
Abstract
Introduction
Although the etiology of PD remains poorly understood, a broad range of studies conducted over the past few decades have collectively identified a number of molecular events that might underlie PD pathogenesis. In particular, the recent identification and functional characterization of several genes, including α-synuclein, parkin, DJ-1, PTEN-induced kinase (PINK1), and leucine-rich repeat kinase 2 (LRRK2), whose mutations are causative of rare familial forms of PD have provided tremendous insights into the molecular pathways underlying dopaminergic neurodegeneration (28, 95). Collectively, these studies implicate aberrant mitochondrial and protein homeostasis as key contributors to the development of PD, with oxidative stress likely acting as an important nexus between the two pathogenic events. Paradoxically, mitochondria are a major source for reactive oxygen species (ROS) by virtue of their function in oxidative respiration, which generates superoxide anion (O2 −, a precursor of other forms of free radical) as an inevitable byproduct (Fig. 1). As a result, mitochondrial components are particularly vulnerable to ROS-induced lesions, the accumulation of which could damage the respiratory chain complexes and lead to a progressive decline in mitochondrial bioenergetics and eventually cell death. Supporting this, several postmortem studies have demonstrated a significant reduction in the activity of mitochondrial complex I as well as ubiquinone (coenzyme Q10) in the SN of PD brains (36, 83, 87). Moreover, mitochondrial poisoning recapitulates PD features in humans and represents a popular strategy to model the disease in animals (15). To protect against potential ROS-induced damage, aerobically respiring cells have developed over the course of evolution an effective antioxidant defense mechanism that consists of plethora of antioxidant enzymes (such as superoxide dismutase [SOD], glutathione peroxidase, and catalase) as well as nonprotein antioxidants (such as glutathione, α-tocopherol, and ascorbic acid) to keep the levels of cellular free radicals in check. For example, ROS byproducts produced by mitochondrial respiration are rapidly detoxified by SOD 2 (which converts superoxide to hydrogen peroxide) so that their basal levels are low and nontoxic. Further, complex mitochondrial dynamics involving organelle biogenesis, fusion, fission, and selective degradation participate to keep the pool of organelle available to the cell bioenergetically competent. Disruption of this dynamism of mitochondrial life cycle has clear impact on cellular (especially neuronal) viability and is an emerging theme underlying PD pathogenesis. Here, we shall provide a brief overview of mitochondrial dynamics and its role in neurodegeneration and discuss some exciting findings from recent studies that support a critical role for parkin in mitochondrial quality control (QC).

Mitochondrial Dynamics: A Primer
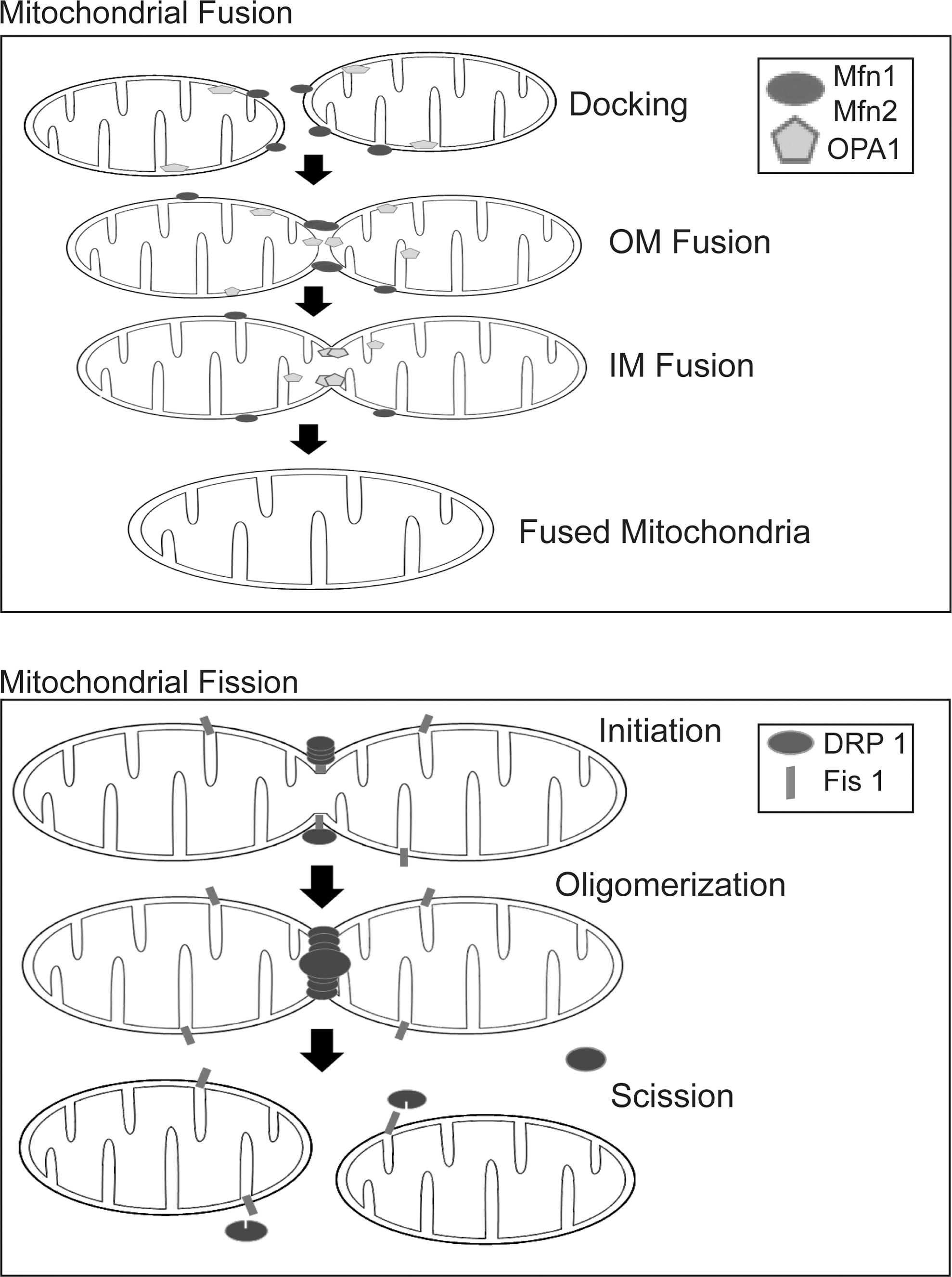
Far from being solitary and static structures as depicted in many textbooks, mitochondria are now recognized to be dynamic and mobile organelles that constantly undergo membrane remodeling through repeated cycles of fusion and fission as well as regulated turnover via a specialized form of lysosome-mediated degradation pathway known as “mitophagy,” a term originally coined by Lemasters (47) (Fig. 2). Collectively, these varied processes help to maintain the quality and thereby optimal function of mitochondria as well as allow the organelle to rapidly respond to changes in cellular energy status. The mammalian GTPases, mitofusins (Mfn1 and Mfn2) and optic atrophy protein 1 (OPA1), constitute the core machinery of mitochondrial fusion and another GTPase, dynamin-related protein 1 (Drp1), is the master regulator of mitochondrial fission. Current evidence suggests that Mfns, which reside on the organelle's surface, initiate outer membrane fusion via the formation of an intermolecular antiparallel coiled coil that tethers two approaching mitochondria together (105). When docked into proximity, lipid bilayer mixing occurs between adjacent mitochondria, leading to the fusion of their outer membrane. This is followed by mitochondrial inner membrane fusion, an event triggered by OPA1 (which resides within the organelle) in a GTP-dependent manner (Fig. 3). Although the two fusion machineries appear to operate in a co-ordinated manner, they are actually independent of each other. Indeed, selective blockade of inner membrane fusion by means of pharmacological dissipation of inner membrane potential or mutant OPA1 overexpression has negligible effects on Mfn-directed outer membrane fusion (54, 59). Unlike Mfns and OPA1, the fission protein Drp1 is predominantly cytosolic but is recruited to the surface of mitochondria during mitochondrial division, where it interacts with Fission 1 (Fis1), a protein whose tail is anchored in the outer membrane. Drp1 docking on Fis1 initiates mitochondrial scission through a self-oligomerization process that leads to the formation of spiral structures that wrap around and eventually sever the organelle (Fig. 3). This Drp1-mediated fission process is highly reminiscent of classical dynamin-mediated vesicular budding. However, although Drp1-Fis1 interaction is essential for mitochondrial fission in yeast (3), it is interesting to note that Fis1 knockdown in mammalian cells impairs the process without affecting the localization of Drp1 to mitochondria (46). Overexpression of hFis1 does not increase Drp1 binding to mitochondria (91). Thus, Fis1 appears to be dispensable for Drp1 recruitment to the mitochondria in mammalian cells. Instead, an alternative protein known as mitochondrial fission factor (Mff ) seems to be required. In an elegant recent study, Otera et al. demonstrated in mammalian cells that Mff knockdown disrupts Drp1 foci on the mitochondrial outer membrane that is accompanied by extension of the organelle network, whereas overexpression of Mff produces opposite effects, that is, stimulates Drp1 foci formation that is accompanied by mitochondrial fission. On the other hand, depletion of Fis1 has no effect on mitochondrial structure (71). The authors further showed that an Mff mutant engineered to localize to the plasma membrane directed Drp1 to the target membrane (71). Together, these findings suggest that Drp1-mediated fission of mitochondria in mammalian cells is dependent on Mff rather than on Fis1.
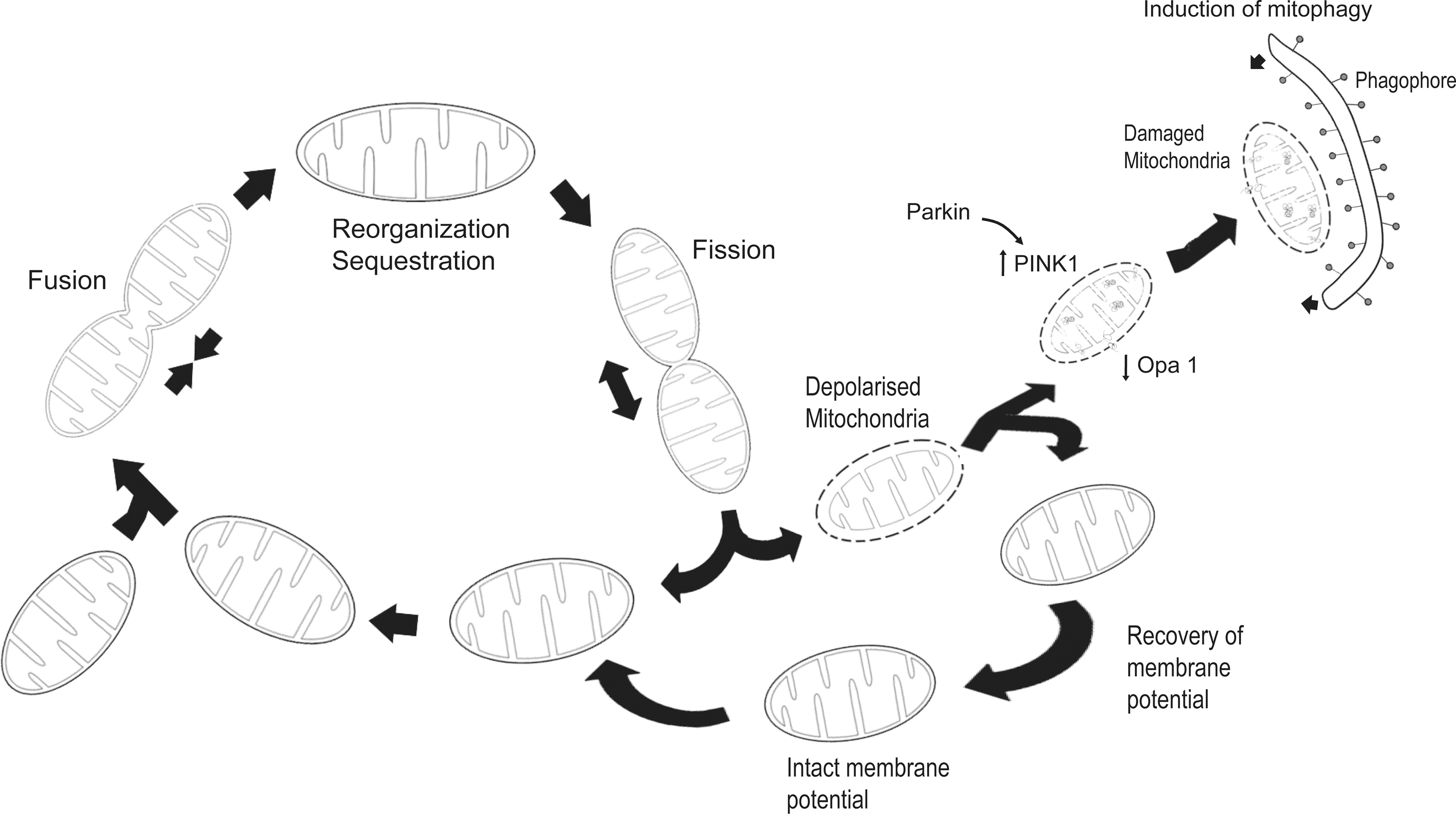
Fusion has been suggested to allow mitochondria to mix their contents (including copies of the genome, respiratory proteins, and metabolic products) and thereby provides an opportunity for a bioenergetically more active organelle to functionally complement its less active counterpart, whereas fission facilitates the segregation of mitochondria into daughter cells during cell division and may also participate in QC by isolating and targeting defective regions of the organelle for clearance via mitophagy (7). In cases wherein the damage sustained by the mitochondria is extensive, en bloc mitophagic degradation could occur. Under such conditions, mitochondrial biogenesis is likely to be co-ordinately enhanced to replace the exited population with newly synthesized organelles. Collectively, mitochondrial biogenesis, fusion, fission, and selective degradation are crucial for cellular survival as they work together to ensure the constant availability of bioenergetically competent organelles required by the cell to support its various energy-dependent activities. Accordingly, imbalance in these events that cause substantial changes to mitochondrial number, biomass, morphology, and bioenergetics could trigger cellular dysfunction leading to apoptosis, an event that mitochondria are also intimately involved (24).
Mitochondrial Dynamics and Neurodegenerative Diseases
The importance of mitochondrial dynamics to cellular function is perhaps best appreciated in neurons, as these postmitotic cells particularly those with vast axonal field require high energy to support their operation, which include the active transportation of components (including mitochondria) toward metabolically demanding synaptic terminals that are distally located. It is noteworthy to highlight that the axonal length of projection neurons such as SNpc dopaminergic neurons can be as long as 470,000 μM or more and each axon in turn can support nearly 400,000 synapses (58). It is therefore not surprising to also note that mitochondrial dysfunction underlies several neurodegenerative diseases including PD. For example, mutations in Mfn2 cause Charcot-Marie-Tooth type 2A (CMT-2A), a classic axonal peripheral sensorimotor neuropathy characterized by degeneration of long peripheral nerves (114). Similarly, mutations in OPA1 promote neuronal loss but of retinal ganglia cells in a condition known as autosomal dominant optic atrophy, the most common form of inherited childhood blindness (1, 16). However, why mutations affecting two proteins that are both involved in mitochondrial fusion should give rise to two phenotypically distinct diseases is unclear, although differences between outer and inner membrane fusion defects may be one explanation. Alternatively, Mfn2 and OPA1 may have other mitochondria-related roles distinct from fusion. Supporting this, Misko et al. recently demonstrated that Mfn2, but not OPA1, interacts with the Miro/Milton complex to regulate mitochondrial transport and that its expression silencing in cultured neurons produced axonal transport deficits that mimic loss of Mfn2 function (60). Their results suggest a mechanism that underlies selective degeneration in CMT-2A. Thus far, a link between Drp1 (or Fis1) and inherited diseases have not been described, save for a recent case of neonatal lethality that is attributed to a dominant negative mutation in Drp1 (104). Autopsy report revealed severe neurological defects in the newborn, including abnormal brain development, microcephaly, and optic atrophy amongst other phenotypic manifestations (104).
Emerging evidence also suggests a role for altered mitochondrial dynamics in the pathogenesis of Alzheimer's disease (AD) and PD. Recently, Wang et al. demonstrated that neuronal cells ectopically expressing wild-type or disease-associated mutant amyloid precursor protein (APP), mutations of which are linked to familial AD, results in altered expression of mitochondrial fusion/fission proteins and consequent increase in mitochondrial fragmentation that is accompanied by reduced organelle availability to neuronal processes (103). Related to this, Cho et al. showed that Drp1 is S-nitrosylated in the AD brain, a redox modification that apparently enhances the fission activity of the protein and thereby mitochondrial fragmentation (9). At around the same time, a landmark paper was published by Richard Youle group that identified parkin as a key mediator of mitophagy (65), suggesting that impaired clearance of damaged mitochondria may trigger the demise of dopaminergic neurons in the PD brain. A flurry of activity to elucidate the precise mechanism underlying parkin-mediated mitophagy ensues to this date, as discussed later. However, several studies also implicate a role for parkin in regulating mitochondrial fusion/fission as well as in biogenesis of the organelle. Thus, parkin appears to be involved in the entire spectrum of mitochondrial dynamics, which is rather amazing and also part of the reason why we have chosen to focus on this protein in our current review.
Parkin, Protein Ubiquitination, and PD
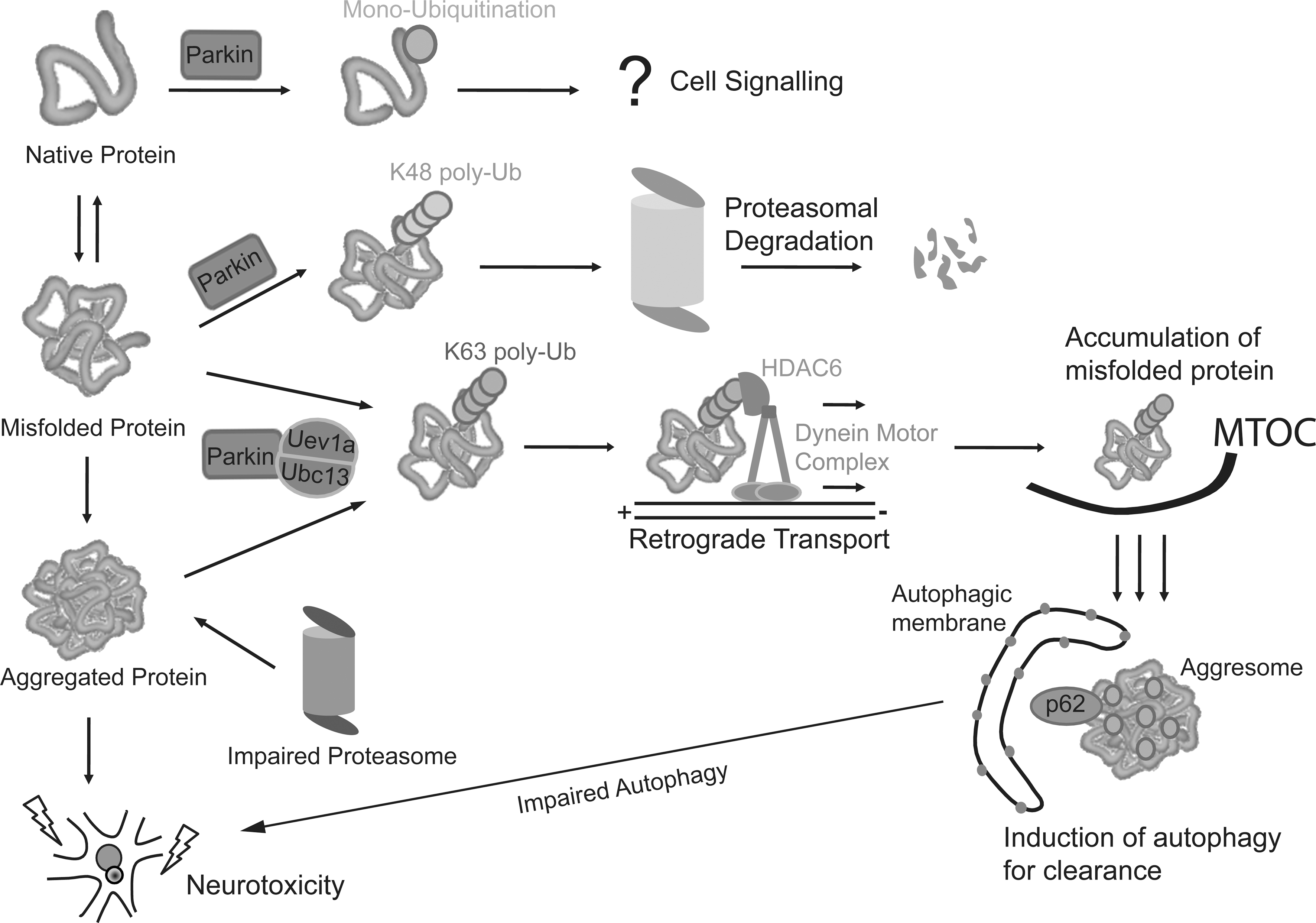
Mutations in parkin, which encodes a ubiquitin ligase, are a predominant cause of autosomal recessive early-onset parkinsonism (39, 52). Further, emerging evidence also support a link between parkin expression variability and sporadic PD (61). Accordingly, a better understanding of parkin function could help elucidate pathways underlying PD pathogenesis. Parkin-mediated ubiquitination was originally linked to proteasome-dependent protein degradation (32, 85, 112), suggesting that loss of parkin function could lead to a toxic accumulation of one or several of its substrates, thereby leading to neurodegeneration. However, we and others have demonstrated that the enzyme (besides catalyzing proteasome-dependent K48 polyubiquitination) is also capable of mediating monoubiquitination and K63-linked polyubiquitination—ubiquitin modifications that are typically considered as “nonproteolytic” (20, 27, 49, 56). Our results would argue that the catalytic function of parkin is not limited to targeting substrate for degradation by the proteasome. A corollary of this is that proteasome-independent pathways may also be relevant to parkin-related neurodegeneration. Supporting this, parkin-mediated monoubiquitination of the adaptor protein Eps15 inhibits its ability to initiate receptor downregulation and leads to enhanced epidermal growth factor (EGF) receptor signaling as well as downstream EGF-induced Akt signaling (21, 30). Loss of parkin function impairs this neuroprotective signaling pathway and, as such, may result in increased vulnerability of neurons toward degeneration. Besides monoubiquitination, parkin-mediated K63 polyubiquitination may also be important for neuronal survival. By virtue of its apparent dissociation from the proteasome, we have originally proposed that parkin-mediated K63-linked ubiquitination may be involved in cargo diversion during proteasomal stress and, accordingly, in the biogenesis of inclusion bodies associated with neurodegenerative diseases (50). Our proposal is consistent with the concept of aggresomes, which are juxtanuclear inclusion bodies formed in the presence of proteasomal stress and that have been suggested to act as staging grounds for the disposal of protein aggregates via the autophagic route (41). Supporting our hypothesis, we found that parkin-mediated K63 polyubiquitination of synphilin-1 [a substrate of parkin (10)] promotes its aggregation into aggresome-like inclusion bodies (49). Corroborating our findings, Olzmann et al. demonstrated that parkin-mediated K63 polyubiquitination of misfolded DJ-1 couples the protein to the dynein motor complex via the histone deacetylase 6 (HDAC6) adaptor, thereby promoting its sequestration into aggresomes (69). Importantly, our recent work identified K63-linked polyubiquitin as a novel cargo selection signal for macroautophagy-mediated clearance of aggresomes (92, 93).
By being capable of mediating both proteasome-associated K48-polyubiquitination and macroautophagy-associated K63-linked polyubiquitination, parkin may potentially act as an important triage between the two major cellular degradation systems. Consistent with this, we observed that the recruitment of Ubc13 (the only E2 known to date to mediate the formation of K63-linked ubiquitin chains) by parkin is selectively enhanced under conditions of proteasomal stress, thus identifying a mechanism by which parkin could promote K63-linked ubiquitin modification in cells undergoing proteolytic stress (Lim and Lim, unpublished observation). This multifunctionality of parkin may in part help to explain its apparent broad neuroprotective properties, as the flexibility of ubiquitin linkage usage would presumably allow the enzyme to adapt to changes in cellular environment. Accordingly, mutations of parkin, particularly those occurring on the RING2 domain that abolish its enzymatic activity (27, 56), would directly compromise its protective function. However, those occurring on other regions of the protein do not appear to affect its catalytic competency but instead tend to promote its insolubility and thereby aggregation in the cell (27, 56, 89, 102). The immobilization of catalytically viable parkin into aggresome-like aggregates is clearly another way by which the normal function of the enzyme can be affected. Notably, several groups, including ours, have shown that normal aging as well as a wide variety of PD-linked stressors, including those that produce oxidative and nitrosative stress, similarly induce parkin's intracellular mislocalization in a manner analogous to that brought about by several of its missense mutations (44, 100, 106). The alteration of parkin solubility induced by various extrinsic and intrinsic stressors thus provides a mechanism for parkin dysfunction that is relevant to the pathogenesis of sporadic PD. A picture summarizing the various roles of parkin-mediated ubiquitination is shown in Figure 4.
Parkin and Mitochondrial Dynamics: Lessons from Flies and Other Models
One of the first hints that parkin may play a role in mitochondrial homeostasis aside from its role as a regulator of protein turnover came from a study in fruit flies. Greene et al. analyzed Drosophila parkin null mutants and observed that these flies are semiviable and display reduced lifespan, locomotor defects, and male sterility (26). However, the most prominent pathology of adult parkin null flies is not in the brain but in the flight musculature, which is plagued by muscle degeneration and pronounced mitochondrial lesions (26). Moreover, mitochondrial pathology appears to be the earliest detectable phenotype in parkin null mutant flies and clearly precedes the onset of apoptosis in the flight muscle by at least several days. Similarly, we observed that muscle-specific expression of an aggregation-prone disease-associated parkin mutant (R275W), but not its more soluble counterpart (G328E), result in muscle defects and mitochondrial abnormalities (101), suggesting that parkin aggregation may also affect mitochondrial dynamics. Why flight muscles in Drosophila appear preferentially sensitive to the loss of parkin function remains unclear, although a feature of insect flight muscles is their high metabolic activity, which is likely to result in significant production of ROS. Consistent with this, Greene et al. reported in a follow-up study that oxidative stress response components are induced in parkin mutants and that loss-of-function mutations in these components enhance the parkin mutant phenotypes (25). In an interesting development, two back-to-back papers reported that pink1 null flies phenocopy their parkin-deficient counterparts (11, 75). Like the latter, pink1-deleted mutant Drosophila exhibit male sterility and are characterized by dramatic mitochondrial defects in muscles and testes, including swelling and fragmentation of the organelle, as well as reduction in ATP levels and mitochondrial DNA content (11, 75). Importantly, parkin overexpression in pink1 −/− flies significantly ameliorated all the mutant phenotypes tested, although the reverse does not happen, that is, pink1 overexpression in parkin null flies does not compensate for the loss of parkin function (11, 75), suggesting that parkin acts in the same pathway but downstream of pink1. These observations were essentially corroborated by a separate but related study conducted by Yang et al. that involved RNAi knockdown of pink in Drosophila (107). Yang et al. also extended the parkin rescue experiment by demonstrating that DJ-1 overexpression has no mitigating effects on pink1 mutant phenotypes. More recently, the same group also demonstrated a role for pink1 as a regulator of mitochondrial dynamics via protein interaction with the mitochondrial fission/fusion machinery (108). The investigators found that pink1 overexpression promotes mitochondrial fission, whereas its inhibition results in excessive fusion of mitochondria (108). Similar findings were reported by Poole et al. and Deng et al. at around the same time, who also showed that the flight muscle degeneration and mitochondrial morphological alterations that result from mutations in pink1 and parkin are strongly suppressed by increased drp1 gene dosage or by heterozygous loss-of-function mutations (or muscle-specific knockdown) affecting the mitochondrial fusion-promoting factors opa1 and Marf (Fly homologue of Mfn) (17, 77). Supporting this, two recent reports identified Mfn as a degradation-associated substrate of parkin in Drosophila (78, 113). Loss of parkin function is thus expected to promote the accumulation of Mfn in vivo and concomitantly enhance mitochondrial fusion. Consistent with this, elongated mitochondria are observed in parkin mutant flies (17, 113). Notably, parkin-mediated Mfn turnover is dependent on pink1, which recruits parkin to the mitochondria, where Mfn resides (78, 113). According to one study in the Drosophila, the recruitment of parkin by pink1 may involve its phosphorylation by the serine/threonine kinase (38), although whether parkin is a bona fide target of PINK1 kinase activity is currently controversial (66). Interestingly, in a recent biochemical screen, Imai et al. isolated a PINK1 binding protein known as protein phosphoglycerate mutase 5 (PGAM5), which is localized to the outer membrane of the mitochondria, and demonstrated that dPGAM5 ablation in flies suppresses mitochondrial degeneration caused by the loss of pink1 activity although not by parkin inactivation (31). Their results suggest that dPGAM5 acts either in between pink1 and parkin, or independent of parkin. However, PGAM5 does not appear to be phosphorylated by PINK1 and the precise mechanism by which PGAM5 regulates the parkin/PINK1 pathway remains to be elucidated.
Consistent with the suggestion that parkin/PINK1 pathway may promote mitochondrial fission, an increase in the number of larger mitochondria was seen in the striatum of PINK1 null mice (22), although no apparent swelling or other morphological changes of the organelle was observed in parkin null mice (72). Further, fibroblasts derived from a patient harboring loss-of-function parkin mutation exhibit marked increase in mitochondrial branching, a phenomenon that can be reproduced simply via SiRNA-mediated silencing of parkin expression (80% reduction) in control fibroblasts (63). In the presence of rotenone treatment, the branches collapsed to form short, irregularly shaped mitochondria, suggesting that parkin-deficient cells can enter fission when stressed, although it remains unclear whether this is secondary to impaired complex I function induced by rotenone, or a precipitating event leading to impaired mitochondrial function (63). Surprisingly, a recent study by Lutz et al. reported that overexpression of parkin or PINK1 in human SH-SY5Y cells does not enhance fission but instead prevents Drp1-induced mitochondrial fragmentation (53). The authors further showed that expression silencing of parkin and PINK1 in these cells induces fragmentation of the mitochondrial network that can be rescued by increasing mitochondrial fusion or reducing fission (53), that is, suggesting that parkin/PINK1 pathway promotes mitochondrial fusion rather than fission at least in SH-SY5Y cells. Corroborating this finding, Cui et al. showed by means of an ecdysone-inducible expression system in N27 dopaminergic neuronal cells (derived from rat fetal mesencephalic cells) that the expression of disease-associated mutant PINK1 (L347P and W437X) mediates an overall fission effect by increasing the ratio of mitochondrial fission over fusion proteins, leading to excessive dysfunctional fragmented mitochondria (12). They made similar observations when endogenous PINK1 is knocked down. In contrast, overexpression of wild-type PINK1 tips the balance toward mitochondrial fusion, which could be restored by parkin expression silencing (12). Further, mitochondrial perturbations induced by mutant PINK1 overexpression can be rescued by treating the cells with mdiv-1, a mitochondrial division inhibitor that inactivates Drp1 (12). Together, these results support the finding by Lutz et al. (53) that both parkin and PINK1 promote events that favor fusion rather than fission.
Several factors might explain the apparent discrepancy between the models by the above two reports (12, 53) and others, including (as highlighted by Lutz et al.) the time of phenotype analysis, species-specificity, and the lack of compensatory mechanisms in their acute RNAi-based gene knockdown cultured cell model compared with those caused by chronic loss of protein function (53). Perhaps another potential confounding factor in such cell-based studies is that cells in culture generate most of their ATP via glycolysis from glucose present in the medium and not via oxidative phosphorylation (80). Notably, mitochondrial morphology can be quite different in cells grown in glucose-containing medium compared with those cultured in medium with glucose replaced by galactose (80). Although we await further clarifications on the role of parkin in mitochondrial dynamics, a fission-promoting function of the protein is attractive particularly in view of recent evidence identifying parkin as a key regulator of mitophagy. Increased mitochondrial fission will probably favor the clearance of damaged mitochondria by mitophagy and, at the same time, promote their replenishment through biogenesis from the remaining pool of functional mitochondria. However, if the level of mitochondrial stress is milder and the damage on the organelle does not necessitate its clearance from the cell, parkin might possibly have a fusion-promoting role to limit the damage and/or repair the damaged organelle.
Parkin/PINK1 Pathway in Mitophagy: Model and Caveats
The degradation of mitochondria by lysosomes, especially upon cellular starvation, has been appreciated for more than half a century. However, whether and how damaged mitochondria are selectively recognized by the autophagic apparatus has been largely unclear until recently. Using the chemical uncoupler, carbonyl cyanide m-chlorophenylhydrazone (CCCP) to induce mitochondrial depolarization in HeLa cells, Narendra et al. from Youle laboratory originally demonstrated in 2008 that parkin plays an essential role in removing damaged mitochondria from the cell via mitophagy (65). Subsequent intense efforts by several groups, including Youle laboratory, have shed much insights into the mechanism by which parkin regulates mitophagy (73, 110). A model that has emerged from these studies is shown in Figure 5. Briefly, a key initial event that occurs upon mitochondrial depolarization is the selective accumulation of PINK1 in the outer membrane of the damaged organelle (presumably after its segregation by fission). Notably, PINK1 accumulation in healthy mitochondria is prevented by a proteolytic event that rapidly cleaves the protein (57, 66, 99). Upon recruitment to depolarized mitochondria by PINK1, parkin then becomes activated (57) and promotes the ubiquitination and subsequent degradation of Mfns (78, 113), the elimination of which will prevent unintended fusion events involving damaged mitochondria and thereby their reentry into undamaged mitochondrial network from occurring (94). Mfn degradation is regulated by p97 (an AAA-ATPase linked to endoplasmic reticulum-associated degradation amongst other cellular processes), whose accumulation on depolarized mitochondria presumably facilitates the extraction of ubiquitinated Mfns (and possibly other parkin substrates) from the outer membrane for degradation by the proteasome (94). Besides Mfns, parkin also apparently mediates the degradation of several other outer membrane proteins such as Tom 20, Tom 40, Tom 70, and Omp 25 of depolarized mitochondria (6, 109). To facilitate this widespread degradation of mitochondrial proteins, parkin activates the UPS upon translocation to the mitochondria (6). This involves the enrichment of K48-linked ubiquitination and recruitment of the proteasome to the mitochondria, which may lead to rupturing of the mitochondrial outer membrane (6, 109). The event is then followed by the induction of mitophagy, which is also parkin dependent. Parkin-mediated K63 ubiquitination, a ubiquitin modification that we have shown to be linked to aggresome formation and clearance by autophagy, is probably important here. Indeed, K63 polyubiquitin modification of several mitochondrial substrates takes place following CCCP-induced uncoupling (23, 68). Consistent with this, stable isotope labeling with amino acids in cell culture analysis revealed a substantial increase (28-fold) in parkin-mediated K63-linked polyubiquitination upon mitochondrial depolarization (6). This may help to recruit HDAC6 and p62, both of which are ubiquitin-binding proteins that preferentially recognizes K63 ubiquitin chains (51, 69, 92) and lead to mitochondrial clustering around the perinucleus region. An important property of p62 is its ability to directly interact with the autophagosome protein, light chain 3, which provides a link between ubiquitinated proteins and the autophagy machinery (2, 74). On the other hand, HDAC6 is able to stimulate autophagosome–lysosome fusion via the recruitment of a cortactin-dependent, actin remodeling machinery to ubiquitinated proteins that is important for their eventual clearance by autophagy (45). The concerted actions of p62 and HDAC6 presumably facilitate the final removal of damaged mitochondria by the lysosome (18, 23, 45), although some reports suggested that p62 may not be essential for parkin-mediated mitophagy (64, 68). Notwithstanding this, it is apparent that the whole process bears striking resemblance to the formation and autophagic clearance of aggresomes. Indeed, we have termed the mitochondrial clustering phenomenon as (formation of ) “mito-aggresomes” (45). Importantly, several groups, including ours, have demonstrated that PD-associated parkin mutants (including the aggregation-prone R275W mutant) are defective in supporting mitophagy because of distinct problem at recognition, transportation, or ubiquitination of impaired mitochondria (45, 57), thereby implicating dysfunctional mitophagy in the development of parkinsonism. Such misfolded parkin-related defects can, however, be rescued by enhancing the levels of molecular chaperones. For example, overexpression of the neuronal Hsp40 chaperone HSJ1a restored mitophagy in parkin C289G (another aggregation-prone mutant)-expressing cells by promoting the relocation of the otherwise mislocalized mutant to depolarized mitochondria (79).
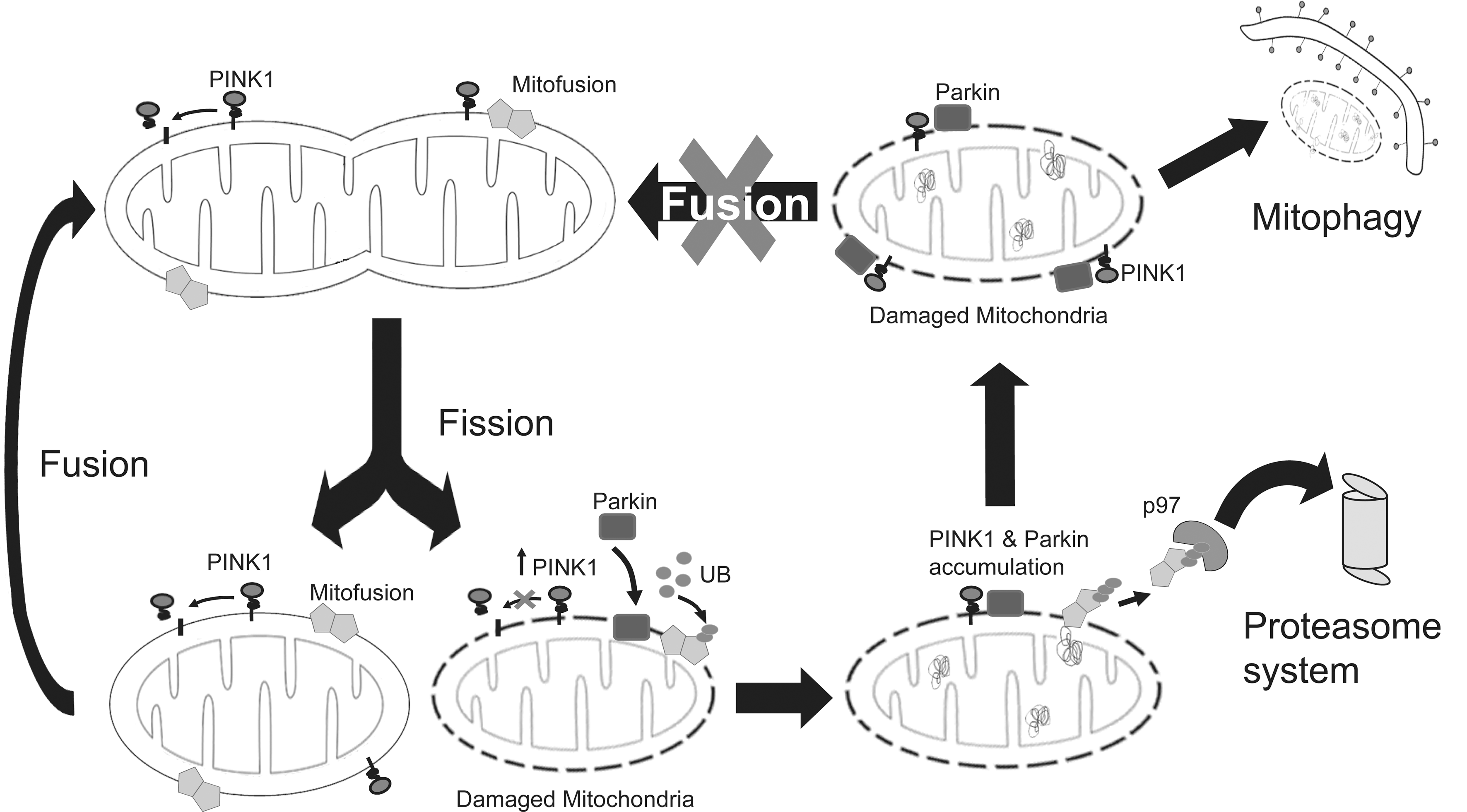
Despite the logic and experimental support for the events surrounding parkin-mediated mitophagy as described earlier, the model is not without controversy. First, whether the recruitment of parkin by PINK1 is absolutely necessary for parkin-mediated mitophagy is debatable, as parkin seems capable of promoting mitophagy in PINK1-deficient cells (13, 14). Second, parkin can also promote the autophagic clearance of damaged mitochondria in Mfn-deficient cells (94), suggesting that these fusion-linked GTPases may likewise be dispensable for mitophagy. Notably, parkin-mediated ubiquitination of another mitochondrial substrate, voltage-dependent anion-selective channel protein 1 (VDAC1), has been previously suggested to be required for mitophagy (23), although like Mfns, VDAC1 ubiquitination may not be an obligatory event for the clearance to occur (64). Third, and importantly, whether mitophagy induced by parkin in cultured cells treated with mitochondrial poisons is relevant to neurons, particularly dopaminergic neurons associated with PD, is currently controversial. According to a recent study by Van Laar et al. (97), the bioenergetics of neurons, which are strictly dependent on mitochondrial respiration, are distinct from other cell types and may as such elicit a different response toward mitochondrial depolarization. Indeed, CCCP-treated primary neurons neither promote the translocation of parkin (both exogenous and endogenous) to the mitochondria nor trigger mitophagy (97). Further, although PINK1 overexpression increased the accumulation of parkin on neuronal mitochondria, it did not sensitize them to depolarization-induced parkin translocation (97). Supporting their theory regarding the relationship between mitochondrial bioenergetics and its response to depolarization, Van Laar et al. showed that parkin's translocation to depolarized mitochondria is similarly attenuated in HeLa cells forced into dependence on mitochondrial respiration (i.e., cultured in glucose-free medium). Interestingly, mitophagy is similarly blocked in yeast even under severe starvation conditions when they are cultured with a nonfermentable carbon source that made them obligatorily dependent on mitochondrial metabolism (35). Thus, parkin-mediated mitophagy appears to be dependent on the bioenergetic status of the cell. Presumably, additional mechanisms are involved in the regulation of mitophagy in neurons and other cell types that derive their energy from oxidative phosphorylation. Notwithstanding this, it is important to highlight that at least two groups have found that parkin accumulation on mitochondria does occur in primary neurons treated with CCCP (65, 99). Further, Seibler et al. have recently demonstrated that parkin recruitment to depolarized mitochondria is impaired in human dopaminergic neurons derived via the induced pluripotent stem cells route from PINK1-related PD patients, a defect that can be rescued by re-introduction of wild-type PINK1 into PINK1-deficient neurons (84). The reason for this discrepancy is presently unclear, but the conundrum obviously needs to be resolved as whether parkin-mediated mitophagy taking place appreciably in neurons has significant implications for its role in PD pathogenesis.
A Role for Parkin in Mitochondrial Biogenesis?
Undoubtedly, it is rather amazing to note that HeLa cells are able to virtually eliminate their whole complement of mitochondria following extended CCCP exposure (65). Treatment of cells with CCCP is of course a “sledgehammer” approach toward promoting mitochondrial depolarization and whether or not such en bloc organelle damage and ensuing clearance are physiologically relevant is questionable. Nonetheless, replenishment of exited mitochondria supposedly occur following extensive mitophagy and it is attractive to speculate, based on a previous report by Kuroda et al. (43), that parkin might have a role in promoting mitochondrial biogenesis after mitophagy. Apparently, parkin overexpression enhances the transcription and replication of mitochondrial DNA by associating with mitochondrial transcription factor A (TFAM) and promoting TFAM-mediated transcription (43). Accordingly, the event is attenuated by parkin expression silencing with siRNA. However, an important caveat is that parkin-induced mitochondrial biogenesis associated with TFAM-mediated transcription only occurs in proliferating but not differentiated cells (43). Hence, its relevance to postmitotic cells such as neurons is unclear. In an interesting recent development, Shin et al. identified the zinc finger-containing protein called PARIS (ZNF746) as a novel substrate of parkin that accumulates in postmortem brain tissues derived from autosomal recessive juvenile parkinsonism and sporadic PD patients as well as in the ventral midbrain region of mice that is conditionally ablated of parkin expression (86). PARIS is a major transcriptional repressor of PGC-1α expression, which in turn regulates the transcription of many genes including those involved in mitochondrial biogenesis. Among these PGC-1α–regulated genes is nuclear respiratory factor-1 (NRF-1), whose activity contributes to the expression of respiratory subunits and mitochondrial transcription factors (82). The findings by Shin et al. thus suggest that parkin can also potentially regulate mitochondrial biogenesis by regulating PGC-1α expression indirectly through its ability to downregulate PARIS (which otherwise represses PGC-1α expression). Notably, both PGC-1α and NRF-1 expression are reduced in the SN of PD brains (86), suggesting that impaired renewal of neuronal mitochondria may contribute to DA neurodegeneration in parkin-related cases. Based on these results, it is also logical to speculate that parkin might assist in the replenishment of exited mitochondria following extensive mitophagy. Thus, parkin appears to be involved in the entire spectrum of mitochondrial dynamics, that is, from biogenesis to clearance.
Other PD-Linked Players That Influence Mitochondrial Dynamics
Although the current review is primarily focused on the relationship between parkin (and PINK1) and mitochondrial dynamics, it is noteworthy to highlight that several other PD-linked genes have been similarly reported to contribute to mitochondrial homeostasis, which we will briefly discuss in this section.
DJ-1
Loss of DJ-1 function, which in humans cause recessive parkinsonism, promotes mitochondrial fragmentation in a variety of cells including lymphoblast cells derived from DJ-1 patients and sensitizes them toward oxidative stress-induced death (33, 42, 96). The mitochondrial phenotype can be rescued by restoration of functional DJ-1 expression or by scavengers of ROS (33, 96). Interestingly, Parkin and PINK1 can also rescue the phenotype of DJ-1–deficient cells (33, 96), which support the earlier suggestion by Lutz et al. (53) that both proteins are promoters of mitochondrial fusion rather than fission, although it is also entirely possible that the rescue by parkin and PINK1 is related to their ability to mitigate oxidative stress. Together, these studies suggest that DJ-1 participates in the regulation of mitochondrial dynamics and works in parallel to the PINK1/parkin pathway to maintain mitochondrial function in the presence of an oxidative environment. However, it remains curious to note that parkin/PINK1/DJ-1 triple knockout mice do not exhibit any evidence of nigral degeneration or dramatic mitochondrial pathology, even at 24 months of age (40).
α-Synuclein and LRRK2
Unlike parkin, PINK1, and DJ-1, mutations of α-synuclein and LRRK2 are associated with dominant parkinsonism. Several groups have previously demonstrated that the accumulation of α-synuclein is a promoter of mitochondrial impairment (29, 70), although the synuclein-mediated effect may be attributed to its ability to enhance oxidative stress. However, a recent study suggested that the presynaptically localized protein may play a more direct role in impairing mitochondrial dynamics (34). Kamp et al. showed that α-synuclein is an inhibitor of membrane fusion and its overexpression in cultured cells or Caenorhabditis elegans results in its localization to the mitochondria, leading to increased organelle fragmentation (34). Conversely, SiRNA-mediated downregulation of α-synuclein expression promotes mitochondrial elongation. Importantly, the mitochondrial phenotype induced by α-synuclein overexpression can be rescued by parkin, PINK1, or DJ-1 coexpression, suggesting a shared pathway among the different PD-linked genes (34). In a parallel development, Chinta et al. also reported the localization of overexpressed α-synuclein to the mitochondria, which in this case happened in SN dopaminergic neurons of transgenic (TH promoter-driven) α-synuclein mice (8). Interestingly, they further observed that synuclein-expressing neurons display enhanced mitophagy (8). Although more work obviously needs to be done, these two reports provide tantalizing evidence for a role of α-synuclein in the regulation of mitochondrial dynamics. Comparatively, a potential relationship between LRRK2 and mitochondrial function is less well characterized, although cells derived from an LRRK2 G2019S patient exhibit reduced mitochondrial membrane potential and total intracellular ATP levels accompanied by increased mitochondrial elongation and interconnectivity (62). Interestingly, the homolog of LRRK2 in C. elegans protects against mitochondrial damage in the organism (81). Moreover, we have demonstrated that parkin protects against LRRK2-induced neurotoxicity in vivo (67) and that the protective effects of parkin may be related to its ability to maintain mitochondrial function in mutant LRRK2-expressing flies (Ng and Lim, unpublished observations). However, it is important to point out that the relationship between parkin and LRRK2 is likely complex, as others have generally found no protective interaction between the two PD-linked proteins in vivo in the context of the Drosophila eye phenotype, wherein the expression of parkin and/or LRRK2 promotes a series of eye defects, that is, bristle loss, holes, rough surface, and loss of pigmentation (although parkin expression inhibits the formation of black lesions in a mutant LRRK2-expressing line) (98). Future studies should help clarify whether there is indeed a role for LRRK2 in mitochondrial dynamics, and if there is, how the kinase interacts with the parkin/PINK1 pathway in regulating mitochondrial homeostasis.
Omi/HtrA2
Although mutations of Omi/high-temperature requirement protein A2 (HtrA2) (90) have also been suggested to cause parkinsonism, their relevance to PD remains contentious, because the purported disease-causing mutation is also present in control population at similar frequencies (e.g., Omi/HtrA2 G399S) (88). On the other hand, a recent study revealed that noncoding heterozygous Omi/HtrA2 mutations occurring in the regulatory regions of the gene are exclusively found in patient populations (4), suggesting that aberrant Omi/HtrA2 expression may increase the risk for PD. Notwithstanding the controversy, the mitochondrial serine-protease is an interesting protein for many reasons. First, Omi/HtrA2 is phosphorylated in a PINK1-dependent manner upon activation of the p38 stress pathway, which upregulates its protease activity (76). Second, loss of Omi/HtrA2 function results in alterations of mitochondrial morphology that include increased mitochondrial fusion leading to elongation of the organelle (37, 55). Apparently, Omi/HtrA2 interacts with Opa1 (37). Third, a recent study reported the ability of Omi/HtrA2 to activate autophagy through digestion of Hax-1, a Bcl-2 family-related protein that represses autophagy in a Beclin-1–dependent pathway (48). However, Omi-mediated autophagy induction facilitates the degradation of aggregation-prone proteins such as mutant α-synuclein (48), whereas its potential involvement in mitophagy has not been yet examined but represents an attractive possibility. Curiously, neither the ablation of orthologous Omi/HtrA2 allele nor the expression of Omi G399S equivalent in Drosophila produces any mitochondrial defects (111). Further, genetic epistasis studies conducted in these flies also failed to reveal any effects of Omi/HtrA2 on mitochondrial homeostasis regulated by parkin/PINK1 pathway (111). The verdict regarding the role of Omi/HtrA2 in mitochondrial dynamics thus remains open.
Concluding Remarks
Unlike Huntington's disease, which is caused by a single gene defect or familial AD wherein the majority of the disease-linked genes are clustered around the APP processing pathway, the genes associated with PD are so disparate in function that, at first sight, they seem to have little things (if at all) in common. Even for parkin-related PD, the laundry list of substrates (or putative substrates) for the ubiquitin ligase is ever growing, and the diverse cellular roles of these targets would point toward a divergence rather than convergence of pathogenic pathways. All these have made the habitual task among biochemists to find a unified pathway for disease pathogenesis really daunting for PD. However, emerging evidence suggests that these seemingly disparate PD-linked proteins may all influence mitochondrial homeostasis, directly or indirectly (Table 1). From our above discussion, it is clear that the activities of parkin, PINK1, DJ-1, α-synuclein, LRRK2, and Omi/HtrA2 all have significant impact on mitochondrial function, which collectively provides strong support to the age-old hypothesis that mitochondrial dysfunction is a key event underlying PD pathogenesis. Nonetheless, several important gaps exist, which need to be resolved, particularly those pertaining to the physiological relevance of findings derived from cell lines. Further, although the emerging field of parkin-mediated mitochondrial QC has proven to be exciting, it is important to recognize that parkin function is not restricted to the mitochondria. The same argument is true for the other PD-linked proteins. Indeed, PD pathogenesis is likely to involve an intricate network of interacting pathways rather than a linear sequence of events. Elucidating the reciprocity of pathways, particularly how other PD-related pathways potentially influence mitochondrial homeostasis, may hold the key to therapeutic development.
Controversial, see main text.
HtrA2, high-temperature requirement protein A2; LRRK2, leucine-rich repeat kinase 2; PD, Parkinson's disease; PARK, PD genetic loci; PINK1, PTEN-induced kinase; ROS, reactive oxygen species.
Footnotes
Acknowledgments
This work was supported by grants from A*STAR Biomedical Research Council, Singapore Millennium Foundation, and the National University of Singapore (to K.-L.L.) and by NIH NS053825 (to T.-P.Y.). L.G.-Y.G. was supported by a graduate scholarship from the Singapore Millennium Foundation.