerating proteolytic processes involved in receptor-ligand binding. As a receptor binds with its ligand, the signal is propagated, and the complex is degraded by a lysosomal-mediated process or by proteolysis via a proteolytic signal. If SOD3 is involved in the proteolytic process, it could accelerate the proteolysis, reduced expression of target genes, specifically inflammatory genes. In addition, considering that SOD3 controls the expression of MHC II and the co-stimulatory molecules, CD80 and CD86, SOD3 may control adaptive immunity by regulating T-helper-cell proliferation and differentiation, and, consequently, may control T-cell-mediated immune disease, including autoimmune disease. Thus, further studies are needed to clarify these events.
In this study, we demonstrated the role of HAF and SOD3 in the skin. We also observed that SOD3 expression was significantly down-regulated in atopic or psoriasis patients, and the results correlated with down-regulation of NFkB signaling (data not shown). Our study revealed that SOD3 plays a critical role in inhibition of HAF-mediated skin inflammation. However, future studies are warranted to clarify the many unknown roles of SOD3.
Materials and Methods
Animals
C57BL/6 wt mice and SOD3 tg C57BL/6 mice were previously described (10). For TLR4-deficient mice, C3H/HeJ and corresponding wt, C3H/HeN mice were obtained from Central Lab Animal Inc. (Woomyun-dong, Seoul, Korea). All mouse care and experimental procedures were performed under specific pathogen-free conditions in accordance with established institutional guidance and approval from the Research Animal Care Committee at Catholic University. To induce skin inflammation, mouse backs were shaved and immediately depilated. Mechanical injury was applied by tape stripping across the back. One day later, HAF obtained from Vacc Tech (Vacc Tech, Daejon, Korea) were applied across the back every day for 5 days. HAF (200 kDa, 50 μl, 50 mg/ml) were applied for the in vivo study. For the control, the same amount of H2O was applied. The mice were the sacrificed, and the skin was removed and prepared for isolation of RNA, protein, hematoxylin and eosin (HE) staining, and immunohistochemistry (IHC).
IHC and immunofluorescence staining
IHC was performed according to standard protocols. Briefly, paraffin-embedded skin section (5 μm) were deparaffinized in xylene twice for 5 min each, followed by rehydration in a graded series of incubation in absolute ethanol (i.e., 95%, 90%, 80%, and 70%) and distilled water. For immunostaining, the sections were microwaved for 20 min in citrate buffer (pH 6.0) for antigen retrieval, and peroxidase activity was blocked using 2% H2O2 solution. The sections were incubated with 5% normal serum of the same species as the individual secondary antibody for 1 h. The antibodies, anti-CD11c (BD pharmingen, San Diego, CA) for DC, anti-F4/80 (Invitrogen, Carlsbad, CA) for macrophages, and NIMP-R14 (Abcam, Cambridge, United Kingdom) for neutrophils, were diluted at 1:100 and incubated for 1 h at room temperature. To detect antigen-specific cells, the sections were incubated with biotinylated Ig G, followed by incubation with horseradish peroxidase-streptavidin complex (Sigma, St. Louis, MO). The sections were visualized with 3-3′-diaminobenzidine (ABC staining system: Santa cruz Biotechnology, Santa Cruz, CA), and hematoxylin staining was performed. Permount (Fisher, Pittsburgh, PA) was used for mounting. Images were taken by fluorescence microscopy (Carl Zeiss, Thornwood, NY).
For immunofluorescence (IF), the paraffin sections were deparaffinized, rehydrated, and subjected to antigen retrieval as just mentioned. Then, the sections were permeablized with PBS containing 0.2% Trition X-100 for 45 min, and incubation with blocking buffer (eBioscience, San Diego, CA) for 1 h at room temperature. FITC-conjugated CD4 (Invitrogen) and PE-conjugated CD11b (eBioscience) antibody were diluted with 1:100 and incubated for 1 h at room temperature. The sections were mounted with a mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA).
Cells line and primary cultured macrophages and DCs
Human keratinocytes cell line, HaCaT, primary cultured human dermal fibroblasts (HDFs), and murine macrophage cell line, RAW 264.7 cells were used. For the preparation of BMDCs or BMDMs, total BM cells from femurs and tibias were flushed out with 2 ml of Roswell Park Memorial Institute (RPMI) 1640 media using a 23-gauge needle. Red blood cell was depleted with hemolysis buffer (Sigma). The cells were purified using a lineage cell depletion kit (Miltenyi Biotec, Bergisch-Gladbach, Germany) and cultured in RPMI 1640 supplemented with 10% fetal bovine serum for 5 days with 10 ng/ml murine rGM-CSF for BMDC or for 10 days with 10 ng/ml murine M-CSF for BMDM. Homogeneity of the CD11c+ for DC and CD11b+ for macrophages was assessed by flow cytometry.
Preparation of recombinant SOD3
Recombinant SOD3 was prepared using mammalian SOD3 overexpression system. Briefly, SOD3 plasmid was transfected in 293 cells. Forty eight hours later, supernatant was collected, followed by purification using a column containing nickel-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen, Valencia, CA) and dialysis. The purified SOD3 activity was measured with an SOD assay kit (Dojindo, Sunnyvale, CA). For treatment, 100 units/ml SOD3 was applied.
Isolation of epidermis cells from the mouse skin
Skin from wt mice was isolated and transferred to a petri dish containing RPMI media. Dispase (5 mg/ml) and collagenase D (0.5 mg/ml) were added in RPMI media for 1 h at 37°C. Skin was then transferred to a new petri dish containing RPMI media to wash away excess enzymes. While holding the skin submerged in medium, the dermis was gently separated from the epidermis with two pairs of curved forceps. Separated epidermis was treated with trypsin for 30 min at 37°C. The tissue collapsed, and cells were released by repeatedly pipeting up and down. The cells were spun down and incubated with RPMI media for 5 h before treatment.
Quantitative real-time PCR
Total RNA was prepared using Trizol (Invitrogen). cDNA preparation and qRT-PCR were performed as described elsewhere (23). The primers used for real-time PCR: mouse IFN-γ, TNF-α, IL-6, IL-1α, IL-1β, MMP-1, MMP-2, and CXCL-1; and human TNF-α, Cox-2, MMP-2, and IL-6 were obtained from Quagen. The other primers used were as follows: mouse TGF-β, 5′-TTGCTTCAGCTCCACAGAGA, 3′-TCGGTTGTAGAGGGCAAGGAC: mouse ICAM-1, 5′-CGATCTTCCAGCTACCATCC, 3′-GGCTGACACAGAGTCACTGC: mouse defensin-β1, 5′-CTCTCTGCTCTCTGCTGCTG, 3′-CAACAGGGGTTCTTCTCTGG: human procollagen, 5′-CTCGAGGTGGACACCACCCT, 3′-CAGCTGGATGGCCACATCGG. The primers used for detection of chemokines and their receptors are as follows: mouse MCP-1, 5′-GAATGGGTCCAGACATACAT, 3′-CCTACAGAAGTGCTTGAGGT: mouse IP-10, 5′-CGGAATCTAAGACCATCA AG, 3′-GAAGACCAAGGGCAATTAG: mouse CCR1, 5′-CAGAGTAAGCAACTGGACCT, 3′-CTAGTTGGTCCACAGAGAGG: mouse CCR2, 5′-GTACCTTTGCAACTGCCTCT, 3′-CAAGACTTCTGTCCCTGCTT: mouse CCR5, 5′-CAGAGACTCTTGGAATGACAC, 3′-GTGGATCGGGTATAGACTGA.
Enzyme-linked immunosorbent assay
Cytokine and chemokine concentrations in supernatants were detected by enzyme-linked immunosorbent assay (ELISA): ELISA for IFN-γ, TGF-β, and TNF-α used mouse antibodies specific for murine IFN-γ, TGF-β, and TNF-α. These antibodies were obtained from BD Pharmingen. ELISA was performed according to the manufacturer's guidelines. Colorimetric changes were measured in an ELISA plate reader and analyzed with a Biotec microplate reader (GMI, Inc., Winooski, VT).
Preparation of lipid raft fraction
Lipid raft fractions were separated as previously described (45). Briefly, the cells treated with HAF, HAF along with SOD3, or SOD3 alone were harvest and lysed in ice-cold HNE buffer (25 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 6.5, 150 mM sodium chloride, and 5 mM ethylenediaminetetraacetic acid (EDTA), supplemented with 1.5% Triton X-100, 1 mM sodium orthovanadate, 5 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/ml each of aprotinin and leupeptin) for 15 min on ice followed by 10 rounds of homogenization. The lysates were adjusted to 4 ml of 40% sucrose by mixing with 2 ml of 80% sucrose and overlaid with 4 ml of 30% sucrose and 4 ml of 5% sucrose in HNE buffer. The samples were subjected to ultracentrifugation at 37,000 rpm for 18 h using a Beckman SW41 rotor at maximum acceleration and no brake conditions and fractionated into 12 sub-fractions. For the immunoprecipitation (IP) of TLR4, lipid raft fractions, 3, 4, and 5 fractions, were pooled.
Immunoblotting and IP
The goat anti-TLR4 was purchased from Santa Cruz Biotechnology, mouse anti-flottlin-1 and gp91 phox were purchased from BD Bioscience (Franklin Lake, NJ), p47 phox was purchased from Upstate Biotechnology (Waltham, MA). IP and sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel electrophoresis was performed as previously described (45).
FACS analysis
PE-conjugated CD11c, antigen presenting cells (APC)-conjugated CD11b, FITC-congjugated-CD4, PE-conjugated MHC II (I-A), FITC-conjugated CD44, FITC-conjugated CD80, and FITC-conjugated CD86 antibody were purchased from BD Pharmingen, PE-conjugated mouse TLR4 antibody was purchased from eBioscience. To detect the cell-surface MHC II, co-stimulatory molecules, CD80 and CD86, CD44 and TLR4, FACS analysis was performed. The data were analyzed with Cell Quest software (Becton Dickinson, Mountain view, CA).
Confocal microscopy analysis
BMDM were seeded onto coverslips for 12 h in serum-free DMEM. Cells were treated with LPS (100 ng/ml) or 200-kDa HAF (100 μg/ml) in the presence or absence of SOD3, or SOD3 alone. Some experiments used SOD1 (ATGen, Seongnam, Gyeonggi-do, Korea) and SOD2 (ATGen). Cells were washed with serum-free DMEM and incubated with 8 μg/ml FITC-conjugated cholera toxin B (Sigma) on ice for 10 min. Cells were fixed and permeabilized, followed by blocking with 5% goat serum (Dako, Carpinteria, CA). To detect TLR4 translocation to the lipid rafts, the samples were incubated for 1 h with 1:100 diluted PE-conjugated TLR4. The samples were washed thrice with BSA solution and PBS and mounted onto glass slides. For ROS analysis, BMDM were pretreated with 100 units/ml SOD3 or 10 mM NAC for 30 min, followed by HAF or LPS treatment for 1 h. The cells were stained with 5 μM H2DCFH-DA (Sigma) for 15 min at room temperature. Confocal microscopy was performed with the Zeiss LSM 510 microscope (Carl Zeiss Microimaging).
ChIP assay
ChIP assay was performed as previously described (23). Briefly, RAW 264.7 cells (1×107 cells) were used for each chromatin preparation. The chromatin sample from cells cross-linked with formaldehyde and sonicated was precleared with salmon sperm DNA and protein A-sepharose. One-seventh of the sample (1.5×106 cells) was used for IP with anti-p65. Immune complexes were collected with protein A-sepharose beads and eluted. After reverse cross-linking and digestion of proteins with proteinase K, the DNA was purified by phenol/chloroform extraction. PCR was performed using the following promoter primers: TNF-α, 5′(-742)-GAGGCTCCGTGGAAAACTCACTTG, 3′(-547)-GCAGAGCAGCTTGAGAGTTGGGAA; IFN-γ, R(-255)-CTTCACACCATTTGGGGTG, R(0)-TCACCTCTCTGGCTTCCAG; IL-1β, 5′(-338)-CCGCACATCCTGACTTAAAATGTA, 3′(+16)-ACCACTGCAGGGTTTGTTGTC; TGF-β1,: 5′(-238)-GTTGGTCACCGGCTTTAG, 3′(+60)-GGCCTGGCTGTCTGGAGGATC; CIITA, 5′-AGCAAACTTGGGTTGCATGT, 3′-TCCTGGCAGCTATCTCACAA; Mouse HPRT, 5′-CTGCCTCTGCCTCCTAAATG, 3′-CTCCCAGAGGATTCCCAGAT.
Footnotes
Acknowledgments
This work was supported by a National Research Foundation (NRF) Grant: Future-based Technology Development Project funded by the Ministry of Education, Science, and Technology (2010-0002058), Republic of Korea.
Author Disclosure Statement
The authors declare no conflict of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.