
Abstract
Introduction
Mitochondrial Dysfunction in Pancreatitis
Mitochondria as a central regulator of cell death
Apoptosis and necrosis are two main types of cell death (26, 44), the classification of which is based on specific morphological features (Fig. 1). In apoptotic cells, these include reduction in cellular volume; chromatin condensation, nuclear fragmentation, and cell fragmentation into apoptotic bodies; and phagocytic clearance of apoptotic bodies by neighboring cells (in vivo). Necrosis is characterized by a rather unorganized dismantling of cellular organelles and the cell's swelling that culminates in rupture of the plasma membrane, leading to spillage of cellular contents into the extracellular space and thus an inflammatory response.
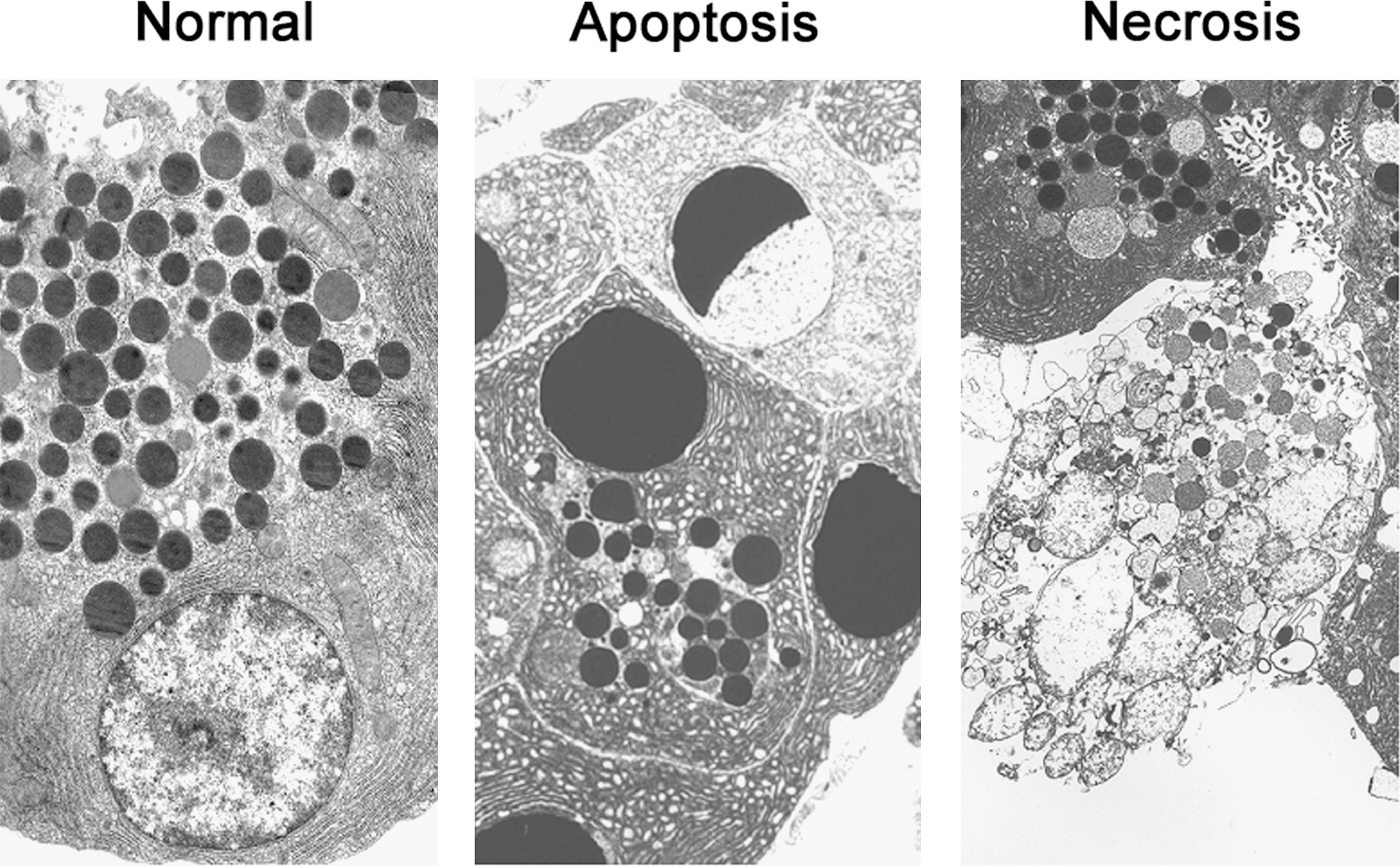
The critical biochemical determinant of apoptosis is activation of a cascade of the cysteine proteases, caspases; other key molecules regulating apoptosis are proteins of the Bcl-2 family (19, 44) and the endogenous inhibitor of apoptosis proteins (IAPs) (49). In contrast to apoptosis, very little is known about molecular mediators of necrosis, apart from the serine-threonine kinases of the receptor-interacting protein (RIP) family (25, 35).
Mitochondria play a central role in regulating cell death (44). Live cells depend on mitochondrial function as these organelles generate most of the cell's energy in the form of ATP supply. Mitochondrial membrane permeabilization (MMP) is a universal trigger of both apoptosis and necrosis and is often considered as the point of no return in the chain of events leading to cell death (21, 24, 44). However, the molecular mechanisms mediating the MMP are not fully understood (21, 24, 44).
Key manifestations of mitochondrial permeabilization triggering apoptotic and necrotic pathways are the release of the mitochondria resident protein cytochrome c (as well as other apoptosis-inducing factors) into the cytosol and mitochondrial depolarization, respectively. Once in the cytosol, cytochrome c interacts with the apoptotic peptidase activating factor 1 and procaspase-9, resulting in the formation of a multiprotein complex (apoptosome) and activation of procaspase-9 (1). Caspase-9, in turn, cleaves and activates effector caspases (e.g., caspase-3), mediating the downstream apoptotic events. On the other hand, loss of the mitochondrial membrane potential, ΔΨm, ultimately leads to ATP depletion and necrosis. Thus, MMP is a central event in both apoptotic and necrotic cell death (Fig. 2).
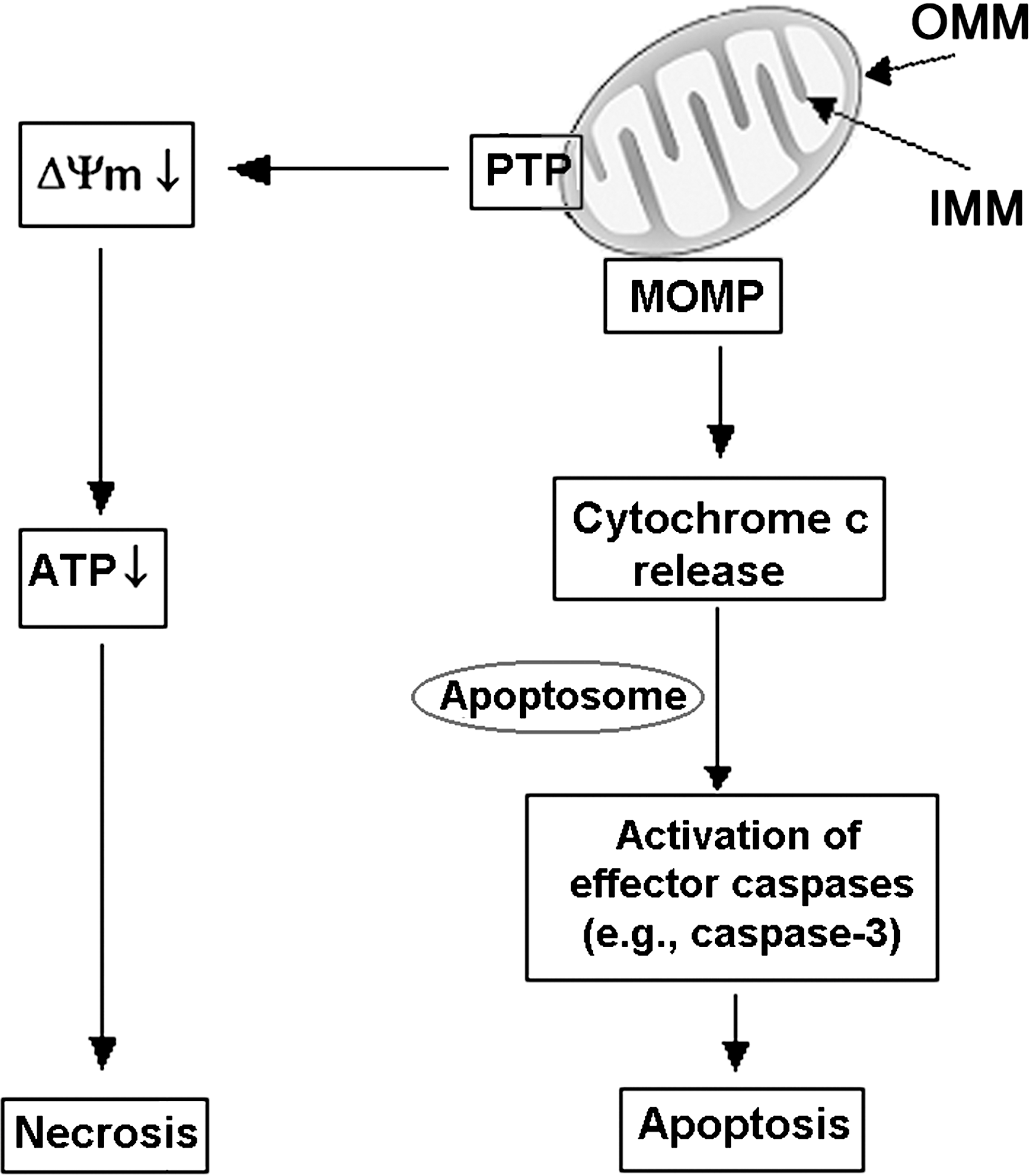
Mitochondria possess two discrete membrane systems, an outer mitochondrial membrane (OMM) in close communication with the cytosol, and an inner mitochondrial membrane (IMM) involved in ion transport, protein import, biogenesis, and energy production (5). The MMP is mediated by two distinct permeability systems (Fig. 3): the permeability transition pore (PTP) and the mitochondrial outer membrane permeability (MOMP) system (44). It is important to discriminate between these two systems as they play different roles in apoptotic and necrotic cell death.

PTP is a multiprotein complex, assembled at the junctions between the inner and outer mitochondrial membranes, that involves proteins of both membranes. The exact composition of PTP is not yet known. The PTP backbone is organized around cyclophilin D (CypD), a resident of the mitochondrial matrix, and likely involves adenine nucleotide translocase (ANT; residing in the IMM) and voltage-dependent anion channel (VDAC; an OMM protein) (44, 45) (Fig. 3A). In response to a number of stimuli (e.g., Ca2+ overload or reactive oxygen species [ROS]), the PTP assumes a high-conductance conformation, allowing unregulated entry of solutes with molecular mass <1500 Da, as well as water, into the mitochondrial matrix. The result is loss of ΔΨm, associated with mitochondrial swelling and rupture of the OMM.
MOMP system represents large channels formed in the OMM by the pro-apoptotic Bax and Bak proteins of the Bcl-2 family; its exact composition and mechanism of activation is yet unknown (Fig. 3B). Although the PTP and MOMP are two distinct permeability systems, their regulatory mechanisms overlap (Fig. 3). Components of PTP, such as ANT or VDAC, regulate MOMP, whereas Bcl-2 proteins regulate PTP (44).
For many years the paradigm for cytochrome c release into the cytosol was that it proceeds through PTP-mediated rupture of OMM. However, recent studies (6, 56) on mice with genetic deletion of CypD, a critical PTP component, revealed that cells isolated from these mice develop normal cytochrome c release and apoptosis in response to various death stimuli. Cytochrome c release in CypD−/− cells occurs in the absence of PTP opening and without OMM rupture. These results convincingly demonstrate that PTP opening is not the only, or even major, mechanism for cytochrome c release. Importantly and in contrast with the lack of any effect of CypD genetic ablation on apoptosis, CypD−/− cells are much more resistant to mitochondrial depolarization and necrosis, compared with the wild-type cells. In particular, neither H2O2 nor Ca2+ (major inducers of necrosis) stimulates mitochondrial depolarization and necrosis in CypD−/− cells. Thus, the current paradigm (24, 44) is that PTP is mostly associated with necrosis, whereas MOMP mediates apoptosis.
Bcl-2 proteins are key regulators of mitochondrial permeabilization
The Bcl-2 family proteins regulate cell death through modulating mitochondrial membrane permeability. Bcl-2 proteins are subdivided into 3 groups on the basis of their Bcl-2 homology (BH) domains (13, 19, 24, 44). The prosurvival members, such as Bcl-2 itself and Bcl-xL, contain four BH domains (BH1–BH4). The pro-apoptotic members, such as Bax and Bak, contain three BH domains, and the BH3-only pro-apoptotic proteins, such as Bad, Bid, or Puma, only contain the BH3 domain.
Each of the three groups of the Bcl-2 family proteins has specific functional roles in the regulation of apoptosis (13, 44). In particular, the pro-apoptotic Bax and Bak selectively promote the formation of MOMP channels through which cytochrome c is released into the cytosol. The prosurvival Bcl-xL and Bcl-2 inhibit apoptosis by sequestering the pro-apoptotic Bcl-2 proteins. The BH3-only proteins play a regulatory role. Some of them activate apoptosis; for example, Bid and Bim directly interact with Bax/Bak, thereby triggering their MOMP-forming function and thus cytochrome c release. BH3 proteins called sensitizers (e.g., Bad) promote MOMP by disrupting the inhibitory interactions between the prosurvival Bcl-xL/Bcl-2 and Bax/Bak. Of note, the role of Bcl-2 proteins in PTP regulation is less studied; it was reported that Bcl-xL and Bcl-2 can inhibit PTP opening, thus preventing the loss of ΔΨm (24, 44). On the other hand, the pro-apoptotic Bax can bind to ANT and VDAC, thus promoting PTP opening (24, 44).
Role of mitochondria in acinar cell death in models of pancreatitis
Apoptosis and necrosis in pancreatitis
Acinar cell death is a major pathological response of pancreatitis. One of the main clinical indicators of outcome in patients with pancreatitis is the amount of necrosis of the gland (81). Greater amounts of necrosis are directly associated with a worse prognosis; thus, in patients with pancreatitis causing detectable necrosis in 50% of the pancreas, the mortality rate approaches 20%. Results in experimental models of acute pancreatitis correlate with the clinical findings. In these models, such as pancreatitis induced in rats or mice by administration of high-dose cerulein (CR) or L-arginine (L-arg), or in mice by feeding them choline-deficient, ethionine-supplemented (CDE) diet, acinar cells have been shown to die through both necrosis and apoptosis (9, 10, 27, 29, 30, 39, 52, 73, 76, 80). Of note, the severity of experimental pancreatitis directly correlates with the extent of necrosis and inversely with that of apoptosis (9, 10, 27, 29, 30, 39, 52, 73, 76, 80). Thus, shifting death responses away from necrosis toward apoptosis may have a therapeutic value.
Characteristics of pancreatic mitochondria
Until very recently, there was little known on the properties of pancreatic mitochondria (77, 90, 91) as compared with other organs, for example, liver mitochondria, the classical object of mitochondriology. We measured in detail the characteristics of isolated pancreatic mitochondria (61) and found, in particular, that they are much more sensitive to Ca2+ than liver mitochondria. Ca2+ at low micromolar concentrations depolarized pancreatic mitochondria through PTP opening; the mitochondria gradually lost ΔΨm even at submicromolar external Ca2+ (see below). Further, we found that the pancreatic mitochondrial PTP has unusual properties, different from the classical (e.g., liver) PTP. A key characteristic of classical PTP opening is mitochondrial swelling, leading to OMM rupture (8, 17, 33). By contrast, Ca2+-induced depolarization of isolated pancreatic mitochondria is not associated with swelling and OMM rupture (61).
Further, Ca2+-induced depolarization causes a dramatic decrease in ROS in pancreatic mitochondria (61), in contrast with ROS burst induced by PTP opening in mitochondria from liver and other organs. Endogenous ROS production in mitochondria is mediated by electrons escaped from the electron transport chain; thus, dissipation of ΔΨm (e.g., by using the protonophore carbonyl cyanide m-chlorophenyl-hydrazone [CCCP]) blocks ROS production (4). The reasons for why the classical PTP opening causes ROS burst, in spite of depolarization, are not clear, but it is likely due to inhibition of mitochondrial antioxidant capacity resulting from swelling (12, 68, 83, 85).
Because of its unusual properties, we termed the pancreatic mitochondrial PTP nonclassical (61).
Mitochondrial permeabilization in experimental acute pancreatitis
We and others showed that MMP is a common and early event in models of pancreatitis, manifest by both mitochondrial depolarization and cytochrome c release (27, 52, 61, 80, 84). In a collaborative study with Robert Sutton's group (Liverpool, United Kingdom), we examined the role of PTP in cell death responses of pancreatitis using CypD−/− mice. CypD genetic ablation inactivated pancreatic mitochondrial PTP and greatly decreased mitochondrial depolarization, resulting in pronounced decrease in necrosis in 3 dissimilar models of pancreatitis (as well as other pathological responses, e.g., hyperamylasemia). These findings (article in preparation) provide compelling evidence that PTP opening is a key mediator of necrosis in pancreatitis. By contrast, the PTP inactivation in CypD−/− mice only partially inhibited cytochrome c release and had little effect on caspase activation and acinar cell apoptosis, indicating that apoptotic cell death in pancreatitis is mostly mediated through PTP-independent pathways.
The role of Ca2+ and ROS in mitochondrial permeabilization and acinar cell death in pancreatitis
Abnormal (global and sustained) increases in acinar cell cytosolic Ca2+, and oxidative stress are key pathological signals associated with acute pancreatitis (16, 46, 70). Elevations of cytosolic Ca2+, which can locally reach low micromolar concentrations (as shown in experimental pancreatitis), are believed to mediate trypsinogen activation and other pathological responses of pancreatitis (70, 74). Oxidative stress is also implicated in the pathogenesis of pancreatitis; however, the roles of ROS and the targets of ROS in the acinar cell have not been elucidated (14, 18, 46).
Both Ca2+ and ROS are major inducers of MMP, leading to cell death in other organs (24, 44). Therefore, we undertook a detailed characterization of the effects of Ca2+ and ROS on ΔΨm and cytochrome c release in both isolated pancreatic mitochondria and the in vitro model of acute pancreatitis, that is, cholecystokinin-8 (CCK)-hyperstimulated acinar cells (61). Elevation of cytosolic Ca2+ in the range from as little as 300 nM to 1–2 μM caused PTP opening in isolated pancreatic mitochondria resulting in loss of ΔΨm. Importantly, the extent of depolarization increased with the duration of mitochondrial exposure to Ca2+ (61). Thus, transient oscillatory increases in cytosolic Ca2+ elicited in acinar cells by physiological concentrations of CCK (or other agonists) have little effect on ΔΨm, whereas sustained elevation of cytosolic Ca2+ associated with pancreatitis should depolarize pancreatic mitochondria. Our findings on isolated pancreatic mitochondria (61) are corroborated by the results in the in vitro model of pancreatitis (27, 61, 80). Supramaximal doses of CCK induce a pronounced loss of ΔΨm, ATP decrease, and acinar cell necrosis; these effects are prevented by chelating the cytosolic Ca2+ with 1,2-bis(o-aminophenoxy) ethoxy-ethane-N′-tetraacetic acid (BAPTA) (61, 80). Similar inhibition of CCK-induced mitochondrial depolarization by BAPTA was also reported by another group (84).
In contrast to Ca2+, exogenous hydrogen peroxide or superoxide-producing system have little effect on ΔΨm in isolated pancreatic mitochondria (61). Mitochondrial antioxidants, such as the inhibitors of complex I, rotenone, and diphenyliodonium (DPI), and the lipid peroxidation inhibitor butylated hydroxytoluene (BHT), all greatly decrease mitochondrial ROS production without affecting ΔΨm (61).
The effects of ROS and Ca2+ on cytochrome c release are drastically different from those on ΔΨm. All the mitochondrial ROS inhibitors applied (e.g., DPI, BHT, and the protonophore CCCP) inhibit cytochrome c release in isolated pancreatic mitochondria (61). Conversely, exogenous superoxide significantly stimulates cytochrome c release from pancreatic mitochondria. Similarly, inhibition of mitochondrial ROS production in intact acinar cells prevents, whereas its stimulation increases, cytochrome c release, with the corresponding inhibitory or stimulatory effects on downstream caspase-3 activation and apoptosis (61). These findings indicate a critical role of ROS in mediating apoptosis in acinar cells (Fig. 4). They are in accord with the recently published study from the Liverpool group (15) that the pro-oxidant menadione greatly stimulates apoptosis in pancreatic acinar cells.

One mechanism whereby ROS may promote cytochrome c release is peroxidation of cardiolipin, a major anionic phospholipid unique to mitochondria, which anchors cytochrome c to the IMM (63, 64, 69). Evidence suggests that dissociation of cytochrome c from cardiolipin is an important first step for cytochrome c release into the cytosol and the induction of apoptosis. Oxidation of cardiolipin by ROS facilitates cytochrome c detachment, increasing cytochrome c level in the intermembrane space and thus its availability for release (64).
Unexpectedly, we found (61) that Ca2+ has a dual effect on cytochrome c release from pancreatic mitochondria, resulting in a nonmonotonous dependence of cytochrome c release on Ca2+ concentration. Lower Ca2+ concentrations (<1 μM) stimulated, whereas greater Ca2+ concentrations inhibited, cytochrome c release. That is, the amount of cytochrome c released from pancreatic mitochondria was maximal at ∼1.0 μM Ca2+ and decreased at greater Ca2+ concentrations (61). We further showed that the inhibition of cytochrome c release at greater Ca2+ concentrations is due to blockade of mitochondrial ROS production caused by the loss of ΔΨm. Thus, the explanation for the dual effect of Ca2+ is that Ca2+ per se stimulates cytochrome c release, whereas Ca2+-induced depolarization inhibits cytochrome c release through inhibiting ROS. In accord with the results on isolated pancreatic mitochondria, we found that Ca2+ stimulates whereas mitochondrial depolarization inhibits cytochrome c release, caspase activation, and apoptosis in intact acinar cells (Fig. 4).
The permeability system that mediates Ca2+-induced cytochrome c release from pancreatic mitochondria is likely the MOMP, involving the pro-apoptotic proteins Bax and Bak. Indeed, as stated above, in pancreatic mitochondria Ca2+-induced cytochrome c release occurs in the absence of OMM rupture.
Our finding that in pancreatic mitochondria, depolarization negatively regulates cytochrome c release (61) is the first demonstration of a negative feedback between mitochondrial signals that promote necrotic (ΔΨm loss) and apoptotic (cytochrome c release) cell death pathways. For example, in liver mitochondria Ca2+ only stimulates cytochrome c release (as well as depolarization), resulting in stimulation of both apoptosis and necrosis (69). As stated above, these differences could be due to the nonclassical properties of the pancreatic mitochondrial PTP. In liver, Ca2+-induced PTP opening causes ROS burst, promoting cytochrome c release, whereas PTP opening in pancreatic mitochondria blocks ROS production, thus inhibiting cytochrome c release.
The implication of this negative regulatory mechanism for cell death responses, in general, is that loss of ΔΨm not only mediates necrosis but at the same time may also limit apoptosis (Fig. 4). Specifically for pancreatitis, the inhibition of cytochrome c release by Ca2+-induced mitochondrial depolarization suggests a molecular mechanism underlying the inverse correlation between necrosis and apoptosis observed in experimental models of acute pancreatitis (9, 10, 29, 30, 39, 52, 73, 76, 80).
Thus, the pattern of acinar cell death in pancreatitis is regulated at the mitochondrial level by interplay between Ca2+, ΔΨm, and ROS. Physiological doses of CCK only induce transient depolarization, with no decrease in ATP and no necrosis. In contrast, pathological, sustained increases in cytosolic Ca2+ cause acinar cell necrosis in pancreatitis through opening of PTP, which results in drop in ΔΨm and, ultimately, cellular ATP. Therefore, stabilizing mitochondria against loss of ΔΨm and ROS decrease may represent a therapeutic strategy to shift the death response of pancreatitis away from necrosis and thus mitigate disease severity.
Role of Bcl-2 proteins in regulating apoptosis and necrosis in experimental acute pancreatitis
To assess the role of Bcl-2 proteins in the regulation of death responses in pancreatitis (80), we first measured changes in pancreatic levels of various Bcl-2 proteins and found that the prosurvival Bcl-xL is markedly upregulated in all models of acute pancreatitis examined, namely, pancreatitis induced by CR in rats and mice, by L-arg in rats, and by CDE diet in mice. Another major prosurvival protein, Bcl-2, is only increased in the rat CR model but not the other pancreatitis models. Upregulation of the pro-apoptotic Bak is mostly in L-arg pancreatitis, and there are no changes in pancreatic levels of Bax, another key pro-apopotic member of the Bcl-2 family. Increases in Bcl-xL occur as early as 30 min after the induction of pancreatitis. Importantly, the increases in total pancreatic Bcl-xL (and Bcl-2) protein are associated with corresponding increases in their levels in mitochondria (80), the principal site of the effects of Bcl-2 family proteins on death responses (24, 44).
To investigate the functional role of Bcl-xL and Bcl-2 in pancreatitis, we next applied (80) small-molecule Bcl-xL/Bcl-2 inhibitors, HA14-1 and BH3I-2′, which recently became a major tool in studying the roles of Bcl-2 family proteins in death responses (20, 86, 87). Of note, the two inhibitors are structurally different (20, 86), and they inactivate both Bcl-xL and Bcl-2 due to similar structures of the catalytic groove of these proteins (67). We also measured the effects of Bcl-xL knockdown with siRNA on death responses in the in vitro model of pancreatitis, that is, CCK-hyperstimulated acinar cells (80).
A key and unexpected finding in these experiments was that Bcl-xL/Bcl-2 inactivation with the pharmacologic inhibitors or siRNA markedly facilitated the CCK-induced necrosis but not apoptosis in the in vitro model of pancreatitis. In accord with the in vitro results on acinar cells, we observed (80) that the extent of Bcl-xL/Bcl-2 upregulation in animal models of pancreatitis inversely correlates with the amount of pancreatic necrosis but not apoptosis (Fig. 5).

These findings are important because, in general, the effects of Bcl-xL and Bcl-2 on necrosis are poorly understood, contrasting their well-established antiapoptotic function (19, 44). On the other hand, as stated above, necrosis is a major determinant of the severity of pancreatitis, whereas apoptosis is associated with milder disease in experimental models (29, 39, 52, 80).
To gain insight into the mechanisms underlying the effects of Bcl-xL/Bcl-2 on cell death responses in pancreatitis, we have shown that Bcl-xL/Bcl-2 inactivation in acinar cells has drastically different effects on ΔΨm versus cytochrome c release and subsequent caspase activation (80). Both the Bcl-xL/Bcl-2 inhibitors and Bcl-xL siRNA potentiate CCK-induced ΔΨm loss and the resulting necrosis, while essentially blocking the CCK-induced caspase-3 activation and apoptosis. Therefore, Bcl-xL/Bcl-2 inactivation shifts the pattern of death response in the in vitro model of pancreatitis toward necrosis. As discussed above, these results can be explained by the negative regulation of cytochrome c release (and subsequent apoptosis) by mitochondrial depolarization.
Thus, our findings indicate that Bcl-xL/Bcl-2 upregulation is a key protective mechanism against necrosis in pancreatitis (Fig. 5).
Interestingly, in cancer cells the effects of Bcl-xL/Bcl-2 inactivation on death responses differ from what we found in pancreatic acinar cells. In various cancer cells, including pancreatic cancer, Bcl-xL/Bcl-2 inhibitors greatly stimulate apoptosis and thus are considered a promising tool for cancer treatment (50, 55, 60). The different effects of Bcl-xL/Bcl-2 inactivation in cancer versus pancreatitis are likely due to the different roles of mitochondria in cancer and normal cells. In cancer cells, ATP production is mostly through glycolysis and, therefore, loss of ΔΨm does not result in severe ATP depletion (66). Further, as we showed for pancreatic cancer cells (82), mitochondrial depolarization does not limit cytochrome c release in cancer cells. Thus, the major effect of Bcl-xL/Bcl-2 inhibitors in cancer cells is increased apoptosis resulting from stimulation of cytochrome c release. Differently, our results demonstrate that the predominant effect of the small-molecule Bcl-xL/Bcl-2 inhibitors in pancreatitis is ATP depletion and necrosis. Thus, Bcl-xL/Bcl-2 inhibition, which is considered a promising strategy to stimulate death of cancer cells, would likely increase necrosis and the severity of pancreatitis.
Molecular mechanisms regulating acinar cell death in pancreatitis
The findings discussed above demonstrate that mitochondrial permeabilization is a central regulator of the pattern of acinar cell death (apoptosis vs. necrosis) and the severity of pancreatitis. In turn, the extent of mitochondrial dysfunction in pancreatitis is determined by the interplay between Ca2+, ΔΨm, ROS, and Bcl-2 proteins. The prediction is that greater mitochondrial depolarization or lower pancreatic Bcl-xL/Bcl-2 levels would facilitate necrosis and limit apoptosis, thus making the disease more severe; in other words, these parameters could serve as a prognostic factor for a more severe, necrotizing pancreatitis. In contrast, minor loss of ΔΨm and higher Bcl-xL/Bcl-2 levels should be associated with a milder disease. Thus, approaches aimed to inhibit PTP opening or upregulate (or stabilize) the Bcl-xL/Bcl-2 proteins may present a novel strategy to prevent or attenuate necrosis in pancreatitis. Indeed, our findings in CypD−/− mice (see above) show that PTP inactivation greatly ameliorates necrosis and other pathological responses of experimental pancreatitis.
The balance between apoptosis and necrosis in pancreatitis is regulated not only at the mitochondrial level (Fig. 4) but also at the level of caspase activation, as we have recently shown (52). In that study we utilized the differences in death responses between the rat and mouse models of CR pancreatitis. The former represents a mild disease characterized by low necrosis and relatively high apoptosis, whereas the mouse model is more severe, with high necrosis and very little apoptosis. We found that caspases are rapidly and greatly activated in rat CR pancreatitis, but not at all in the mouse model. We further showed (52) that the caspase activation block occurs downstream of cytochrome c release, and that one mediator of this block is X-linked inhibitor of apoptosis protein (XIAP), a major member of the family of endogenous IAPs (49), which negatively regulate caspase activities. XIAP is rapidly and fully degraded in rat CR pancreatitis but remains intact in the mouse model. [Interestingly, we did not observe a significant difference in properties of rat vs. mouse pancreatic mitochondria, e.g., in their responses to Ca2+ overload (Ref. (61) and data unpublished).]
We further showed (52) that specific caspase inhibitors not only inhibit apoptosis (in the rat model) but also increase necrosis and worsen CR pancreatitis; conversely, XIAP inhibition (in the mouse model) removes caspase activation block, stimulates apoptosis, and decreases the amount of necrosis. These findings provide strong evidence that caspases protect from acinar cell necrosis in pancreatitis and that it is possible to modulate disease severity by shifting the necrosis/apoptosis balance.
In addition to mitochondria and capases, other factors, undoubtedly, are involved in the regulation of acinar cell death in pancreatitis. For example, our data (52) suggest the involvement of serine-threonine kinases of the RIP family, important mediators of necrosis-like cell death (25, 35). Genetic ablation of RIP3 was recently reported (35) to greatly decrease necrosis in CR pancreatitis. The pronecrotic effect of RIP3 is likely mediated through mitochondrial permeabilization. Another regulator of cell death responses of pancreatitis is p53; we have recently shown (57) that in CR pancreatitis, neutrophils infiltrating the pancreas suppress acinar cell apoptosis (and thus facilitate necrosis) through down-regulating p53/caspase-2 pathway. There is also some evidence that acinar cell death in pancreatitis is regulated by lysosomal pathways (7, 32, 75, 88).
Lysosomal Dysfunction and Impaired Autophagy in Pancreatitis
The lysosomal/autophagic pathway
The lysosome is the principal digestive organelle of eukaryotic cells serving both to eliminate obsolete components of the cell itself (autophagy) and to degrade material taken up from outside (endocytosis and phagocytosis) (48, 72). Lysosomes contain many different types of hydrolytic enzymes, including proteases, lipases, nucleases, glycosidases, phospholipases, phosphatases, and sulfatases, that usually exert their maximal enzymatic activity at low pH. The acidic milieu of lysosomes (pH ∼5) is maintained by a vacuolar ATPase that pumps protons from the cytosol into the lysosomal lumen (48). The lysosomal membrane protects the rest of the cell from hydrolases and is itself protected because integral lysosomal membrane proteins, such as LAMP-1 and -2, are heavily glycosylated and hence resist digestion (72).
Cathepsins represent a major class of lysosomal proteases (22, 37, 43, 71). They are synthesized as inactive proforms that are activated through proteolytic processing (maturation) during cathepsins' transit along the endolysosomal pathway. Procathepsins first undergo partial cleavage into an intermediate, single-chain form, which is further cleaved in late endosomes and lysosomes to form fully mature, double-chain cathepsins (22, 37, 43, 71). Defective cathepsin processing may lead to inefficient lysosomal protein degradation and impaired autophagic flux (see below).
Autophagy (more precisely, its major pathway, macroautophagy) is a multistep, lysosome-driven, adaptive process by which cells degrade cytoplasmic organelles and long-lived proteins (40, 47, 53, 54). This process begins when an isolation membrane (phagophore) wraps around material to be sequestered, for example, mitochondria, forming a double-membrane vacuole, the autophagosome (Fig. 6). Autophagy progression/resolution (flux) proceeds through fusion of autophagosomes with lysosomes, generating autolysosomes in which sequestered material is degraded (Fig. 6). Autophagy is controlled by a series of evolutionary conserved ATG genes (40, 47, 53). The product of the Atg8 gene, LC3 protein (microtubule-associated protein 1 light chain 3), is present in cytosol in the LC3-I form; upon autophagy induction it is modified (lipidated) to LC3-II form, which uniquely localizes to autophagosomal membranes (38, 40, 53). Thus, the LC3-I to LC3-II conversion (measured by immunoblot) or the number of LC3-positive vacuoles (measured by immunofluorescence) is used to monitor autophagy (41, 54). The efficiency of autophagic process is measured by the rate of degradation of long-lived proteins (41, 54).
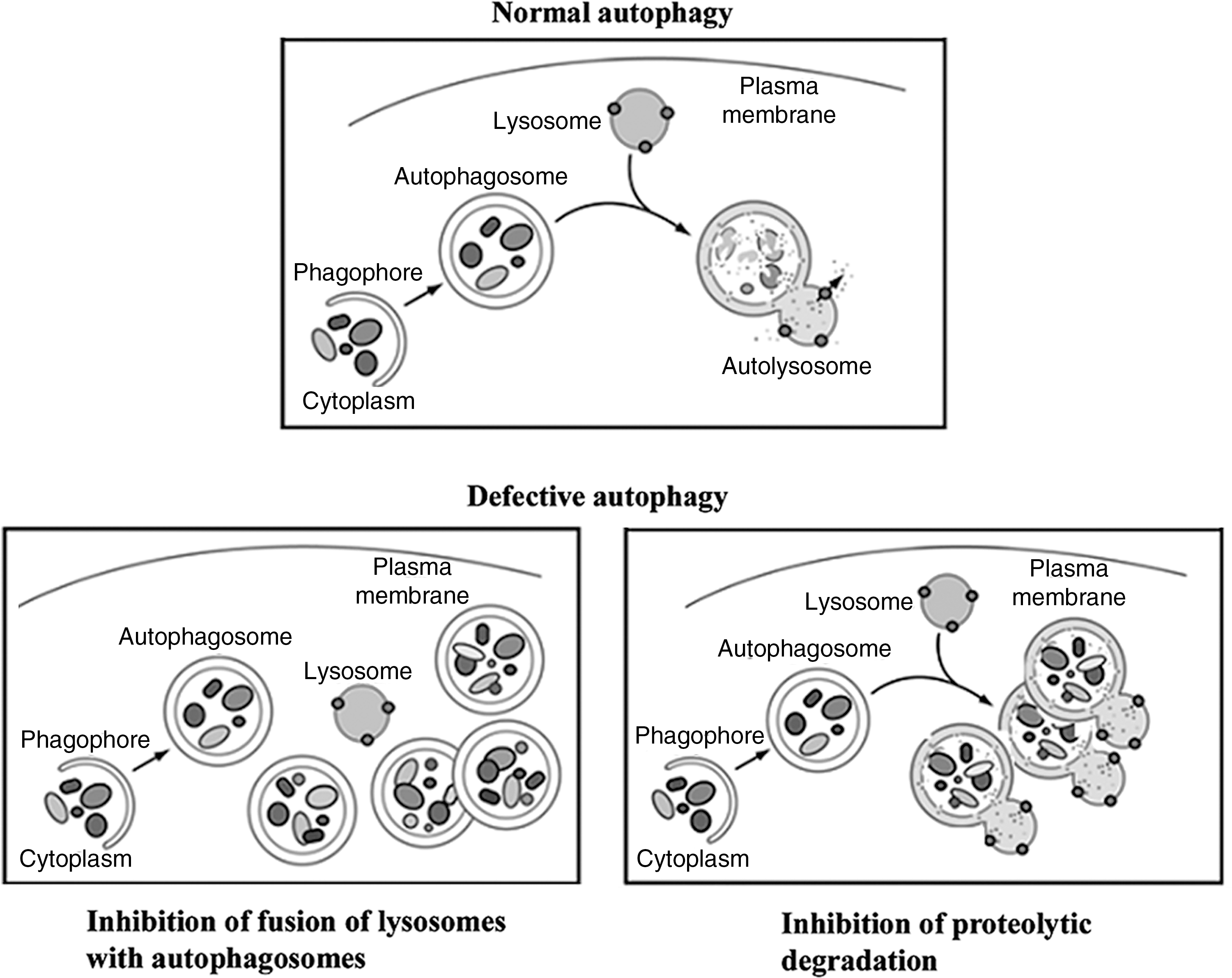
Starvation is a strong inducer of normal, physiological autophagy, which generates vital nutrients (primarily amino acids) critical for cell survival. Physiological autophagy triggers efficient turnover of autophagosomes into autolysosomes and stimulates protein degradation. Many diseases are associated with impaired autophagic flux, which is manifest, first of all, by vacuole accumulation (47, 59). Impairment or retardation of the autophagic flux could be caused by a defective fusion of autophagosomes with lysosomes or by inefficient degradation of cellular constituents in autolysosomes (Fig. 6) that may result from defective cathepsin activities (47, 59).
Lysosomal dysfunction, resulting in impaired autophagic flux, mediates acinar cell vacuolation in experimental pancreatitis
Accumulation of large vacuoles in acinar cells is a long-noted feature of both experimental and human pancreatitis (2, 3, 11, 36, 42, 58, 89); however, the mechanism of their formation and relation to other pathological responses of pancreatitis remained unclear. In a recent study (31, 51) we showed that these vacuoles have characteristics of autophagic vacuoles (e.g., a double membrane and the presence of LC3-II). Compared with starvation, not only does pancreatitis induce many more vacuoles, but they are also strikingly larger. Most contain partially digested material, in particular, zymogen granules (ZG). These features suggest that autophagic flux is impaired in pancreatitis.
To prove this we used several approaches (51). We found that the rate of long-lived protein degradation is markedly stimulated in starved acinar cells but is ∼40% inhibited in the in vitro pancreatitis, providing direct evidence for inefficient autophagy. The decrease in autophagic protein degradation is associated with accumulation of LC3 dots in acinar cells. Another manifestation of retarded autophagic flux is the accumulation of lysosomal markers, such as Rab7 and LAMP-2, in the ZG-enriched (i.e., heavier) pancreatic subcellular fraction. This is consistent with accumulation of heavy autolysosomes (containing incompletely degraded material) due to impaired, retarded autophagic flux. In contrast, starvation does not affect the subcellular distribution of lysosomal markers in pancreas.
We considered two mechanisms that might underlie autophagic flux impairment in pancreatitis (51). One possibility could be a blockade of autophagosome fusion with lysosomes, which occurs in lysosomal diseases. However, the presence of a great number of vacuoles with partially degraded material in pancreas of animals with pancreatitis (as well as in human tissue) argues against this possibility. Indeed, colocalization of Rab7 or LAMP-2 with LC3-II increases in models of pancreatitis, which would not occur if autolysosome formation were blocked.
Therefore, we next considered decreased activity of lysosomal hydrolases, that is, cathepsins, as a mechanism for the retarded autophagic flux in pancreatitis. To this end, we measured (51) the effects of starvation and pancreatitis on the processing and activities of two major lysosomal hydrolases, cathepsins (Cat) B and L. Both cathepsins are relevant for pancreatitis: CatB proteolytically converts trypsinogen to trypsin (32, 75, 79), whereas CatL degrades both trypsinogen and trypsin (51, 88). We found that starvation has little effect on pancreatic activities of CatB and CatL and their processing into fully mature, active forms. In contrast, pancreatitis causes a dramatic decrease in enzymatic activities of both cathepsins in the lysosome-enriched pancreatic subcellular fraction, and accumulation of their immature forms. These findings indicate that defective cathepsin processing and activities mediate the inefficient lysosomal degradation in pancreatitis, resulting in retarded autophagic flux. In fact, pharmacologic inhibition of CatB or CatL, or both is sufficient to cause acinar cell vacuolation (51).
It is possible that pancreatitis, in addition to impairing the autophagic flux, also affects the initial stages of autophagy, that is, autophagosome formation; this remains to be investigated.
Lysosomal/autophagic dysfunction mediates the pathological, intra-acinar accumulation of active trypsin in pancreatitis
Our findings (51) also provide potentially important insights into the mechanism of the pathological, intra-acinar trypsinogen activation, considered a critical initiating event in both human and experimental acute pancreatitis (65, 75, 79). The immunofluorescence data (51) show that autophagic vacuoles represent one site of trypsinogen activation, as its marker partially colocalizes with both LC3-II and LAMP-2. Trypsinogen activation in acinar cells is completely prevented by 3-methyladenine, a blocker of autophagy induction; moreover, we notice that a number of dissimilar pharmacological inhibitors of trypsinogen activation are all inhibitors of various stages of autophagy. The link between autophagy and trypsinogen activation has been first indicated by a recent study from Ken-ichi Yamamura's group on Atg5 null mice (34), which proposed enhanced autophagy as the underlying mechanism. However, our data (51) show that it is not the autophagy induction per se but its impairment (due to defective lysosomal degradation) that is responsible for the pathological manifestations of pancreatitis. Indeed, there is no increase in pancreatic trypsin activity even after prolonged starvation of rats or mice, which greatly stimulates autophagy.
For many years, the paradigm for the pathological, intra-acinar trypsinogen activation has been the colocalization hypothesis (75, 79) stating that in pancreatitis, CatB becomes missorted and thus colocalizes with trypsinogen in some (undefined) ZG-containing compartment(s). However, our immunoblot data show no CatB redistribution from the lysosome-enriched to the ZG-enriched pancreatic subcellular fraction. Instead, our data suggest that the intra-acinar accumulation of active trypsin is a result of deficient lysosomal protein degradation, in particular, a disbalance between CatB and CatL in pancreatitis (Fig. 7). Indeed, we (51) and the groups of Markus Lerch and Walter Hallangk (32, 88) showed that these cathepsins have opposite roles in trypsin accumulation: whereas CatB converts trypsinogen to trypsin, CatL does not activate trypsinogen but, on the contrary, degrades both trypsin and trypsinogen. We propose (51) that in pancreatitis, because of defective lysosomal function, CatL activity is not sufficient to degrade activated proteases (such as trypsinogen), resulting in trypsin accumulation in autophagic vacuoles (Fig. 7). Indeed, a specific CatL inhibitor markedly increases trypsin activity in acinar cells (51).

Our findings indicate that colocalization of trypsinogen and CatB is not in itself pathological, as it occurs during physiological autophagic process both in normal conditions and with starvation; moreover, our immunoblot data (51) indicate that there is no increase in CatB missorting in the experimental models of pancreatitis tested. Alternatively, we suggest that deficient lysosomal degradation caused by impaired cathepsin processing [rather than CatB missorting (75, 79) or excessive autophagy (34)] is a dominant mechanism for the intra-acinar accumulation of active trypsin in pancreatitis, thus representing a paradigm shift. It may be more appropriate to refer to the hallmark response of pancreatitis as intra-acinar trypsin accumulation rather than trypsinogen activation.
Thus, our findings indicate that autophagic flux is impaired in acute pancreatitis, mediating two key pathological responses of this disease: acinar cell vacuolation and the intra-acinar trypsin accumulation (Fig. 8). Further, autophagy impairment may be a manifestation of a more general phenomenon, indicating that the endolysosomal traffic is disordered in acute pancreatitis. Indeed, trypsinogen activation has been recently reported (78) in endocytic vacuoles in response to CCK hyperstimulation of acinar cells. A number of characteristics of pancreatitis are similar to those observed in lysosomal disorders (which are mostly caused by mutations that inactivate lysosomal hydrolases or affect their delivery to lysosomes)—such as block of autophagy, impaired maturation of cathepsins, decreased protein degradation, cell vacuolation and death, and the inflammatory response (59). Thus, our findings indicate that acute pancreatitis has features of a lysosomal disease, which should be taken into consideration in designing strategies to treat or mitigate pancreatitis.

Conclusion
Our recent findings discussed in this review show the importance of mitochondrial and lysosomal dysfunctions for the mechanism of acute pancreatitis, mediating key pathological responses, that is, apoptosis, necrosis, trypsinogen activation, and acinar cell vacuolation. These results need to be followed by detailed investigations into the molecular events that underlie the disorders described here as well as how these events are connected to each other and other pathological processes that occur during pancreatitis, for example, endoplasmic reticulum stress and inflammation. Such studies are necessary to develop potential strategies for therapy that are currently absent for this serious disease.
Footnotes
Acknowledgments
Department of Veterans Affairs; NIH R01 grants DK59936 and AA19730; Southern California Research Center for Alcoholic Liver and Pancreatic Diseases and Cirrhosis (NIH P50AA11999).