
Abstract
Alzheimer's disease (AD) is the most common cause of dementia and a progressive neurodegeneration that appears to result from multiple pathogenic mechanisms (including protein misfolding/aggregation, involved in both amyloid β-dependent senile plaques and tau-dependent neurofibrillary tangles), metabolic and mitochondrial dysfunction, excitoxicity, calcium handling impairment, glial cell dysfunction, neuroinflammation, and oxidative stress. Oxidative stress, which could be secondary to several of the other pathophysiological mechanisms, appears to be a major determinant of the pathogenesis and progression of AD. The identification of oxidized proteins common for mild cognitive impairment and AD suggests that key oxidation pathways are triggered early and are involved in the initial progression of the neurodegenerative process. Abundant data support that oxidative stress, also considered as a main factor for aging, the major risk factor for AD, can be a common key element capable of articulating the divergent nature of the proposed pathogenic factors. Pathogenic mechanisms influence each other at different levels. Evidence suggests that it will be difficult to define a single-target therapy resulting in the arrest of progression or the improvement of AD deterioration. Since oxidative stress is present from early stages of disease, it appears as one of the main targets to be included in a clinical trial. Exploring the articulation of AD pathogenic mechanisms by oxidative stress will provide clues for better understanding the pathogenesis and progression of this dementing disorder and for the development of effective therapies to treat this disease. Antioxid. Redox Signal. 16, 974–1031.
I. Introduction
A. Overview of Alzheimer's disease and scope of the review
Population demographics are rapidly changing as a result of the increasing life expectancy with a fast increase of aged population, favoring prevalence of dementing disorders. As a group, the main risk factor for AD is age (14, 196, 256, 412, 617). In fact, the incidence of sporadic AD increases exponentially as function of age after the sixth decade of life (256). Epidemiologic data identify two different AD populations depending on age of appearance. The named late onset or sporadic AD is the most prevalent group accounting for over 95% of cases (256). According to the Alzheimer's Association, 11–16 million cases of AD are projected just in the United States by 2050, and 95%–99% of AD cases, besides the rare familial AD, will be observed in the older population, with prevalence above 30% for individuals older than 85 years.
There is near consensus that AD is not a unique nosological entity, as considerable heterogeneity exists in its risk factors, pathogenesis, and neuropathological findings (294). Even more, a mixture of AD and a second pathological identity is present in most dementia patients; being the most frequent, the coexistence of AD and cerebrovascular disease. For that reason, at present days, the recommended preventive measures for AD are similar to those for cerebrovascular diseases (165, 254, 421).
Despite intensive research in neuropsychological evaluation, biomarkers, and imaging techniques, AD still remains as an exclusion diagnosis. Currently, clinical evaluation allows for the diagnosis of probable AD. However, the unequivocal AD diagnosis requires confirmation by anatomopathological analysis, in which the observation of amyloid β (Aβ) plaques and fibrillary tangles in defined regions of the brain, in conjunction with the clinical evolution of the patient, constitute the gold standard (294, 569). AD patients are identified by neuropsychological testing to evaluate neurocognitive performance in various areas and by assessing their cognitive decline and impairment on everyday life activities. During the last decade, an early stage of cognitive impairment without dementia has been defined. On the basis of histopathology, imaging, and cognitive evaluation scores, mild cognitive impairment (MCI), an intermediary stage between a cognitively intact person and AD, is considered an early stage of AD progression (501). MCI is further divided into two broad subtypes: amnesic MCI (affects memory) or nonamnesic MCI (602). The rate of amnesic MCI conversion to AD is roughly 10%–15% per year.
As a neurodegenerative disease, the pathology of AD is characterized by the presence of senile plaques, a heterogeneous set of aggregated Aβ, and by the presence of neurofibrillary tangles (NFTs), constituted by highly phosphorylated tau proteins. In addition, the histopathology involves synapse loss, neuronal loss (hippocampus, entorhinal, and temporoparietal cortex being the most affected brain regions) (656), and glial cell activation (257, 294, 319, 669, 674). Already in his original proposal, Alzheimer stated that these histopathological lesions were a marker of an upstream process instead of the disease cause (163). However, the most popular hypothesis for AD, the amyloid cascade hypothesis, still views Aβ as the cause of the neurodegenerative changes, stating that the first event of the pathogenic cascade of AD depends on Aβ accumulation.
On the 1980s, Aβ and the whole gene of amyloid precursor protein (APP) were characterized on the chromosome 21 (628). Once defined as one of the main components of plaques, Aβ was quickly associated to the early-onset, inherited autosomal dominant dementia (308, 546) familial AD. Later, mutation on two other genes encoding for enzymes processing APP, presenilin 1 (PS1) (441) and presenilin 2 (PS2) (714), were also shown to cause familial AD. PS mutations affect APP processing, resulting in an increasing amount of Aβ available to aggregate (214, 560). The most prominent feature of familial AD is its young age of appearance, as early as the fourth decade of life. Finally, the presence of apolipoprotein E4 (ApoEɛ4) was associated to sporadic AD, which could result in an increase of production or reduction of removal of Aβ (Fig. 1). In addition to these four original genes involved in AD, recent studies show that other genes could be also involved, including ADAM10 (316), ATXN1 (718), CD33 (447), as well as polymorphisms of various receptors, like alpha(2)-macroglobulin (270) and Vitamin D receptors APA1 and TAQ1 (349) and cytokine polymorphisms, such as interleukins (ILs), IL1β, IL1α, IL-6, and tumor necrosis factor α (TNFα) (219, 229, 518, 715).
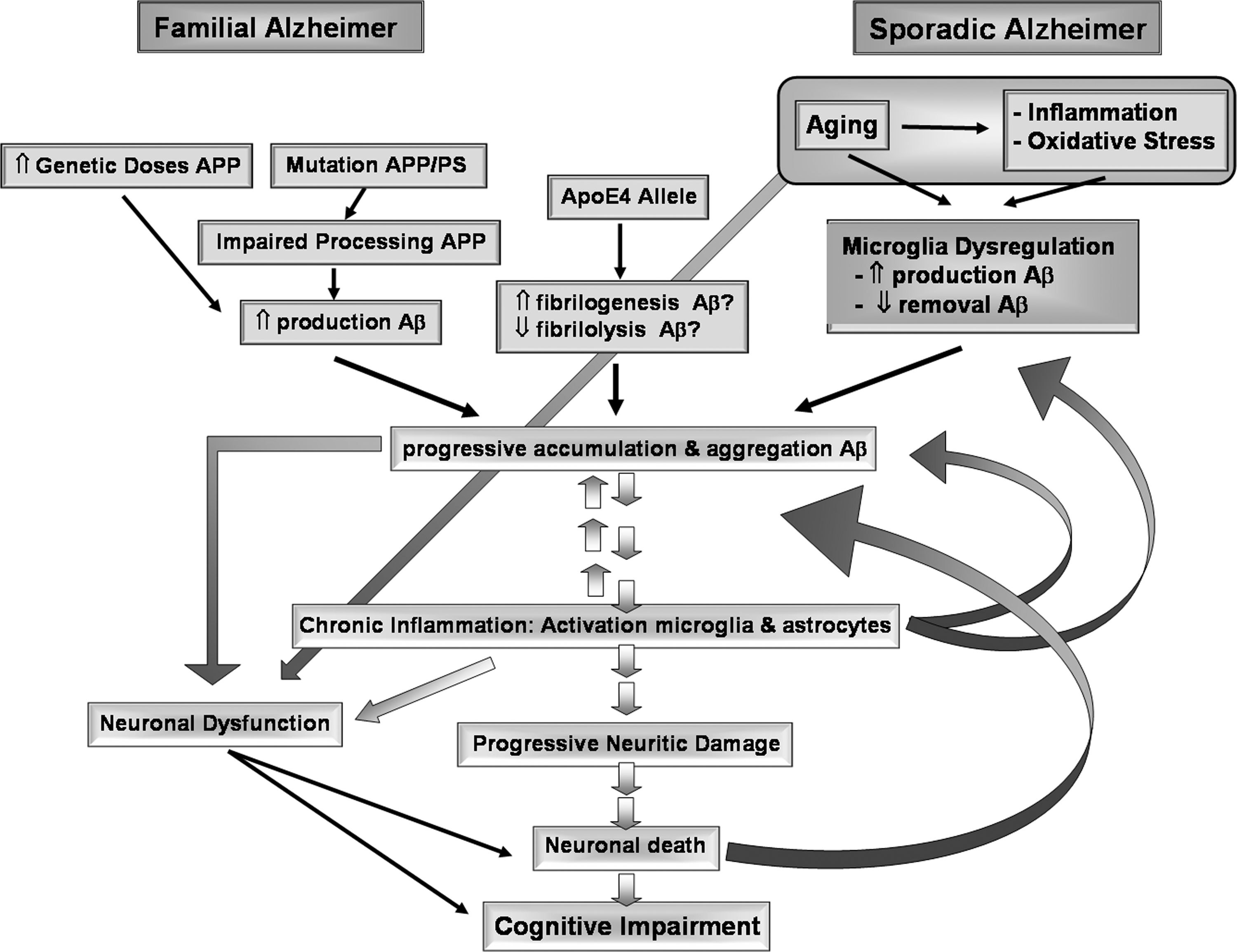
Similarities between familial and sporadic AD prompted the notion that both diseases share a common pathological mechanism dependent on Aβ. However, familial AD comprise around 2% of patients, and appears unreasonable to extrapolate this etiological mechanism to the sporadic form dismissing the possibility that such amyloidosis could be a secondary event to a totally different etiology (617). Indeed, familial AD and sporadic AD could represent the final clinical stages of different diseases (294, 421).
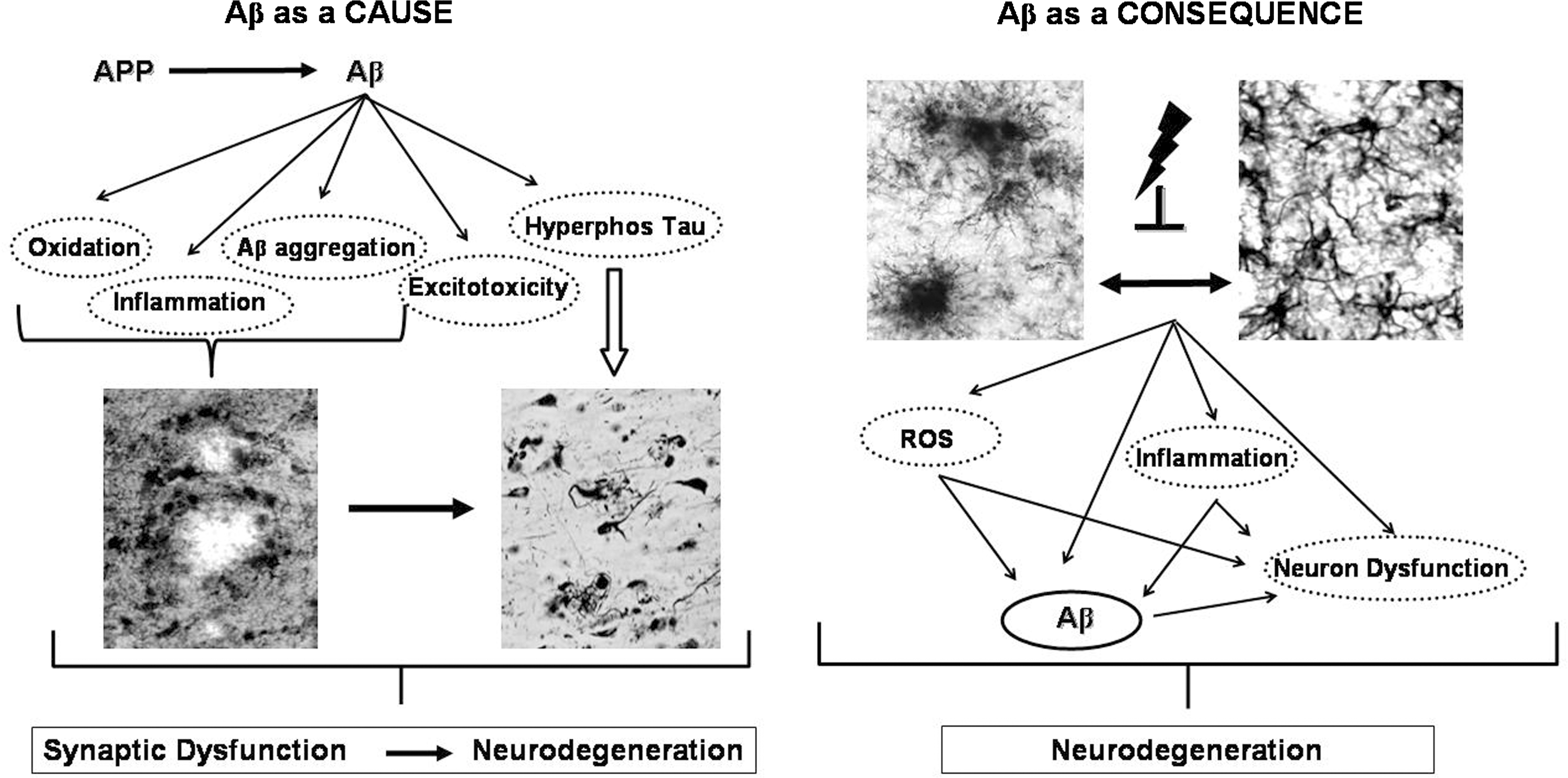
The precise mechanism by which Aβ produces synaptic impairment and neurodegeneration is still under intense discussion. Previous studies led us to propose that glial cell dysfunction is involved in the pathogenics of AD, through the generation of a cytotoxic environment (669) that would favor both Aβ aggregation and neuronal dysfunction and neurodegeneration. Over the years, other hypotheses have been proposed to explain the pathogenesis of AD. In addition to glial cell dysfunction (section I.C), these theories include oxidative stress (section II.A); protein misfolding/aggregation (section II.F), which can result in both Aβ-dependent senile plaques and tau-dependent NFTs (section II.B); mitochondrial dysfunction and impairment of calcium metabolism (section II.C); and neuroinflammation (section II.D), among others. Most of these hypotheses are not mutually exclusive, but could co-participate. Moreover, instead of the highly sequential view regarding the progression of AD traditionally held, an emerging view is that several mechanisms co-exist simultaneously affecting each other at multiple levels, establishing triggering loops that feedback on the cascade of degenerative changes.
On the other hand, despite such mechanistic diversity, common factors can be articulated in a convergent way for generating the disease, one of the axes being glial cell dysfunction (Fig. 2) (669) and another, oxidative stress. In this review, we discuss evidence supporting the role of oxidative stress in the pathogenesis and progression of AD, focusing on elements that allow recognition of oxidative conditions as a common factor for several biological mechanisms. Such recognition is especially relevant for therapeutic strategies capable of targeting effectively on common mechanisms responsible for disease evolution.
B. Oxidative stress: General concepts
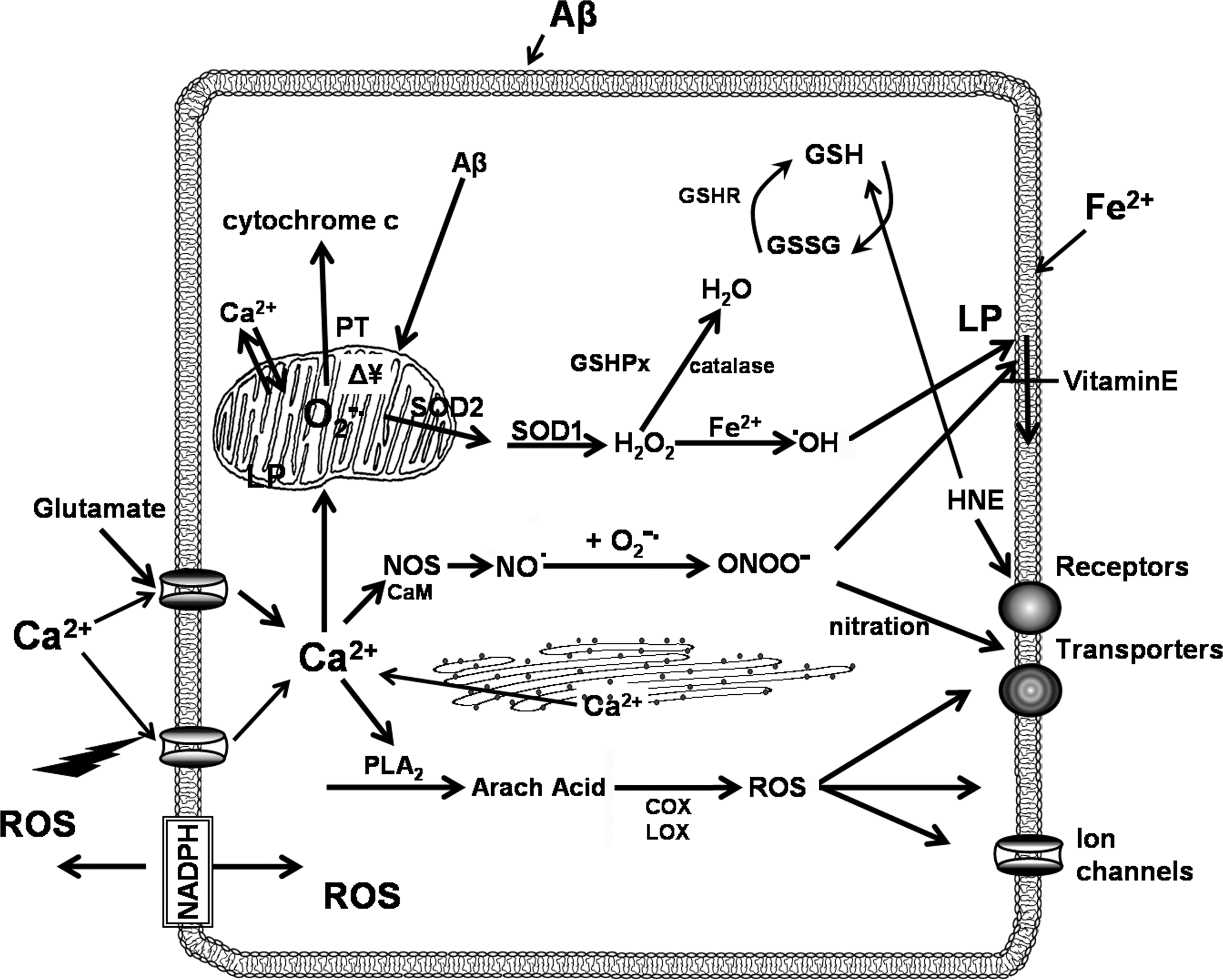
Oxidants, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS), are normally produced, although at low concentrations, as part of aerobic metabolism. ROS include superoxide radical anion (O2 ·−), hydrogen peroxide (H2O2), and hydroxyl radical (·OH), among others. O2 ·− can be dismutated to H2O2 and oxygen by superoxide dismutase (SOD), an antioxidant enzyme. These reactive species can accomplish functions as intracellular messengers. Nevertheless, reactive species, especially when produced at high concentration, can result in oxidative stress associated with cell damage (Fig. 3). O2 ·− can act as an intermediate in the generation of more reactive ROS, like hypochlorous acid and ·OH; by reaction with nitric oxide (NO·), it can form the highly oxidizing agent peroxynitrite (ONOO−). NO· is a free radical gas that participates in the physiological regulation of cerebral blood flow and neurotransmission, but also exhibits pro-oxidative, cytotoxic effects by inducing oxidative and nitrosative stress (95, 157). Both NO· and ONOO− are relatively long-lived diffusible oxidants, thus RNS/ROS-mediated damage may spread over relative long distances. At physiological pH, ONOO− can be protonated to form peroxynitrous acid (ONOOH), which then generates ·OH and nitrate (NO2 ·), leading to lipid peroxidation and nitration of aromatic amino acids (157). In addition, in the presence of SOD, or upon interaction with carbon dioxide (CO2), ONOO− can also generate nitrosonium cations (NO+) that causes the nitration of tyrosine residues in proteins. Finally, since NO· is capable to release iron from the Fe-binding protein ferritin, it may also facilitate the production of ·OH via the Fenton reaction (690).
Oxidative stress is caused by the imbalance between production of ROS and breakdown of the chemically reactive species by reducing agents and antioxidants enzymes. This imbalance may be due to environmental factors, stressors, or disease. Thus, in practical terms, oxidative stress is determined by excessive exposure to oxidant molecules when there is insufficient availability of antioxidant mechanisms (238).
1. Sources of oxidative stress
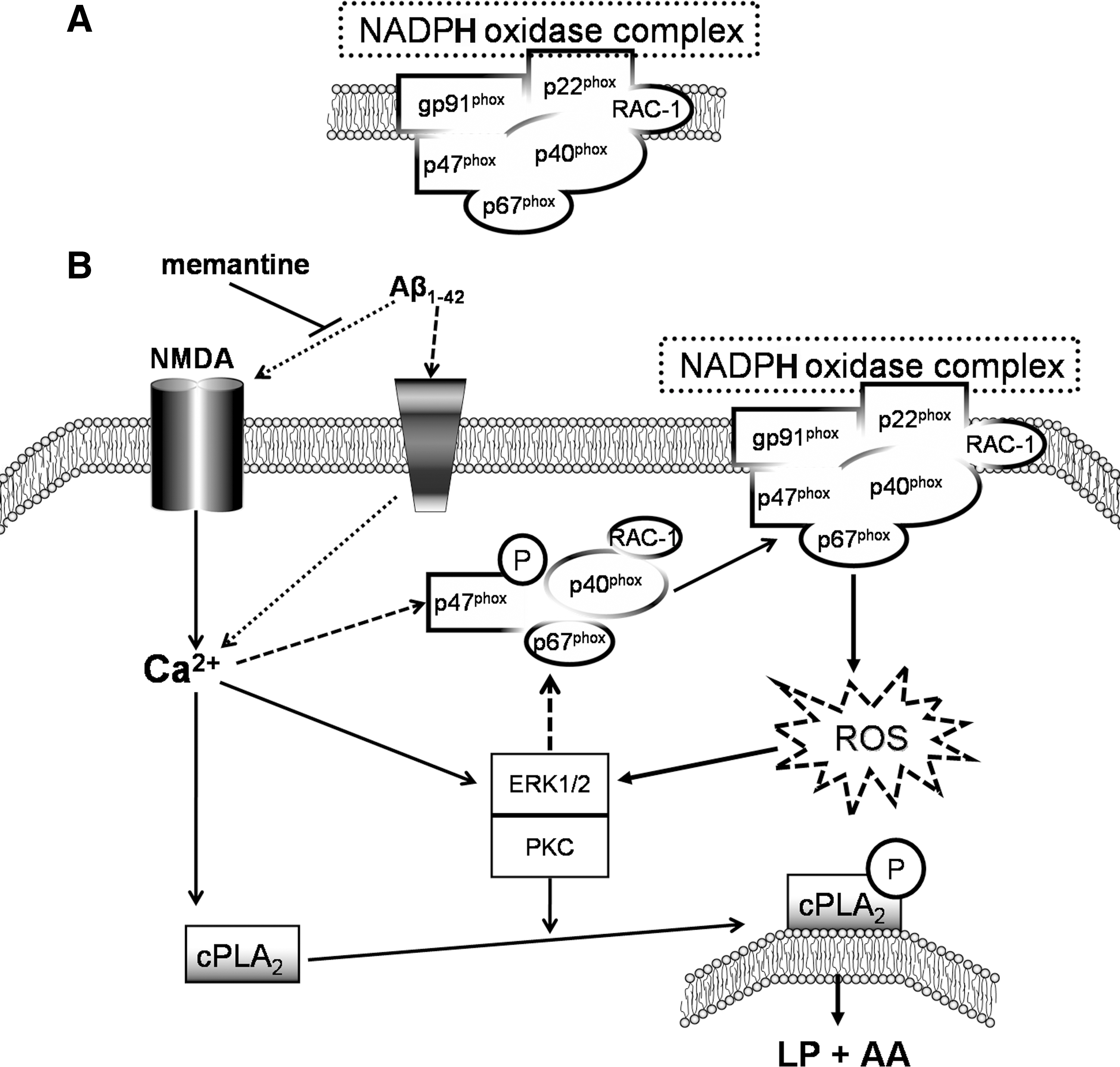
There are two major sources of oxidative stress, one associated with the chronic formation of ROS derived from the mitochondrial electron transport chain (34), and another related to the acute and high output formation of ROS derived from nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a mechanism especially relevant during the activation of the innate immune system, including microglial cells in the central nervous system (CNS) (285). NADPH oxidase is a transmembrane protein complex that transports electrons across biological membranes for accomplishing the reduction of oxygen to O2 ·−. NOX2, the prototype homolog for the six members NOX family of NADPH oxidases, also known as phagocytic oxidase gp91phox, is activated in macrophagic cells, such as microglia, to catalyze the production of O2 ·− during host defense (see section I.C). NOX2 is activated when the cytosolic subunits are phosphorylated and Rac is activated in the cytosol (Fig. 4a), resulting in their translocation to the membrane and formation of the active NADPH oxidase complex with cytochrome b558 (24, 25).
Other enzymatic sources of ROS are xanthine oxidase and uncoupled endothelial NO· synthase (eNOS). Uncoupling refering to the fact that, in the absence of critical cofactor for NO· synthesis like tetrahydrobiopterin, NOSs (including eNOS) produce ROS rather than NO·, with the resulting increased oxidative stress and reduced levels of NO·. An usual way to measure oxidative stress in biological samples is to determine the level of oxidative stress markers, such as protein carbonyls, 3-nitrotyrosine (3-NT), thiobarbituric acid reactive substance (TBARS), free fatty acid release, iso- and neuroprostane formation, acrolein, 4-hydroxy-2-nonenal (HNE), carbohydrate-mediated advanced glycation end products (AGEs), and 8-OH-2′-deoxyguanosine (8-OHd6) and 8-oxo-7,8-dihydroguanosine (8-OHG) and other oxidized bases, and altered DNA repair mechanisms (79, 405, 529).
2. ROS in physiology and pathology
ROS, at low concentrations, can serve physiological functions (571). Cells produce ROS at low levels largely due to monoelectronic reduction of oxygen, generating O2 ·− at the mitochondrial respiratory chain (296, 321, 350, 658). ROS play a necessary role in cell signaling cascades (571). For example, small amounts of ROS participate in synaptic signaling, with ROS acting as a messenger in long-term potentiation (LTP), a well-known model for synaptic plasticity (536). Likewise, H2O2 traverse biological membranes and can oxidize cysteine residues exerting actions on intracellular protein tyrosine phosphatases and lipid phosphatases as part of their reversible modulation (536).
Overabundance of ROS is observed during neuronal development, as well as in neuropathological conditions associated with chronic oxidative stress, such as Parkinson's disease (PD) (295) and AD (503), and in more acute settings such as ischemic reperfusion injury after stroke (131). Neurons are highly susceptible to oxidative stress because they are intrinsically ill-equipped to defend themselves against an increase in ROS, having low levels of antioxidants compared with those in other mammalian cells. Nonetheless, glial cells, including astrocytes, play a key supplementary role in antioxidant defense of neurons (264)—an important defense mechanism that will be discussed at the neuroinflammation section (section II.D). Nevertheless, particularly in the brain, even small imbalances can be deleterious. Abnormally elevated ROS is implicated in age-related LTP impairment (571).
NADPH oxidase plays a role in N-methyl-D-aspartic acid (NMDA)-induced O2 ·− production (71), linking oxidative stress with excitoxicity. Neurons in culture and in mouse hippocampus responded to NMDA with a rapid increase of O2 ·− production, followed by neuronal death. ROS production and neurotoxicity were inhibited by the NADPH oxidase inhibitor apocynin and in neurons lacking the p47phox subunit, needed for NADPH oxidase assembly (Fig. 4). O2 ·− production was also blocked by inhibiting regeneration of the NADPH substrate, and by inhibiting the protein kinase C (PKC) zeta, which activates the NADPH oxidase complex (71). Activation of NMDA receptors is associated with intracellular free calcium increase, which again will participate in both signaling and O2 ·− production (55, 535) as well as potentiating NMDA-induced neurotoxicity (284).
Because ROS can be simultaneously useful as well as deletereous, it has become quite problematic to experimentally establish a cause-and-effect relationship of their involvement in various pathophysiological processes (525). For example, expression of ectopic catalase (CAT) in mitochondria in combination with overexpression of manganese superoxide dismutase (MnSOD), which results in a decreased mitochondrial H2O2 production, was found to shorten rather than prolong the life span of Drosophila (39). In consequence, enhancement of antioxidant defenses will not be necessarily protective. Similarly, although the role of inducible nitric oxide synthase (iNOS) and high levels of NO· in neuropathology have been associated with neuronal damage and death (72), there is strong evidence that iNOS activity can be neuroprotective (114, 586, 627), and NO· under conditions of long-term injury or disease reduces functional loss and pathological changes of the brain (5, 476), through the ability of NO· (i) to block apoptosis via inhibition of caspases (317, 318); (ii) to interact and regulate two signaling pathways that are key to cell survival, extracellular signal-regulated kinase/mitogen activated protein kinases (ERK/MAPK) and the phosphatidylinositol 3-kinase/Akt pathways (638, 704); and (iii) to up-regulate other cytoprotective proteins such as MnSOD and Bcl-2 (75, 114). Furthermore, NO· is an effective antioxidant, preventing oxidative modification of proteins and lipids caused by other oxidizing species such as H2O2 (191, 399). All these factors could explain the greater cognitive impairment after head injury observed in iNOS KO mice than in wild-type animals (586) and that iNOS-deficient mice have increased brain oxidative stress (38). Similarly, aged hypertensive rats with poor cognitive performance have a fourfold decrease in hippocampal levels of iNOS compared with cognitively normal animals (279). Again, these rats showed decreased iNOS levels, which were associated with increased oxidative stress.
There is an important controversy. Tg2576 APP transgenic mice crossed with iNOS KO mice develop extensive NFT-like pathology, showing evidence of neurodegeneration (120). APPSwDI/iNOS KO mice displayed impaired spatial memory and significant neuron loss in the hippocampus and subiculum compared with the APPSwDI mice, but had unaltered levels of Aβ. These results show that removal of iNOS from an APP transgenic mouse results in development of a more robust AD-like pathology and behavioral impairments (694). In contrast, Nathan et al. (452) reported that in APP/PS1-double transgenic mice with iNOS ablation, deficiency of iNOS resulted in decreasing premature mortality, substantially protecting mice from cerebral plaque formation and from increased Aβ levels, protein tyrosine nitration, astrocytosis, and microgliosis. Thus, for this Aβ accumulation model, iNOS appears to favor Aβ deposition and disease progression (452).
Oxidative stress is implied in the pathogenesis of diseases, including ischemia, cancer, and neurodegenerative disorders, among many others (658). Diverse molecular machinery converges to generate highly reactive species to oxidize target molecules that include proteins, nucleic acids, polysaccharides, and lipids. Oxidative modification of proteins may lead to defects in their structure and function that consequently will result in the progression of degenerative modifications. Among nucleic acids, RNA, being single stranded, is more susceptible to oxidative damage because, unlike DNA, its bases are not protected by hydrogen bonding or histones, and their oxidation will result in impairments on translation or on its regulatory functions. Oxidation of lipids (glycolipids, phospholipids, and sphingolipids) will affect membrane properties, and several complex functions mediated by specific receptors will be also affected. In contrast to the oxidative attack occurring inside the endocytic vacuole, reactive species released to the interstitial space are unable to discriminate between friend and foe, and could result in self-damage. Important self-targets are mitochondrial components, for instance, mitochondrial DNA (mtDNA) (502), which in turn leads to dysfunction of mitochondrial oxidative phosphorylation and increased production of ROS, potentially triggering an oxidative vicious cycle (26, 204).
3. Metal ions and oxidative stress
Accumulation of metal ions, especially iron (Fe), has been observed in the aged CNS in humans (18) and animal models (237) as well as in AD (12, 269). Strong evidence indicates that dyshomeostasis of redox-active biometals, copper (Cu) and Fe, and oxidative stress contribute to the neuropathology of AD (12). Metals can interact directly with Aβ, modulating several physicochemical properties of Aβ. They can promote the in vitro aggregation of Aβ. Moreover, Aβ toxicity is linked with the presence of redox metals, mainly Cu and nonredox zinc (Zn) (76, 78, 659). Studies have confirmed that amyloid plaques in postmortem AD brain are abnormally enriched in Cu, Fe, and Zn (134, 281). Conversely, metal chelators dissolve these plaques from postmortem AD brain tissue and attenuate cerebral of Aβ burden in APP transgenic mice (77, 263, 589). The role of magnesium (Mg) in neurodegenerative disorders and its effect in AD have also been investigated. Mg appears to be decreased in AD patients in clinical and autopsy studies (15, 116).
The generation of free radicals is tightly linked with redox-active trace metals (299). The redox state of the cell is maintained within strict physiological limits and is associated with Fe and Cu redox couples. Normally, there is no free intracellular Fe; however, under stress conditions, O2 ·− in excess acts as an oxidant of Fe-containing enzymes releasing free iron (372). The released Fe2+ can participate in the Fenton reaction, generating highly reactive HO·. NO· also readily binds certain transition metal ions, and many physiological effects of NO· are exerted as a result of its initial binding to Fe(II)-Heme groups in the enzyme guanylate cyclase (168).
Metals are not only associated to oxidative stress mechanisms, but are also essential components for multiple antioxidant defense systems. Glutathione peroxidase (GPx), cytoplasmic SOD, and CAT enzymes contain selenium (Se), Cu–Zn, and Fe metals as cofactors, respectively. Essential trace elements play a major role in metabolic pathways, and they have been studied in many diseases, including autoimmune, neurological, and neuropsychiatric disorders, including AD, suggesting that both excess and deficiency of these elements could be related to the pathophysiology of AD (133).
4. Redox state and redox buffering
The redox state depends upon the ratios of reduced and oxidized forms of redox couples, which include glutathione/oxidized glutathione (GSH/GSSG), NADH/NAD+, NADPH/NADP+, cysteine/cystine, thioredoxinred/thioredoxinoxid, and glutaredoxinred/glutaredoxinoxid. The GSH/GSSG ratio is the primary determinant of the cellular redox state because it is 1000 to 10,000-fold more abundant than other redox couples, and because of its low standard redox potential (Eo=−240 mV). GSH is the most abundant intracellular nonprotein thiol, reaching concentrations of up to 10 mM in mammalian cells, with astrocytes being the major source (266). Furthermore, GSH is also tightly linked to other redox couples, either metabolically or because of its ability to form thiol-GSH mixed disulfides (130, 525). GSH acts as an oxyradical scavenger by scavenging NO· and other oxidants, thereby protecting cells against oxidative damage by reducing oxidized or nitrosylated protein thiols (241). In addition, GSH is involved in the elimination of toxic oxidation products such as HNE, a product of lipid peroxidation (324, 689), and is able to detoxify various oxidants by directly scavenging free radical or acting as coenzyme in GSH-peroxidase-catalyzed reactions (375). Within the cell, GSH is kept in its thiol-reduced form (>98%) by GSSG reductase, an NADPH-dependent enzyme; additional amounts of GSH are present as GSSG and as GSH conjugates. Maintaining optimal GSH/GSSG ratios in the cell is critical because GSH is a major endogenous antioxidant defense system of the brain, removing hydrogen- and lipid-peroxides (418). Activation of several signaling pathways, including PKB, calcineurin, nuclear factor jB, and MAPK, is associated with changes in redox status of neurons after oxidation of GSH (698).
Peroxiredoxins (Prxs), also called thioredoxin (Trx) peroxidases, is a family of antioxidant enzymes that reduce H2O2, ONOO−, and a range of organic hydroperoxides using reducing equivalents provided by Trx/thioredoxin reductase (TrxR) system (697), which have been studied as a stress responsive system (21). Several Prx isoforms are induced in the brain in response to insults (330), as well as the presence of aberrant patterns of Prx expression in the CNS of patients affected by neurodegenerative disorders (463), being suggested that they can serve neuroprotective functions.
Trx is an ubiquitous thiol oxido-reductase system that regulates cellular redox balance, undergoing reversible oxidation of the cysteine pair while reducing disulfide bridges of proteins (21), and appears to mediate signal transduction elicited by several growth factors and cytokines (536, 537). Trx plays an essential role in cell function by limiting oxidative stress directly via antioxidant effects and indirectly by protein–protein interactions (158). In fact, cellular redox regulation for many processes is provided in mammalians by interaction between the Trx and GSH systems (147). Together, they form a powerful system controlling redox regulation of gene expression, signal transduction, cell proliferation, protection against oxidative stress, anti-apoptotic functions, growth factor and co-cytokine effects, as well as regulation of the redox state of the extracellular environment (614). Besides the role as a source of reducing equivalents, Trx by itself acts as antioxidant or ROS scavenger (159).
In mitochondria, the main redox buffering systems are GSH, glutaredoxin, and Trx systems. GSH, besides acting as reducing agent and antioxidant, also acts as mediator of several physiologic reactions, including metabolism of xenobiotics, thiol disulfide exchange reactions, and cell signaling (cell-cycle regulation, proliferation, and apoptosis). In the nucleus, GSH maintains critical protein sulfhydryls (PSHs) that are necessary for DNA repair and expression (658). Cerebellar granule neurons in culture exhibit marked functional deterioration and die in response to loss of both mitochondrial and cytoplasmic GSH, but not after cytoplasmic GSH loss alone (585, 700). Additionally, although mtGSH depletion does not affect astrocytes' viability, they become more susceptible to NO· exposure (444).
The brain is particularly vulnerable to oxidative stress due to (i) its high demand of oxygen, showing the highest oxygen metabolic rate of the economy, consuming around 20% of the total amount of oxygen in the body, (ii) its dependence on oxidative metabolism for obtaining metabolic energy, (iii) its high content of iron, which can catalyze the generation of ROS and RNS, (iv) its content of antioxidant enzymes, which is lower than in other organs (198, 412), and (v) the postmitotic nature of neurons, which make them especially vulnerable by accumulating mutations and impairments that are not removed by cell replacement. In particular, aged, AD or injured brains of any sort, show oxidative modifications in nucleic acids, proteins, lipids, and sugars (Fig. 5); oxidative damage; and changes deriving in loss of function (239, 381, 405). Most of cells have protective mechanisms, responsible for enzymatic breakdown or scavenging of ROS (Fig. 3). For example, H2O2 is converted to water and O2 by CAT or glutathione peroxidases (GPxs). However, in the brain, antioxidant systems are reported to be less functional, which can lead to further increase of ROS and RNS reacting with the various target molecules (239).

C. Microglia, astrocytes, and oxidative stress
Microglia are the brain-resident macrophages (255, 522, 539) representing its innate immune system, and providing the first line of defense for injury or disease. They colonize the brain at late stages of prenatal life, before the development of the blood–brain barrier (BBB) (522). In the healthy adult brain, microglia display a characteristic ramified morphology, and serve a surveillance role, dynamically monitoring brain microenvironment (161, 458). Microglia can sense a wide range of stimuli, including CNS trauma, ischemia, infection, toxic insult, and autoimmune injury (319, 332, 385, 566, 603, 674), recognizing a wide range of targets, such as peptides, lipoproteins, glycolipids, and nucleotides (450, 496, 661). In addition to abnormally processed, modified, or aggregated proteins (e.g., Aβ), stimuli include inflammatory cytokines, which can trigger microglia activation, damaged neurons, which provide the strongest signal for microglia proliferation, inducing reactive microgliosis, membrane breakdown products, altered molecules (e.g., active forms of matrix metalloproteinase), impaired neurotransmitter function (e.g., elevated glutamate concentration), and cytosolic compounds (240, 385, 450, 522, 563).
When stimulated, microglia become activated presenting structural and morphological changes, enlarging their size (200, 458) as well as modifying their functional properties (374, 385, 671). Microglial cell can undergo at least three activation pathways (and probably several intermediate ones) starting from the quiescent, surveillance state (222, 407, 434): (i) classical activation (M1 type activation), which under certain conditions will tend towards cytotoxicity, (ii) alternatively active/phagocytic/neuroprotective (M2) (222, 407), or (iii) regulatory (434). In fact, activation of microglial cells, far from a single phenotype, represents a continuum change from innate to adaptive activation with the expression of different cytokines and cytokine receptors (645).
Activation of interferon-regulatory factor 5 (IRF5), member of the IRF family, defines commitment to the M1 macrophage lineage (558). IRF5 activates genes encoding for type I interferon, inflammatory cytokines (including TNFs, IL6, IL12, and IL23) and tumor suppressors (331, 473). In mice, IRF4 controls M2 polarization by stimulating the expression of specific M2 macrophage markers (331, 558). For M2 macrophages, activation of nuclear factor-kappa B (NF-κB) subunit p50 has been associated with the inhibition of M1-polarizing genes (500), whereas CREB-mediated induction of transcription factor C/EBPβ has been shown to up-regulate M2-specific genes (548). M2-type induction, through secretion of IL4, IL10, and transforming growth factor β (TGFβ), secrete IL4, IL6, IL10, and IL13, cytokines that promote humoral immune responses and down-regulate M1-mediated responses, inhibiting numerous macrophage inflammatory functions (645). M2 polarization is further subdivided according to the production of IL10 or IL12, depending on specific associations, like wound-healing, tumor-associated, or heterogeneous macrophages (434). A priori, it may appear that M2 activation is the one associated with protective functions. However, there is strong evidence that chronic allergic inflammatory processes like asthma depend on expression of M2 cytokines IL4, IL5, IL9, and IL13 (230, 701). Finally, regulatory macrophages can arise during the later stages of adaptive immune responses, and their primary role appears to limit inflammation (433). There are many different ways to generate regulatory macrophages, although ERK/MAPK has emerged as a potential candidate to be the mediator (384, 434).
Microglia are activated in virtually all CNS diseases (240, 332, 453). Activated microglia produce and secrete a spectrum of inflammatory mediators, such as eicosanoids, cytokines (319, 450, 642), chemokines, ROS, NO·, small metabolites acting as mediators, proteases like α-antichymotrypsin and α-antitrypsin, and inflammatory markers, such as serum amyloid P and C-reactive protein (45, 361, 385, 450, 453, 603, 642). These inflammatory mediators both regulate innate immune defense and act on neuronal properties, specially modifying synaptic function (10, 11, 173, 570). Furthermore, initially intended to destroy pathogens and injured brain cells as part of their protective response, they are also responsible for bystander damage of neurons at later stages, depending on the environmental context (361, 669). In fact, their cytotoxic activation is often associated to neuronal loss and decline of cognitive function in literature (61, 93, 319). Nevertheless, in addition to deleterious effects, microglia are also capable of secreting trophic factors and modulator cytokines, and in that sense, they have the repertoire to mediate neuroprotection.
As we discuss on section I.D, the inflammation hypothesis stresses the notion that hyperactive microglia are the primary cause of neurotoxicity in AD. In contrast, we propose that neurotoxicity is not a consequence of hyperactive but rather of mis-active, dysfunctional microglia (669). Thus, the innate immune response, initially protective, becomes a chronic mis-activation contributing to brain cytotoxicity (Fig. 6) (455, 559, 669, 703). The distinction is not trivial when interested in the development of therapeutical approaches. Adequately activated microglial cells are needed as scavenger cells in the CNS; however, by not responding to normal regulatory feedback and/or by having an impaired ability to clear Aβ (479, 669), glial cells could develop a predominantly cytotoxic feature. Thus, the aim of therapy should be oriented to potentiate a certain mode of microglial cell function rather than functionally inhibiting the whole microglial cell population, an approach that likely has a major bearing in the limitations of past and current thinking about how one might use immunoinhibitory drugs simply to turn microglia off for therapeutic benefit of neurodegenerative diseases.
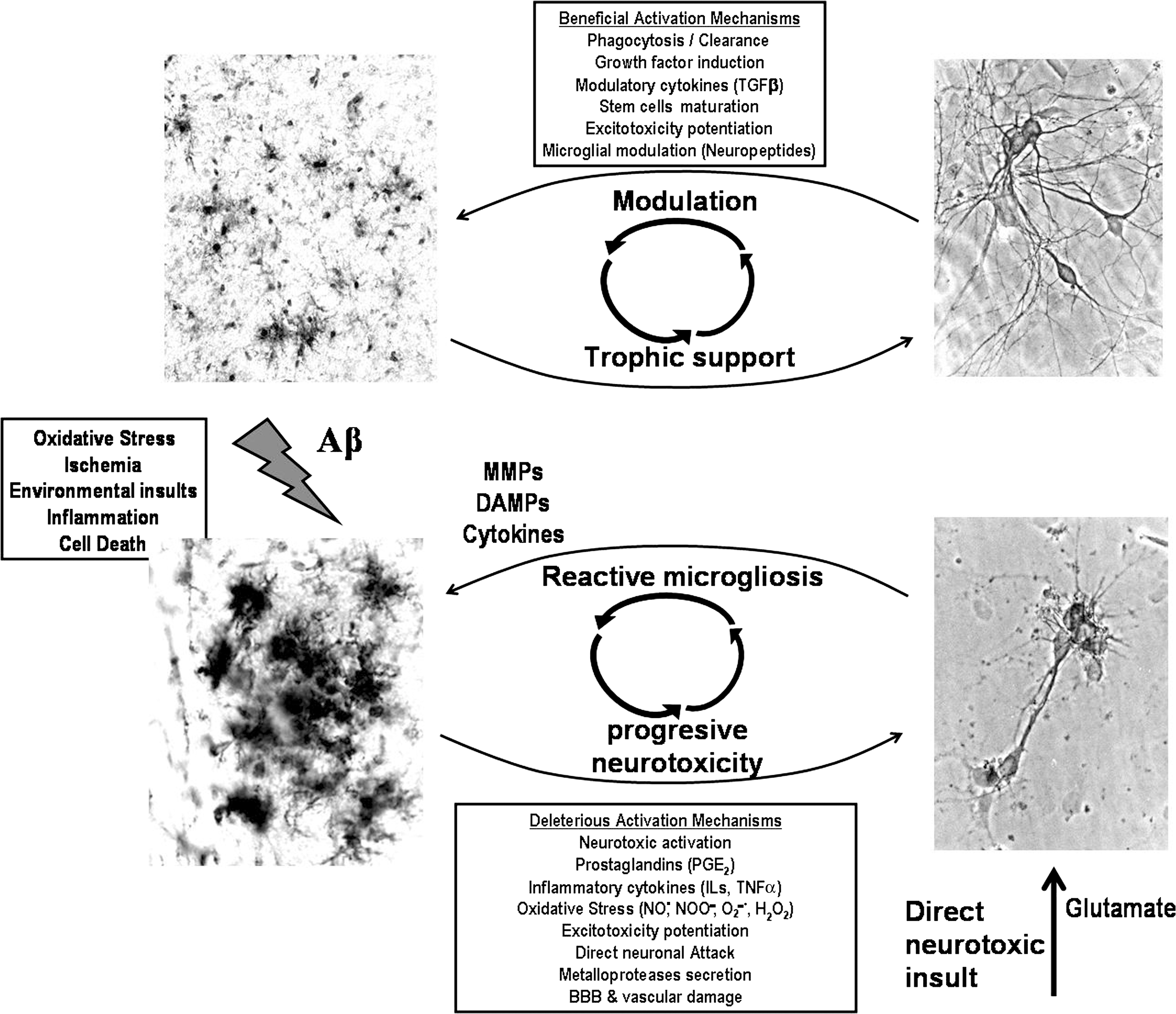
Regarding AD, the activation of microglia induced by Aβ (249, 326, 584) results in several cell transformations (282) making glia highly competent for phagocytose Aβ plaques (31, 541, 584). Microglia are intimately associated with Aβ plaques in AD, but not with the diffuse Aβ plaques characteristic of aging (249, 262, 287, 669, 672). The trigger for activation in AD is unclear, but the invasion of plaques by morphologically active microglia is seen in AD transgenic mice models, as well as when Aβ is injected into the brain of otherwise healthy mice or in in vitro experiments (9, 462, 528, 635, 672). Multiple receptors have been reported to mediate this process: for instance, the receptor for advanced glycation end products (RAGE) (706), scavenger receptors (9, 123, 635), the G-protein-coupled chemo-attractant receptor (341), the formylpeptide receptor 2 (262), and toll like receptors (528). Cytotoxic activation of glia can be associated with the activation of reduced NADPH oxidase (665) or the induction of the novo expression of inducible NO· synthase (iNOS); both critical enzymes responsible for the generation of oxidants. Importantly, Aβ is also clearly indicated as a source of oxidative stress (662), as Aβ activates microglia to produce extracellular O2 ·− (30, 510) as will be discussed in section II.B.
Astrocytes and neurons play a critical role in propagating and modulating the nature of neuroinflammatory activation, signaling reciprocally to microglia to either accelerate or slow microglial components of autocrine neuroinflammatory cycling. There is an intense cross-talk between neurons and glial cells manifested not only in the regulation of normal neuronal function but also in disease mechanisms (173, 559, 668). Microglia and astrocytes are strongly activated in AD, producing an array of inflammatory mediators and fulfilling phagocytic functions (9, 45, 453, 541, 669, 672, 674). Such robust activation in AD includes (i) the activation of complement pathway (3, 540), (ii) the up-regulation of cytokines, including IL1, IL6, IL8, TNFα, TGFβ, and chemokines like the macrophage inflammatory protein 1 (MIP-1) (6), and (iii) a strong innate immune cellular system activation (257, 385). Indeed, immunohistochemical studies reveal that activated microglia and astrocytes gather around senile plaques in AD (249, 287, 413) and appear to facilitate amyloid plaque clearance (7, 262).
At the cellular level, it is well established that astrocytes modulate production of cytokines, proteases, phagocytosis, and microglial cytotoxic activation (642, 671). The astrocytic modulation of microglial cell production of RNS and ROS can be lost under certain conditions of pathological astrocytic activation (562). Furthermore, whether microglial cell cytotoxic activation occurs or not will depend also on the interplay of cytokines that are released by microglia and astrocytes (Fig. 7). TNFα acts as a positive feedback signal, helping to potentiate glial cell activation, and as a consequence, enabling them to produce IL1β, and promote the feed-forward activation of astrocytes. This escalating cascade, however, is also under systemic inhibitory control, for example, by IL6, which is known to exert a negative feed-back on TNFα signaling. Inhibition of the stimulatory signals and maintenance or reinforcement of the inhibitory signal loops can be a strategy to prevent escalating microglial cytotoxicity. Another alternative would be the immunomodulation of reactive microglia that brings them back into a more protective activation state.
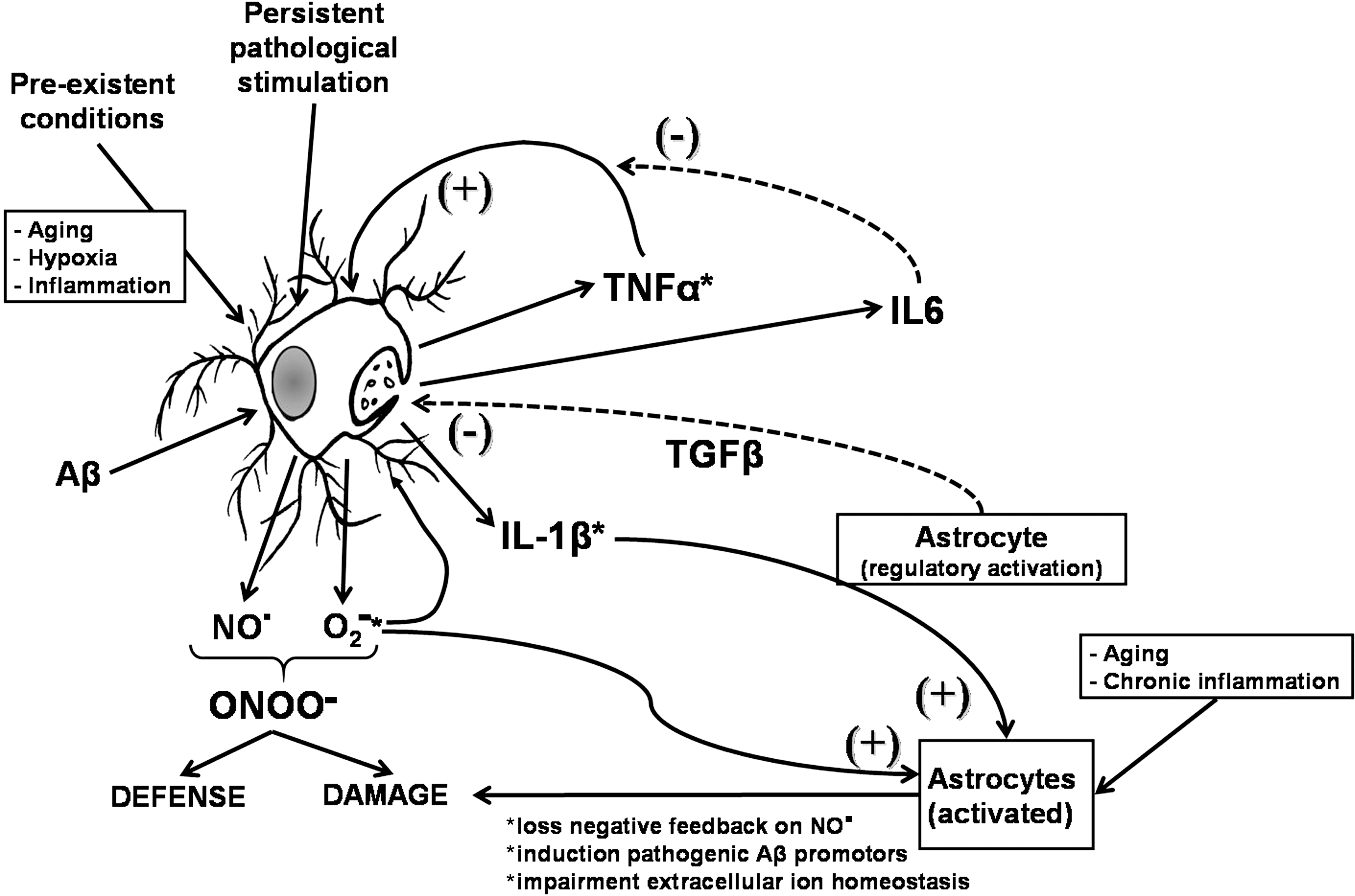
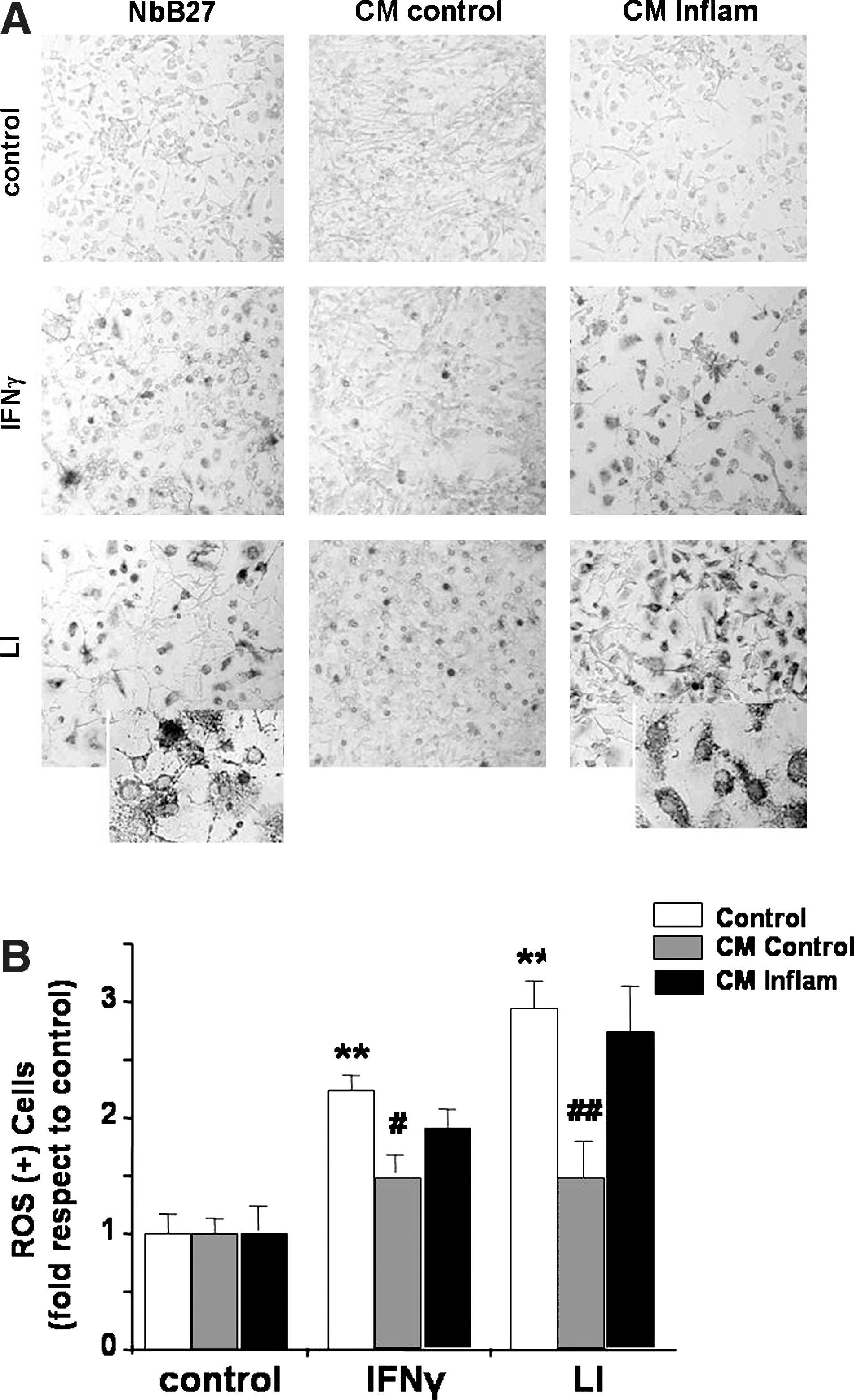
Astrocytes interact with neurons by nourishing, protecting, and modulating growth and function, and providing a robust antioxidative potential (397), favoring survival of neurons in conditions of oxidative stress (187). Regardless the strong antioxidative potential of astrocytes, hippocampal neurons show a more robust effect than astrocytes on the regulation of cytotoxic activation of microglia (259) modulating the production of RNS and ROS (Fig. 8), an effect that was shown to be mediated by TGFβ (259, 655). However, under conditions of strong inflammatory activation, their capacity of regulating Aβ- and APP-induced microglia-mediated cytotoxicity and phagocytic activity of microglia are reduced. These results support the hypothesis that, in neuroinflammation, microglial cell scavenger function is impaired and reactivity is enhanced as an initial step for neurodegeneration (673). Furthermore, under inflammatory conditions, neurotoxicity induced by Aβ or APP is greatly enhanced in the presence of cytotoxic microglia. The observed cytotoxicity could be consequence of a persistent mis-activation, in which elevated concentrations of IL1β and radical species support chronic cytotoxic activation of glial cells and cell damage (519). In agreement with our hypothesis, hippocampal cells exposed to inflammatory stimuli do not retain their ability to modulate production of ROS by microglia (Fig. 8).
D. Aging and oxidative stress
ROS are an unavoidable by-product of cell metabolism. A small percentage (∼1%–4%) of electron transfer normally goes toward basal production of O2 ·− (40). Oxidative stress is increased at basal conditions in aging (61, 350, 571, 581, 631). The increased production of radical species is proposed by the free radical theory of aging. Electron transport chain activity in mitochondria declines with age (470), whereas basal oxidative stress increases with age (350). This means that there is a decrease on electron transfer efficiency, resulting in an increase on the production of ROS, relevant for the accumulation of mitochondrial oxidative damage. In fact, accumulation of ROS and RNS appears to be the most important causal factor for aging (40).
Robust evidence indicates that the magnitude of the imbalance between antioxidant defenses and oxidants, or the level of oxidative stress, widens during the aging process. The age-associated increase in oxidative stress appears to depend on the fact that antioxidant defenses do not keep pace with the age-related increases in ROS production (525). Thus, the balance shifts progressively toward a more pro-oxidant state with aging.
In this context, an age-related loss of GSH can be predicted to have at least two potentially deleterious consequences: (i) increase of basal levels of H2O2, leading to a rise in the formation of ·OH, and in consequence, in greater molecular structural damage. Enhanced lipid peroxidation would result in increased level of malondialdehyde and hydroxynonenals, which can form adducts with DNA or proteins, modifying their structure and function (652). (ii) Protein oxidation can interfere with the catalytic efficiency of enzymes, which could reduce their ability to mount adaptive responses under stress conditions (181). Both types of alterations indeed occur during the aging process (524). Thus, maintenance of an optimal GSH redox state is imperative for limiting the level of macromolecular oxidative damage.
Aging, especially of the nervous system, is also characterized, among other changes, by an inflammatory status that induces functional changes of the immune responses (345, 414, 669, 674, 702) and increased oxidative stress state (525). So, through oxidative stress, increased level of inflammatory cytokines and glial cell activation (674) set the stage for a neuroinflammatory environment that could facilitate progression of neurodegenerative changes. Microglia become activated in a more cytotoxic fashion as function of age. Aged microglia increase their production of inflammatory cytokines (see section II.D) and reduce some of their protective functions, like induced phagocytotic activity—condition that could lead to their failure to efficiently clear Aβ (669). These changes represent a shift in basal cell reactivity and defense activity that can result in a regulatory impairment that can favor the genesis and progression of neurodegenerative processes (563). The role of aged microglia is also relevant to understand therapy failures in AD. It is possible that anti-inflammatory drugs may not work in AD patients because, in the brain, neuroinflammation is mediated by microglia rather than by activation of an eicosanoid pathway. As we mentioned in section I.C, the inflammation hypothesis stresses the notion that hyperactive microglia are the cytoxicity entity, whereas we propose that AD is rather a consequence of dysfunctional microglia (669). Neuroprotective microglial cells are needed in the CNS. However, when glia are dysregulated, cytotoxic activation could predominate.
During aging there are numerous documented changes at cellular, tissular, and organ levels (Fig. 9). There are well-known biomarkers of aging, such as cross-linking of collagen, the accumulation of AGEs, lipofuscin, and accumulation of damage induced by ROS and RNS (40) at the DNA and protein level. Accumulation of AGEs is also observed in neurological disorders such as multiple sclerosis and AD. The receptor for AGEs, RAGE, is increased after oxidative stress, immune and/or inflammatory responses, and upon altered cell functions, suggesting that AGE accumulation is not a process privative of aging (598). Engagement of RAGE induces the release of inflammatory cytokines and free radicals, thus perpetuating a cycle of damage. RAGE is increased in AD, where it is found to be expressed on neurons and astrocytes. In astrocytes, AGE-conjugated proteins form granules, suggesting that astrocytes uptake and degrade glycated proteins (598).
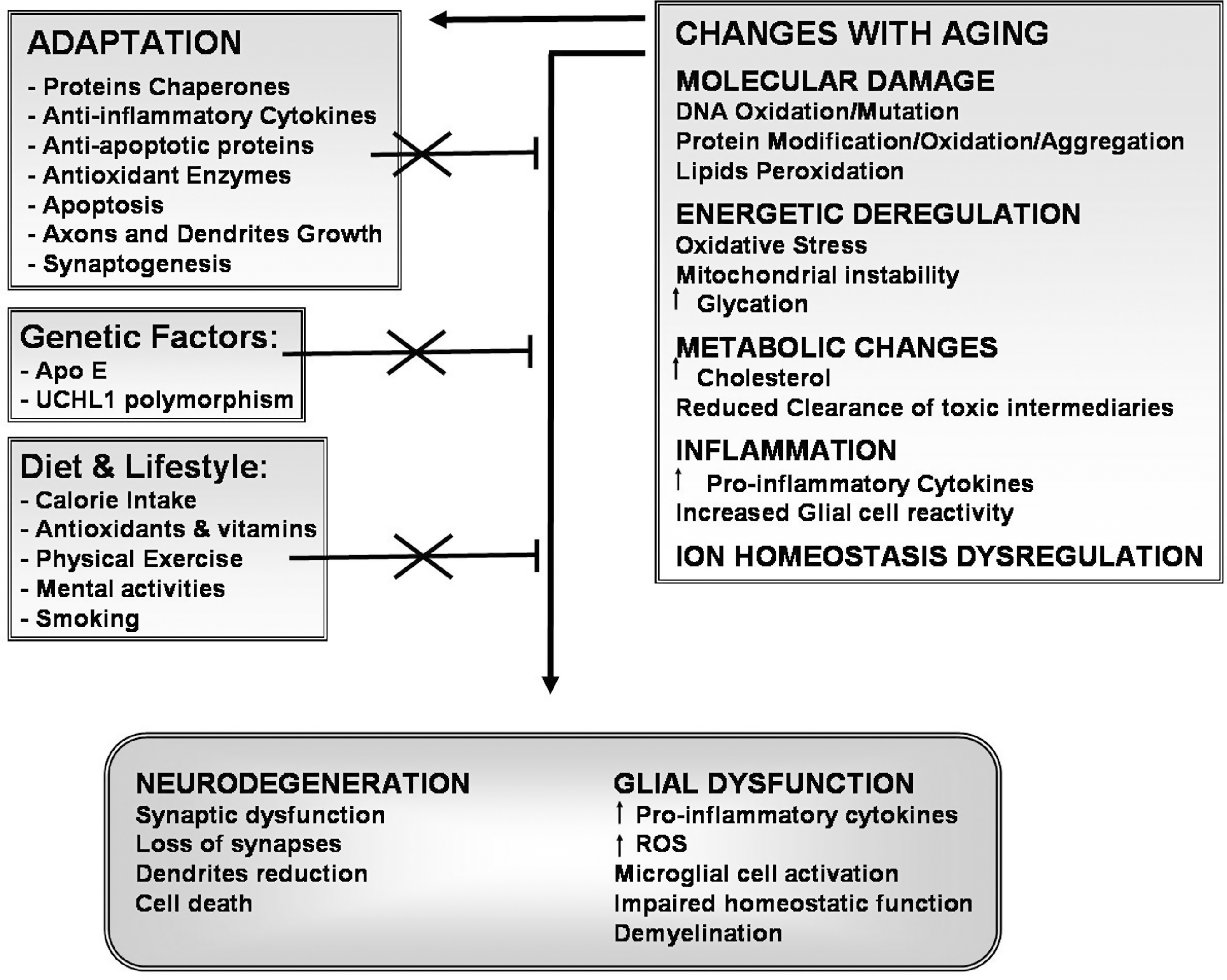
When assessing the effect of aging on CNS function, it is necessary to keep in mind that at every level of complexity, the nervous tissue depends on a finely tuned cross talk of glial cells and neurons, as well as on its capacity to respond to information being delivered from outside of the CNS, via endothelial cells and sensory neurons input. Glia secrete growth factors, such as nerve growth factor (NGF), basic fibroblast growth factor, glioma-derived growth factor, and insulin-like growth factor, as well as cytokines, including ILs, TNFα, and TGFβ. Glial cells and neurons maintain a reciprocal trophic and regulatory interaction, allowing for a regulated activation of the immune response, including limited production of radical species when needed (Fig. 10A). Under this view, cytotoxic activation could depend on the impairment of any of the interacting members of this regulatory cross-talk. In aging, there is a decrease on trophic support, as well as in the modulatory effect exerted by neurons and astrocytes (Fig. 10B). Microglial cell reactivity is greatly increased, which in turn increases the production of inflammatory cytokines, NO·, and ROS both at basal conditions and in response to any activating stimulus.
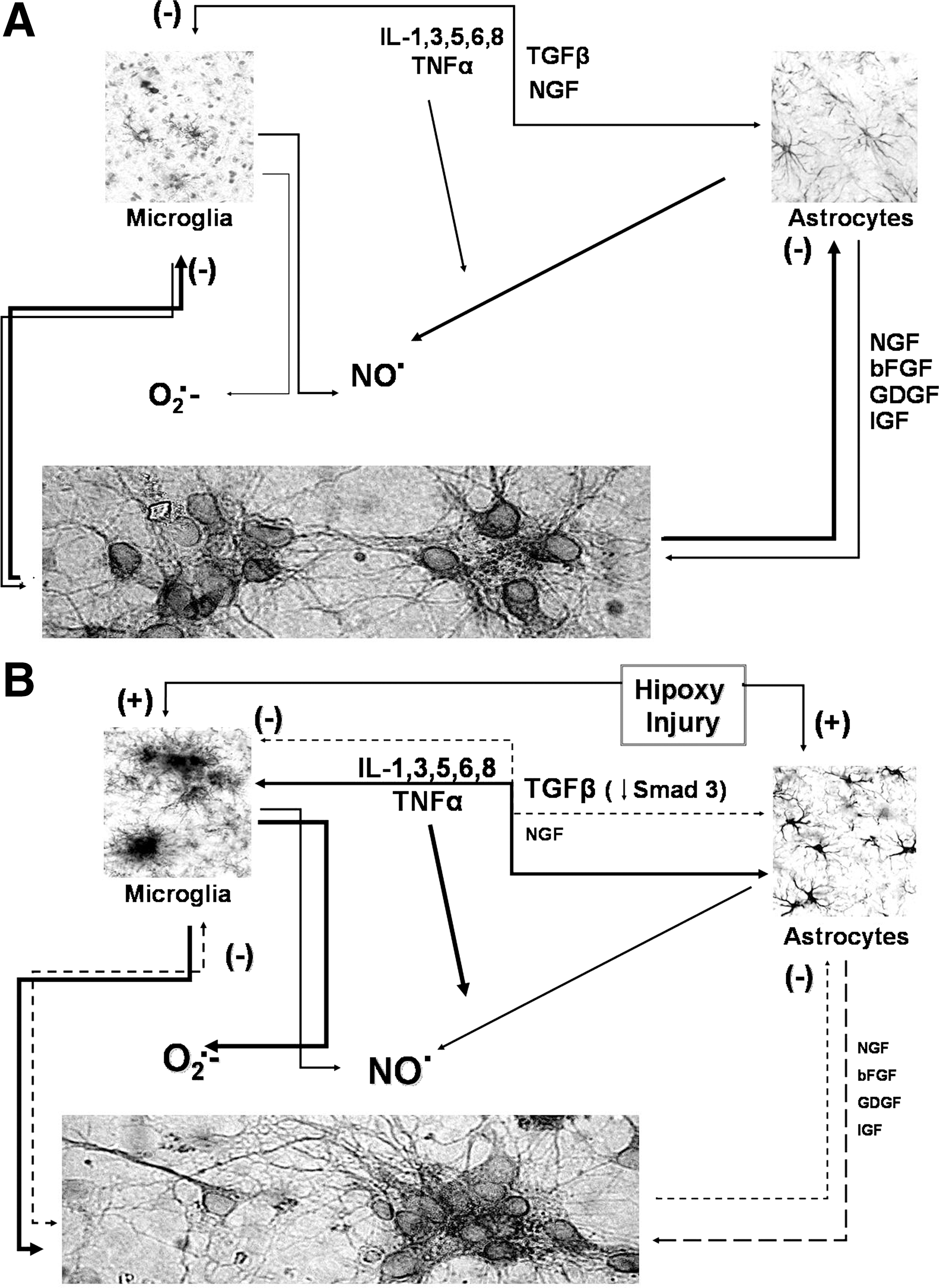
Age-dependent mitochondrial oxidative stress involves controversy and there are detractors questioning its significance (see section II.C). In any case, oxidative stress appears to influence life span extension. Life span extension by caloric restriction appears to depend on the reduction of superfluous oxidative metabolism. Furthermore, mitochondrial dysfunction is involved in apoptosis associated to mitochondrial membrane instability occurring when mitochondrial depolarization, oxidative stress, or energetic failure surpass a certain threshold level, with subsequent activation of cell death cascades (264, 371, 445).
II. Oxidative Stress in AD
Available data are consistent with the oxidative stress hypothesis of AD. Oxidative stress appears to be an early event in AD (491, 581), and deficiency in antioxidant enzymes occurring in AD, such as SOD, exacerbates the disease's phenotype (356), increasing amyloid and tau aggregation (356), phosphorylation (419), and accelerating the onset of behavioral impairment (192). Oxidative damage, observed in brains of AD patients and animal models, is significantly more intense than that observed in brains of age-matched control individuals. In experimental models, severity and early onset of AD shows an inverse correlation with the presence of antioxidant enzymes. Conversely, overexpression of antioxidant enzymes attenuated AD phenotype (183).
In addition to the role of oxidative stress secondary to Aβ neurotoxicity, excitotoxicity, accumulation of aggregated proteins, and impaired metabolism of calcium (226, 333, 377, 554) in neuron dysfunction and degeneration, oxidative stress appears to be a common mediator unifying the spectrum of cellular mechanisms leading to AD. Studies in in vivo and in cell culture models strongly support the notion that AD, as well as other chronic neurodegenerative diseases, involve several converging disease mechanisms, generating a functional interplay between neurons and glial cells (Fig. 11). It is clear that glia actively promote neuronal dysfunction and neurodegeneration (669) through oxidative stress mechanisms. Their deleterious activity (Fig. 11) could be exerted (i) by exacerbating the production of ROS (4, 722) and as consequence of this, by modifying intracellular proteins and lipids (239, 381, 722); (ii) by inducing mitochondrial dysfunction, which will increase production of ROS, and will further activate caspases, promoting cell death pathway (27, 370, 371) and ATP depletion (27); (iii) by facilitating the formation of ubiquitinated aggregates due to protein misfolding (468) as consequence of the impairment of energy-dependent cellular processes, like the ubiquitin–proteasome pathway necessary for the elimination of aberrant or misfolded proteins and abnormal phosphorylation of cytoskeleton components (20); (iv) by inhibiting the activity of the glial cell excitatory amino acid (glutamate) transporter 2 (641) or by inducing liberation of glutamate by astrocytes (338); overactive glutamate receptors will increase enormously the intracellular free calcium, which can cause mitochondrial toxicity (310, 395) and affects several calcium-dependent enzymatic pathways leading to dysfunction of the cell machinery and the potential initiation of apoptosis (411); and (v) by activating microglia and astrocytes to produce and release inflammatory cytokines (4, 348, 669, 674) and other potential toxic mediators (NO·, ROS) (4, 59, 669, 674, 722). These factors activate several inflammatory signaling pathways, as well as other inflammatory pathways depending on eicosanoids produced by cyclooxygenase (COX)-2 (647, 683).
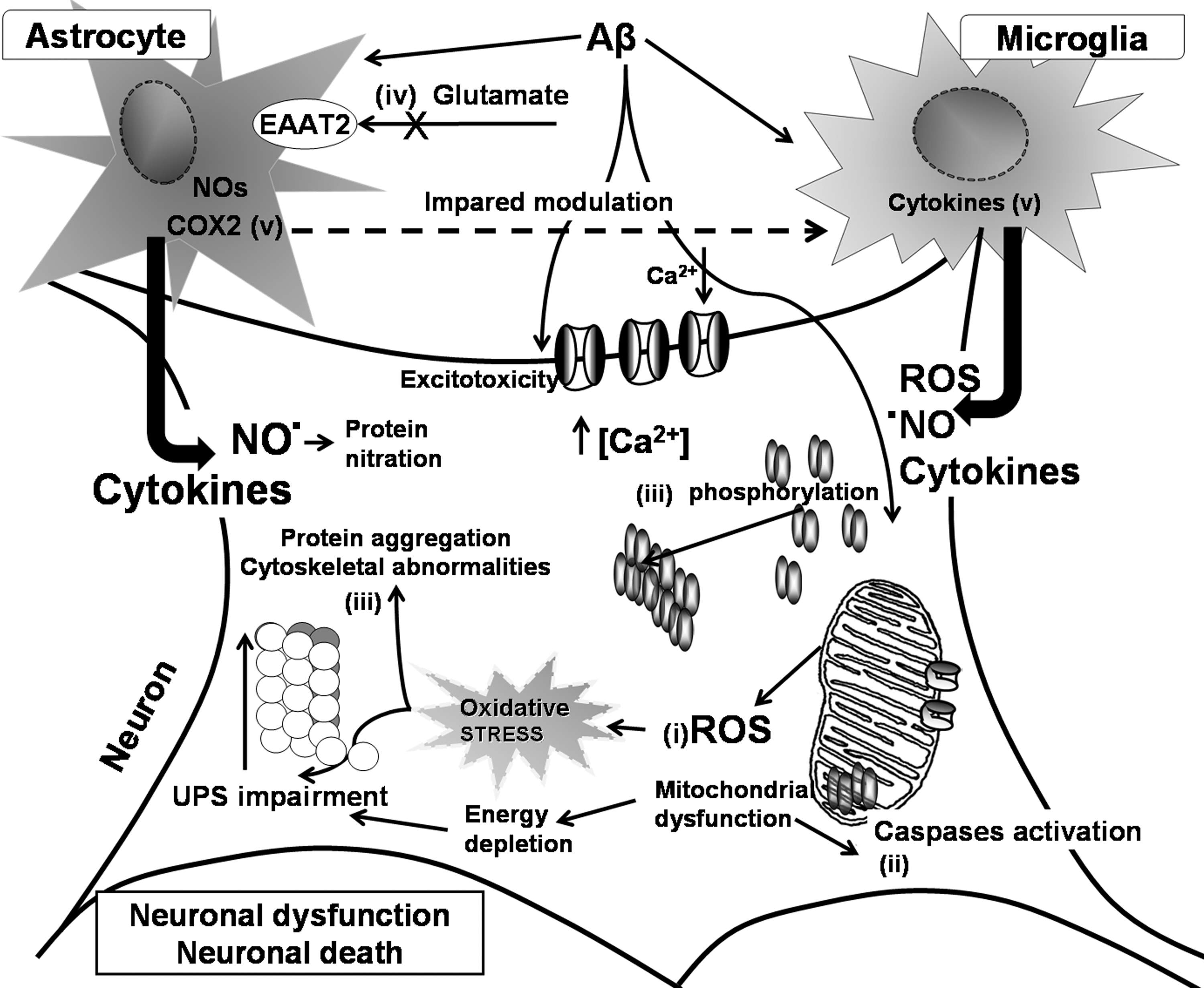
Among additional factors that could induce oxidative stress in AD, recent evidence shows that the risk of AD is increased by diabetes (54), which is also closely linked with inflammation. Hyperinsulinemia promotes inflammation and the formation of ROS, inhibits the degradation of oxidized proteins, and increases the risk for lipid peroxidation (127). Insulin acts synergistically with endotoxins to raise inflammatory markers like cytokines and C-reactive proteins (42). Chronic hyperglycemia is associated with an increased production of ROS (42, 290), and neuronal and mitochondrial calcium influx. ROS also potentiate the inflammatory activation, and together with chronic hyperglycemia, are associated with increased production of AGEs, which further favor neuroinflammation (290).
Epidemiologic observations identified midlife hypercholesterolemia as another major risk factor for AD (16, 695). In vitro studies (304) support this notion showing that membrane cholesterol promotes the amyloidogenic processing of APP. Recent studies with genetic mouse models of brain cholesterol accumulation, such as Tg-SREBP-2 (118, 195), expanded the role of cholesterol and mtGSH in AD, indicating that, in addition to fostering Aβ-production, mitochondrial cholesterol modulates Aβ neurotoxicity through selective mtGSH depletion. In addition to GSH depletion and altered GSH redox cycle described in AD (67, 533), mtGSH depletion specifically can account for the increased susceptibility to Aβ. First, mtGSH levels determine the sensitivity of brain mitochondria to Aβ-mediated oxidative stress and release of apoptotic proteins. Second, treatment with GSH ethyl ester, which recovers the depleted pool of mtGSH, prevents the enhanced neuroinflammation and neuronal damage observed in Tg-SREBP-2 mice after Aβ intracerebroventricular infusion (195). Collectively, these results suggest that a possible therapeutic approach to slow disease progression could be to replenish mtGSH (404).
The GSH:GSSG ratio could be a determinant factor in AD pathogenesis. In AD brains, this ratio becomes pro-oxidizing during aging due to an elevation in the GSSG content with a concomitant reduction in GSH content (8). Decrease in GSH concentrations is the result of a decline in the ability for de novo GSH biosynthesis due to the increase during aging of the Km of glutamate-cysteine ligase, the rate-limiting enzyme for de novo GSH biosynthesis. (376). Reduction of GSH is associated with an increasing vulnerability to oxidative stress (135, 593), manifested by the intensification of several markers of oxidative stress damage in AD brains, like 4-hydroxynonenal, indicative of oxidative damage to lipids, and 3-NT, indicative of ONOO− production (380). During chronic inflammation and oxidative stress, as seen in AD, astrocytes, the main source of GSH, switch from a neurosupportive to a cytotoxic and neurodegenerative role by releasing inflammatory cytokines and free radicals (61, 205). In fact, under oxidative stress, intracellular reduced or oxidized GSH is actively secreted (180, 266, 564). Furthermore, depletion of GSH during aging or neurological diseases may not only lead to cytotoxic activation of glia, but may also render neurons sensitive to cell death.
In aging and AD there are also changes in other enzymes involved in GSH metabolism [for a complete view on GPxs, see (644)]. GST activity is decreased in the AD amygdale, hippocampus, parietal lobe, and nucleus basalis of Meynert (380). Decreased levels of glutathione S-transferase family members mu (GSTM1) in cerebrospinal fluid (CSF) (380) and decreased transcript levels of GSTM3 were reported in AD-affected hippocampus (58), suggesting a role for GSTs in the pathogenesis of AD. Moreover, glutathione S-transferase omega-1 (GSTO1), whose transcripts are down-regulated in the hippocampus of AD patients (363), can be involved in the activation of IL1β (336), a fundamental component in the inflammatory response involved in the pathogenesis of AD (227, 229). Since GSTO1 can control the calcium efflux through ryanodine receptors present in the endoplasmic reticulum (ER) of pre- and postsynaptic terminals, it could contribute to the modulation of synaptic transmission and structural plasticity (182).
Decrease of the GSH:GSSG ratio results in oxidation that may induce reversible formation of mixed disulfides between PSH groups cysteine side chains and GSH originating S-glutathionylated proteins (PSSG) of a large number of proteins (155, 156, 324). S-glutathionylation is a reversible posttranslational modification from which the release of GSH can be catalyzed enzymatically by glutaredoxin (129, 579). Redox proteomics has allowed identification of specific targets of protein S-glutathionylation in AD, including proteins involved in glucose and energy metabolism (454). In AD, deoxyhemoglobin, α-crystallin B, glyceraldehyde phosphate dehydrogenase (GAPDH), and α-enolase are S-glutathionylated (136, 138). S-glutathionylation often results in a reduction of activity of proteins (454). For example, GAPDH that would act as an NO· sensor shows drastic reduction in its activity when nitrosylated (109, 242, 610). Activity is recovered with the addition of GSH (36). So, S-glutathionylation could be a form of redox regulation of protein function and activity (104, 155, 156, 324). Even more, S-glutathionylation could serve as a temporary shield from irreversible, more dangerous oxidation of cysteine residues (37, 155) allowing GAPDH to resume function once the redox state normalizes. Thus, S-glutathionylated p53 can also protect the integrity of its sulfhydryl groups from irreversible modification that occurs in the progression of AD, leading to permanent inactivation (171, 664). Selective glutathionylation of p53 in AD brain could prevent aggregate formation involved in oxidative stress conditions and neurodegeneration (171). On the other hand, S-glutathionylated tau undergoes rapid polymerization in vitro. Future studies are needed to address the formation and role of S-glutathionylated tau during neuronal oxidative stress (176).
NADPH is essential for maintaining GSH and Trx in a reduced state through reactions catalyzed by GSH reductase and TrxR, respectively (21). In AD patients, decreased Trx and increased TrxR levels have been reported in the amygdala and hippocampus (379). Several studies have shown that Prx (Trx peroxidases) expression is elevated in neurodegenerative diseases such as AD, PD, Creutzfeldt-Jakob disease, and amyotrophic lateral sclerosis (ALS) (100, 330). Thus, it has been suggested that Trx might play a protective role in AD, and Trx deficit might eventually contribute to increased oxidative stress and subsequent neurodegeneration in AD (379). However, it is not known if increased Prx expression occurs as a general response to nerve cell death or is a protective mechanism elicited by disease-specific stimuli. Exposure of cortical neurons to Aβ causes a time-dependent increase in Prx oxidation that is countered by a thiol-specific antioxidant (137). Aβ-resistant neurons not only survive exposure to high concentrations of Aβ but are also less sensitive to H2O2, in part due to increased activity and expression of the antioxidant enzymes GPx and CAT (551), as well as high levels of NADPH (596). Noteworthy, nerve cells selected for Aβ-resistance exhibit increased expression of specific Prx isoforms that are less susceptible to oxidative inactivation. Concomitant with enhanced TrxR activity, other main antioxidant enzymatic activities such as GPx, GSSG reductase, CAT, and SOD1 were also found elevated in regions of AD brain, where lipid peroxidation and protein denaturation were most pronounced. This elevation of antioxidant enzyme status was suggested to reflect a compensatory response to counteract increased oxidative stress characterizing this pathology (379). These observations strongly support a role for nitrosative stress in the pathogenesis of AD and indicate that the stress responsive genes, such as TrxR, may represent important targets for novel cytoprotective strategies (100).
In neurodegenerative diseases, like AD, there are often increased generation of RNS and ROS, which can contribute (discussed in section II.E) to neuronal cell injury via a series of redox reactions. In addition, NO· can react with cysteine residues of target proteins to form S-nitrosothiols. A key determinant of the specificity in NO· signal transduction is the interaction between NOS enzymes and proteins that are targets of S-nitrosylation (261). S-nitrosylation, the coupling of a NO+ moiety (nitrosonium cation, generated by the oxidation of NO·) to a cysteine, has emerged as a ubiquitous protein posttranslational modification. In a manner akin to protein phosphorylation, S-nitrosylation switches the on-off functions of receptors, GTPases, and transcription factors, among other proteins. In that sense, cysteines are critical in driving protein folding, metal ion chelating, posttranslational modifications, such as palmitoylation and prenylation, and thiol-based redox regulatory switches (117).
Nitrosative modifications can result in changes in mitochondrial function. For example, NO· reversibly inhibits Complexes I and IV (117), thus inducing release of ROS by mitochondria. Nitrosative and oxidative stress can also elicit dysfunction of mitochondrial dynamics (35, 65). Physiological and chemical evidence shows that S-nitrosylation modulates GTPase activity of the mitochondrial fission protein dynamin-related protein 1 (Drp1), thus contributing to altered mitochondrial dynamics, synaptic damage, and eventually neuronal death (148) as discussed on section II.C. Other examples follow: (i) The S-nitrosylation of protein-disulphide isomerase (PDI), an enzyme assisting the maturation and transport of unfolded secretory proteins. Its S-nitrosylation abolishes PDI-mediated attenuation of neurodegeneration triggered by ER stress, or misfolded proteins, or proteasome inhibition (653), and (ii) the S-nitrosylation of ApoE, which is discussed in section II.B, resulting in changes of its interaction with low-density lipoprotein (LDL) receptors (1).
Oxidative damage of the brain of AD patients and AD animal models includes peroxidation of lipids (83, 504), and oxidation of proteins and nuclei acids (465, 466). The oxidation of RNA and DNA could impair protein synthesis, DNA repair, and transcription, and could eventually lead to cell death (175). Furthermore, oxidation of mtDNA is ∼10-fold higher than nuclear DNA (nDNA). High levels of mtDNA oxidation could support the reported mitochondrial abnormalities in the AD brain, which may contribute to the increase of O2·− leakage, ultimately leading to elevated oxidative stress (see section II.C), one on the new hypothesis for the pathogenesis of AD (617, 618).
Protein carbonyls are increased in the hippocampus and parietal cortex, the brain regions that are most severely affected in AD, but not in the cerebellum, where little pathological damage is found. Increased levels of dityrosine and 3-NT levels are found in hippocampus, inferior parietal lobule, neocortical regions, and in the CSF of AD patients (109, 610). High levels of free HNE, an α,β-unsaturated hydroxyalkenal metabolite derived from lipid peroxidation, are reported in amygdala, hippocampal structures, and CSF (504). In addition, in vitro studies using Aβ1–42- or HNE-treated synaptosomes show an antioxidant-inhibited loss of lipid asymmetry, suggesting a role for Aβ and its associated oxidative stress in the loss of lipid asymmetry and consequently in neuronal loss of AD (198, 570).
Increased levels of HNE-bound proteins were also reported in AD brain (83, 86, 489). Increased levels of 8-hydroxyaguanine (8-OHG) have been reported in frontal cortex of familial AD subjects (465). The elevated level of 8-OHG in RNA of AD hippocampus correlates with the Aβ load, suggesting that RNA damage is an early event in AD (378, 466, 573). Markers of DNA damage like 8-oxo-7,8-dihydro-2-deoxyguanosine (8-OHdG), 8-hydroxyadenine, and 5-hydroxyuracil are also elevated in temporal, parietal, and frontal cortex in AD (207). 8-OHdG is also increased in CSF from AD patients and appears to be associated to the age-associated decrease of hydrolytic enzymes (594).
Protein oxidation affects many proteins and lipids in AD. However, a relatively few protein appears to be especially relevant for the pathology (609). Several irreversible protein modifications correspond to protein nitration and HNE modification. During progression of AD, there is an increased protein nitration and HNE modification that lead to decreased enzymatic activity, especially affecting energy metabolism, mitochondrial dysfunction, and cholinergic neurotransmission, in addition to changes on cytoskeleton components (90, 499). Several of the up-regulated and oxidized proteins in aging are known to be oxidized in neurodegenerative diseases as well, suggesting that these proteins may be particularly susceptible to processes associated with neurodegeneration (499) and can be relevant for the pathogenesis and progression of AD (87, 90).
A. Oxidative stress and progression of AD
Although the exact mechanism by which Aβ produces synaptic and neuronal loss is highly controversial, there are reports showing that antioxidant compounds block Aβ-induced neurotoxicity in culture and in in vivo and ex vivo AD models, consistent with a role of oxidative stress in AD pathogenesis and disease progression (611, 710).
The important number of proteins oxidatively modified in the MCI brain support the involvement of oxidative stress in dysfunction of glucose utilization and oxidative-dependent impairment leading to energy dysfunction, neuritic length, excitotoxicity, lipid peroxidation, cholinergic dysfunction, pH buffering and CO2 transport, regulation of the cell cycle, APP processing, tau hyper phosphorylation, antioxidant defense, cell signaling, and protein synthesis (88, 529). The proteomics to identify oxidized brain proteins show that most changes are common for MCI, early and late AD, suggesting that certain key pathways are triggered early and are maintained during the progression of AD (609).
An example of a protein oxidatively modified in both MCI and AD is provided by peptidyl prolys cis/trans isomerase (Pin-1). In addition to its role for the cell cycle, Pin-1 also regulates other biological functions such as protein assembly and folding, intracellular transport, intracellular signaling, transcription, and apoptosis (80). An additional important function of Pin-1 in the specific case of AD is its ability to regulate APP function (484), by regulating the production of Aβ (80, 484). Proteomics have facilitated the identification of of other proteins oxidatively modified that are involved in energy and glucose metabolism. The content of oxidatively modified energy-related proteins in MCI and AD brains correlates well with the decrease of glucose utilization and altered activity of enzymes involved in glucose metabolism (88, 272), including oxidation of proteins involved with glycolysis and tricarboxylic acid (TCA) cycle, and significant decreases in TCA enzyme levels (91, 607). Positron emission tomography (PET) studies showed that glucose uptake is reduced in early AD brains (164, 596). Alpha-enolase, triosephosphate isomerase, and phosphoglycerate mutase 1 are enzymes contributing to the glycolytic pathway, and together with ATP synthase-alpha, are common targets of oxidation in AD brains (109, 489, 608, 610). Alteration of the function of the above-mentioned enzymes may lead to increased production of methyl glyoxal (MG), a small ketoaldehyde compound, derived from triosephosphate, a glycolytic intermediate. MG can react with lysine, arginine, histidine, and cysteine residues and glycate them to form AGEs products and can ultimately lead to altered structure and function of proteins (226, 707). The proteins enolase, neuropolypeptide h3, ATP synthase, and aldolase appear to be oxidized in brain as early as MCI and at every stage of AD (88, 109, 489, 608, 610).
In MCI brain, there are decreased activity and levels of enzymatic and nonenzymatic antioxidants (231), which can lead to increased production of free radicals during the progression from MCI to AD. Coherent with a decreased antioxidant activity, there are elevated protein carbonyls, protein-bound HNE, free HNE, TBARS, and MDA and 3-NT in the brain of MCI subjects compared to age-matched controls (88, 89, 313). MCI subjects also show high levels of isoprostanes in plasma, urine, and CSF compared with those of healthy subjects (406). Further, in MCI, increased oxidative damage in nDNA and mtDNA, with increased levels of 8-OHdG,2,6-diamino-4-hydroxy-5-formamidopyrimidine, 8-OHG, 4,6-diamino-5-formamidopyrimidine, and 5-hydroxycytosine is reported (679). Augmented 8-OHG, reported in cytosol of AD brains, decreases when Aβ and NFT burden increases, suggesting that the oxidative damage to RNA is an early event in the progression of AD (466). Brains from early AD patients also show increased levels of protein nitration, indicative of increased levels of RNS, and elevated protein-bound HNE (530). Interestingly, it has been proposed that Aβ, at low concentrations, has protective effects (112, 342), on the basis that Aβ could be part of the antioxidant defense in conditions of aging and disease (467). This notion is especially relevant considering the therapeutical strategies based on decreasing Aβ levels, which could worsen the disease (492, 590).
Genes whose products participate in reducing oxidative stress, inflammation, and accumulation of toxic metabolites contain the antioxidant response element (ARE). ARE-containing gene promoters include GST, coenzyme Q10 (Q10), NAD(P)H:quinone oxidoreductase, SOD1, TrxR, and HO-1 [also referred to as heat-shock protein (Hsp)32], and GPx (292, 343). The ARE promoter element is bound by several transcription factors, being the nuclear factor E2-related factor 2 (Nrf2) responsible for activating transcription in response to oxidative stress. Nrf2 coordinates the up-regulation of cytoprotective genes via ARE.
Nrf2 is expressed in both the nucleus (predominant expression) and the cytoplasm of neurons in normal hippocampi. In AD hippocampus, Nrf2 levels are reduced (521) and it is not translocated to the nucleus, resulting in a significant reduction in Nrf2 nuclear content. The opposite occurs in PD, where Nrf2 label remains strongly nuclear in nigral neurons (521). These findings suggest that in AD neuronal Nrf2-mediated transcription is not induced despite the presence of oxidative stress.
The ability of Nrf2 to up-regulate the expression of antioxidant genes suggests that increasing Nrf2 activity may provide a useful system to combat oxidative insults. In fact, overexpression of Nrf2 in cell culture protects against oxidative damage elicited by H2O2, NO·, and glutamate (170, 582), whereas neurons from Nrf2 KO mice show an increased vulnerability to neurotoxic insults. Boosting the activity of the Nrf2-ARE pathway by tert-butylhydroquinone treatment or adenoviral Nrf2 gene transfer protects against Aβ toxicity, a neuroprotection associated with increased expression of Nrf2 target genes and reduced phosphorylation of p66Shc, a marker of increased susceptibility for oxidative stress (307, 329). On the other hand, in transgenic AD mice model APP/PS1, the Nrf2-ARE pathway is attenuated at the time of Aβ deposition (307). Increasing levels of Nrf2 in the brain of aged APP/PS1 mice significantly reduce their spatial learning deficits (306).
Nrf2 gene transfer is associated with a robust reduction in astrocytic, but not microglial activation and induction of Nrf2 target gene HO-1. Similarly, whereas Nrf2-ARE regulated gene expression attenuates inflammatory responses, disruption of Nrf2 expression can result in autoimmune disease and enhanced oxidative stress-induced inflammatory response (360). The association between neuropathological abnormalities and changes in expression of ARE-driven genes suggests that Nrf2-ARE signaling pathway could represent an endogenous protective response aimed to limit oxidative neural injury and to restore the redox-balance. Interestingly, recent cDNA profiling studies performed in Nrf2 KO mice, and various in vitro models have led to the discovery of several Nrf2-dependent genes that could be implicated in neuronal protection and survival (327, 344, 358, 359, 582).
There are additional processes that are regulated by cellular stress responses. The brain detects and overcomes oxidative stress by a complex system of longevity assurance processes. Hsps are highly conserved and facilitate correct protein folding. HO-1/Hsp32, an inducible redox-regulated enzyme and its interplay with TrxR, appears to have an important role in cellular antioxidant defense (96). Hsp serve as chaperones that bind to other proteins and regulate their conformation, preventing protein misfolding and oligomerization, protein movement across membranes, availability of receptor, or enzyme activity (497). Hsps are induced under conditions of cellular stress (396), and have been reported in a variety of disorders and injuries, including stroke, epilepsy, cell trauma, aging, and neurodegenerative disease (97, 99). Molecules that activate this defense mechanism are therefore possible candidates for cytoprotection (498). Some of the Hsps include ubiquitin, Hsp10, Hsp27, Hsp32, Hsp47, Hsp60, Hsc70, Hsp70, Hsp90, and Hsp100/105 (100, 172, 306).
Hsp response is also involved in cellular homeostasis during brain development, brain differentiation, cell cycle, apoptosis, growth factors action, mRNA, and protein half-life. Hsp can also be activated by non-noxious stimuli, such as nutritional antioxidants or acetylcarnitine. Acetylcarnitine, through activation of the redox-sensitive transcription factor Nrf2, and its consequent binding to the ARE in the HO gene, up-regulates HO-1 and TrxR, thus counteracting nitrosative stress and NO·-mediated neurotoxicity (96). Mitochondria are specific targets of the protective effect of Hsp against oxidative injury (394).
The antioxidant protein HO could sense NO· and thus protect against ROS and RNS insults. This notion is supported by the fact that NO· and NO·-related species increase HO activity and induce HO-1 expression, and besides, that cells pretreated with NO·-releasing molecules acquire increased resistance to H2O2-mediate cytotoxicity in association with HO activation (436). In fact, NO· could act as a signaling molecule that, by triggering expression of cytoprotective genes such as HO-1 and Hsp70, may lead to adaptation and resistance to subsequent more severe nitrosative and oxidative stress (435). Acetylcarnitine treatment of astrocytes exposed to cytokine-induced nitrosative stress restores GSH/GSSG ratio and complex IV inhibition, an effect associated with up-regulation of HO-1 and nuclear translocation of Nrf-2. In in vivo experiments, GSH is a critical factor for induction of cytoprotective Hsps (98).
Hsp60, Hsp70, and Hsp90 alone or in combination protect against intracellular Aβ stress through the maintenance of mitochondrial oxidative phosphorylation and functionality of tricarboxylic acid cycle. Notably, Aβ inhibition of complex IV activity is selectively neutralized by Hsp60. The combined effect of HSPs reduces free radical burden, preserve ATP generation, decrease cytochrome c release, and prevent caspase-9 activation, all important mediators of Aβ-induced neuronal degeneration (663).
Oxidation of specific brain proteins in common for MCI and AD indicates that certain key pathways are affected early and become triggers for additional pathological changes, thus affecting the progression of AD. Similarly, there are also protective changes being induced by oxidative stress. Exploring these deleterious and protective pathways in detail will provide clues for better understanding the pathogenesis and progression of AD and also for the development of effective therapies to treat or delay this disorder.
B. Aβ, Tau, ApoE4, and oxidative stress
1. Amyloid β
As already mentioned, the amyloid hypothesis (244, 245) proposes that the deleterious effects of Aβ and amyloid plaques are the causative agent of neuronal dysfunction and death in AD (355). The main component of senile plaques are peptides of approximated 4 kDa, being Aβ1–40 and Aβ1–42, the most abundant components. In vitro and in vivo studies indicate that Aβ1–42 is both more amyloidogenic and more toxic than Aβ1–40 (66, 81, 426). The self-association of Aβ is dynamically structured from monomers passing through dimers and protofibrils to organized oligomeric and multimeric fibrils (167, 169, 262, 570, 574, 677). Almost every Aβ structural entity (169, 236, 244, 351, 574, 671) can lead to the impairment of various neuronal functions ranging from in vitro hippocampal neuron LTP (677, 681), or synaptic pruning (94, 574) to reduced learning ability in vivo (274, 351), and to excitotoxicity and progressive synaptic damage and neuronal death. The progressive accumulation of Aβ adds chronic inflammatory activation to the neuronal dysfunction (especially for some oligomeric forms of Aβ). In sporadic AD, both increased production and reduced removal of Aβ can be linked to aging, oxidative stress, and the inflammatory status of the nervous tissue, which can also influence neuronal function and eventually neuronal viability (206, 669). In fact, signal transduction cascades activated by Aβ can be linked with cell injury and neurodegenerative changes (364). However, aggregated Aβ fails to show this correlation with pathological lesions or clinical compromise (29, 294, 517, 602). In contrast, recent evidence shows that the level of nonfibrillar Aβ appears to be directly correlated with the severity of the pathology (386, 416), suggesting that small Aβ oligomers are the actual toxic species rather than fibrillar Aβ (667).
As many as 30% of cognitively normal individuals in their 70s have Aβ plaques in their brains as revealed by PET scans, without showing signs of cognitive impairment or dementia (193, 431). Similarly, autopsy studies also highlight that Aβ plaques are present in cognitively normal individuals (14, 294). In an autopsy-based cross-sectional study, it was found that Aβ plaques accumulated before the development of cognitive impairment; thereafter, the amount of Aβ in the brain remained nearly constant and was similar across a wide range of patients showing different clinical severity. In contrast, atrophy, neuronal and synaptic loss, and tangles increase with severity of illness (94, 570, 667).
Three broad types of plaques can be recognized. The first type is nonamyloid Aβ-deposits, which can be further distinguished as diffuse and focal. The other two have amyloid structure (i.e., Congo red or thioflavin S positive), a focal type of deposits and the neuritic plaque, which is surrounded by a neuritic corona of neurofibrillary degeneration. In addition, other categories can be recognized depending on structure, morphology, and staining [for a detailed description, see (184, 632)]. Diffuse Aβ-deposits are poorly immunoreactive and ill-limited. There is no association with glial cells and show no inflammatory activation. Thus, diffuse plaques appear to be associated to the aging process and have no correlation with AD. In contrast, in neuritic plaques, the focal deposit constitutes the core, which commonly shows microglia directly associated with it. Indeed, focal deposits with and without amyloid aggregation (234) are associated with morphologicaly activated microglia (19), which is a hallmark of AD pathology (217, 577), although this point is not devoid of controversy (555). The corona surrounding the Aβ deposit contains neuritic and astrocytic components. Neuritic plaques are always associated with congophilic deposits (420). This characteristic could depend either on the fact that microglia themselves are necessary for the transformation of the diffuse deposit into amyloid (691) or because the amyloid conformation is what activates microglia. Furthermore, it is unclear if amyloid plaques originate from diffuse plaques or if they arise by independent pathways (568, 599, 705).
There are also other special plaques, like the cotton wool plaque, formed by N-truncated Aβ (423); the inflammatory plaque, characterized by a strong microglial reaction, surrounding an amyloid core without Aβ reactivity, observed in PS1 mutations (580); and the vascular deposits of Aβ in the vessel walls, of the hereditary cerebral hemorrhages Dutch type, related to a mutation of the APP gene (354). It should be emphasized that plaques are not pathognomonic of AD. They have been described in chronic head trauma (297, 464) and prion disease (478), and they can be also observed in cognitively normal population, and MCI population (23, 68, 505), although the density of neuritic plaques and of NFTs is, on average, higher in early AD cases than in MCI patients (43, 221). The major difference of Aβ deposits found in AD is their distribution (22, 68, 633). In cognitively normal individuals, parenchymal and vascular Aβ deposits are usually restricted to the cerebral cortex, the basal ganglia, the thalamus, and the hypothalamus. In contrast, in AD patients, Aβ deposits are found in the midbrain, brain stem, and cerebellum, in addition to the brain regions potentially affected in cognitively normal individuals (633, 634).
Some propose nowadays that AD can be a two-stage process, in which Aβ initiates a cascade of early damage, which when progresses, becomes independent of Aβ. This independence of Aβ would explain the mild efficacy of therapies targeting Aβ after AD is well established, because in this later stage biological processes like calcium elevation, calcineurin and caspase activation, mitochondrial dysfunction, impairment of axonal transport, neuronal impairments, and inflammation predominate (333). An alternative view held by others is that once Aβ has aggregated in plaques, the damage to the brain is done, and cannot be reverted. Furthermore, plaques represent a source of persistent insult for the brain (either by its mere presence, by its effect on neuroinflammation and oxidative stress, or by being a source of soluble oligomers). Under this view, even if the downstream effects of plaques are targeted therapeutically, if the plaque is not neutralized, the insult persists and the therapy fails to be efficacious (669, 671, 672). In this way, depending upon the hypothesized pathogenic process of AD, the plaque has a different meaning for a therapy to be effective.
Although the mechanism of damage induced by Aβ remains elusive, there is evidence suggesting that oxidative stress is at least partially responsible for Aβ-induced damage (Fig. 12), as it appears to be a mechanism shared with several neurodegenerative diseases (215, 218, 456, 596). In particular, markers of oxidative stress in AD brains co-localize with Aβ plaques (477, 591), situation also observed in animal AD models (591).

It has been reported that Aβ exerts toxicity that correlates with the generation of cellular H2O2 (41), and becomes abolished by activation of SOD (639), O2 ·−/H2O2 scavengers (73), and vitamin E (247). Conversely, oxidative stress increases Aβ levels (707), implicating the perpetuation of the damage induced by oxidative stress. One current controversy is whether or not Aβ fibrils are required for toxicity. Interestingly, oxidative stress significantly precedes the appearance of Aβ plaques and neurofibrillar tangles in AD brains (427, 429). However, in vitro studies indicate that exposure to aggregate Aβ leads to cytotoxicity due to calcium influx and the induction of oxidative free radical damage (409). Several different hypotheses have been proposed to explain this toxic effect, including the formation of ion channels in cell membranes (516), the spontaneous fragmentation of Aβ to generate peptidyl radicals, and the direct formation of H2O2 by Aβ (623), by mechanisms at least partially dependent on a Fenton-type reaction (276).
Respiratory burst of microglia is triggered by several factors, and Aβ also appears to be a potent trigger, indirectly contributing to oxidative stress (119). Aβ is partly targeted to mitochondria and, under pathologic conditions, appears to induce mitochondrial dysfunction and oxidative stress. Oxidative stress, in turn, can induce proteins to adopt insoluble β-pleated sheet conformations. Some authors propose that Aβ is not toxic in absence of redox metal ions (277). Because Cu and Fe are required for electron transport, their chelation could inhibit indirectly oxidative phosphorylation. Accordingly, in vitro studies show that chelation of Cu and Fe inhibited the formation of ROS (77, 263, 589). Thus, the oxidative damage of Aβ would be linked with the presence of redox metals, Cu, Zn, and Fe abundant in mitochondria. Aβ has unusual high affinity for both transition metal ions Cu and Fe and has the capacity to reduce these metals and, subsequently, produce H2O2 (134) and OH· (232). These readicals will contribute to the generation of oxidized amyloid, cross-linked forms of Aβ, which in turn, will promote the oligomerization of monomer Aβ and the formation of insoluble precipitates. Precipitates, in turn, will sequester the ions that enable their aggregation (76, 178, 252, 278). Subsequent extracellular secretion of metal-Aβ complexes would give rise to insoluble Aβ plaques, which presumably would promote microglial cell cytotoxic activation and invasion. In turn, aggregated Aβ triggers ROS production by microglia, thereby potentiating deposition of Aβ. Both Aβ plaques and NFTs are enriched in Zn, Cu, and Fe. Furthermore, Fe, by activating the Fe-response element (IRE) in the 5′ UTR of APP mRNA, or by modulating the activity of α-secretase, can promote the generation of Aβ (543). In concordance with those observations, concentration of metal ions, such as Fe, Zn, and Cu, are markedly elevated in AD patient brains, particularly in those areas most severely affected (76). Hence, Aβ aggregation and oxidative stress appear to be potent reinforcing phenomena (278).
Cu(II) is known to bind to Aβ with a high affinity via histidine and tyrosine residues (281). Cu(II) promotes Aβ-dependent neurotoxicity by promoting the formation of H2O2 (134) and influencing its aggregation. Moreover, Cu–Aβ produces·OH (232), at least partly by inducing conformational changes that enable SOD-like activity (111). Furthermore, Cu appears to play a role in generating ROS through its binding to APP, which reduces Cu(II) to Cu(I) through an electron-transfer reaction that could enhance the production of·OH (722).
The role of Zn in the etiology of AD is very intriguing. Several reports indicate that Zn in micromolar concentration inhibits Aβ-induced toxicity (134, 212). The exact mechanism of the protective effect of Zn against Aβ toxicity is unclear; however, one of the hypotheses is that Zn competes with Cu (or Fe) for Aβ binding. Binding of Zn changes Aβ conformation and Cu ions cannot reach its metal-binding sites.
Despite the overwhelming correlations between oxidative stress and Aβ, the fine tune mechanism is still a matter for active research. In addition, Aβ-induced oxidative stress and neurotoxicity can involve methionine-associated formation of ROS (82). Finally, there are also reports that Aβ can increase oxidative stress through its effects on mitochondria, which will be described in the next section.
2. Tau
The hypothesis stating that tau pathology causes AD has been the main alternative to the amyloid hypothesis (440). As mentioned, intraneuronal tangles containing hyperphosphorylated tau are a hallmark of AD pathology (328) and tau has been considered a mediator for Aβ cytotoxicity (588). Tau is a protein that binds to tubulin, stabilizing microtubules (MTs). In AD, tau is abnormally phosphorylated, resulting in its aggregation as NFTs, which are toxic to neurons. Cytotoxic activation of microglia may also participate in tau phosphorylation and formation of NFTs (223). Nevertheless, as mentioned for Aβ plaques, neurofibrillar tangles are also present in various kinds of dementia besides AD (243, 283), suggesting that they are, more than a cause of neurodegenerative disease, a sign of degenerative changes.
The toxicity of tau aggregates depends on the loss of physiological functions and the gain of pathological functions (213, 382). In the first case, tau aggregates are unable to bind and stabilize MTs, while in the second, soluble oligomeric tau appears to be more toxic than aggregated tau (48). Early tau oligomers appear to be constituted by highly phosphorylated or truncated tau (724). Tau oligomers are rich in β-sheet structures and promote formation of fibrils once they reach a size of 20 nm (392). In AD, during the transition of an unfolded conformation to the aggregated state, formation of oligomers plays a key role in the formation of intracellular aggregates of tau. In vitro assays suggest that tau oligomerization is required for stabilization of MTs through neuronal development (398) and oligomers act as an electrostatic zipper during MT stabilization (545). Furthermore, tau oligomers appear to be able to induce neurodegeneration and memory impairment in the absence of Aβ plaques (311, 425, 552).
Other factors also affect tau aggregation. Pin-1, one of the proteins we mentioned that is oxidized in AD, regulates the function of proteins involved in cell cycle regulations such as cell cycle-dependent protein kinase 5, which is involved in the phosphorylation of tau and the genesis of NFTs via its action on both kinases and phosphatases (80). Thus, decreased levels and oxidative dysfunction of Pin-1 could promote tangle formation and Aβ production and lead to neuronal dysfunction (80, 88, 608).
Acting together, increased calcium concentration, oxidative stress, and mitochondrial dysfunction induced by Aβ plaques (333) can activate two interactive cell effector systems, calcineurin and caspases (154). One of the targets of caspases is calcineurin, generating a constitutively active form of the enzyme. Activated caspases also cleave tau, generating a shortened molecule that aggregates and also can lead to calcineurin activation, oxidative stress, and mitochondrial dysfunction, as it will be discussed in the next section.
Oxidative modification and altered function of tau can affect cytoskeleton protein properties that are crucial for maintaining neuronal structure, connections, and axonal transport, which in turn could also contribute to the reported loss of synaptic and neuronal structural integrity in AD brain (597). Cytoskeleton proteins are also oxidatively modified in in vitro and in vivo Aβ models of AD, suggesting again that Aβ could have a role in triggering the oxidation of these proteins and in the consequent development or progression of AD (66).
3. Apolipoprotein E4
ApoE is the major component of lipoprotein particles in the brain that mediate transport of cholesterol and other lipids between neurons and glial cells. The only difference among various ApoE isoforms is the number of cysteine residues (two in ApoE2, one in ApoE3, and none in ApoE4) that may serve as potential targets for S-nitrosylation. Data reported by Abrams et al. (1) suggest that S-nitrosylation of ApoE3 can result in changes of its binding and/or substrate specificity for LDL receptors. However, the authors also note that direct measurement of binding affinities is still missing (1). S-nitrosylation affecting ApoE3 can be involved in regulating lipid metabolism and play important roles in the pathogenesis of neurodegenerative disorders such as AD and PD (148, 153, 653).
Among the several potential risk factors for AD, the most consistently associated risk gene is ApoE (294). This suggests that cerebral lipid metabolism may be involved in the pathogenesis of AD. Individuals with two ApoEɛ4 alleles have a more than seven times increased risk of developing AD compared with those with ApoEɛ3 and ApoEɛ2 alleles. How ApoE4 increases the incidence and lowers the onset age of AD is not well understood. ApoE4 could be more vulnerable than other isoforms to degradation, limiting lipid mobilization for neuronal repair, neuroplasticity, and cognitive reserve in response to the neurodegenerative process (2); β-amyloid clearance is less efficient, and deposition of the neurotoxic peptide more pronounced in the brains of aging ɛ4 carriers (44). The O2-mediated interaction between ApoE4 and soluble Aβ peptides suggests that the reaction of tissue O2 with ApoE4 may catalyze oxidative changes of nonaggregated Aβ, a process that is normally slow and results in formation of aggregates (246). Thus, when H2O2-induced microglial cell activation occurs, as observed in neuroinflammatory activation, the time required to complete the transformation is reduced. Also, it has been proposed that enhanced formation of C-terminal-truncated fragments of ApoE4 stimulates tau hyperphosphorylation and formation of NFTs (246). ApoE4 has been linked with increased levels of LDL and as a risk factor for cardiovascular disease, with increased levels of atherosclerosis (188, 225), which could have detrimental effects on brain function through decreased blood flow and altered metabolic properties (246). Indeed, ischemia and hypertension-related vascular lesions are more frequent in ɛ4-positive patients (166), which can contribute to an altered oxidative stress environment. Nondemented ApoE4 patients show a similar regional pattern of hypometabolism before the onset of disease that correlates with changes seen in the AD brain, suggesting that an ApoE-related decrease in brain metabolism could contribute to development of AD (531, 532, 587). Finally, ApoE genotype may indirectly impact patterns of protein synthesis in AD by influencing the expression of microRNAs (561). In AD, there is up-regulation of microRNAs that correlates with down-regulation of target genes grouped in functional categories like synapse activity, transcription, redox homeostasis, and DNA damage (393, 561).
C. Mitochondrial impairment, mitophagy, and oxidative stress
In neurons, synaptic transmission, cellular remodeling, and cell death are processes tightly associated with mitochondrial function (445, 565). On the other hand, mitochondrial dysfunction, oxidative stress, and cellular death are common denominators in many neurodegenerative diseases (371). The mitochondrial cascade hypothesis proposes that the life span of mitochondria is finite and, when it is overpassed, mitochondrial functions are impaired; this propasal offers interesting insight for understanding the biochemical, histological, and clinical features of AD (619). Evidence supporting this hypothesis is multiple and include altered morphology, depressed metabolic activities, and the release of mitochondrial pro-apoptotic protein in both animal models and cell culture assays of neurodegeneration (Fig. 13) (164, 428). Electron microscopy analysis of mitochondria in AD brains revealed significant morphological alterations (27). Mitochondria are vital organelles and the main energy source for cells; they are also critical regulators of intracellular Ca2+ level, and cell death (194, 713). Nevertheless, they are also one of the main intracellular sources of ROS (350). Overexpression of mitochondrial uncoupling proteins would reduce production of ROS and would provide neuroprotection (122, 315, 408, 612). However, a direct link between respiratory uncoupling and these two activities has not been conclusively provided (296).
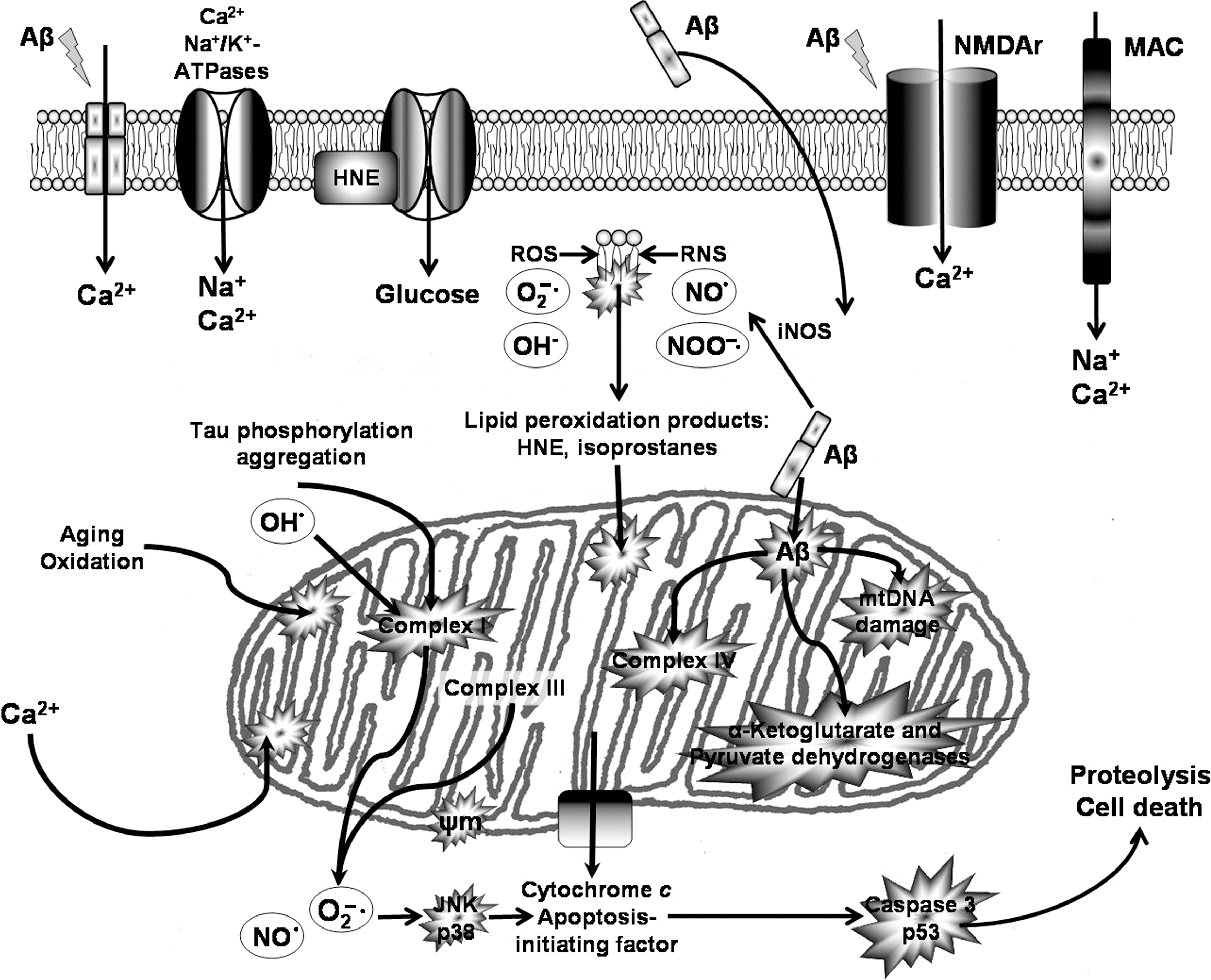
For neurons to generate the large amounts of energy they require, mitochondria undergo fission and fusion (referred as mitochondrial dynamics) to generate smaller or elongated organelles (684). Normal mitochondrial fission and fusion facilitate biogenesis of mitochondria, repair of defective mtDNA, and redistribution of mitochondria to sites requiring high-energy production (141, 203, 325). The complex morphology of neurons adds additional complexity to mitochondria dynamics, which should be moved into different neuronal regions for satisfying local demands of energy and calcium buffering. Indeed, mitochondria are highly mobile in neurons and become abundant in neuronal segments with high metabolic demands such as active growth cones and synapses (305), where mitochondrial biogenesis is particularly required for synaptic transmission and maintenance of synaptic structure (141, 357, 365, 388). The dynamic balance of mitochondrial fission and fusion not only controls mitochondrial morphology, length, size, and number but also regulates mitochondrial function and distribution, having profound impact on neuronal function (139, 684). In AD, mitochondria accumulate in the somata and are reduced in neuronal processes of some neuron populations (684) likely contributing to the observed synapse dysfunctions. Impaired mitochondrial dynamics is manifested in altered mitochondrial morphology and reduced cell bioenergetics and thus contributes to neuronal injury in many neurodegenerative disorders, including AD, PD, and Huntington's disease (HD) (35, 65, 325). Mitochondrial abnormalities correlate with dystrophic neurites, the loss of dendritic branches, and the pathological alteration of the dendritic spines in brains of AD patients (28, 619, 620).
The mitochondrial cascade hypothesis establishes that in sporadic AD, Aβ deposition, NFT formation, and neurodegeneration may be consequences of mitochondria malfunctioning. Mitochondrial fragmentation is an early event during apoptosis that precedes cytochrome c release and caspase activation (201). Excessive mitochondrial fission is correlated with increased ROS production (684, 685, 716), but there is no total agreement on what causes mitochondrial malfunction in sporadic AD. It is possible that accumulation of oxidative stress-induced mtDNA damage during aging and subsequent mitochondrial dysfunction may serve as a trigger. There is increased oxidation in both mtDNA and nDNA in frontal, parietal, and, especially, in temporal lobes of AD cases compared with age-matched controls, with a 10-fold predominance of mtDNA oxidation over nDNA oxidation (680). There are also mtDNA mutations associated with increased incidence of AD (128). It has been found that AD brains show increased unique mtDNA mutations compared with control cases. These mtDNA mutations are enhanced in an age-dependent fashion and preferentially occur in mtDNA regulatory elements, such as the control region (128).
Therefore, in neurodegeneration, abnormal mitochondrial morphology is frequently observed (27, 265, 684), and patients in early stage of AD exhibit declining mitochondrial energy metabolism and ATP production (366, 526, 686). Posttranslational modification of mitochondrial fission and fusion proteins contributes to altered mitochondrial dynamics. Phosphorylation, ubiquitination, sumoylation, and proteolytic cleavage of the mitochondrial fission protein Drp1 (also referred as DLP1), which is found in AD brains (148, 684), regulate mitochondrial fission by affecting its activity in culture (140, 448, 688, 711). Excessive accumulation of nitrosative stress triggers abnormal mitochondrial morphology in brains of neurodegenerative patients via S-nitrosylation of Drp1 (148). Exposure of neurons to aggregated Aβ25–35 induces an acute impairment in mitochondrial axonal transport (550) and striking mitochondrial fragmentation before neuronal demise. In fact Aβ is associated to S-nitrosylation and hyperactivation of Drp1 (148), which leads to mitochondrial fragmentation (684, 685), bioenergetics impairment, and synaptic damage in cell models of AD (449). Besides, Aβ treatment is associated to abnormally distributed mitochondria (35, 685), suggesting that Aβ may trigger excessive mitochondrial fission in AD patients, which can lead to an increase in NO·. Aβ-induced axonal and dendritic swelling and Aβ-decreased axonal transport in hippocampal neurons (572) can contribute to altered mitochondrial distribution. Other studies also suggest that overexpression of APP and treatment with amyloid-beta-derived diffusible ligands induce mitochondrial fragmentation and abnormal distribution without cell death (684, 685), suggesting that Aβ-induced abnormal mitochondrial dynamics may play a role at early of AD pathogenesis. In fact, synaptic dysfunction is one of the early and most robust correlates of AD-associated cognitive deficits (684).
Aβ inhibits enzyme complex IV, a major source of ROS generation, even in in vitro preparations lacking biological membranes (105). In vivo, Aβ interacts with several proteins belonging to the outer mitochondrial membrane and affects the transport of nuclear-encoded mitochondrial proteins, such as subunits of the electron transport chain complex IV, as well as inner mitochondrial membrane and mitochondrial matrix proteins. Reduction of the complex IV reduces the amount of hydrogen that is translocated, thus lowering mitochondrial membrane potential. These events cause abnormal mitochondrial electron activities, resulting in the reduction of complex V activity, the decrease in ATP levels, and the increase in ROS generation [reviewed in (186)]. The 50% inhibition of complex IV by Aβ is, likely, the cause for a similar reduction in ATP generation. However, inhibition of the respiratory chain and depletion of ATP could result in oxidative stress through the production of ROS from electron leak at complexes III and I or through the previous formation of ROS damage complex IV. Moreover, ROS induce peroxidation of mtDNA and mitochondrial lipids. The complex of Aβ bound to alcohol dehydrogenase changes mitochondrial membrane permeability and reduces activities of respiratory enzymes, exacerbating ROS production and leading to mitochondrial failure. As already discussed, Aβ binding also activates Fis1 (fission protein) and promotes increased mitochondrial fragmentation, and favors the opening of the mitochondrial permeability transition pore (PTP) (186).
As we age, we accumulate wear-and-tear from oxidative mitochondrial damage, especially accumulation of toxins that reduce cell metabolic activity, favoring neurotoxicity and ROS-induced oxidative stress. This triggers the 3-R response (Fig. 14) characterized by Reset of neurons trying to repair themselves by manufacturing Aβ, which behaves as a signaling molecule that reduces energy production; removal of neurons by undergoing programmed cell death when faced with oxidative stress; and replacement of cells that faced with metabolic stress undergo cell division (619). Neurons, however, are terminally post mitotic and die if they try to divide. In fact, one of the proposed mechanisms for the generations of NFTs (others are discussed in the previous section) is this attempted remodeling of the cytoskeleton, furthering neuronal dysfunction (179).
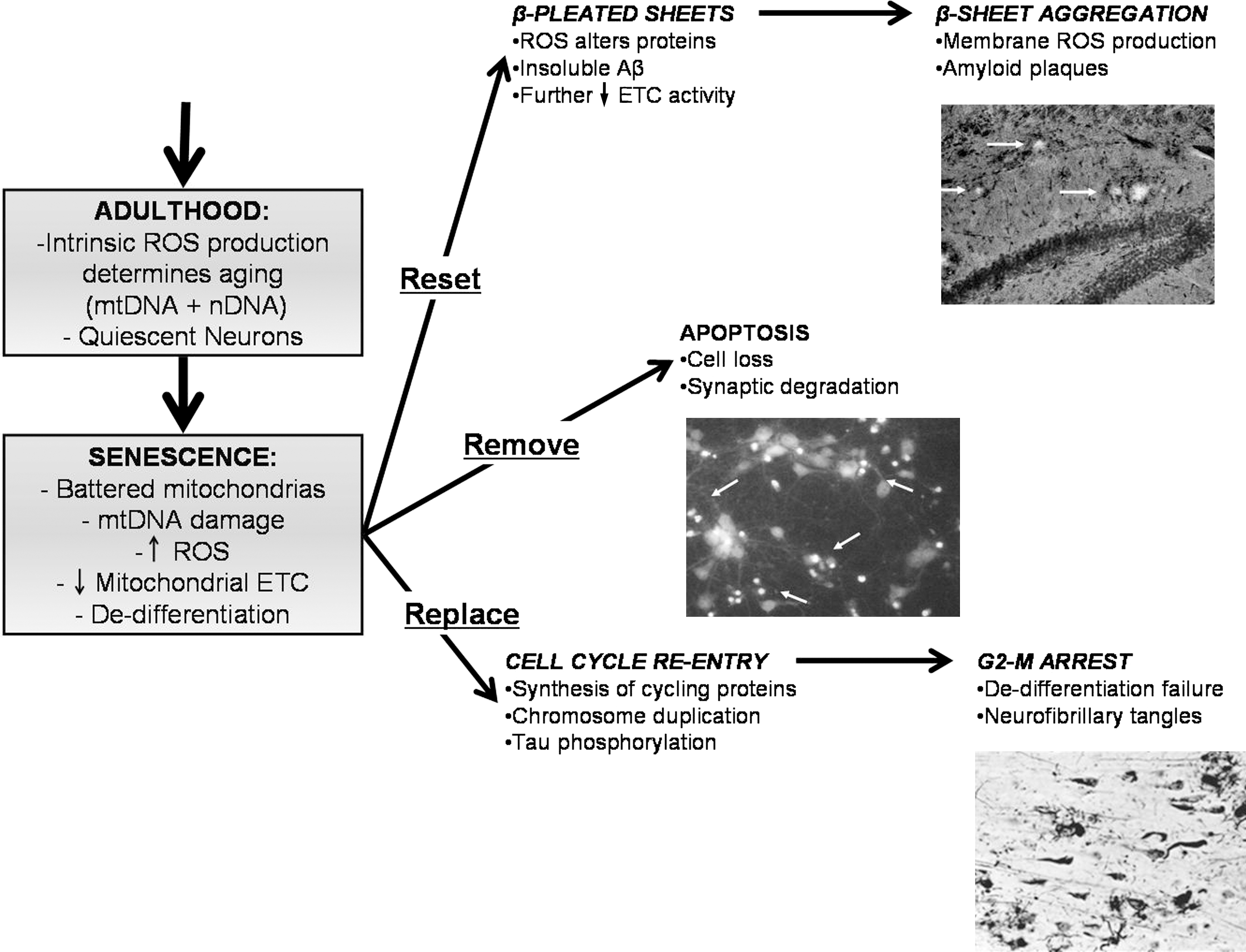
Mitochondria are both source and target of ROS, because an important part of oxidative damage occurs near the site of their production (658). Exposure to high levels of oxidants, especially in presence of calcium, can also induce the opening of the mitochondrial PTPs, a voltage-sensitive, nonselective ion channel that opens between the mitochondrial matrix and cytoplasm (126, 719). The opening of the mitochondrial PTPs appears to be a mediator of cell death due to an inhibition of the electron transport chain. This in turn results in oxidative stress and increased leakage of Ca2+ currents that induce apoptosis, by the release of cytochrome c (264, 300). In situ hybridization to mtDNA, immunocytochemistry of cytochrome c oxidase (Cyt-c), and electron micrographs of brain biopsies confirm that mitochondrial abnormalities are present in AD brains (110, 265). These studies also show that in AD patients, neurons with increased oxidative damage had increased levels of mtDNA and Cyt-c in the cytosol, which correlated with the significantly reduced number of mitochondria in these tissues.
In some AD cases, decreased Cyt-c activity was accompanied by increased free-radical generation and reduced energy metabolism (27, 265, 370). Analysis of mitochondria, isolated from platelets of AD patients, showed a decrease in Cyt-c activity and in ATP levels, associated with an increase in ROS (101). In transgenic AD mice with mutations of the mitochondria-specific antioxidant enzyme MnSOD, the partial enzymatic deficiency of mitochondrial MnSOD exacerbated AD phenotype, increasing amyloid deposition (356), tau phosphorylation (419), and accelerating behavioral impairment (192). Conversely, MnSOD overexpression attenuated the AD phenotype (183).
Inhibition of the electron transport chain (208) or enhancement of beta secretase (BACE) activity induced by oxidative stress (626) increases APP processing toward production of Aβ, strengthening the notion that not only Aβ lays at the origin of AD. Distinct mitochondrial enzyme deficiencies have been reported in AD brains, including decreased activity of pyruvate dehydrogenase complex and the enzymatic step linking the glycolysis metabolic pathway to the citric acid cycle (490, 709). As mentioned in section II.A, glucose metabolism impairment is part of the constellation of mechanisms involved in the pathogenesis of AD.
Interestingly, decreasing tau levels prevented deleterious Aβ-induced effects in hippocampal neurons (675). Notably, Aβ injections amplify tau pathology (64, 224), whereas absence of tau abolishes Aβ toxicity (288, 289), indicating that mitochondria show enhanced vulnerability toward Aβ insult in tau transgenic mice (162, 185), and suggesting the existence of a synergistic action of tau and Aβ pathologies. When expression of truncated tau was combined with Aβ at sublethal concentrations, a decrease in moving mitochondrial population (401, 637), a decrease in oxidative stress levels, and an enhancement of Aβ-induced mitochondrial potential loss were observed in neurons (514).
It is also proposed that reduced cell energy promotes tau phosphorylation in both sporadic and autosomal dominant AD (618). Accordingly, induction of tau phosphorylation is produced by prolonged fasting, complex I inhibition, carbon monoxide (CO) inhibition, mitochondrial uncoupling, and hibernation (state associated with mitochondrial uncoupling) (189, 621, 708). Tau may also contribute to mitochondrial fragmentation in AD brains. Tau is an in vitro substrate for the mitochondria caspase-3 (152, 209). Caspase-cleaved tau and tau truncated at Asp-421 to mimic caspase cleavage (T4C3) induced mitochondrial fragmentation and mitochondrial fission in a calcineurin-dependent manner (515). The expression of caspase-cleaved tau fragments in neurons can impair mitochondrial function, leading to changes in mitochondrial membrane potential, dysregulation of mitochondrial calcium level, mitochondrial integrity, and increase in mitochondrial O2 ·− (515). Conversely, it has been shown that antioxidant treatments inhibit glycogen synthase kinase 3β, the major kinase for tau phosphorylation (624), and that MnSOD overexpression can reduce tau phosphorylation back to normal, indicating the role of mitochondrial O2 ·− in tau dysfunction and, by extension, in AD.
Aβ and NFTs induce deficits in the function of the respiratory chain: acting independently as well as synergistically on mitochondrial dysfunction—dysfunction that is intrinsically tied to oxidative stress. Both events occur early in aging and in the pathogenesis of aging-related neurodegenerative diseases. In triple transgenic mice (triple AD) that combine P301L tau transgenic pR5 mice and APPswPS2N141I double-transgenic APP152, it was possible to determine functional consequences of the combined Aβ and tau pathologies. Proteomic and functional analysis showed deregulation of 24 proteins of which one-third were mitochondrial proteins mainly related to complexes I and IV. Deregulation of complex I was tau dependent, whereas deregulation of complex IV was Aβ dependent. Synergistic effects of Aβ became more robust as animals aged (538). A similar synergistic effect of aging and Aβ pathology was also observed in double Swedish and London mutant APP transgenic mice. Pronounced mitochondrial dysfunction in adult Thy-1 APP mice, with reduction of mitochondrial membrane potential and reduced levels of ATP, was already observed at 3 months of age, when elevated intracellular, not extracellular, Aβ deposits are present. Mitochondrial dysfunction was associated with higher levels of ROS, an altered Bcl-xL/Bax ratio, and reduction of complex IV activity (251). Aberrant expression of proapoptotic molecules probably contribute to neuronal loss in the late stages of AD pathology. Up-regulation of mitochondrial apoptotic molecules, including Bax and other pro-apoptotic members of the Bcl-2 familiy, is observed in AD (323, 391, 446). It has been proposed that mitochondrial protection and reduction of oxidative stress are important components of a neuroprotective treatment (352). Addition of Vits C and E effectively scavenged intracellular Aβ-mediated ROS generation, but was less effective in protecting the activity of oxidative phosphorylation enzymes and ATP generation (663). These data argue that Aβ directly interferes with oxidative phosphorylation resulting in oxidative stress.
In addition, Aβ appears to inhibit directly or indirectly key mitochondrial enzymes in the brain and isolated mitochondria, such as Cyt-c (107, 250, 527). It also reduces the activity of the respiratory chain (102, 488), pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and decreasing NADH, and, when entering the mitochondria, compromises mitochondrial function by inactivation of MnSOD, with subsequent increase of reactive species (13, 335). Other proposed target for Aβ is the PTP (105, 106). Additionally, a mitochondrial dehydrogenase/reductase, the amyloid-β peptide alcohol dehydrogenase (ABAD), that interacts with Aβ has been identified (400). Interactions between mitochondrial functions and Aβ were also validated in AD patient brains and in a transgenic mouse model overexpressing APP/ABAD, which also has elevated levels of oxidative stress and impairment of memory (387). In vitro, it has been shown that Aβ impairs mitochondrial movement inside dendritic spines (549) in cultured hippocampal neurons (550).
1. Mitophagy
Mitophagy is the specific autophagic elimination of mitochondria. Autophagy is discussed in section II.F. The fact that mitochondria are central to the cell death process has also relevance for autophagic cell death, specifically in relation to the process of mitophagy (264, 445, 643). Mitochondrial damage can induce selective mitophagy (712), but beyond quality control, to remove damaged mitochondria is also a normal constitutive process, being required for steady-state turnover of mitochondria (625), for the adjustment of mitochondrion numbers to match changing metabolic requirements (322), and during specialized developmental stages in mammalian cells, such as during red blood cell differentiation (334, 567). However, it is not well understood how mitophagy is able to selectively target mitochondria, and whether mitophagy is beneficial or detrimental for cells under pathological conditions. In mammalian cells (650), mitophagy is preceded by mitochondrial fission (693), which divides elongated mitochondria into pieces of manageable size for encapsulation and also serves as quality control of damaged mitochondria for selective removal by mitophagy.
D. Neuroinflammation and oxidative stress
Neuroinflammation is a choreographed process involving numerous cells, including neurons, astrocytes, macrophages (mainly microglia), and lymphocytes. In contrast to the classical inflammation involving the peripheral immune system (hot, painful, swollen, and red being its key features, reflecting intense vascular reactivity), neuroinflammation is defined mainly by the activation of microglia and astrocytes, as well as by the increased levels of many molecular mediators, including cytokines like IL1β, TNFα, and TGFβ, all of which increase with aging (248, 414, 669, 674). Microglial cell cytotoxic activation and elevated levels of inflammatory cytokines further induce the secretion of more cytokines and ROS (674).
Herrup proposes that age-dependent changes gradually take their toll on the brain (260, 674), changing, among several things, glial cell reactivity (669). Under those conditions, stimulatory event or injury will be capable of initiating the disease process. The injury triggers a protective response, but the age-related impairment of normal homeostatic mechanisms determines persistance of responses, associated, for example, to a robust induction of oxidative stress that determines progression of degenerative changes (260, 669).
Neuroinflammation emphasizes crosstalk between neurons and glia and establishes a complex interaction with oxidizing agents through redox sensors localized in enzymes, receptors, and transcription factors. The redox status modulates the participation of cytokines in signaling processes, which are critical mediators of oxidative stress, neuroinflammation, and neurodegeneration in the CNS (314, 439). Oxidative stress, in turn, results in increased inflammatory cytokine production. Additionally, neuroinflammatory cytotoxic activation induces oxidative changes, creating a vicious cycle between oxidative stress and neuroinflammation [comprehensive review in (544)]. This close relationship between neuroinflammation and altered redox state has physiological implications in the maintenance of the cellular homeostasis and survival (557).
Neuroinflammation, whereas cause or consequence of neurodegeneration, can be visualized as a consequence of oxidative stress, depending on its various origins (534). Neuroinflammation promotes increased formation and aggregation of Aβ and NFT. Increased Aβ plaque and NFT then exacerbate inflammatory stress, establishing a dysregulated activation cascade with a positive feedback loop that rapidly and irreversibly progresses toward cytotoxicity (649). Besides, microglial activation combined with impaired ability to cope with increased oxidative stress in the aging brain becomes deleterious (121), producing a free-radical-mediated oxidative stress that results in neuronal injuries (61).
In the 1990s, findings of activated complement factors, cytokines, and a wide range of related receptors in the brain of AD patients led to the concept of neuroinflammation and the inflammation hypothesis for AD, which states that the innate immune response of the brain is likely to be involved in the pathology of degenerative diseases of the CNS (57, 455, 674). This new pathogenic mechanism determined the re-consideration of the role of Aβ aggregation in the pathology of AD at least for some groups, which considered glial cells to be a leading factor for this disease (669). However, for most scientists studying AD who adhere to the amyloid hypothesis, neuroinflammation is still considered as a consequence of the activation of microglia by Aβ, which initiates an inflammatory cascade, leading to degenerative changes of neurons (6) in AD (257), PD (267), and HD (57). However, the exact role of inflammation in the pathology of AD and its mechanisms in terms of the cell type that are involved—microglia, astrocytes, or other cells—are still debated.
1. Interleukin 1
There are two distinct isoforms, IL1α and IL1β, but their actions are regulated through the type I IL1receptor, IL1RI, which elicits similar effects after binding of both isoforms, and also binds the IL1RI antagonist, IL1ra. In the brain, IL1 is primarily released by activated microglia. However, IL1 is expressed by and targeted to many other CNS cell types, including astrocytes, endothelial cells, neurons, and oligodendrocytes. In turn, IL1 stimulates back specially on microglia, astrocytes, and endothelial cells (216), presenting a broad response to IL1 constituted by mediators favoring the propagation of neurodegenerative diseases such as inflammatory cytokines, chemokines, adhesion molecules, prostaglandins (PGs), ROS, RNS, and matrix metalloproteases (439).
IL1 is thought to be an influential cytokine in AD, as suggested by the high concentration of IL1, over control, found within brain lesions from AD patients, in reactive microglia surrounding amyloid plaques (228, 600). High concentration of IL1 is also observed in AD animal models (46, 368). In Down's syndrome that predisposes to AD neuropathological changes, IL1 elevation and neuroinflammation precede plaque formation (438). Furthermore, specific polymorphisms in the IL1 genetic loci appear to be associated with increased risk for AD (414), although a meta-analysis has not supported such association (49).
IL1β up-regulates expression of APP in vitro (74, 367, 542) and when it was injected into rat brain (576). Furthermore, IL1ra KO mice demonstrate enhanced Aβ-induced neuropathology (132). Also, after neuronal injury, increased IL1 levels can induce overexpression of APP. Reciprocally, increased APP and Aβ can lead to up-regulation of IL1 in vitro (32). In addition to association with modulation of APP processing, IL1 activity can also favor NFT development (578). These studies suggest that neuronal injury can influence IL1 levels at the CNS. These increased levels contribute to the Aβ burden in the CNS, which also contributes to IL1 overexpression. In consequence, increased release of IL1 can result in a self-sustaining cycle of IL1 production, which can lead to cell injury and death.
Finally, in addition to data implicating IL1 in deleterious effects, there is evidence for beneficial effects of IL1 (702), being associated with neuroprotective mechanisms. Activation of microglia by intratecal injection of lipopolysaccharide, which up-regulates IL1β and microglial activation, induces reduction of Aβ load (258). Also, IL1β reduces excitotoxicity in pure mouse and rat neuronal cultures (103, 604), whereas in mixed glia/neuron cultures, IL1β induces neurotoxicity through the release of free radicals (640).
2. Tumor necrosis factor
TNFα is one of the inflammatory cytokines significantly increased in AD, and it plays a central role in initiating and regulating the cytokine cascade during inflammatory responses (6, 197). TNFα is produced in response to oxidative stress and the presence of Aβ. TNFα is produced by microglia and its overproduction has been linked with neuronal cell death (226, 562, 647). It exists in two main forms, TNFα and TNFβ. TNFα exerts its biological functions via two distinct receptors: TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2). TNFR1 is a membrane-receptor that is expressed in most tissues. It binds both membrane-bound and soluble forms of TNFα. Its functions range from mediating cell death to activation of NF-κB-mediated cell survival (273). Similar to TNFR1, the 75 kDa TNFR2 is also a membrane-receptor, but with low affinity for soluble TNFα, being fully activated only by membrane-bound TNFα. The functions of TNFR2 are not understood in all its detail. However, they appear to be strongly dependent on the cell type. Thus, TNFR2 amplify apoptotic signals from TNFR1 in cancer cell lines (676), whereas it mediates neuroprotection in glutamate-induced excitotoxicity (403). TNFR2 exerts its protective properties when prestimulated with TNFα, which suggests a neuroprotective role in the CNS (177, 403).
In AD patients, TNFR1 levels are high (197), whereas TNFR2 levels are low (197). Animal models of AD show TNFα up-regulation. Spinal levels of TNFα are increased up to 25-fold in AD patients compared with control subjects, and the increase correlates with clinical deterioration of AD. TNF pathway activation in the brain influences APP processing, Aβ deposition, immune activation, and cell survival (462), leading to increased Aβ and NFT formation. In correspondence with that effect, TNFR deletion prevents Aβ formation and diminishes plaque formation. Although TNF has been suggested to play a protective role after acute brain insults, sustained or chronic perturbations of the brain can determine that TNF role shifts to one of the agent inductors of damage.
Extrinsic signals can trigger degeneration via proapoptotic receptors, including some members of the TNFR super family like p75NTR, Fas, and TNFR1 (235). Expression of TNF-related apoptosis inducing ligand (TRAIL) seems to form a direct link with AD pathogenesis and represents a promising venue in development of future therapeutic targets. Binding of TRAIL with some receptors of the TNF/NGF family, like death receptors (e.g., DR4 and DR5), can induce cellular apoptosis (651). After Aβ exposure, expression of the DR5 has been shown to be increased (651). Furthermore, inhibition of TRAIL signaling pathway can protect human neuronal cells from Aβ toxicity (651). Similarly, DR6 (a.k.a. TNFR super family member 21, one of eight members possessing a cytoplasmic death domain) and APP activate a widespread caspase-dependent self-destruction program (457). DR6 is widely expressed by neurons as they differentiate and become pro-apoptotic. In transfected cells, it triggers cell death in a Jun N-terminal kinase-dependent manner (474). It is reported that DR6 links passive and active degeneration mechanisms (457). After trophic deprivation, DR6 triggers neuronal degeneration. DR6 signals via Bax and caspase-3 in cell bodies, Bax, and caspase-6 participate in axonal degeneration. Nevertheless, DR6 is also activated by a pro-degenerative ligand that is associated to the cell membrane but released in active form upon trophic deprivation. The candidate ligand appears to be APP, a transmembrane protein that undergoes regulated shedding associated to cell damage and stimulation, in which trophic deprivation leads to its cleavage by beta-secretase (BACE1), followed by further cleavage by an as yet unidentified mechanism generating an ∼35-kDa fragment (N-APP) (457). Interestingly, DR6 is unregulated in injured neurons (146), raising the question whether overexpressed DR6 can trigger ligand-independent degeneration, as reported for p75NTR (495).
As a consequence of innate immune activation in many neurodegenerative disorders, the levels of the inflammatory cytokines TNFα and IL6, and of the chemokine CXCL8, are increased. Whereas IL6 could block inflammatory response by inhibiting TNFs and IL1R, or promote the inflammatory response by influencing chemotaxis, there is little understanding of the role of IL6 in the development of neuroinflammation in AD. Downstream effects, including an increase in caspase activity, in intracellular calcium levels, and in the production of ROS have been implicated in AD, traumatic brain injury, and Huntington's chorea (615).
3. Transforming growth factor β
The hippocampus, the principal region affected by neurodegeneration in AD, is also one of the most densely populated by microglia. Differences in microglial cell cytotoxicity could depend on multiple factors related to cell identity and environmental conditions. One of the factors could be the differential secretion of TGFβ. TGFβ is found at low concentrations in normal brains, whereas its expression is increased in the injured or diseased brain (62). IL1β, TGFβ, and inducible COX-2 are elevated in CSF and the brain tissue of AD patients. The induction of TGFβ1 by inflammatory molecules could limit the temporal and spatial extent of the inflammatory response (520, 559). Interestingly, TGFβ1 produced by neurons modulates several glial cell functions, including microglial cell activation, inhibiting production of inflammatory molecules like IL1β and TNFα, and NO· release. Unregulated neuroinflammation, due to impairment of normal control processes, can ultimately result in cellular damage. In late-onset AD unregulated neuroinflammation likely contributes to amyloid plaque and NFT formation, and ultimately to clinical deterioration.
4. Prostaglandins
The inflammatory state is recognized as a risk factor for AD, and it is also supported by epidemiological retrospective observations that patients regularly taking nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids because of suffering from chronic, systemic inflammatory diseases have a 45%–60% reduction of risk for suffering AD (70, 601, 622). However, clinical trials in AD patients have failed to show the benefit of taking anti-inflammatory drugs. Some small reports have suggested that indomethacin and ibuprofen could be beneficial, but most of studies show little, if any, beneficial effect. PGs are ubiquitous signaling molecules with pleiotropic effects. Synthesis depends on COX, enzyme that exists in constitutive (COX-1) and inducible (COX-2 and COX-3) forms, all of which are inhibited by NSAIDs. In the brain, PG formation is regulated by COX activity primarily in astrocytes and microglia. COX-2 activity has been found to be elevated in AD patients compared with controls, and it has been linked to promotion of APP processing and cognitive decline in AD animal models (415, 647). Although there is not an abundance of clinical data regarding the action of PGs in AD, evidence from animal models suggests that prostaglandin E2 (PGE2) has both pro-inflammatory and pro-amyloidogenic effects (683). As a whole, these results suggest that anti-inflammatory benefits can be reached as a prophylactic method before AD is already installed, but not as a treatment for patients already presenting dementia.
5. Oxidative stress mediators
As described in section I.B, NADPH oxidase is associated with neurodegenerative disorders, being activated in the brain of AD patients (583). Several inflammatory stimuli activate microglial cell NADPH oxidase to cause neuronal damage, including Aβ (53, 512), APP (508), rotenone (210), paraquat (PQ) (699), substance P (60), and α-synuclein (721). ROS generated by NADPH oxidase mediates Aβ-induced cerebrovascular dysfunction (483), and it also plays a role in the mechanism by which reactive microgliosis causes additional neuron damage (211). Neurotoxicity observed in inflammatory microglial activation is significantly decreased in both NADPH−/− mice (in vivo) as well as in in vitro assays using cell cultures derived from NADPH−/− mice (512). Similarly, inhibition of NADPH oxidase by diphenyliodonium (506), some peptides, like dynorphin and leucine enkephalin (509, 511), the antibiotic minocycline (150), or small molecules such as dextromethorphin, statins, and naloxone (124, 507, 509), have an anti-inflammatory and by extension, a neuroprotective effect in culture, strongly suggesting that NADPH oxidase may be a common pathway of microglia-mediated inflammatory neuronal damage.
Furthermore, within the mammalian brain, in conditions of inflammatory stimuli, or in response to various forms of injury, NO· is produced predominantly by the inducible form of NOS, iNOS (233). NO· can activate cellular signaling cascades. However, under conditions of high concentration of NO· and ROS, as observed during inflammatory processes, their interaction generates peroxynitrites, which can oxidize proteins. Localized increases in carbonylated-, 4-hydroxynonenal-, and 3-NT-modified proteins have been reported in hippocampus and parietal cortex of AD brains, demonstrating disease-specific modifications due to RNS (84, 109, 610), including tau nitration (271). Those results indicate that nitrosative stress secondary to an increase in RNS (61, 455) also plays a role in AD pathology.
6. Oxidative stress signal transduction pathways
In addition to various trophic factors and hormones molecules, ROS regulates several signal transduction pathways. Hypoxia inducible factor α (HIFα) is regulated by ROS (125, 408). Inhibition of ROS through pharmacological inhibition and antioxidant treatments leads to increased HIFα stability. Recent evidence demonstrates that HIF activation plays a protective role in neurodegenerative diseases (125).
NF-κB is a transcription factor presenting both pro-survival and deleterious effect that is activated by ROS and inflammatory mediators. NF-κB regulates several important cellular defense mechanisms, including the activation of genes regulating cellular survival, growth, differentiation, inflammation, and cell death. At basal, nonstimulated conditions, NF-κB is bound and held inactive by inhibitor of κB (IκB) in the cytoplasm. Moderate ROS levels lead to phosphorylation, polyubiquitination, and degradation of IκB, allowing the activation of NF-κB. Once activated, NF-κB regulates the transcriptional activation of anti-apoptotic proteins, such as X-chromosome-linked inhibitor of apoptosis and growth arrest and DNA damage-inducing protein β (GADD45β), and genes involved in decreasing mtROS, especially MnSOD (486). NF-κB also plays a pro-survival role by inhibiting the c-Jun N-terminal kinases/stress-activated protein kinase (JNK) and caspase cell death pathways. In contrast to the effect of moderate ROS levels that activate NF-κB, high ROS levels inactivate NF-κB through oxidation of the cysteine in position 62 of its p50 subunit, inhibiting its DNA binding.
However, NF-κB activation can be also detrimental and associated with decreased cell survival. In fact, robust activation of NF-κB and MAPK pathways are paramount signals in neuronal cell death induced by oxidative stress (143, 151) and Aβ (595). NF-κB also plays a central role in the initiation and amplification of inflammation by responding to inflammatory stimuli such as TNFα or IL1 and lead to the expression of a large variety of cytokines and chemokines, which could be in part responsible for neurotoxicity.
TNFα also activates NF-κB associated with neuroprotection against Aβ toxicity in cultured neurons (33). In fact, Aβ can be a potent inducer of NF-κB in neuronal cell death via the induction of intracellular ROS (347, 657). Thus, inhibition of these pathways could be beneficial in the treatment of neurodegenerative diseases, including AD (346, 347, 442, 482, 682). The effect appears to involve attenuation of Aβ-induced activation of ERK1 and p38 MAPKs, which are upstream NF-κB signaling pathways (475, 657). Aβ has been also shown to activate NF-κB through the TNFR1 signaling, which results in neuronal apoptosis (362, 657). However, there are also reports that NF-κB activates anti-apoptotic responses and protects neurons from excitotoxicity and ischemic injury in the mature brain (51, 410, 487). Furthermore, NF-κB is not the only transcription factor activated under inflammatory conditions. Activation of other transcription factors such as peroxisome proliferator-activated receptor gamma (PPARγ) and signal transducers and activators of transcription 1 (STAT-1) has also been implicated in AD (149, 556).
There are interesting differences on NF-κB response depending on age. TNFα activates signaling involving NF-κB, with a beneficial or detrimental response depending on age and the type of stimuli. For example, compared with treatment with Aβ alone, TNFα plus Aβ is toxic for 24-month-old rat neurons, whereas the same stimuli are protective for 10-month-old neurons (485). Patel and Brewer hypothesize that age-related down-regulation to the TNFR1 and TNFR2 signaling results in defective NF-κB activation and fails to provide a neuroprotective response against Aβ toxicity by TNFα (485). NF-κB accumulates in old neuron nuclei—an effect that is mimicked by blocking TNFR2. An explanation for failure of NF-κB to activate protective pathways could depend on high ROS production (480), and an oxidized redox state in aged cells (481). The redox state of NF-κB could be a control mechanism regulating the amount of NF-κB (606). Available data indicate that NF-κB activation is associated to both protective and deleterious effects, depending on the context of stimulation and the co-activation of various signaling pathways.
A different aspect of ROS-activated transcription factors is represented by the p53 tumor suppressor, because it appears to promote both cell survival and death (337, 373). Cellular damage activates p53, leading to the inhibition of cell cycle or initiation of apoptosis. In addition to these classical roles, p53 also presents a pro-survival role in response to increased ROS levels by up-regulating several antioxidants, including GPx, MnSOD, and aldehyde dehydrogenase 4. In addition, p53 also regulates TP53-induced glycolysis and apoptosis regulator (TIGAR). TIGAR inhibits glycolysis and directs glucose to the pentose phosphate pathway producing NADPH, which is required to reduce GSH and thus lower ROS levels (301, 302). This antioxidant function of p53 is activated during low cellular stress, playing an integral role in pro-survival pathways, whereas high stress and ROS concentrations result in p53-mediated apoptosis through activation of several pro-apoptotic genes and p53-induced genes (PIGs). Although the exact mechanism by which p53 senses ROS and responds via either pro-apoptotic or anti-apoptotic functions is not well known, these differing functions possibly depend on the level, posttranslational modification, and cellular localization of p53 (373).
E. Neurotoxicity, excitotoxicity, and oxidative stress
Atrophy, neuronal loss, synaptic loss, and morphological alterations by NFT, in addition to functional impairment, increase with severity of AD. Many of the pathophysiological mechanisms discussed here are involved directly or indirectly with neurodegenerative changes of the disease. Aβ at its various molecular forms, inflammatory mediators, and radical species induce functional changes and damage, either locally, at process terminals, or along the neuron as a whole, leading to loss of dendritic spines, remodeling of neurites, and inflammatory changes. Cytoskeleton changes also have a key role in neuron energetic and in the conformation and alteration of synapses and dendritic spines. Mitochondrial dysfunction, energetic impairment, calcium mishandling, and accumulation of aggregated proteins impose additional burdens, further promoting neuronal loss, with generalized neurodegeneration, ultimately leading to brain atrophy.
1. Neurotoxicity
Neurotoxicity initial events probably include activation of macrophages and microglia as a result of inflammatory stimuli mediated by pattern recognition receptors, such as Scavenger receptors (9, 443), key molecules that drive the innate immune responses. Neuronal death may occur via several mechanisms, including necrosis, apoptosis, and autophagy. Necrosis is generally observed in acute injury as a result of the release of L-glutamic acid (glutamate), NO·, ROS, and calcium (169, 309). There are diverse mechanisms associated with neurodegeneration, like glutamate-induced excitotoxicity that can be facilitated by aging. For example, age-dependent changes, such as accumulation of mutations associated to misfolded or aggregated proteins, macrophage and microglia activation stimulating ROS production, or induction of mitochondrial dysfunction, facilitate that neurons exposed to excess glutamate could follow a route to excitotoxicity and succumb to a neurodegenerative process.
2. Excitotoxicity
Ionotropic glutamate receptors, especially the NMDA subtype, have been implicated in AD (570). Glutamate, the major excitatory neurotransmitter of the mammalian CNS playing important roles in development, when massively released, induces neuronal death. Necrotic death of neurons caused by glutamate excitotoxicity in AD and PD depends on increased Ca2+ influx, activation of Ca2+-dependent enzymes, induction of ROS and NO· (Fig. 15), and oxidative stress. There is evidence that Aβ can induce glutamate release from primary cortical neurons and, in turn, alter signaling cascades that are required for long-term potentiation (646). As an additional mechanism, there is also evidence that activation of NMDA receptor is linked to ROS production from NADPH oxidase (Fig. 4b), which in turn leads to altered synaptic plasticity and memory (321).
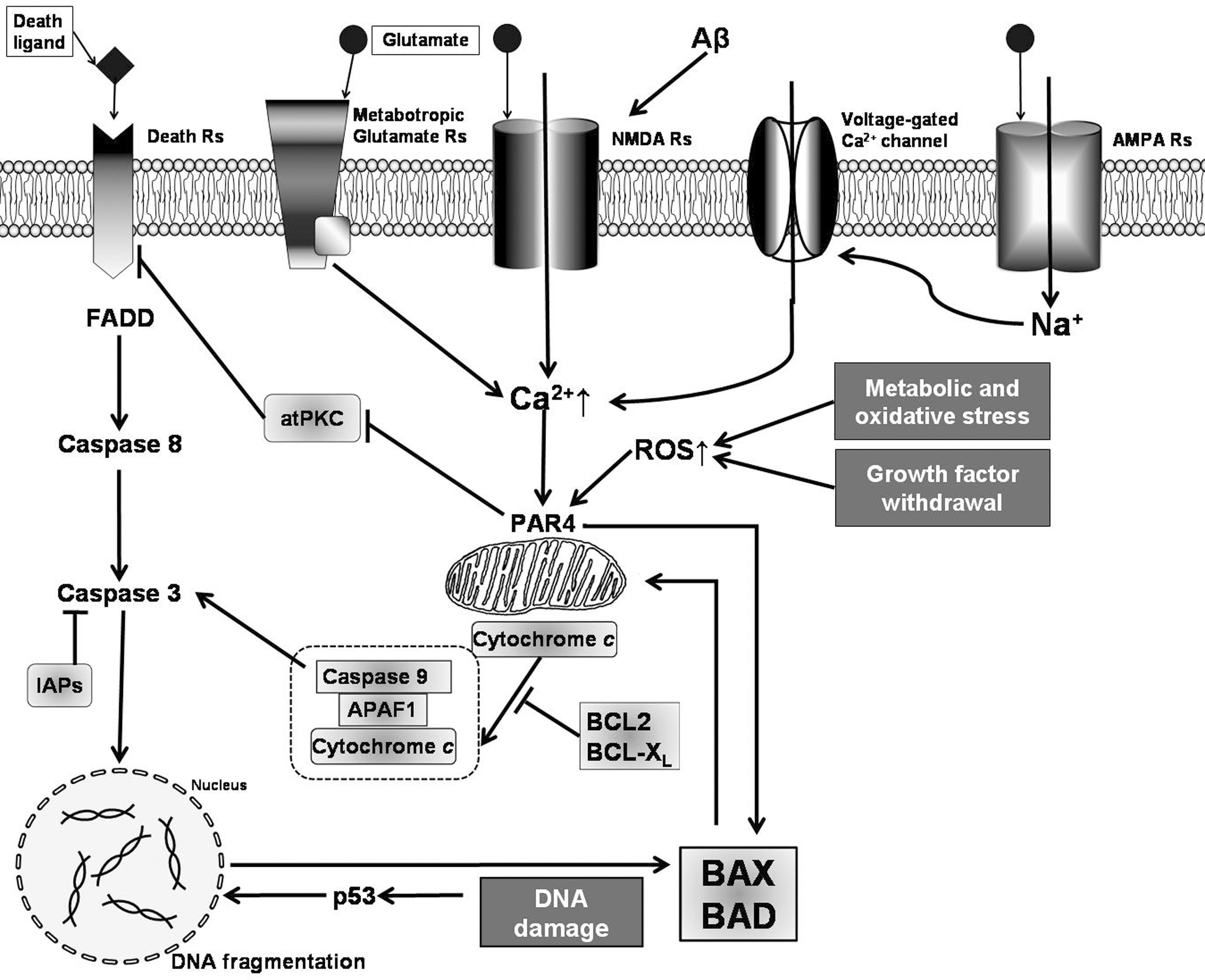
Neuronal excitation secondary to the stimulation of NMDA receptors is linked to the activation of the Ca2+-dependent cytosolic phospholipase A2α (cPLA2α) and release of arachidonic acid (AA), which besides being a precursor for prostanoid synthesis, is also a retrograde messenger implicated in memory formation. Activation of cPLA2α depends on phosphorylation by MAPK and PKC. Overactivation of cPLA2α is involved in different forms of neurodegenerative diseases, including AD (613).
Increasing evidence supports a link between Aβ and the NMDA receptors (592). This link involves a novel oxidative mechanism for Aβ-mediated neurotoxicity through crosstalk with the NMDA receptor contributing to the oxidative stress hypothesis and synaptic failure that underlies the pathogenesis of AD. Similar to NMDA, oligomeric Aβ can induce ROS production from cortical neurons through activation of NADPH oxidase (575). ROS derived from NADPH oxidase triggers the activation of ERK1,2, phosphorylation of cPLA2α, and AA release (Fig. 4b). In addition, Aβ-induced AA release can be inhibited by memantine, an NMDA receptor antagonist, suggesting that Aβ triggers these effects through the NMDA receptor (575).
An alternative mechanism suggested to contribute to the loss of neuronal cells by excitotoxicity induced by oxidative stress is based on the observation that in vitro and in vivo exposure to Aβ increases the content in protein carbonyls, HNE, and 3-NT in synaptosomes and neuronal cells (338). In both MCI and AD brain, the glutamate-related enzyme, glutamine synthetase (GS), is found to be oxidatively modified (88, 108), which reduces its enzymatic activity. GS is important in the conversion of extracellular glutamate into glutamine. Hence, oxidative modification and altered function of GS may acutely exacerbates the excitation of postsynaptic neurons, Ca+2 accumulation, and free radical formation, all contributing to synaptic dysfunction and excitotoxic neuronal cell death (85).
F. Ubiquitin proteasome system, unfolded protein response, and autophagy in oxidative stress
1. Ubiquitin proteasome system
Studies in animal models have demonstrated participation of ubiquitin proteasome system (UPS) in normal synaptic function (56, 253), including UPS-mediated degradation of numerous substrate proteins to phenomena as synaptic plasticity. On the other hand, dysfunction of UPS has been connected to several neurodegenerative diseases (253, 654). Abnormal deposition of highly insoluble protein aggregates or inclusion bodies within nerve cells is commonly observed in association with chronic neurodegenerative diseases, including AD (115), suggesting that impairment of UPS function could be involved in AD pathogenesis. These inclusion bodies show ubiquitin immunoreactivity, along with immunoreactivity to other proteins, like Aβ (199, 340). In AD brains, ubiquitin co-localizes with both plaques and tangles (468) being both tau (312) and Aβ aggregates capable of inhibiting the proteasome function (469, 648). Several aggregated proteins can inhibit the UPS (298, 339), an effect that could be relevant for the genesis and progression of neurodegenerative disorders, in which protein aggregates are a common trait. In the AD context, diverse studies have shown that Aβ directly binds and selectively inhibits the activity of the proteasome, similar to the effect of a known proteasome inhibitor (469). Aggregation of Aβ appears to have an especially robust inhibitory effect on proteasome activity—an inhibition that is rescued by Aβ immunotherapy (648).
Protein oxidation, which can be abundant in AD brains as we have previously described, also inhibits proteasome activity (722). In particular, the oxidization of ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), an enzyme that removes ubiquitin from ubiquitinated proteins, is down-regulated in brain regions affected in early AD cases (108). Remarkably, different brain regions show different UPS susceptibility. Consistently, there is a selective decrease in proteasome activity in the hippocampus, which is a main area damaged in AD brains, whereas another brain regions, like the cerebellum, maintain their proteasome activities (108).
2. Unfolded protein response
Associated to the accumulation of abnormal proteins, additional cellular processes capable of inducing cell damage can be activated. In fact, several stimuli known to be present in neurodegenerative diseases, such as perturbation in calcium homeostasis or redox status, expression of misfolded proteins, altered glycosylation, overloading of cholesterol, and proteasome dysfunction, can disrupt ER homeostasis, imposing stress to the ER, and activating a highly specific signaling pathway response named the unfolded protein response (UPR) (309).
In general terms, proper protein folding, essential for cell function and viability, is a delicate and energy demanding process. The failure of any of the aforementioned mechanisms inducing ER stress results in the accumulation of misfolded proteins (523, 720). Unfolded or misfolded proteins in the ER lumen are retro translocated to the cytoplasm, where they are ubiquitinated and degraded by the proteasome (692). Proteasomal degradation of ER-associated misfolded proteins is required for protection from the UPR activation. Proteasomal inhibition is sufficient to activate the UPR, and if the noxious condition is not properly resolved, the UPR can lead to cell death (445).
In familial AD, mutated PS1 affects the ER stress response by reducing the activation of the ER stress transducers IRE1, PERK, and ATF6. The reduced global ER response to stress augments the vulnerability of AD patient cells (445). Likewise, in sporadic AD, the spliced isoform of PS2 induces the expression of the high mobility group A1a protein, and down-regulates the UPR signaling pathway in a manner similar to that of PS1 mutant in familial AD, increasing ERstress-induced neuronal cell death in AD (309).
3. Autophagy
Autophagy or programmed cell death-type II is a homeostatic turnover of cellular contents, organelles, and misfolded proteins through the lysosomal system (643). The brain normally shows basal low levels of autophagy, which is tightly regulated. Due to the postmitotic nature of neurons, it is thought that a robust autophagy mechanism maintains homeostasis of functional organelles, such as mitochondria (630, 631). The prominence of autophagy in neurons, cells that do not divide, depends on the build-up of dysfunctional mitochondria and protein aggregates (i.e., Aβ or alfa synuclein [αSN]) and chronic oxidative stress conditions (445, 553, 643).
Autophagy is up-regulated under trophic stress or with the formation of misfolded protein aggregates that cannot be degraded by the UPS (190, 459). Misfolded proteins are problematic because they impair normal cell function when they accumulate. The deregulation of autophagy has been linked to the pathophysiology of neurodegenerative diseases. Autophagy involves the delivery of organelles (including mitochondria; discussed in section II.C) or cytoplasmic proteins to lysosomes for degradation. Depending on the mechanism of cargo delivery, several types of autophagy are identified, namely, macroautophagy, microautophagy, and chaperone-mediated autophagy. Various pathways are implicated in the turnover of proteins that are prone to aggregation [see (145)]. The autophagosome fuses with endosomes to form an amphisome, which combines with lysosomes for degradation of its cargo, or it can fuse directly with lysosomes to form an autolysosome and become degraded by lysosomal enzymes (424, 547).
In aging, many mitochondria undergo enlargement and structural disorganization, while lysosomes gradually accumulate the nondegradable, polymer lipofuscin. It is believed that this is a result not only of oxidative stress, causing oxidation of mitochondrial constituents and autophagocytosed material, but also of the inherent inability of cells to completely remove oxidatively damaged structures. Lipofuscin accumulation may diminish lysosomal degradative capacity, further limiting mitochondrial recycling. Terman and Brunk proposed the mitochondrial–lysosomal axis theory of aging, according to which mitochondrial turnover progressively declines with age, resulting in increased oxidative damage, accumulation of damaged organelles and lipofuscin, decreased ATP production, release of apoptotic factors, and cell death (629).
Cells showing induction of autophagy share several features with neurodegenerative diseases, including the presence of apoptotic neuronal loss, excitotoxicity, oxidative stress, intracellular protein aggregates, synaptic abnormality, and, more recently, deregulation of the autophagic pathway (69). Autophagosome–lysosome pathway is compromised in AD (460). In fact, there is accumulation of autophagic vacuoles in the brain of AD, PD, and HD patients (353, 461, 547, 717) and autophagy activation is elevated after Aβ stimulation and in the AD mouse model APP/PS1 (280, 678, 717). In AD, autophagy seems to have a dual role in Aβ accumulation. It is implicated in degradation of APP and Aβ (92, 291, 666), but may also have a role in Aβ formation and aggregation (275), as Aβ also appears to be generated in autophagic vacuoles, suggesting that autophagy could exacerbate AD pathogenesis by increasing Aβ levels (461, 717). Autophagy has also been implicated in the modulation of tau level and fragmentation, and its inhibition elevates tau aggregation and toxicity (47, 687). Moreover, clearance of autophagic vacuoles is impaired in AD brains (63).
The autophagic key regulator protein beclin1 appears to be decreased in affected brain regions of AD patients (494). APP transgenic mice with reduction of beclin1 show an increase in intraneuronal Aβ accumulation, extracellular Aβ deposition, and neuronal abnormalities. Increase of beclin1 reduced Aβ pathology (494), indicating a strong correlation between autophagy and APP metabolism. Studies of AD patient brains have shown an elevated incidence of apoptosis as compared to age-matched controls. DNA fragmentation is associated with increased expression of JNK pathway, an early pro-apoptotic signaling pathway, decreased expression of anti-apoptotic Bcl-2, an increased expression of pro-apoptotic Bax in neurons with NFTs, and increase in active caspase-3 expression levels as compared to age-matched controls (515). In addition, reduced complex II, III, IV, and cytochrome oxidase activities have been found, indicating mitochondrial involvement in the apoptotic cascade, as well as the presence of activated caspase-3 within autophagic granules (113).
III. Therapeutic Approaches for AD Aimed to Oxidative Stress
The existence of converging disease mechanisms demands that any therapeutic proposal for the treatment of AD should be multi-targeted, aiming at the simultaneous management of pro-oxidant, pro-inflammatory, and death-inducing changes, which in turn depend on impairment of proteasome system, induction of stress because of protein aggregation, mitochondrial dysfunction, and excitotoxicity, among other factors.
In addition to the symptomatic treatment targeting cholinergic and glutamatergic neurotransmission presently used for AD patients, efforts to modify the progressive course of AD have been mostly based on altering the production or clearance of Aβ, including the most recent attempts with immunotherapy (670). Results have been mostly disappointing, possibly because our disease animal models—mostly based on the rare familial form—may not be applicable to the much more common sporadic form. On the other hand, because of the close association of sporadic AD with vascular disease and type 2 diabetes mellitus, efforts focused in the treatment and prevention of these conditions may be an interesting approach for reducing the incidence of AD. In that sense, management of risk factors linked to metabolic syndrome and cerebrovascular disease, as well as treatment for neuropsychiatric symptoms, if needed, should be used for improving clinical practice standards. However, there is insufficient overall evidence from epidemiological studies to support with certainty any association between dietary or supplementary intakes of antioxidants or vitamins with augmented risk or higher incidence of dementia, although several cohort studies have reported the potential of a Mediterranean diet for reducing the risk of incidence of AD.
A. Nonsteroidal anti-inflammatory drugs
Therapeutic approaches with NSAIDs, regardless the fact that neuroinflammation is involved in the pathogenic mechanisms of AD, has proven to have some effect in prevention but is ineffective for patients who already have AD. However, therapies with COX-2 inhibitors target a single pro-inflammatory factor (PGE2) among a large number of inflammatory factors released by activated microglia, which could explain why an anti-inflammatory monotherapy could be inefficacious.
B. Modulation of microglial cell activation
Existing evidence indicates that microglia is important for maintenance, repair, and defense, and even that microglial overactivation is deleterious. Therefore, an effective therapy should target early attenuation of the microglial response to levels that are beneficial, rather than the elimination of the microglial response. Because microglial function, as well as their potential deleterious effects are regulated by NADPH oxidase and the production of intracellular and extracellular ROS (60, 61), this enzyme complex appears as an ideal therapeutic target. Recently, several peptides, antibiotics, and small molecules have been identified as inhibitors of NADPH oxidase and, hence, potential neuroprotective agents (150, 509). Inhibition of NADPH oxidase activation might be a more efficacious approach because it targets the predominant enzyme complex by which microglia produce high amounts of ROS. Its inhibition would down-regulate multiple inflammatory factors, including PGE2 (683). In addition, although they are not discussed here, because they are outside the scope of this review, some cytokines released by microglia have been considered as possible targets of drug therapy for AD patients (226, 257, 348).
C. Activation of antioxidant pathways
An alternative approach for decreasing the production of ROS would be the activation of antioxidant pathways, an approach that could be particularly important for the brain, an organ that has relatively weak antioxidant defenses. Moreover, aging leads to some loss of the free radical scavenging ability depending on endogenous mechanisms. Even more, reduced cellular expression and activity of antioxidant proteins are fundamental triggers for AD. Among cellular antioxidant defenses, Hsps have been regarded as cytoprotectors from oxidative damage in neurodegenerative diseases. HO-1 polymorphisms have been considered associated with increased AD susceptibility. Deregulation of the HO system has been associated with the pathogenesis of both AD, and brain aging (405, 477). Among the stress proteins, HO-1 is encoded by the HO-1 gene that is regulated by redox, and its activation represents a protective system potentially active against brain oxidative injury. Its expression in AD patient brains is significantly increased in neurons and astrocytes of the hippocampus and cerebral cortex. In individuals with AD, HO-1 co-localizes in senile plaques and NFTs being its spatial distribution, similar to that found in the pathological expression of tau. There are reports that strongly suggest that activation of HO-1 is strongly protective against oxidative damage and cell death in neurons. Thus, modulation of HO-1 should represent a potential pharmaceutical strategy for the treatment of neurodegenerative disorders (517).
D. Mitochondrial antioxidants
Considering the important component of cell death observed in AD and the fact that mitochondria also have a key role in production of ROS and in apoptosis signaling, therapeutic strategies aimed to reduce mitochondria damage appears as an interesting approach for the treatment of neurodegenerative diseases. Several compounds targeting mitochondria are currently undergoing clinical trials. Mitochondrial antioxidants appear to be especially efficient at preclinical level. Coenzyme Q10 (CoQ10, an electron carrier embedded in the inner mitochondrial membrane, part of the electron transport chain of oxidative phosphorylation), has been shown to have neuroprotective effects in several neurodegenerative diseases by attenuating mitochondrial dysfunction. However, the poor capability of these oral antioxidants to cross the BBB has slowed down their therapeutical use, focusing the investigation on more soluble, shorter chain antioxidants of the family of CoQ10 derivatives, such as idebenone [6-(10-hydroxydecyl-2,3-dimethoxy-5-methyl-1,4-benzoquinone], decylubiquinone, and the ubiquinone derivative MitoQ10, which appears to be the most successful. MitoQ10 has the advantage of being accumulated within mitochondria, where it is reduced into its active ubiquinol form, which can prevent mitochondrial oxidative damage (383).
Szeto-Schiller (SS) peptides represent another class of mitochondrial antioxidants that offer the advantage of localizing within mitochondria at all mitochondrial membrane potential; a characteristic that may improve their therapeutic use. In vivo experiments have revealed that SS peptides are protective in a mouse model of ALS, increasing survival and motor performance, and decreasing cell loss (430). SS peptides also protect dopaminergic neurons against 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine neurotoxicity, both in vitro and in a PD animal model (430).
A drug rising high expectations over the last few years has been latrepirdine (dimebon), known antihistaminic drug, with a novel mitochondrial mechanism of action, associated to the improvement of mitochondrial stability by preventing mitochondrial permeability pore transition, in addition to effects as an NMDA receptor inhibitor. However, a phase III AD trial of Dimebon in 598 patients failed to show any significant improvement in primary or secondary outcomes (50).
E. Free radical scavengers
Another area of therapeutic development has been the use of free radical scavengers, in order to slow neuronal damage associated with Aβ-induced oxidative stress, acting as neuroprotective agents. Various herbs and fruits contain active phenolic substances endowed with potent antioxidant and chemopreventive properties, which have been studied for potential neuroprotective effects. A promising antioxidant already being tested in clinical trials is curcumin, a polyphenolic substance derived from the root Curcumin longa. Epidemiological studies have suggested that curcumin, one of the most prevalent nutritional and medicinal compounds used in India, appears to be responsible for the significantly reduced (4.4-fold) prevalence of AD in India compared with the United States. Consistent with these data, convincing evidence has been provided that chronic (6 months) dietary curcumin given to an AD transgenic mouse model resulted in a suppression of indices of inflammation and oxidative damage in the brain of these mice and reversed Aβ-induced cognitive deficits (220, 723). Curcumin attenuates oxidative damage in cortical neurons by reducing the intracellular production of ROS and protecting mitochondria (723). Curcumin inhibits lipid peroxidation and effectively intercepts and neutralizes ROS (superoxide, peroxyl, and hydroxyl radicals) and NO·-based free radicals (NO · and ONOO−). In addition, it has the potential to reduce cytochrome c release, block caspase-3 activation, and alter the expression of Bcl-2 family proteins (412, 517, 723). Of particular interest is the capacity of curcumin to inhibit COXs and to reduce the activation of nuclear transcription factor NF-kB, key in inflammatory signaling pathway. As for its ability of increasing antioxidant capacity, low concentrations of curcumin show a robust induction of the expression and activity of HO-1 in vascular endothelial cells, in rat astrocytes, and in cultured hippocampal neurons (412, 517, 723). Such ability of curcumin to induce HO-1 has been associated to its strong antioxidant and anti-inflammatory properties (220). Preincubation of neurons with a low concentration of curcumin in culture enhanced cellular resistance to oxidative damage. Also, depending apparently on a different mechanism, curcumin is effective protecting cortical neurons against apoptotic death induced by Aβ.
Curcumin is highly lipophilic and might cross the BBB, and, regardless its low bioavailability, because it is rapidly metabolized, curcumin may reach high brain concentrations to decrease Aβ aggregation at least in animal models. Nowadays, there is a randomized, controlled trial study of curcumin (ClinicalTrials.gov Identifier: NCT00528151), but it is oriented to a different degenerative disease and not to AD patients.
Curcumin is safe, is well tolerated, and appears to lack side effects at doses up to 12 g/day (220, 268). However, there are reports showing some effects that potentially could be deleterious. Inhibition of NF-κB by curcumin (17) could affect survival mechanisms associated to inflammatory activation. Similarly, curcumin is a specific inhibitor of iNOS (369), effect that also can depend on its inhibition of NF-κB, responsible for initiating the expression of iNOS. As inhibition by curcumin attenuates induction of NO production, this effect leads to an overall increase in ROS levels due to a failure in the scavenging of superoxide by NO.
Moreover, in PD animal models, exposure of rat mesencephalic cells to curcumin induces the expression of LRRK2 at the mRNA and protein level; LRRK2 overexpression is strongly associated with pathological cellular inclusions found in several neurodegenerative disorders (472). Curcumin has also been identified as an activator of apoptosis. Subtoxic concentrations of curcumin in a PD model sensitize mesencephalic cells to PQ-mediated apoptosis (471). Apoptosis appears to be enhanced via a mechanism that is dependent on the generation of rROS (52, 160, 303, 605, 636, 696), although other reports show that curcumin inhibits ROS, and consequently apoptosis, through its well-known antioxidant properties (142, 144).
Curcumin, although a potent antioxidant with ROS scavenging properties, is able to increase ROS production in different models (293, 451). Interesting enough, although cell death after the PQ+curcumin exposure was an ROS-dependent event, antioxidants were able to significantly protect against cell death generated by the exposure of cells to PQ alone, but were unable to prevent cell death in PQ+curcumin co-treatment, suggesting that the co-incubation sets cells into a no-way-back cascade leading to cell death that could not be retrieved by antioxidants (471).
Other plant-derived phenolic agents, with analogous chemical structures to curcumin, produce also a strong activation of HO-1 expression and enhancement of cellular defense against oxidative stress. On the same line of antioxidants, in principle, iNOS inhibitors could be used for AD treatment. However, there are no specific drugs being tested (476). There is evidence of beneficial effects of resveratrol on Aβ cytotoxicity in culture. However, protection by resveratrol was not associated exclusively to inhibition of iNOS expression, but it also attenuated PGE2 release, suppression of COX-2 expression, and suppression of nuclear translocation of NF-κB. The multiplicity of anti-inflammatory effects precludes the assignment of resveratrol protective effect to iNOS inhibition (320). There are also reports on the in vivo effect of three NSAIDs, lysine clonixinate, indomethacine, and meloxicam on NO production and iNOS expression in rat cerebellar slices (174). However, as resveratrol, reduction of iNOS and nNOS levels is only one of several anti-inflammatory effects of these NSAIDs (174). Nevertheless, the decrease in NOS expression after in vivo treatment with NSAIDs could represents a novel mechanism of therapeutic action of these NSAIDs, and may also be of importance in the prevention of NO·-mediated neuronal injury and as new strategies to reduce the risk of AD. Currently, a phase-III, randomized, double-blind clinical trial oriented to AD patients (ClinicalTrials. gov identifier: NCT00678431) to evaluate primary efficacy of resveratrol for disease progression is being done. Results in terms of the disease outcome are not available yet.
Antioxidant treatment with vitamin E (α-tocopherol) has been pursued with phase II and III clinical trials. Despite initial promise, recent randomized, controlled trials in MCI fail to report any benefit (286, 493). A problem of the antioxidant studies is the dissimilar nutritional status of patients, as well as the potential need of using co-administration of several antioxidants. Furthermore, α-tocopherol may also have different effects within any given population of AD patients (390), recognizing groups of responders and nonresponders α-tocopherol. In a recent meta-analysis of 19 clinical trials, only small trials show either an increase or a decrease in all-cause mortality and that the overall effect is near zero (422). In an effort to understand the lack of effect, AD patients were stratified into α-tocopherol respondents and nonrespondents (389), based on measures of plasma GSSG, the oxidized form of the common antioxidant GSH. Even at high doses of α-tocopherol, half of the patients failed to lower the plasmatic levels of plasma-GSSG, and showed a continuous deterioration in their cognitive performance (389). The other half of the patients, those showing decrease of plasmatic levels of plasma-oxidized GSH with α-tocopherol treatment, had no significant difference in the cognitive performance compared with the first group.
Considering the increase of oxidized macromolecules with aging and AD, one wonders why the antioxidant α-tocopherol is ineffective. The answer may be that a lipid-soluble antioxidant like α-tocopherol, with a high membrane/water partition, could be ineffective protecting against oxidation of aqueous phase nucleic acids and proteins. In addition, if the oxidized α-tocopherol is not removed from the membrane, it will either accumulate or pass the electron to another lipid, inducing further damage to the membrane. Because oxidized α-tocopherol can be recycled by passing the extra electron to a water-soluble electron acceptor such as vitamin C or other compounds, combining antioxidants could be a good strategy for improving their antioxidant effects. There are some reports showing better results with the combined use of vitamins E and C for enhancing the reduction in lipid peroxidation seen in AD patients after treatment with α-tocopherol (513). Effects may depend on the intrinsic antioxidant potential of vitamin C or on its interaction with α-tocopherol, as it is known to recycle α-tocopherol radical to maintain its antioxidant properties. However, several trials have failed in showing improvement of cognitive performance. Failure could depend on the fact that even more complex mixtures of antioxidants, including flavonoids and polyphenols—which have shown efficacy in animal models of aging—may be needed, or on the fact that aging persons have an increasingly oxidized plasma redox potential, which can interfere with removal of radicals (302).
A recent study showed that serum α- and γ-tocopherol levels were not associated with the risk of developing AD (616). However, the use of various tocopherol forms, other than just α-tocopherol, may be key conditions for AD (432). A dementia-free cohort was followed for 6 years to detect incident AD, focusing on α-, β-, γ-, δ-tocopherols and α-, β-, γ-, δ-tocotrienols. The authors found that the risk of developing AD was reduced only for high plasma levels of β-tocopherol, while the other subunits had only a small effect (402).
Another possibility for the failure of simple antioxidants to reverse damage could be that we are aiming at the wrong target: ROS signaling is required for processes as diverse as transcription factor activation, insulin signaling, endothelial function, or LTP model of memory. Also, ROS damage could depend on a variety of co-morbidities. We may need a dose–response targeted reductive shift in the metabolically oxidized redox potential seen in many aging individuals. Thus, vitamin E therapy trials are needed, with a special focus on individual monitoring of redox potential to avoid toxicity and assess biomarkers of efficacy (389).
F. Cyto- and neuroprotective therapies
As previously outlined, Nrf2-induced ARE-Nrf2 signaling leads to a broad cytoprotective response associated with enhanced antioxidant capacity and attenuation of neuroinflammation (660). In view of the multiple mechanisms of its protective effects, Nrf2-ARE pathway could represent a potential therapeutic target for AD treatment (306). In vitro and in vivo studies show that monofunctional inducers exhibit profound neuroprotective effects in an Nrf2-ARE-dependent manner. In contrast, in Nrf2 KO mice and after Nrf2 disruption, the chemically induced ARE-mediated transcriptional response, as well as the chemopreventive efficacy of monofunctional inducers, is completely abolished (344, 417). In addition, it has been shown that curcumin and pyrrolidine dithiocarbamate, capable of inducing Nrf2, alleviate cognitive defects in AD transgenic models (202). Because clinically safe compounds are widely available in the form of chemopreventive agents, the Nrf2-ARE signaling pathway can become a key drug target for neurodegenerative disorders.
IV. Concluding Remarks
Aging populations is increasing all over the world, and many countries are facing an increased prevalence of age-related diseases, such as AD, with escalating material and emotional burden and health-care costs. Understanding AD pathophysiology has a prominent role in developing new therapeutic strategies, especially considering that prevention and treatment are important public health goals for the disease. As discussed here, events contributing to the initiation and progression of AD are numerous, complex, and intermingled.
The data reviewed here provide evidence that oxidative stress is present in conditions of Aβ accumulation, mitochondrial impairment, cytotoxic activation of microglia, proteasome dysfunction, and protein misfolding, all of which appears to be involved in AD pathogenesis. Participation of oxidative stress can be both as a trigger and as a consequence of these processes, and often is part of both, contributing to the interaction and potentiation of these otherwise seemingly independent disease mechanisms. Combined age-dependent changes, including increased microglia and neuroinflammatory mis-activation, production of ROS, and decreased proteasome activity, could favor the establishment of the necessary grounds for microglial cell dysfunction that may lead to both cytotoxicity and the accumulation of Aβ. Additionally, once oxidative stress, cytotoxicity, and Aβ aggregation are established, they further decrease proteasome activity creating a vicious circle leading to more Aβ and tau aggregation. Whereas each of the age-dependent changes are mild and part of a normal aging process, their combined effect, in addition to some characteristics of the genetic background and environmental stimuli mechanism, may initiate this vicious circle of cytotoxic activation, reflecting an abnormal function of the innate immune system.
Innate immune responses, with microglia as the key player, but in an intimate interplay with astrocytes, neurons, and other brain cells, and often in collaboration with the adaptive immune system (not discussed in this review), are crucial for the elimination of abnormal proteins, damaged cells, and pathogens for clearing debris and stimulate tissue repair in the CNS. In this sense, microglial cell responses in the CNS are primarily beneficial. However, chronic activation and dysregulation of microglia can lead to deleterious effects that induce malfunction and damage of the CNS cells.
Although triggered by different initiating events, most neurodegenerative diseases share the chronic glial cell activation as a common feature. What drives chronic inflammation is not fully understood in most cases. Possibly, it is the incapability of the innate response against certain stimuli, such as misfolded and/or aggregated proteins, or the impairment of their regulatory mechanisms that triggers cytotoxic activation with production of ROS, high levels of inflammatory cytokines, and activation of damage signaling that contribute to the establishment of a final pathogenic pathway. Conceivably, a vicious cycle is established when the initial response of glial cells is unable to fully process the original trigger, and keep being stimulated by its reappearance, either directly or indirectly, in an increasingly vulnerable environment. For that reason, despite the undeniable potential of microglial cell responses to become pathogenic, it must be kept in mind that the innate immune responses in the CNS have a profound immune-modulatory and reparative potential. Instead of turning off microglial cell activation as a whole as it has be proposed, identification of protective and modulatory pathways of immune activation, to potentiate them while inhibiting cytotoxic activation, may well constitute the way to treat neurodegenerative disorders. A better understanding of these mechanisms is needed for the identification of new pathways that may decrease the impact of microglial cell dysfunction, thus breaking the abovementioned vicious circle.
As efficient antioxidants become available, there are a several matters to consider. First of all, antioxidant treatments could inhibit ROS-dependent cell death, but be ineffective for the underlying mitochondrial defects and may therefore result in other cell death pathways that are ROS independent. Second, and extremely important for the use of a therapy, since ROS are involved in physiological cell signaling, including some pro-survival signaling pathways, it will be necessary to avoid blockage of this effect by these treatments.
A. Future directions of research in this field
The role of microglia in AD has received increasing attention as possible avenues for therapies that modify disease outcome in AD. For example, minocycline studies point out the complexity of the effects of microglial manipulation in AD models. Various effects of minocycline on microglial activation and cognitive function predominate during different phases of the life spans of experimental animals, suggesting that microglial inhibition may be beneficial at one phase of disease progression but detrimental at others. A critical area of research involves the dissection of these mechanisms that may lead to the development of drugs with more clearly defined profiles of efficacy and fewer side effects. It is clear that causes and consequences of microglial cell activation are complex, and pharmacologic manipulation should be directed to selected properties of microglial cell function oriented to the promotion of protective functions while minimizing oxidative and neuroinflammatory damage.
An interesting combination for immunomodulatory therapy would be the use of antioxidants. Once again, antioxidant therapy should take into account that low concentrations of radical species serve physiological functions, including important roles in cell signaling. Thus, any treatment should be oriented to modulate ROS and RNS production rather to abolish them. Furthermore, if it probes possible to target microglia and oxidative stress modulations as an effective therapy for AD, other neurodegenerative diseases may prove similarly susceptible to their benefit.
Footnotes
Acknowledgments
The authors gratefully acknowledge the financial support from grant FONDECYT 1090353. We acknowledge bibliographic input of Dr. Andrea Vecchiola and Dr. Dagoberto Soto and technical help of Ms. María Gonzales and Ms. Laura Eugenín-von Bernhardi with the preparation of figures.