
Abstract
Introduction
Aβ's upstream designation is consistent with the amyloid cascade hypothesis (107 –109), which postulates the Aβ (99) byproduct of amyloid precursor protein (APP) degradation (135) causes AD. Outside of rare familial autosomal dominant forms, though, it is unclear why Aβ dynamics change in AD. After all, Aβ is constantly produced in brains of young and old people. Extracellular Aβ levels rise during the day and fall during sleep (136). Interstitial Aβ falls after severe closed head injuries, and rising levels signal clinical recovery (27). Clearly, Aβ production is a regulated process and the simple presence of Aβ in the brain does not necessarily initiate AD. What, then, could possibly constitute the “upstream” regulator of brain Aβ? This review argues mitochondria and cell bioenergetics (Fig. 1) regulate Aβ, and that in sporadic AD, changes in mitochondrial function and cell bioenergetics occur upstream to Aβ changes.
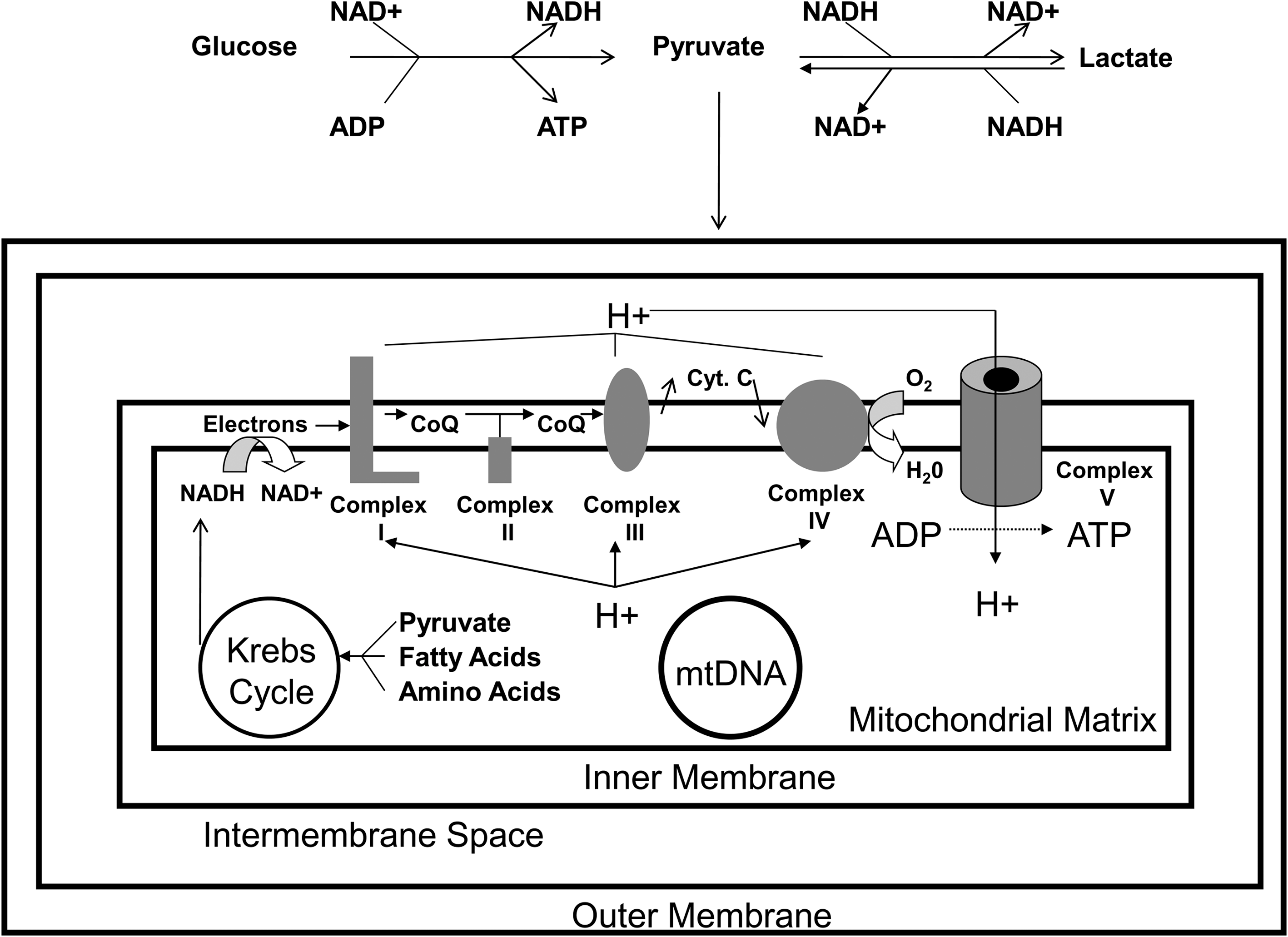
Mitochondria Are Increasingly Implicated in AD and AD Models
Altered oxidative metabolism in AD was reported in the 1960s (89), and abnormal glucose utilization was noted throughout the 1970s and beyond (25, 67, 87, 90, 126, 243, 255). During this time, changes to the main bioenergetics organelle, the mitochondrion, were observed on several levels (263). Mitochondrial ultrastructure was perturbed, and activities of several mitochondria-localized enzymes (including pyruvate dehydrogenase complex and α-ketoglutarate dehydrogenase complex) were reduced (97, 128, 212, 247, 291). Mitochondrial oxygen consumption in AD subject frontal cortex homogenates was shown to differ from control subject homogenates (243). Reduced brain oxygen utilization was demonstrated using oxygen-15 positron tomography (88, 92). Interestingly, bioenergetics and mitochondrial changes were found to extend beyond the brain to nondegenerating tissues such as fibroblasts and lymphocytes (20, 22, 96, 97, 214, 215, 241, 242). Pioneering investigators postulated energy metabolism might constitute an important feature of AD (22, 97, 242), but interest in this line of investigation was largely restricted to the field's periphery.
In 1990, reduced activity of the electron transport chain (ETC) enzyme complex IV (cytochrome oxidase; COX) was demonstrated by Parker et al. (203). The AD COX defect in this study was identified through studies of platelet mitochondria. A similar finding was subsequently demonstrated in independent studies of brain, platelet, and fibroblast mitochondria (24, 31, 40, 62, 144, 165, 176, 189, 204, 205, 240, 280, 281, 284, 292). Some researchers attributed the COX activity reduction to declining COX protein or COX subunit mRNA levels (42, 43, 111, 145). Others reported the enzyme's kinetic properties, and therefore the enzyme structure itself, were altered or that the enzyme was improperly assembled (206, 284). For example, in AD brain mitochondria, COX activity was reduced even when referenced to its own aa3 subunit (206), thus suggesting reduced AD brain COX activity is not simply or solely a consequence of less COX holoenzyme.
Mitochondrial mass, number, and content were shown to be changed in oxidatively stressed hippocampal pyramidal neurons from AD brains (113). These neurons showed increased levels of mtDNA, mtDNA molecules containing a specific 5 kb deletion, and the mtDNA-encoded COX1 protein subunit. This was not due, however, to increased numbers of intact mitochondria or, at least in the case of mtDNA, increased amounts of mtDNA within intact mitochondria. Rather, increases were driven by an expanded pool of autophagocytized mitochondria. While this mitochondrial pool increased, the number and mass of normal mitochondria actually fell. Because the amount of PCR-amplifiable mtDNA also drops in AD brains (28, 55, 65, 113, 220), it is presumed mtDNA within autophagosomes is not extracted or amplified using standard techniques. Additional AD brain morphometric studies further found reduced mitochondrial number, mass, and size; disrupted cristae; and altered intracellular distributions (9). Moreover, these changes were not limited to cells and regions that manifest concomitant Aβ or protein aggregation changes.
Other studies quantifying AD brain mitochondrial mass and mitochondrial COX protein content emphasize that the stage of disease, or at least the stage of disease a given neuron is in, influences what is observed. Using immunochemical and mitochondrial dye approaches, de la Monte et al. noted that, despite an overall reduction in mitochondrial mass, especially in terms of COX content, considerable heterogeneity between individual brains and neurons within brains exist (65). The authors postulated less severely affected neurons mount ultimately unsustainable compensatory increases in COX protein and mitochondrial mass. This interpretation is consistent with the findings of Nagy et al., who found “healthy-appearing” neurons in AD brain hippocampi had increased COX protein levels, while tangle-bearing neurons lacked COX protein (190).
During the 1990s, mitochondrial defects were demonstrated in other neurodegenerative diseases, and it was proposed the brain might be particularly susceptible to mitochondrial dysfunction (259). Oxidative stress was documented in AD brains (138, 160, 170, 172, 199, 245, 246) and it was postulated that mitochondria, a well-recognized source of reactive oxygen species (ROS) production (39), could be responsible (11, 264). Mitochondria were discovered to play a central role in programmed cell death (115, 148, 216, 301), a process felt possibly to mediate neuron loss in AD (57, 65, 66, 151, 194, 222, 231, 244, 251). Age-dependent increases in mtDNA damage, either in the form of oxidative modifications or frank mutations, were documented (54, 179). The age-dependent nature of these changes offered a potential explanation for why AD prevalence increased so much with advancing age. Cytoplasmic hybrid (cybrid) modeling of AD mitochondrial dysfunction further suggested AD-specific mtDNA signatures existed and were physiologically relevant (257). AD cybrid studies are discussed in detail in a following section.
As interest in mitochondria grew, speculation arose over the possible presence of an Aβ-mitochondria nexus. Initial attempts to address this involved exposing cultured cells or isolated mitochondria to Aβ. These studies found Aβ impaired mitochondrial respiration and inhibited COX (30, 35, 61, 207, 210), thereby providing a potential mechanism through which AD brain mitochondrial dysfunction could arise. Subsequently, reduced brain respiration was demonstrated in multiple Aβ-producing transgenic mouse models (74, 76, 112, 298, 299). Related studies from transgenic mice reported expression of genes encoding mitochondrially-located proteins and respiratory chain subunits increased (218), and that brain regions that do not accumulate Aβ may actually show elevated COX activity (249). This suggested Aβ-induced mitochondrial dysfunction triggers a compensatory response. Consistent with this finding, Diana et al. reported that in cultured cells low concentration Aβ exposure decreases mitochondrial mass, fragments mtDNA, and initiates an apparent compensatory increase in mtDNA synthesis (73).
Adding Aβ to cell cultures, though, does not inhibit all bioenergetic pathways. Allaman et al. found exposing cultured astrocytes to Aβ increased glucose uptake as well as glycolysis, pentose phosphate shunt, Krebs cycle, and glycogen synthesis fluxes (4). These flux increases may reflect the fact that from a metabolism perspective the brain is not a homogeneous entity. Instead, astrocytes and neurons likely constitute functionally interactive units (3, 163, 209, 253).
In 2001, Cardoso et al. reported Aβ exposures that were toxic to human teratocarcinoma cells were not toxic to teratocarcinoma cells depleted of endogenous mtDNA (ρ0 cells) (33). Because mtDNA encodes subunits belonging to complexes I, III, IV, and V, these holoenzymes are not fully functional in ρ0 cells. With proper metabolic support, though, ρ0 cells remain viable and expand in culture. In the native teratocarcinoma cells, Aβ decreased MTT reduction, increased extracellular LDH, inhibited ETC enzymes, reduced ATP levels, and depolarized mitochondria. These effects were not observed in the Aβ-exposed ρ0 cells. In the native cells, antioxidant pretreatments minimized some of the Aβ-induced changes, including decreased MTT reduction. While this study did not explicitly demonstrate a physical interaction between Aβ and either mitochondria or the ETC, it showed in cell culture that some aspects of Aβ toxicity are mediated through direct or indirect mitochondrial effects. Following this, a number of studies reported APP and Aβ co-localize with mitochondria in both AD autopsy brains and AD transgenic mice (7, 8, 36, 61, 72, 74, 75, 106, 157, 162, 167, 296, 298). Interestingly, the APP-cleaving gamma secretase complex is also present within mitochondria (105, 271).
Over the last decade, investigators have reported several specific Aβ–mitochondria interactions. In studies involving AD autopsy brains and Aβ-producing transgenic mice, Aβ was found to bind a redox enzyme, Aβ-binding alcohol dehydrogenase (ABAD), within the mitochondrial matrix (162). Aβ-binding to ABAD deformed the enzyme, prevented NAD binding, and interfered with enzyme activity. Blocking the Aβ–ABAD interaction minimized cognitive decline in this model. In a subsequent publication, this team reported Aβ also interacted with cyclophilin D, a constituent of the mitochondrial permeability transition pore, and that binding of Aβ to cyclophilin D activated a mitochondrial permeability transition (75).
Despite the fact and perhaps because the number of intact mitochondria is reduced in degenerating neurons, in AD brains the balance between mitochondrial fission and fusion is shifted in favor of fission (168, 287 –289). The proteins that mediate mitochondrial fusion, Opa1, Mfn1, and Mfn2, are reduced. Fis1, which mediates mitochondrial fission, is increased. Drp1, which also facilitates fission, was reduced in one study but increased in another (168, 288). In the study where Drp1 was increased, Drp1 mRNA expression was similarly elevated (168). This study also reported a physical association between Aβ and Drp1. In the studies where Drp1 was reduced, exposing cultured neuronal cells or primary neurons to Aβ fragmented mitochondria and altered mitochondrial fission–fusion protein levels (288). The intracellular mitochondrial distribution shifted from neurites and axons and towards the perinuclear region. Overexpressing Opa1 and Drp1 mitigated Aβ-induced mitochondrial fragmentation and restored a normal mitochondrial distribution pattern.
Overall, multiple cell and animal-based studies show Aβ functionally and structurally alters mitochondria. While the sequence of this relationship is consistent with the amyloid cascade hypothesis, it is important to note the experimental models used for these studies (adding Aβ to cell cultures, and transgenic mice whose mutant APP expression facilitates Aβ production) do not address whether a converse relationship also exists.
This point is critical, as data suggest mitochondrial function affects Aβ, and mitochondrial dysfunction promotes amyloidosis. One study found exposing APP-expressing COS cells to a combination of sodium azide, a COX inhibitor, and 2-deoxyglucose, a glycolysis inhibitor, profoundly affected APP processing (93). This treatment, which presumably reduced ATP levels, shifted cell APP processing to the endoplasmic reticulum (ER) and yielded an “amyloidogenic” derivative. As defined by the authors, this new amyloidogenic derivative consisted of an 11.5 kD APP fragment that contained the entire Aβ amino acid sequence. The sodium azide-2 deoxyglucose treatment therefore appeared to shift APP processing away from its α-secretase cleavage. Similar effects were seen in neuroglioma cells and following treatment with CCCP, a respiratory chain uncoupler.
Other studies have focused on whether bioenergetic manipulation affects APP α-secretase cleavage. One report tested the effect of 2-deoxyglucose and oligomycin, an ATP synthase inhibitor, on differentiated PC12 cell (a rat adrenal pheochromocytoma cell line) APP processing (290). This intervention decreased α-secretase-mediated APP cleavage. The authors speculated reducing α-secretase activity might shift APP processing towards the amyloidogenic direction. Another study performed using COS cells found glucose deprivation, 2-deoxyglucose, and sodium azide independently decreased levels of the soluble α-secretase-derived APP product (94). APP mRNA levels remained unchanged, so diminished amounts of the α-secretase-produced APP product was probably not an artifact of reduced APP. Also, glutathione restored the α-secretase-generated product to normal and even supranormal levels suggesting ROS, conditions that occur in conjunction with ROS, or some other glutathione-sensitive parameter shifts APP processing from the α-secretase cut. In this context, data that show ROS activates the APP β-secretase (BACE) and increases Aβ production are potentially relevant (47, 268, 269).
Fukui et al. evaluated how bioenergetic manipulation affects mouse brain amyloidosis (91). The authors crossed transgenic mice expressing mutated human APP and PSEN1 genes with mice that had a Cre–loxP-mediated knockout of the neuron COX10 gene. COX10 synthesizes a heme component that constitutes part of the COX holoenzyme. The brains of the crossed mice showed reduced numbers of amyloid plaques. Oxidative stress, BACE activity, and Aβ42 levels were also lower. It is unclear why COX10 knockout reduced brain oxidative stress and COX1 levels, since these changes are opposite to what is observed in the AD brain (113). It is perhaps conceivable COX10 knockout lowered levels of an otherwise normal COX holoenzyme, lowered COX holoenzyme levels reduced the overall respiratory flux, a reduced respiratory flux lowered free radical production, lowered free radical production deactivated BACE, and less Aβ was produced. If correct, the Fukui et al. report could join other APP transgenic mouse studies that found manipulating oxidative stress alters brain Aβ dynamics or plaque accumulation. Pertinent studies include those showing paraquat-induced oxidative stress increased brain Aβ levels, increasing periredoxin 3 decreased brain Aβ levels, increasing MnSOD decreased plaque accumulation, and reducing MnSOD increased plaque accumulation (47, 78, 155).
Khan et al. determined Aβ levels in SH-SY5Y cybrid cell lines expressing AD and control subject mtDNA (141). AD cybrid lines had reduced COX activity and increased ROS production. Intracellular and extracellular levels of the 40 and 42 amino acid Aβ species were elevated in AD cybrid cell cultures. These and other data that argue mitochondria play a key role in AD are discussed below.
Could Mitochondria Cause AD?
The first demonstration of reduced AD subject COX activity came from studies of platelet mitochondria (203). Although it is currently recognized that systemic biochemical and clinical changes associate with AD, and therefore AD may not be a brain-limited disease (22, 260), in 1990 this was not a generally acknowledged view. Why, then, did Parker et al. decide to assay ETC activities in platelet mitochondria? In this case, the decision to assay mitochondria from a nondegenerating tissue was based on epidemiologic and mitochondrial genetic principles. Specifically, the authors recognized that most AD cases, and especially late-onset AD cases, typically do not demonstrate recognizable Mendelian inheritance patterns. Parker, who was at that time developing a genetic theory of sporadic disease that was based on mitochondrial DNA (mtDNA) inheritance (201, 202), postulated mtDNA might contribute to the apparently sporadic late-onset form of AD. Immediately prior to his AD platelet study, Parker and colleagues had successfully shown that subjects with Parkinson's disease, another neurodegenerative disorder in which only a minority demonstrate Mendelian inheritance, have reduced platelet and muscle mitochondria complex I activity (17, 202). If his sporadic disease hypothesis was correct, Parker predicted, a systemic ETC defect should also exist in AD.
Parker's sporadic disease hypothesis was based on several mtDNA principles including maternal inheritance, heteroplasmy, threshold, and mitotic segregation (Fig. 2). Because mtDNA is essentially all inherited maternally (98), inherited mtDNA mutations cannot produce Mendelian disease patterns. Maternal inheritance might arise from inherited mtDNA mutations, but only in situations where the mutations are present in adequate amounts in both the germ line and somatic tissues. Due to heteroplasmy, threshold, and mitotic segregation, these conditions might not be met. For example, a heteroplasmic mutation might only be present in a limited number of the mtDNA molecules of an ovum. Because during mitosis mtDNA molecules are stochastically distributed to daughter cells, different tissues may end up with different mtDNA mutation burdens. A relatively high level of mutation, therefore, may end up in the nervous system but not the germ line, in which case a carrier female's chances of developing a neurologic disease would exceed her chance of transmitting a neurologic disease. Alternatively, if a relatively high level of mutation ends up in the germ line but not the nervous system, a carrier female's chances of transmitting a neurologic disease to her offspring would exceed her chances of developing the disease. This latter scenario takes into account the concept of threshold, which assumes that in order to cause disease, the burden of mtDNA mutation in an affected tissue must exceed the threshold at which the mutation or mutations produce biochemical consequences.

Next, the question of whether mtDNA contributes to low platelet mitochondria COX activity in AD subjects needed to be addressed. COX is a 13-subunit holoenzyme, and three of its subunits are encoded by genes on mtDNA. The transcription and translation of these mtDNA subunits also depends on the integrity of a set of mtDNA rRNA and tRNA genes, as well as a regulatory region that influences mtDNA replication and expression. To evaluate mtDNA's contribution to the AD platelet mitochondria COX defect, a cybrid approach was used.
AD Cybrids
Cybrid cells are generated by mixing the contents of non-nucleated cells, or cytoplasts, with nucleated cells (257) (Fig. 3). This approach was developed during the 1970s to explore the functional consequences of unique mtDNA species (29, 286). In its earliest permutation, investigators created cybrid cell lines with mixed mtDNA populations; the mtDNA in these lines consisted of a nucleated cell's endogenous mtDNA plus the cytoplast's mtDNA. In the 1980s, investigators discovered how to fully deplete cell lines of endogenous mtDNA (70, 71, 181). These lines were called ρ0 cell lines, since prior to its identification as mtDNA cytoplasmic DNA was classified as “ρ” DNA (83). As ρ0 lines became available, investigators began using them to produce cybrid lines whose mtDNA derived entirely from a cytoplast mitochondrial donor (143). In 1994 it was shown that platelets, which contain mitochondria but not nuclei, function effectively as cytoplasts when mixed with ρ0 cells to produce cybrid cells (49).
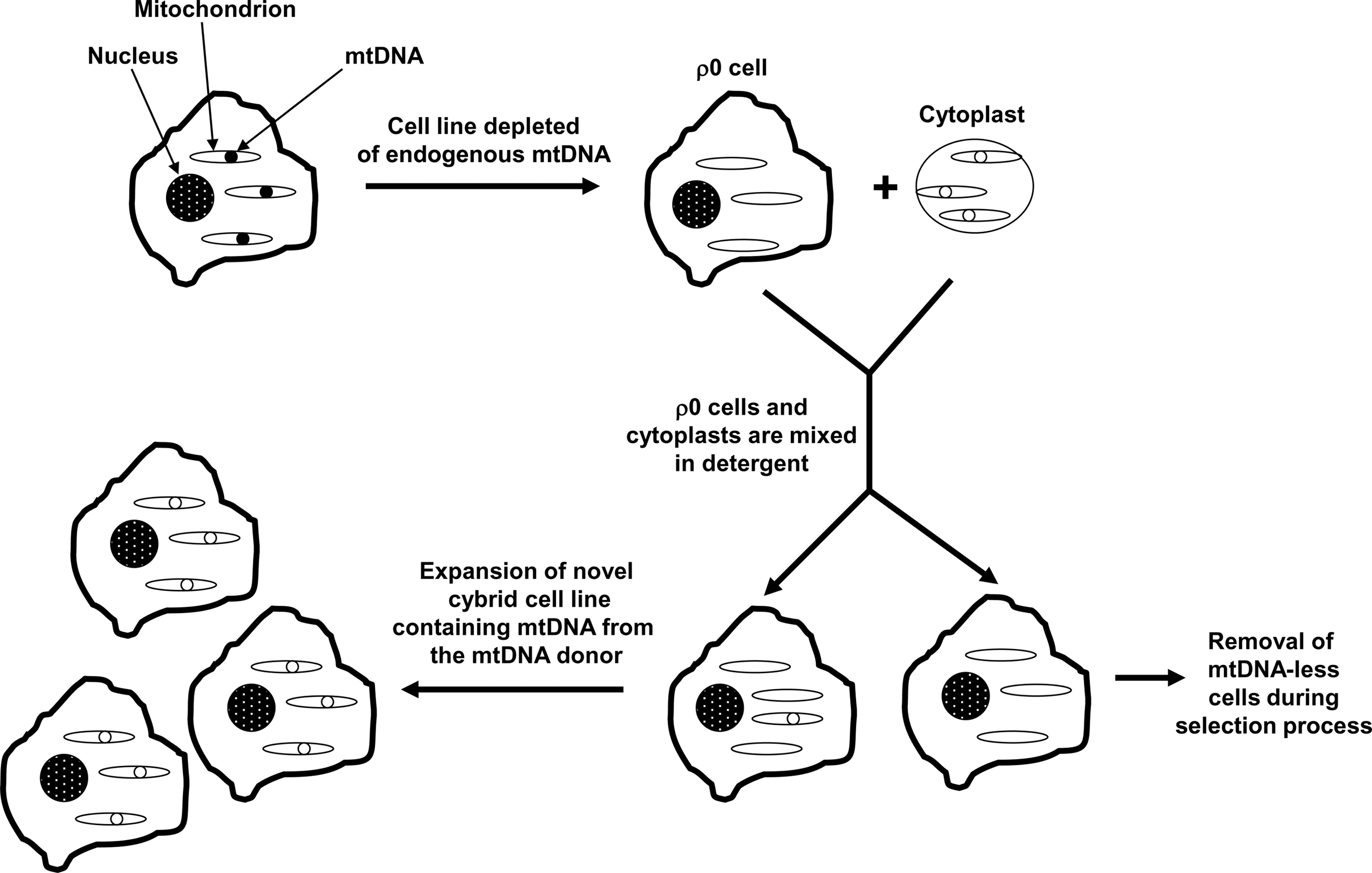
The first studies of cybrid cell lines containing AD subject mtDNA were published in 1997 (64, 233, 264). These cell lines are referred to as “AD cybrid lines” or simply as “AD cybrids”. To produce the AD cybrids, blood samples were taken from subjects diagnosed with AD. Platelets were isolated from each individual blood sample, and the platelet mitochondria from each platelet preparation were incorporated into ρ0 cells derived from either human neuroblastoma SH-SY5Y cells or teratocarcinoma NT2 cells. At the same time that the AD cybrids were generated, platelet mitochondria obtained from non-AD, age-matched control subjects were also used to generate “control cybrids”. In both the neuroblastoma and teratocarcinoma-based studies, mean COX Vmax activities were lower in the AD cybrid group than they were in the corresponding control cybrid group (64, 233, 264). Reduced COX activity was subsequently observed in several independent neuroblastoma and teratocarcinoma AD cybrid series (32, 37, 95, 195 –197, 276, 277). Across studies the magnitude of this reduction has ranged from about 15% to 50%. The defect is greatest when assayed in enriched mitochondrial fractions, and least when studied in whole cells. This bioenergetic lesion, either directly or indirectly, is further associated with a reduced cell ATP level; in the NT2 AD cybrid series of Cardoso et al., the ATP level was lowered by 28% (32). This effect is presumed to be COX mediated, since complex I activities between groups, when analyzed, have been comparable (64, 95, 233, 277).
It is important to note that cybrid cell lines are created not through the transfer of isolated mtDNA, but rather through the transfer of whole mitochondria to ρ0 cells. An assumption is made that all transferred cytoplast components that cannot perpetuate independently of the host cell nucleus degrade over time and dilute over the course of repeated cell divisions. Therefore, any transferred cytoplast component that cannot perpetuate itself independent of the cell nucleus should not have a sustainable molecular or biochemical effect. Theoretically, a cytoplast's only self-perpetuating component is its mtDNA. For this reason, the most straightforward explanation of these cybrid studies is that the AD cybrid COX activity reduction is determined by mtDNA.
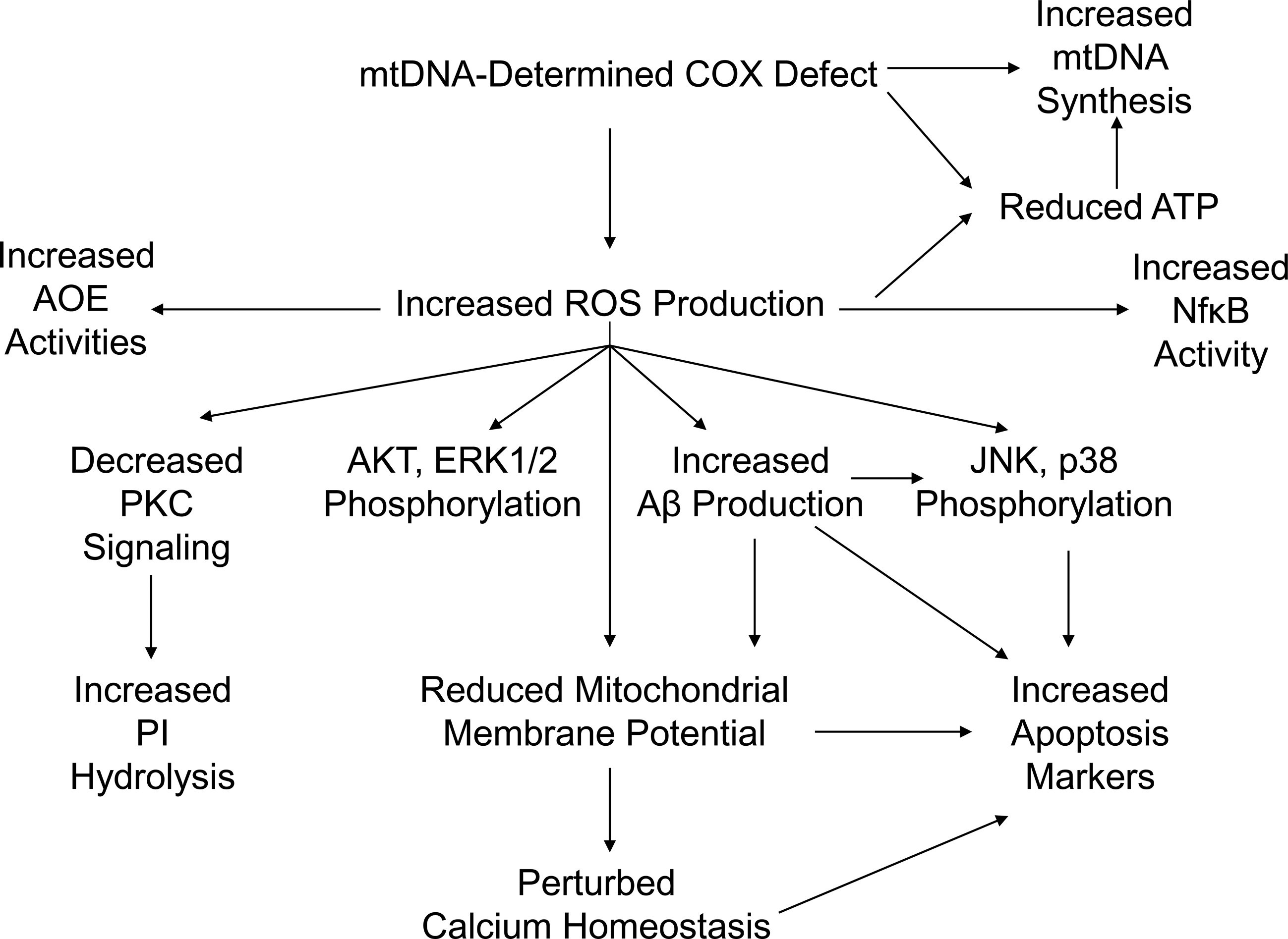
In AD cybrids, the mtDNA-derived COX defect produces other features that recapitulate phenomena observed in AD subject brains (Fig. 4). Altered mitochondrial morphology, membrane potential, free radical production, calcium handling, movement, and mtDNA synthesis rates are observed. Cell transcription factor activities, calcium homeostasis, intracellular signaling, energy levels, apoptosis markers, and APP processing are perturbed.

Electron microscopy (EM) reveals AD cybrids have increased numbers of enlarged mitochondria with swollen pale matrices; disrupted, reduced cristae; broken outer membranes; and crystal-like inclusions (276, 277, 302). Despite this, relative to control cybrids, the AD cybrid average mitochondrial size is reduced. The overall cell mitochondrial mass is equivalent between AD and control lines, though, because AD cybrid cells appear to contain more mitochondria (276). Preserved mitochondrial mass has been visually demonstrated through EM, and by biochemical assays that find AD cybrids maintain normal citrate synthase and complex I activities (31, 64, 95, 233, 276, 277). Mitochondrial degradation rates in AD cybrids have not been directly characterized, but mitochondrial mtDNA synthesis is increased in AD cybrids and may represent part of a compensatory response to mitochondrial dysfunction (276). Some of these findings reflect changes observed in human AD brains, which show an increased frequency of disrupted mitochondria and generally smaller mitochondria that potentially reflect a shift in the mitochondrial fission–fusion balance towards fission (9, 113, 168, 288). While preservation of mitochondrial mass is not typical of the AD brain, several caveats are worth noting. First, Hirai et al. found that when cell mtDNA is quantified so that intact mitochondria, disrupted mitochondria, and phagosome mtDNA are accounted for, the AD neuron total mtDNA content is actually elevated (113). Second, total cell COX protein is increased in what are apparently healthier AD brain neurons (65, 113, 190). Third, during early AD stages, COX subunit gene expression is increased while complex I subunit gene expression is not (169). Fourth, early in life AD transgenic (tg) 2576 mice show upregulated expression of genes that encode mitochondria-localized proteins (218). Therefore, by increasing their mtDNA synthesis rate and maintaining their mitochondrial mass, AD cybrids are possibly demonstrating a compensatory response that seems to initially occur (but ultimately fail) in AD brain neurons.
Compared to differentiated control cybrids, differentiated SH-SY5Y AD cybrids have a higher percentage of sedentary mitochondria (275). The mitochondria that are moving have a slower mean velocity. Direct studies of AD brain also suggest mitochondrial transport abnormalities occur, and that mitochondria are directed away from axons and neurites (254, 288).
Past fibroblast and lymphocyte studies demonstrate that calcium dynamics are altered in AD (96, 124, 150, 214, 215). The ability of mitochondria to regulate calcium levels is also perturbed in AD cybrids. The total amount of calcium that mitochondria can store, as well as the rate at which they can store it, is reduced (233). This may partly explain why AD cybrids have increased cytosolic calcium levels under basal conditions (233), and why AD cybrid mitochondria, when subjected to cyclosporin A-mediated hyperpolarization, fail to show the typical membrane potential oscillation response (i.e.,“flickering”) that occurs in cyclosporin-treated normal cells (272). Reduced mitochondrial calcium sequestration is also consistent with a relative depolarization of the mitochondrial membrane potential, a phenomenon that has been shown in several AD cybrid studies (37, 141, 276, 277).
Under physiologic conditions, mitochondrial superoxide production is associated with mitochondrial membrane potential hyperpolarization (26). AD cybrid mitochondria, which are relatively depolarized, also generate excess superoxide (16, 32, 68, 195, 233, 264, 302). Mitochondrial ROS overproduction in the setting of a low membrane potential suggests that AD cybrids have a fundamentally defective ETC. Additional evidence of AD cybrid oxidative stress includes increased markers of lipid peroxidation, including thiobarbituric acid reactive substances (TBARS) and 4-hydroxynonenol (4-HNE) protein adducts, and increased protein carbonylation (32, 196). Similar changes are also observed in AD brains (139). AD cybrids also show upregulated activities of several antioxidant enzymes (AOEs), including catalase, copper-zinc SOD, MnSOD, glutathione peroxidase, and glutathione reductase (264). This most likely represents a compensatory, yet ultimately unsuccessful, response to enhanced mitochondrial ROS production. Increased oxidative stress is of course a well-documented feature of the AD brain (113, 193, 213, 245, 250).
In AD cybrids, oxidative stress mediates, at least in part, the observed mitochondrial membrane potential depolarization. Administering trolox, a water-soluble vitamin E analog and antioxidant, to AD cybrids increased their mitochondrial membrane potential (141). Other cybrid data provide potential mechanistic insight into how oxidative stress affects the membrane potential, and also influences other phenomena typical of the AD brain. One particularly interesting finding is that AD cybrids overproduce both Aβ40 and Aβ42. Khan et al. reported a series of SH-SY5Y AD cybrids secreted Aβ40 and Aβ42 at approximately twice the rate of a control cybrid group (141). This study also found AD cybrids contained more intracellular Aβ40 than the control cybrids; intracellular Aβ42 levels trended towards an increase but this difference was not significant. Subsequently, Onyango et al. reported that, compared to SH-SY5Y control cybrids, AD cybrid intracellular and conditioned medium Aβ42 levels were increased (195). Since oxidative stress is believed to activate BACE, the enzyme that processes APP to its Aβ derivative (47, 268), an ROS-mediated increase in AD cybrid Aβ production might be expected. Increased basal Aβ levels may also explain why exposing AD cybrids to exogenous Aβ40 causes an exaggerated decline in their mitochondrial membrane potential (32), and why exposing them to Aβ25–35 exacerbates their already impaired ability to handle an inositol triphosphate (IP3)-mediated ER calcium release (233).
AD cybrids show profound alterations in intracellular stress signaling pathways. Relative to control cybrid lines AKT phosphorylation is increased, as is phosphorylation of the p38, JNK, and ERK1/2 serine-threonine kinases (195, 197, 302). The status of AKT in the AD brain is unclear, as its intracellular distribution and absolute amount seem to change, but available data suggest the AKTser-473 phosphorylated to nonphosphorylated ratio increases (101, 159, 208, 219). ERK, p38, and JNK phosphorylation is also increased in the AD brain (101, 304, 305). AD cybrid studies suggest these activating phosphorylations arise as a consequence of increased oxidative stress, since they are reduced by the isoflavone antioxidant puerarin and other antioxidants (195, 302). Further, in AD cybrids, oxidative stress-mediated p38 and JNK activation may confer harmful consequences, as p38 and JNK inhibition protects these cells from externally-induced oxidative stress (192, 196). AKT pathway and ERK1/2 activation, on the other hand, appear to play a protective role as the specific inhibition of either AKT or ERK1/2 reduces AD cybrid cell viability (196, 197, 228). These viability-reducing and promoting associations are consistent with existing mitogen activated protein kinase (MAPK) data (192, 228). Tyrosine kinase activity, which is also activated by ROS (133), is additionally increased in AD cybrids (195, 197).
AD cybrids manifest evidence of decreased neurotrophin signaling. TrkA and p75NTR receptors are depressed, and the activity of the remaining TrkA receptors is reduced (195). Despite this, exposing AD cybrids to nerve growth factor (NGF) normalizes their elevated cytosolic calcium level and improves their ability to recover from an IP3-mediated ER calcium release. This has led some to argue NGF treatment may benefit AD patients (234). TrkA levels are also decreased in the basal forebrain and cortex of AD brains (23, 60, 226).
The phosphoinositide (PI) hydrolysis-dependent signal transduction system is perturbed in both AD brain (129) and AD cybrids (68). PI signaling occurs when phospholipase C (PLC) cleaves a phosphorylated inositol (an inositol phosphate such as IP3) from a membrane phosphatidylinositol phospholipid; phosphatidylinositol phospholipids are collectively called PIs. De Sarno et al. assessed the integrity of this system in AD and control cybrid lines under basal conditions and following exposure to carbachol, a cholinergic agonist that binds M1 metabotropic receptors and activates PI hydrolysis (68). Although G-protein and PLC levels were equivalent between groups, in AD cybrids basal and carbachol-stimulated PI hydrolysis rates were higher, the G-protein activator sodium fluoride had an overly robust effect, as did the PLC activator ionomycin, and glutathione depletion had a blunted effect. These findings suggest AD cybrids have an overactive PI hydrolysis system. Since acute oxidative stress typically reduces carbachol-induced PI hydrolysis (156), and AD cybrids have elevated PI hydrolysis and chronic oxidative stress, it was postulated PI hydrolysis hyperactivity in these cells represents a direct or indirect adaptive response to chronic oxidative stress.
In addition to producing a phosphorylated inositol molecule, PI hydrolysis by PLC also generates diacylglycerol (DAG). DAG activates protein kinase C (PKC), which promotes AP-1 transcription factor DNA binding; AP-1 regulates aspects of cell differentiation, proliferation, and apoptosis. Although PKC levels are equivalent between groups, less AP-1 DNA binding occurs in AD cybrids treated with carbachol or phorbol 12-myristate 13-acetate (PMA), a DAG substitute that directly activates PKC (68). This may also represent a consequence of oxidative stress, since oxidative stress reduces carbachol-induced AP-1 DNA binding (156). In any case, in AD cybrids, oxidative stress appears to uncouple PKC signaling from PI hydrolysis. It is therefore possible that PI hydrolysis hyperactivity represents an attempt to compensate for this uncoupling. PKC function is reduced and its physical status is altered in the AD brain (51, 175, 236).
In addition to perturbed AP-1 DNA binding, AD cybrids also show altered function of other transcription factors. Activity of the oxidative stress-sensitive NFκB transcription factor (127, 133, 229, 230) is likely increased. This is supported by the finding that phosphorylation of its inhibitor, IκBα, is elevated; IκBα.phosphorylation targets it for proteosomal degradation, which frees NFκB and allows it to promote the expression of generally pro-inflammatory genes (196). NFκB activity is also increased in AD patient brain neurons and astrocytes (132). In AD cybrids, NFκB activation likely plays a protective role, since NFκB inhibition reduces cell viability (195 –197). In another study, HSF-1 DNA binding was found to be reduced in AD cybrids (16). It was further shown that in glutathione-depleted cybrid cell lines, hydrogen peroxide exposure induced a less robust HSF-1 activation in AD cybrid lines than it did in control cybrid lines (16). Because acute hydrogen peroxide exposures typically induce HSF-1 DNA binding, these data suggest reduced basal and peroxide-stimulated HSF-1 DNA binding in AD cybrids reflects a compensatory response to chronic oxidative stress.
A final downstream consequence of the AD cybrid COX defect is a reduction in “viability” markers and an increase in apoptosis markers. Relative to control cybrids, AD cybrids show less XTT and MTT reduction, a phenomenon that is partly reversed by antioxidant treatment (195 –197, 302). AD cybrid cultures have increased numbers of cells with condensed nuclei, annexin V positive-propidium iodide negative staining, and annexin V positive-propidium iodide positive staining (195 –197, 302). AD cybrids show increased PARP cleavage and increased H2A.X phosphorylation; reducing Aβ production through gamma secretase inhibition lowers levels of these apoptosis markers (195 –197). The BAX/Bcl2 ratio is increased due to higher BAX and lower Bcl2 levels, while treating AD cybrids with the antioxidant puerarin reduces BAX and increases Bcl2 (302). BAX overexpression in AD neurons has been reported, while Bcl2 levels seem to fluctuate depending on the overall health of the neuron being analyzed (252, 273). AD cybrids have reduced mitochondrial cytochrome c levels, elevated cytosolic cytochrome c levels, and excess caspase 3 activity (32, 141, 302). The greater frequency of swollen mitochondria observed in AD cybrids may also reflect increased apoptosis-related activity, since mitochondrial swelling is associated with mitochondrial permeability transition and permeability transition occurs during apoptosis (114, 276 –278, 302).
It is important to note there is one published AD cybrid report in which AD cybrids prepared on a HeLa cervical carcinoma nuclear background were studied (125). In this report, four AD cybrid lines were prepared from platelet mitochondria, three control cybrid lines were prepared from platelet mitochondria, and two control cybrid lines were prepared from fibroblast mitochondria. Only semi-quantitative data in the form of bar graphs are provided, but the bar graphs do indicate COX activity in each cybrid line is comparable to or at least not dramatically different from the COX activity of the native HeLa cell line. Also in this study (125), HeLa ρ0 cells were mixed with synaptosomes prepared from a single AD autopsy brain following a 20 h postmortem interval. After this mixing, three cell colonies that contained mtDNA were later identified and the COX activity from each colony was determined. The data from this experiment, which also are presented only as part of a semi-quantitative bar graph, show the COX activity from these three colonies resembled the COX activity of the native HeLa line. The authors concluded that mtDNA is equivalent between AD and control subjects and that AD and control cybrid lines are functionally equivalent. Because the methods used in this negative AD cybrid study dramatically differ from those of the 17 published positive AD cybrid studies, the degree to which this single study contradicts the positive studies is unclear. The 17 positive studies used SH-SY5Y and NT2 neuronal cell nuclear backgrounds, and in aggregate the positive studies have now assayed over 200 cybrid lines prepared on three continents. To date, the positive studies have compared over 100 AD cybrid lines to over 100 control cybrid lines. Findings from the 18 reported AD cybrid studies are summarized in Table 1.
Three articles by Onyango et al. all report reduced COX activity, but are only counted once since the same cell lines are used in each paper. Similarly, the study of Trimmer and Borland includes a subset of previously reported cell lines, and so is not independently counted. AD, Alzheimer’s disease.
Where Are the mtDNA Mutations?
Cybrid data imply mtDNA can account, at least in part, for low COX activity in AD subjects. They further imply that by influencing ETC function, mtDNA can induce important AD-associated biochemical and molecular phenomena.
While cybrid data do not identify the critical mtDNA feature or features, several inferences are possible. Because complex I activity is not reduced, at least in AD subject platelet mitochondria wholesale mtDNA degradation is unlikely. Similarly, preserved complex I activity as well as preserved mtDNA levels indicate low COX activity in AD cybrids occurs independent of reduced mtDNA levels.
Investigators have argued that somatic acquired mtDNA mutations may contribute to AD (285). An attractive feature of this hypothesis is it provides a mechanism that could account for the striking correlation between AD incidence and advancing age (130). Mitochondrial DNA mutations certainly do accumulate with age, and in animal models somatic mtDNA mutations can drive an aging phenotype (149, 274). Substantial evidence that somatic mtDNA mutations accumulate in AD subjects already exists. Several groups report levels of the 4977 base pair “common” mtDNA deletion, which increases with age in postmitotic tissues such as the brain, is further elevated in the AD brain (54, 104, 113). This particular deletion encompasses mtDNA-encoded COX genes, and so when present in high enough amounts reduced COX activity should result (58, 59). Mitochondrial DNA deletions such as the common deletion, though, are not known to accumulate in blood (18) which suggests it is unlikely to account for findings from AD cybrids.
Lin et al. systematically catalogued low abundance heteroplasmic point mutations in brain mtDNA from young control subjects, aged subjects without dementia, and AD subjects (158). Correlations between subject age, mutation burdens, and brain COX activity were observed. Specifically, the number of low-abundance mutations found in the mtDNA COX1 subunit gene increased with advancing age, and a greater number of mtDNA mutations corresponded with a lower COX Vmax activity. However, between the AD and age-matched control groups, the absolute number of detected COX1 mutations was similar. To date, no study has reliably catalogued low abundance heteroplasmies or compound heteroplasmies in the other two mtDNA-located COX genes (COX2 and COX3).
Other studies, though, have argued that heteroplasmic point mutations are more common in AD. In AD brain but not AD lymphocytes, Chang et al. found a three-fold elevation of a particular mtDNA displacement (D) loop C to T transition (45). The authors felt the nature of the sequence change could represent a consequence of increased AD brain oxidative stress, a view compatible with several studies finding excessive levels of mtDNA oxidative adducts in the AD brain (65, 113, 170, 179). Coskun et al. also reported the frequency of several specific D-loop mutations differed dramatically between AD and control subject brains (55). This group subsequently found that compared to controls, D-loop mutations were also much more frequent in AD subject serum and lymphoblastoid cell line mtDNA (56).
Investigators have also argued inheritance of particular mtDNA sequence variations influences AD risk. One of the earliest deviations to be reported was an A4336G transition in the mtDNA tRNAGln gene (81, 122, 227, 237), although several other studies found no association (48, 80, 221, 294, 306). This transition is characteristic of particular mtDNA haplogroup H subgroups (164, 227). To date, some studies have found haplogroup H is relatively over-represented in AD cohorts (86, 173, 174, 227). As a potentially related finding, haplogroup H may represent a particularly well-coupled haplogroup, which infers that individuals with haplogroup H may produce more ROS than individuals with other haplogroups (171). Other studies, though, have reported associations between AD and less-coupled mtDNA haplogroups (34, 86, 123, 152, 266, 283). Associations between AD and particular rare polymorphisms or polymorphism combinations may also exist (41, 270).
Sequencing of AD subject mtDNA thus far has not revealed any particular high-abundance “smoking gun” mutation that “causes” this disease. This makes sense, since in some demographics AD is an incredibly common disorder. Almost half of those over 85 qualify for a diagnosis of AD or its frequent prodrome, mild cognitive impairment (MCI), as do more than half of those beyond the age of 90 (85, 295). Because AD prevalence is so high, any particular causal variation would probably not qualify as a mutation. This has led some to further consider whether mtDNA and also nuclear COX gene polymorphisms may account for some degree of AD risk.
To assess population variation in COX genes, Lu et al. sequenced 13 COX subunit genes, the three mtDNA COX subunit genes, and ten nuclear COX subunit genes, from 50 nondemented individuals (161). Approximately 20% of individuals carried a nonsynonymous mtDNA COX gene polymorphism. Synonymous mtDNA COX gene polymorphisms were even more frequent. Interestingly, the synonymous polymorphisms were not evenly distributed, but rather were clustered in the less conserved COX3 gene. This suggests that even synonymous polymorphisms could have a functional consequence, a possibility consistent with existing data that indicate synonymous polymorphisms, by changing the rate of protein translation, can alter protein folding and therefore protein function (142, 146, 147). Aside from a common single nucleotide polymorphism (SNP) in the nuclear COX4I1 gene nonsynonymous nuclear changes were rare, but synonymous polymorphisms and especially 5` and 3` untranslated region (UTR) polymorphisms were extremely common. The nonsynonymous COX4I1 SNP and a frequently detected hexanucleotide deletion in the COX7A1 5` UTR were both found to have functional consequences. Expression of the COX4I1 SNP was associated with a lower COX Vmax activity, and in a reporter assay the COX7A1 deletion reduced COX7A1 expression. Underscoring the tremendous degree of genetic variation that was found, when synonymous and UTR polymorphisms were taken into account, no two individuals shared an identical COX holoenzyme genotype.
A considerable degree of inherited and acquired inter-individual COX subunit gene variation, and especially mtDNA COX subunit gene variation, has thus already been demonstrated. Some association studies further report potential differences between AD and non-AD cohorts. In these positive studies, odds ratios are similar to or even exceed those of other genes associated with AD through large nuclear gene genome wide association studies (110, 131, 153, 232). Therefore, instead of asking “where are the mutations”, a more reasonable question is “how much do demonstrable mtDNA sequence variations influence AD risk?”
Data from AD endophenotype studies to some extent indirectly address this question. When a particular trait or biomarker typically found in conjunction with a disease is detected in persons who do not have that disease, the presence of that trait or biomarker is said to constitute an endophenotype. An endophenotype state does not indicate a carrier will develop the full-blown disease, although it infers that compared to persons without the endophenotype, those with the endophenotype carry an increased risk. To date, a number of studies, several of which are neuroimaging-based, have found the adult children of AD-affected mothers are more likely to express AD endophenotypes than the adult children of AD-affected fathers. The first AD endophenotype study, which analyzed fluorodeoxyglucose positron emission tomography (FDG-PET) scans, found the cerebral metabolic rate of glucose (CMRglu) of subjects with AD mothers showed AD-characteristic changes while subjects with AD fathers did not (184). Over time, CMRglu decline rates are greater in subjects with AD mothers (187). Subjects with AD mothers have more atrophy in AD-affected brain regions, as well as faster rates of atrophy progression (15, 118, 119). Pittsburgh compound B (PIB) PET reveals a greater degree of Aβ plaque deposition in those with AD mothers (186, 188). Cerebrospinal fluid (CSF) analysis reveals a lower CSF Aβ42/Aβ40 ratio and CSF isoprostanes, a marker of oxidative stress, are higher (186). On memory tests, APOE4 carriers with AD mothers do not perform as well as APOE4 carriers with AD fathers (69). Platelet mitochondria COX activity is lower in those with AD mothers than it is in those with AD fathers (185).
Cybrid, endophenotype, and positive mtDNA–AD association studies are consistent with epidemiology data that suggest an AD maternal inheritance bias does exist (10, 77, 79, 183). One such study, which took into account that women outlive men and could therefore have a higher lifetime risk of dementia, found greater female longevity did not account for this relationship (79). It therefore seems that although both parents influence an individual's AD risk, mothers have a greater impact.
Synthesizing the Data: The Mitochondrial Cascade Hypothesis
Various explanations for reduced AD subject COX activity have been proposed (Table 2). Some, such as the possibility that reduced AD brain synapse activity induces COX downregulation (293), could account for low brain COX activity but do not explain reduced platelet, fibroblast, or cybrid COX activity.
In general, AD cybrid studies suggest mtDNA gives rise to a COX defect, the COX defect causes oxidative stress and reduced ATP levels, and this produces multiple other AD-typical phenomena, including increased Aβ production (Fig. 5). All these features, in turn, influence the overall health of the cell. Because direct correlations between AD cybrid cell line defects and specific mtDNA sequence features are not yet established, this interpretation of the cybrid data assumes that only differences in mtDNA can explain persistent specific differences between individual cybrid cell lines. At this time, no alternative mechanism that causes sustained, inter-cell line biochemical differences has been demonstrated.
An individual's AD risk is influenced by whether they have an affected parent or parents (137). Although nongenetic factors can and probably do affect risk (103, 198), this strongly implies a genetic contribution. Very rare autosomal dominant forms of AD are recognized (100, 154, 235), but persons with autosomal dominant, familial AD (FAD) tend to present at younger ages than those with sporadic AD; most FAD patients are symptomatic prior to 60 years of age, while most sporadic AD patients develop symptoms after the age of 60 (256). Regarding sporadic AD, which accounts for over 99% of cases, several nuclear DNA nondeterministic polymorphisms are currently believed to influence AD risk. The most strongly associated and extensively studied risk factor gene is APOE on chromosome 19q13.2 (52, 211). AD risk is increased in APOE4, reduced in APOE2, and intermediate in APOE3 carriers. How and why apolipoprotein E, the protein encoded by the APOE gene, modifies AD risk is unknown. One hypothesis is that apolipoprotein E degradation products may directly interfere with mitochondrial function (44, 46, 191). Also, one study reported that between APOE4 carriers and noncarriers, posterior cingulate cortex COX activity was lower in the APOE4 carriers (282).
Interestingly, polymorphic variations in a neighboring gene, TOMM40 (translocase of the outer mitochondrial membrane 40 kDa subunit homolog), were recently shown to track with AD risk (12, 13, 217, 224, 267, 300). Recent studies suggest particular TOMM40 polymorphisms may affect the age of AD onset more stringently than APOE polymorphisms, leading some to speculate this mitochondrial protein is potentially more relevant to AD than apolipoprotein E (223). Several studies have also associated variation in the TFAM gene with AD risk (5, 14, 102, 303). The TFAM gene encodes transcription factor A of the mitochondria, which plays a major role in mtDNA replication and expression (50, 82, 134, 200).
The possibility therefore exists that nuclear genes directly or indirectly related to mitochondrial function mediate a large proportion of a person's nuclear DNA-determined AD risk. As discussed in the previous section, maternal inheritance bias and maternally-defined endophenotypes also suggest mtDNA inheritance modifies risk. If these observations and interpretations are correct, then the inheritance of mitochondrial genes, in conjunction with the inheritance of nuclear genes that specifically influence baseline mitochondrial function and durability, could have a profound impact on whether and when an aging individual develops AD.
Somatic mtDNA mutations have also been shown to accumulate in both the aging and AD brain (53, 54, 158). These mutations appear to influence mitochondrial function (158). The burden of somatic mutation required to affect function is not entirely clear and may depend on multiple factors, such as the type of mutations accumulated, their location on the mitochondrial genome, and the functional baseline upon which they are superimposed (Fig. 6). An important possibility to consider further is that an mtDNA molecule's primary sequence may actually influence the rate at which somatic changes accumulate (117, 279).
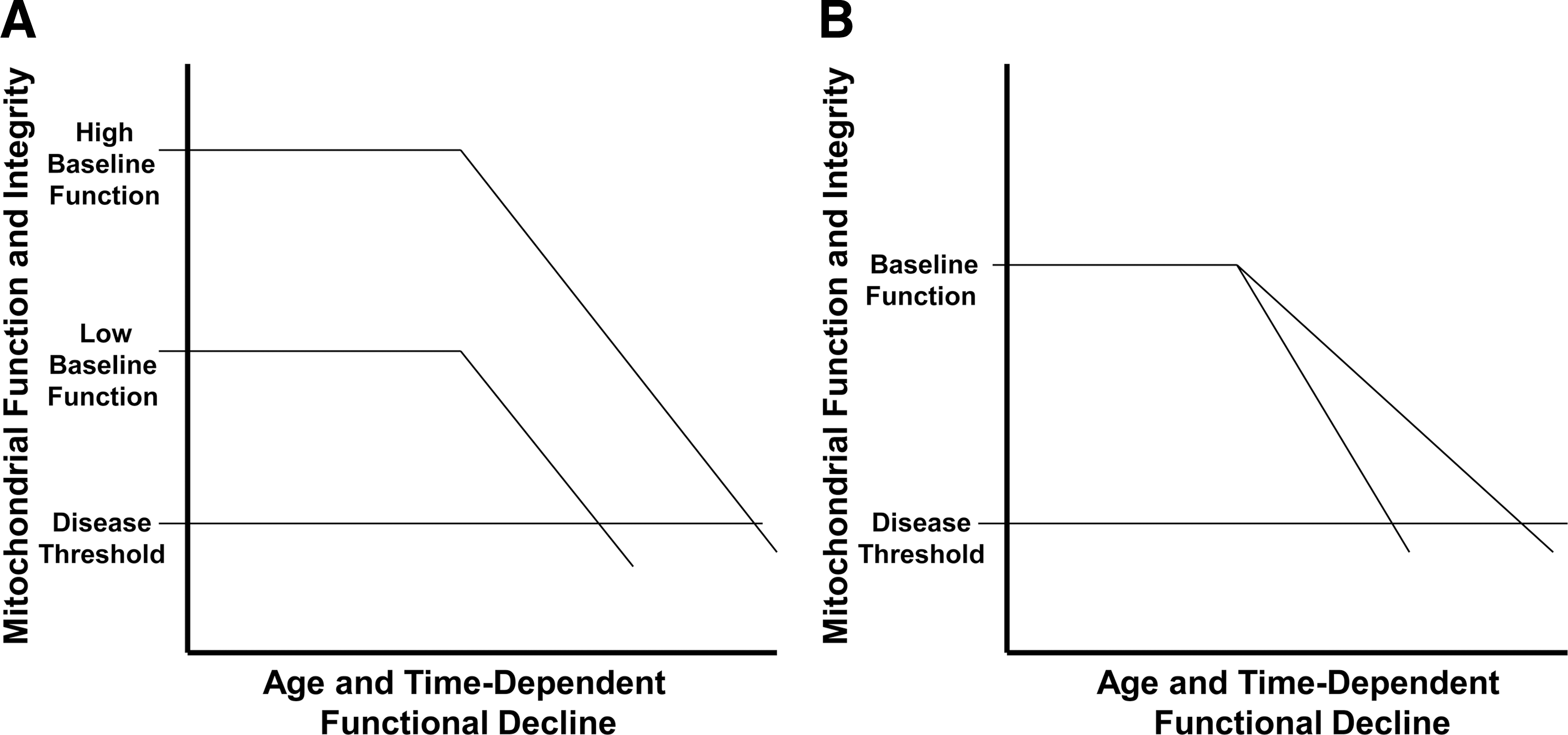
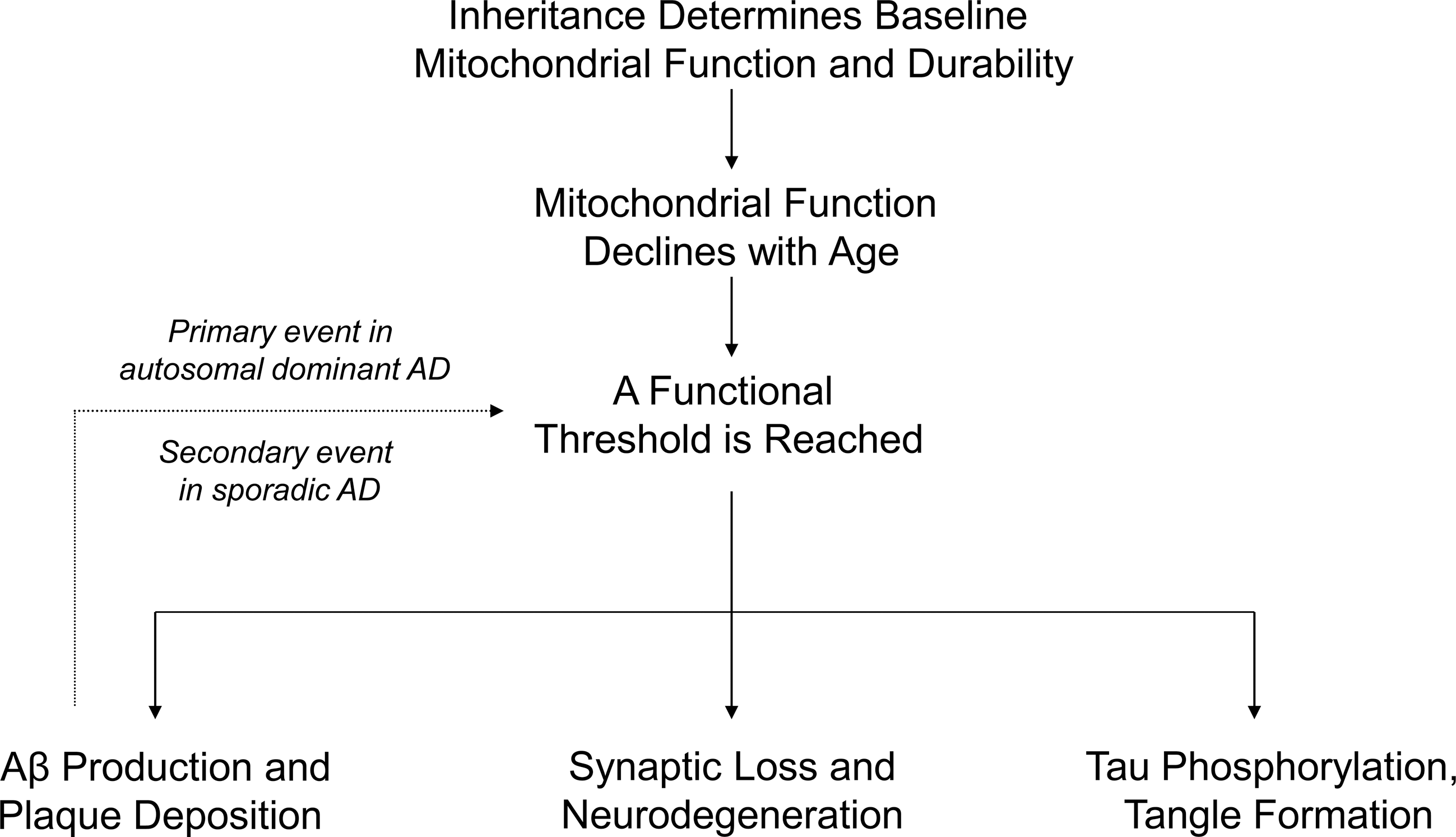
When a threshold is reached and biochemical consequences manifest, various AD histology and molecular phenomena, as well as clinical signs and symptoms, would result. This rationale constitutes the core assumptions of the mitochondrial cascade hypothesis (Fig. 7), a bioenergetics-centric scheme that places mitochondrial dysfunction at the apex of the AD pathology pyramid. The mitochondrial cascade hypothesis is reviewed in detail elsewhere (258, 260 –262). It is important to note other authors have also speculated mitochondrial and bioenergetic dysfunction may represent the primary cause of AD (11, 21, 22, 38, 63, 120, 121, 166, 182, 201, 203, 225, 285).
Conclusion
Because the literature supporting a role for mitochondrial and bioenergetic dysfunction in AD has become so extensive, many relevant studies could not be discussed. For instance, mitochondrial uncoupling induces tau-paired helical filament formation (19), in mice ETC inhibition and fasting robustly induce neuron tau phosphorylation (84, 116, 265, 297), and mitochondria may mediate Aβ–tau relationships (238, 239). Apologies are offered to those whose valuable contributions to this field were not cited.
Data from AD cybrids, which effectively model numerous AD phenomena, suggest mtDNA may at least partly account for reduced COX activity and other biochemical changes in nonbrain tissues. An mtDNA contribution is also compatible with the growing number of AD epidemiology, association, endophenotype, and gene analysis studies that implicate a mitochondrial and possibly even mtDNA role in this disease. In an attempt to synthesize these data, and to also acknowledge the central role aging plays in late-onset, sporadic AD, the mitochondrial cascade hypothesis was proposed. The mitochondrial cascade hypothesis postulates mitochondrial dysfunction represents the most upstream pathology in AD.
Footnotes
Acknowledgments
The author is supported by the Morgan Family Foundation and NIH P30AG035982.
Author Disclosure Statement
The author reports no conflicts of interest.